
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe unrevealed potential of elemental sulfur for the synthesis of high sulfur content bio-based aliphatic polyesters†
Martín E.
Duarte‡
a,
Birgit
Huber‡
 a,
Patrick
Theato
ab and
Hatice
Mutlu
*a
a,
Patrick
Theato
ab and
Hatice
Mutlu
*a
aSoft Matter Synthesis Laboratory, Institut für Biologische Grenzflächen, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Karlsruhe, Germany. E-mail: hatice.mutlu@kit.edu
bPreparative Macromolecular Chemistry, Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstraße 18, 76128 Karlsruhe, Germany
First published on 2nd October 2019
Abstract
We introduce a novel sulfur-containing polyester derivative based on a renewable monomer bearing secondary disulfide groups. Specifically, we demonstrate that this secondary disulfide moiety, placed in each repeating unit within the polymer, enables the introduction of high sulfur content by virtue of an organocatalytically triggered sulfur exchange reaction using elemental sulfur, thereby facilitating the synthesis of sustainable aliphatic polyesters with tailored functional properties.
Commodity polymers are ubiquitous to human life, and the current state of the art reveals a tremendous number of studies devoted to the development of new approaches for the synthesis of novel synthetic polymeric materials with elaborate structures and functions.1 Albeit, these powerful methods have enabled transformative advances, most of them entail the use of depleting petroleum-based commodity chemicals as monomers, which also has adverse effects on the environment. Recently, taking advantage of the structural diversity of alternative inexpensive resources (e.g. elemental sulfur,2 as a by-product of the oil and gas industry, and diverse biomass3 derivatives), significant interest has been shown towards the design of novel sustainable polymers. On the one hand, carbon dioxide-neutral renewable monomers have shown immense potential as a natural analogue replacement for fossil-based chemicals.4 In this context, vegetable oils and their corresponding fatty acid derivatives have proven to be valuable resources.5,6 Nevertheless, modern applications of these well-known resources require more research to introduce additional functionalities, which in terms of chemical functional groups exhibit higher reactivities compared to the available double bonds in fatty acids. Indeed, many examples disclose the application of sulfur7 chemistry (explicitly, thiol–ene/yne reaction)8 as a promising and efficient strategy for the introduction of reactive functional groups into methyl oleate to deliver precursors for the synthesis of diverse aliphatic and aromatic polymers (e.g. polyurethanes, polyamides and polyesters amongst others). However, from a chemical structural perspective, all of the aforementioned thiol-containing sustainable polymers are composed of carbon–sulfur single bonds, i.e. –S–, per repeating unit, and hence are inadequate to address the demand of the growing complexity of modern polymeric materials. On the other hand, elemental sulfur (S8) as a cost-effective raw material9 is readily available as a by-product of a process called hydrodesulfurization10 and has re-gained increasing attention in the last decade. Explicitly, S8 has been employed as a reaction medium (e.g. solvent) and comonomer in the presence of a variety of crosslinkers for the synthesis of chemically stable and processable high sulfur content11 polymeric materials via a simple, scalable, and highly atom efficient method named inverse vulcanization.12 Indeed, this new method, as an excellent example of green chemistry,13 has delivered polymeric materials with radically different properties (e.g. electrochemical, optical, and redox-regulated structural dynamic properties) in comparison with their merely carbon containing counterparts. For instance, Theato and colleagues reported on the sustainable preparation of polymeric, high sulfur content-based vegetable oil composites from S8 and vegetable oils, which were further evaluated as cathode materials in Li–S batteries.14 Despite the tremendous progress that has been made in inverse vulcanisation for improving the synthesis of polymeric materials with high content of sulfur, there are still issues to be addressed, such as the restriction of the type of crosslinker solely to polyene derivatives. Despite the nearly unlimited plethora of polyene derivatives ranging from industrial feedstock derivatives (such as divinyl benzene, dicyclopentadiene, etc.)12,15,16 to the ones based on renewable resources (e.g. limonene, squalene, diallyl disulfide and triglycerides amongst others),14,17,18 their direct use in inverse vulcanisation reactions is still energy intensive (elevated reaction temperatures, e.g. above 140 °C), and requires long reaction times to drive the reaction towards high conversion. Furthermore, very accurate comonomer stoichiometry is essential to obtain high-molecular-weight products. Critically, during the process, addition of S8 to polyenes results in branching of the polymeric chains, ultimately leading to hyperbranched or insoluble crosslinked materials. Therefore, in order to overcome the above-mentioned limitation and further optimize the reaction conditions, catalytic inverse vulcanisation methods have been recently postulated.19 Although effective, the latter examples still do not fully exploit the potential advantages of S8 within the field of synthetic polymer chemistry. Indeed, to the best of our knowledge, there exists no example of a linear high sulfur content polymer, particularly a polyester derivative, derived via inverse vulcanisation. Thus, going beyond the conventional aliphatic polyester based materials and with the idea to unlock the full potential of S8, we exploit the organocatalytically triggered reaction of S8 with secondary disulfide bonds, which were present in each repeating unit of the sustainable polyester derivative, to deliver high sulfur content aliphatic polyester derivatives with tailored properties (Fig. 1).
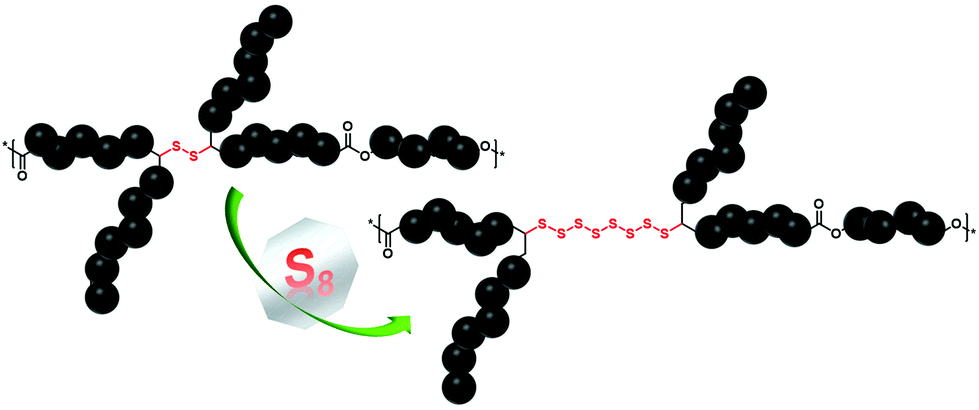 | ||
| Fig. 1 Organocatalytically triggered reaction of S8 with secondary disulfide bonds, which are placed in each repeating unit of the sustainable polyester derivative, to deliver high sulfur content linear polyester derivatives with tailored properties. | ||
Accordingly, we designed a bio-based aliphatic monomer that is functionalized with internal secondary disulfide bonds. The disulfide renewable monomer dimethyl 9(10),9′(10′)-dithiodistearate, M1 (depicted in Scheme 1), was synthesized in a consecutive three-step synthesis starting from the methyl ester derivative of oleic acid (MO), an abundantly available renewable feedstock.20 As a side note, the utilization of bio-based raw materials (such as MO) is in the spotlight of the chemical industry, particularly due to the fact that vegetable oils are one of the most prominent platform chemicals for the synthesis of sustainable polymers (e.g. polyesters) by virtue of their high availability, inherent biodegradability and low price, as mentioned above.20 Furthermore, in technically relevant polyester derivatives, the aliphatic segments are relatively short, typically amounting to six atoms or shorter linear carbon chains; hence M1 is considered an attractive monomer for the synthesis of a bio-based polyester derivative that contains a longer aliphatic segment –(CH2)n– as a structural feature along with an n-alkyl dangling side chain and a secondary disulfide bond.3 Critically, the dynamic nature of the latter, i.e. the secondary disulfide bond, would also account for unique properties (e.g. stimuli triggered adaptability, stress resistance, self-repair, degradability or sulfur exchange reaction) in the resulting polyester.7
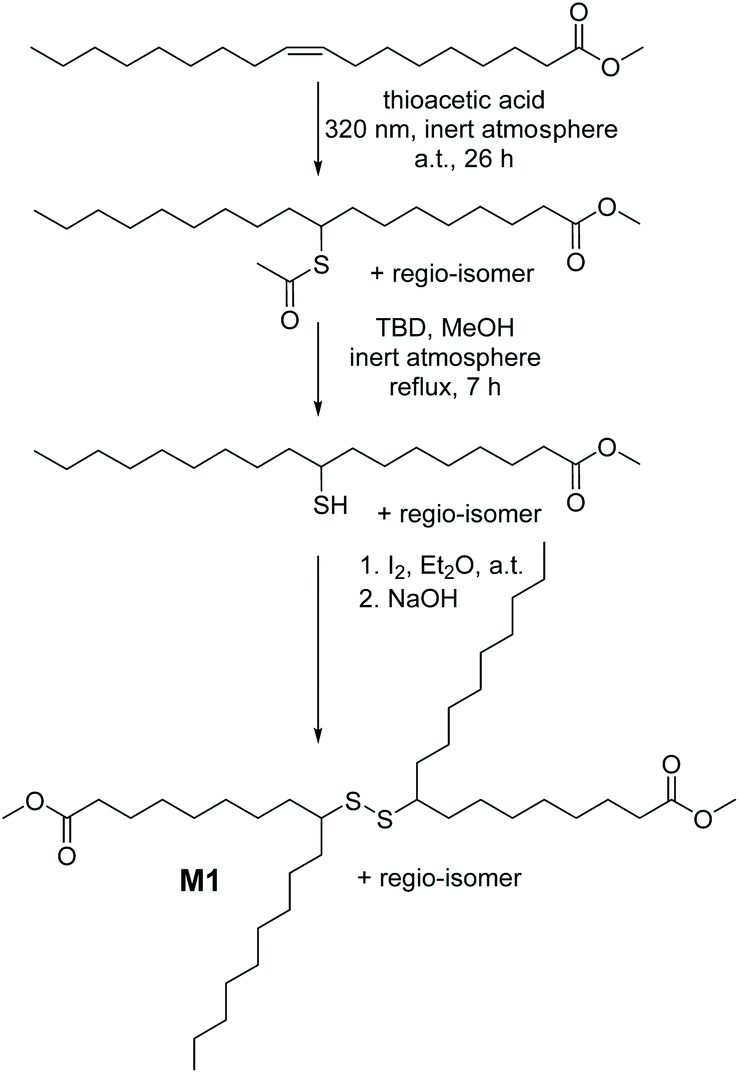 | ||
| Scheme 1 Synthetic route for the preparation of renewable monomer M1 (dimethyl 9(10),9′(10′)-dithiodistearate) bearing secondary disulfide groups, suitable to undergo the organocatalytic sulfur exchange reaction. | ||
Because of the possibility of adding virtually any functional group to olefins, the photochemically induced thiol–ene addition of thioacetic acid to MO under UV irradiation (λ = 320 nm) was initially performed, by adopting slightly modified reaction conditions of a previously reported procedure (refer section A.2 in the ESI†).21 Intriguingly, after the reaction of the internal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond, a dangling alkyl chain remains in the structure of the so-formed molecule. In fact, these dangling chains can interfere with efficient packing of the derived polymers, and lead to less dense polymers compared to the linear ones. Nevertheless, the protected thiol intermediate, methyl 9(10)-(acetylthio)stearate (Scheme 1), was isolated as a mixture of two regio-isomers in reasonable yields (∼90%) after column chromatography. Subsequently, this intermediate compound was converted into the corresponding monomer dimethyl 9(10),9′(10′)-dithiodistearate (M1) as follows: first the free-thiol derivative, methyl 9(10)-mercaptostearate, was obtained by the base-catalysed alcoholysis22 of methyl 9(10)-(acetylthio)stearate, which was further oxidized in a straightforward manner with elemental iodine to yield the disulfide M1 (for preparative details refer section A.2.3 in the ESI†). Our efforts for ensuring the environmentally benign nature of M1 were not limited to biomass as a source. We employed 1,5,7-triazabicyclo[4.4.0]dec-5-ene, TBD, as a safe and non-toxic sustainable organocatalyst for the hydrolysis of the protected thiol derivative.23 Nevertheless, each of the above-mentioned transformations was evidenced by nuclear magnetic resonance (NMR) in addition to mass spectrometry (either by gas chromatography–mass spectrometry, GC-MS, or high-resolution electrospray ionization mass spectrometry, ESI-MS) analyses (Fig. S1–S8 in the ESI†). While the 1H NMR spectrum of methyl 9(10)-(acetylthio)stearate in Fig. S1† shows no magnetic resonances associated with the olefin region (5.38–5.30 ppm) of MO, a new signal appeared at 3.48 ppm corresponding to the protons of the α-carbon (i.e. the methanetriyl group (>CH–), 9a in Fig. S1†), to which the acetylthio group is also attached. Besides, the resonances arising from the hydrogens (8a and 10a, respectively) of the α-methylene groups adjacent to the methanetriyl functionality shifted to the region between 1.63 and 1.54 ppm. Concomitantly, the synthesis of the free-thiol derivative methyl 9(10)-mercaptostearate was confirmed by the disappearance of the resonance at 2.30 ppm (arising from the methyl functionality of the thioacetyl group, 21, compare Fig. S1 and S2 in the ESI†), in addition to the shifting of the resonances associated with the protons of the methanetriyl group (9b) from 3.48 ppm to the regions of 2.80 and 2.72 ppm. Attributable to the instability of free thiols that makes them oxidizable under ambient conditions, the oxidative formation of M1 was also detected during the purification step of methyl 9(10)-mercaptostearate. In particular, the resonances of the methanetriyl group (9c) of M1 shifted to the regions of 2.62 and 2.50 ppm (Fig. S3†). Nonetheless, in addition to the latter resonance, the 1H NMR spectra of M1 showed a singlet at 3.67 ppm associated with the terminal methyl ester group, and the shifted resonances in the regions of 1.60 and 1.48 ppm arising from the hydrogens (8c and 10c, respectively) of the α-methylene groups adjacent to the methanetriyl functionality. The important features of the 13C NMR spectrum of M1 in Fig. S4† are magnetic resonances at 174.14 ppm, and at 52.32 and 34.06 ppm, which are associated with the carbon atom of the ester unit in addition to the carbon atoms of the methanetriyl group and the α-methylene groups adjacent to it, respectively. Last but not least, conventional two-dimensional NMR (COSY and HSQC) experiments were employed for the determination of the exact structure of M1 possessing a unique secondary disulfide bond (Fig. S5 and S6 in the ESI†). Additional analysis of a solution of M1 in THF
C double bond, a dangling alkyl chain remains in the structure of the so-formed molecule. In fact, these dangling chains can interfere with efficient packing of the derived polymers, and lead to less dense polymers compared to the linear ones. Nevertheless, the protected thiol intermediate, methyl 9(10)-(acetylthio)stearate (Scheme 1), was isolated as a mixture of two regio-isomers in reasonable yields (∼90%) after column chromatography. Subsequently, this intermediate compound was converted into the corresponding monomer dimethyl 9(10),9′(10′)-dithiodistearate (M1) as follows: first the free-thiol derivative, methyl 9(10)-mercaptostearate, was obtained by the base-catalysed alcoholysis22 of methyl 9(10)-(acetylthio)stearate, which was further oxidized in a straightforward manner with elemental iodine to yield the disulfide M1 (for preparative details refer section A.2.3 in the ESI†). Our efforts for ensuring the environmentally benign nature of M1 were not limited to biomass as a source. We employed 1,5,7-triazabicyclo[4.4.0]dec-5-ene, TBD, as a safe and non-toxic sustainable organocatalyst for the hydrolysis of the protected thiol derivative.23 Nevertheless, each of the above-mentioned transformations was evidenced by nuclear magnetic resonance (NMR) in addition to mass spectrometry (either by gas chromatography–mass spectrometry, GC-MS, or high-resolution electrospray ionization mass spectrometry, ESI-MS) analyses (Fig. S1–S8 in the ESI†). While the 1H NMR spectrum of methyl 9(10)-(acetylthio)stearate in Fig. S1† shows no magnetic resonances associated with the olefin region (5.38–5.30 ppm) of MO, a new signal appeared at 3.48 ppm corresponding to the protons of the α-carbon (i.e. the methanetriyl group (>CH–), 9a in Fig. S1†), to which the acetylthio group is also attached. Besides, the resonances arising from the hydrogens (8a and 10a, respectively) of the α-methylene groups adjacent to the methanetriyl functionality shifted to the region between 1.63 and 1.54 ppm. Concomitantly, the synthesis of the free-thiol derivative methyl 9(10)-mercaptostearate was confirmed by the disappearance of the resonance at 2.30 ppm (arising from the methyl functionality of the thioacetyl group, 21, compare Fig. S1 and S2 in the ESI†), in addition to the shifting of the resonances associated with the protons of the methanetriyl group (9b) from 3.48 ppm to the regions of 2.80 and 2.72 ppm. Attributable to the instability of free thiols that makes them oxidizable under ambient conditions, the oxidative formation of M1 was also detected during the purification step of methyl 9(10)-mercaptostearate. In particular, the resonances of the methanetriyl group (9c) of M1 shifted to the regions of 2.62 and 2.50 ppm (Fig. S3†). Nonetheless, in addition to the latter resonance, the 1H NMR spectra of M1 showed a singlet at 3.67 ppm associated with the terminal methyl ester group, and the shifted resonances in the regions of 1.60 and 1.48 ppm arising from the hydrogens (8c and 10c, respectively) of the α-methylene groups adjacent to the methanetriyl functionality. The important features of the 13C NMR spectrum of M1 in Fig. S4† are magnetic resonances at 174.14 ppm, and at 52.32 and 34.06 ppm, which are associated with the carbon atom of the ester unit in addition to the carbon atoms of the methanetriyl group and the α-methylene groups adjacent to it, respectively. Last but not least, conventional two-dimensional NMR (COSY and HSQC) experiments were employed for the determination of the exact structure of M1 possessing a unique secondary disulfide bond (Fig. S5 and S6 in the ESI†). Additional analysis of a solution of M1 in THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH (3:2, v/v) using a Q-Exactive Orbitrap mass spectrometer evidenced the expected compound with m/zexp 681.4915 [M + Na]+ (refer Fig. S8 in the ESI†).
:MeOH (3:2, v/v) using a Q-Exactive Orbitrap mass spectrometer evidenced the expected compound with m/zexp 681.4915 [M + Na]+ (refer Fig. S8 in the ESI†).
Having demonstrated the successful synthesis of M1, we sought to leverage its melt polycondensation with 1,6-hexanediol (M2) to deliver the aliphatic polyester derivative P1 (depicted in Scheme 2). Industrially, 1,6-hexanediol is prepared by the hydrogenation of adipic acid, and since the first pilot plant producing adipic acid via the fermentation process using non-food, plant-based feedstock has been recently set up, M2 can be considered (potentially) a biomass derived compound,24 thus contributing to the sustainability of P1.
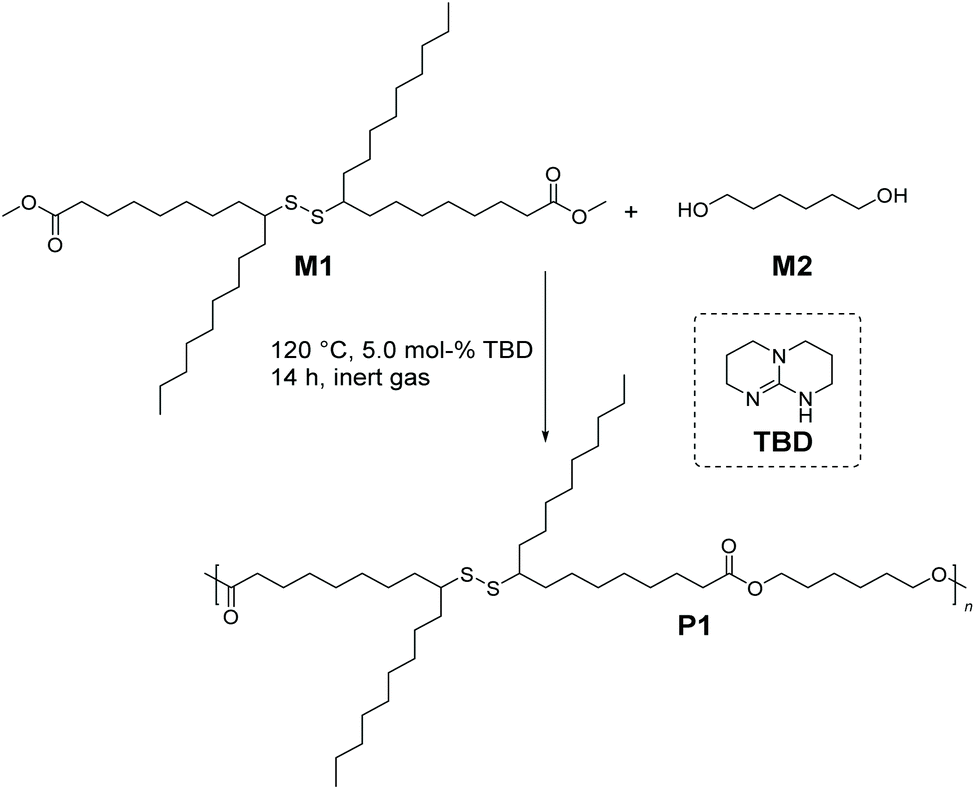 | ||
| Scheme 2 Synthetic route for the preparation of bio-based aliphatic polyester P1via organocatalytic melt polycondensation of M1 (dimethyl 9(10),9′(10′)-dithiodistearate) with M2 (1,6-hexanediol). | ||
On the one hand, differential scanning calorimetry (DSC) analyses confirmed that M1 was thermally stable up to 200 °C, and thus it could be polymerized under solvent-free conditions. On the other hand, the DSC chromatogram of M1 showed a unique thermal behaviour attributed to the fact that M1 is a long chain alkyl monomer composed of mixtures of regio-isomers with different lengths of n-alkyl side chains (e.g. n is either 8 or 9). In other words, M1 exhibits a glass transition temperature, Tg, of −63.4 °C attributed to the n-alkyl dangling side chain, and two melting points with onset temperatures at −24 and −14 °C, respectively (see Fig. S9 in the ESI†). Interestingly, although disulfide bonds are known to be temperature responsive,25 no homolytic scission or any other side reactions were observed at elevated temperatures during the DSC measurements of M1. Hence, test polymerizations of M1 with M2 were carried out at 120 °C, since the catalyst of choice, i.e. TBD, has been recognized as a highly effective and sustainable transesterification catalyst within this temperature range for the synthesis of fatty acid derived polycondensates with a vast array of chemical functionalities.26–31 Different loadings of TBD (1.0, 5.0 and 10.0 mol%) per ester group of M1 were tested (refer section A.3 in the ESI†). Even though M1 is thermally stable under the abovementioned reaction temperatures, hydroquinone (2.0 μmol%) was added to the polymerization mixture as a radical quencher to avoid any plausible cleavage of secondary disulfide linkages during melt polycondensation. The size exclusion chromatography (SEC) and NMR spectroscopy results of these preliminary polymerization experiments disclosed that 1.0 mol% catalyst yielded only oligomers (see Fig. S10 in the ESI†), while a higher amount of TBD (10.0 mol%) resulted in undesired side reactions as depicted explicitly by 1H NMR analyses (see Fig. S11 in the ESI†).
Accordingly, P1, with an apparent molar mass of 13500 g mol−1 and with a dispersity of Đ = 1.7 (as determined by SEC relative to polymethacrylate standards, Scheme 2 and Fig. 2), was synthesized under the most optimal conditions for polymerization, which were determined to be 120 °C, 5.0 mol% catalyst and a reaction time of 14 h under a continuous flow of an inert gas.
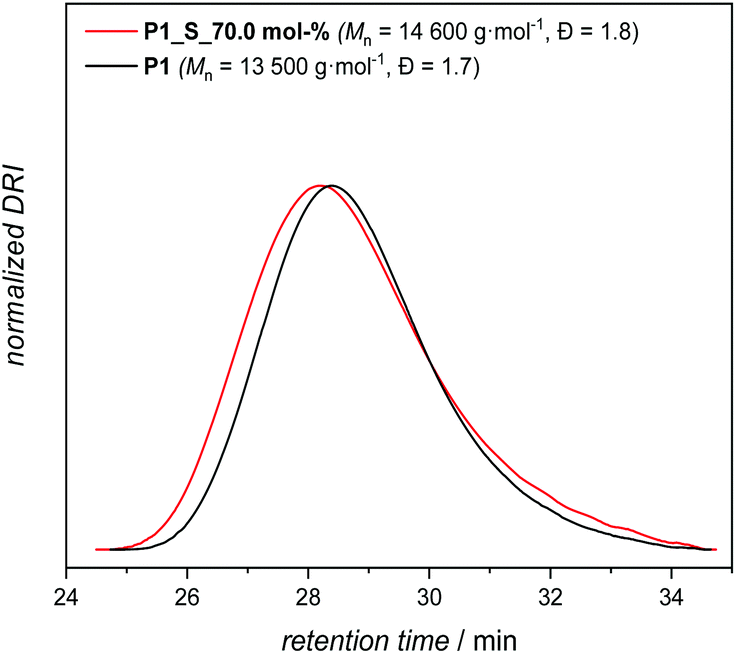 | ||
| Fig. 2 SEC trace of the parent polymer P1 (black line) and P1_S_70.0 mol% (red line). | ||
The 1H NMR spectrum of P1 in Fig. 3 displays the proton resonances of the methanetriyl unit in the region between 2.62 and 2.50 ppm (h), which remained intact during the polymerization process. Furthermore, the proton resonances of the α-methylene units adjacent to the ester and methanetriyl groups were identified at 4.06 (a′), 2.28 (a), and in the range between 1.63 and 1.54 ppm (g and i), respectively. Critically, the singlet corresponding to the terminal methyl ester (CH3O–) group of M1 at 3.67 ppm was significantly lower in intensity, also leading to the success of polymerization. Furthermore, it is worth highlighting that along with 1H NMR, 13C NMR analysis also demonstrated that the secondary disulfide functionality remained intact and did not interfere with any other functional group, indicating the compatibility of melt polycondensation chemistry with this specific functionality (Fig. S12 in the ESI†). In addition, an attenuated-total-reflectance IR (ATR-IR) assay provided supplemental spectroscopic proof for the successful synthesis of polymer P1, although secondary disulfide bonds lack a distinctly strong IR handle (Fig. 4). Nevertheless, the subsequent disappearance of the symmetrical and antisymmetrical –O–CH3 deformation bands (1435 cm−1), along with the respective shift of the characteristic CO stretching band of the ester functionality from ∼1740 cm−1 to a lower wavenumber (1736 cm−1) underpinned the successful transformation of the monomeric methyl ester groups of M1 into the polymeric ester derivatives of P1 (zoomed-up inset in Fig. 4).
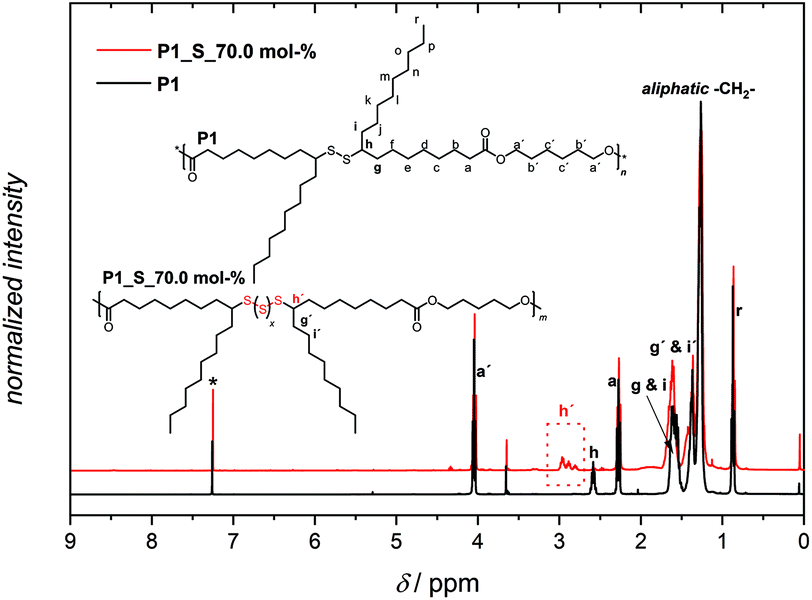 | ||
| Fig. 3 Analysis of P1 and P1_S_70.0 mol% by 1H NMR spectroscopy, indicating the success of each transformation, the synthesis of the parent polymer P1 and the subsequent organocatalytic sulfur exchange reaction with 70.0 mol% elemental sulfur added incrementally. | ||
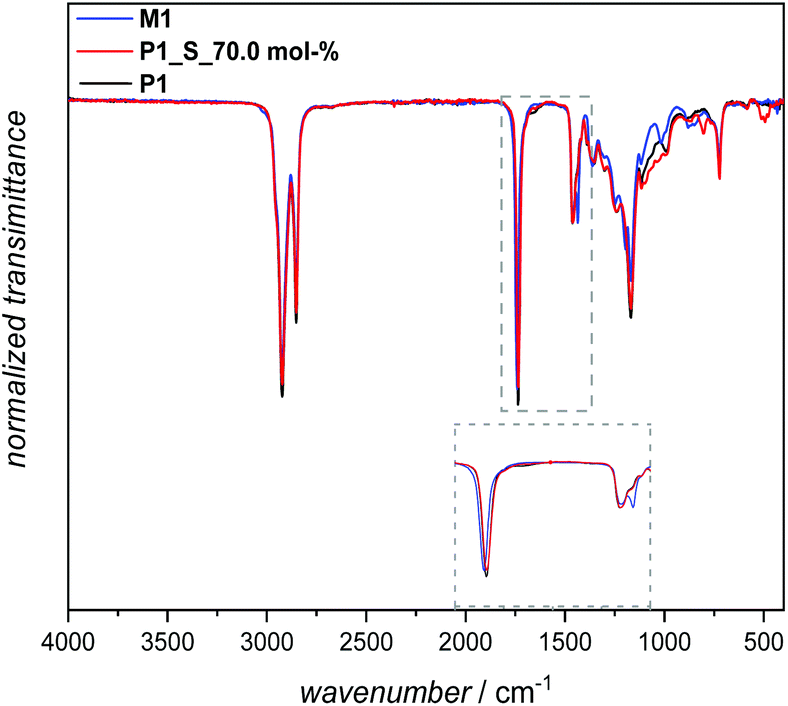 | ||
| Fig. 4 Analysis of M1, P1 and P1_S_70.0 mol% by attenuated total-reflectance infrared spectroscopy, indicating the success of each transformation, i.e., synthesis of the parent polymer P1 and the subsequent transformation into high sulfur content polyester P1_S_70.0 mol%. The inset shows the zoomed-up region from 1800–1400 cm−1, respectively. | ||
Next, in order to evaluate the efficiency of the anticipated reaction (i.e. the organocatalytically (i.e. TBD) triggered sulfur exchange reaction, Scheme 3a) between the secondary disulfide bonds of the new polymer system P1 and S8, a proof-of-concept molecule (M1_Sx, where x indicates the number of sulfur atoms introduced after the sulfur exchange reaction) was designed, and further assessed by diverse analytical methods. Equimolar amounts of M1 and S8 were reacted for 1 h at 135 °C (i.e. the temperature at which S8 starts to melt) under an inert gas atmosphere with and without the catalyst, respectively. For the first case, 2.0 mol% of the strong nucleophile TBD (pKa,MeCN of 26)32 was utilized as the catalyst. Interestingly, quite recently, it has been shown that nitrogen-based nucleophilic activators such as pyridine derivatives are capable of ring-opening S8 under various conditions and enhancing the reactivity of sulfur exchange reactions at low reaction temperatures within short reaction times.19 Similarly, DBU, another well-known guanidine derivative, was utilized for the direct cleavage of disulfide into the corresponding thiol derivatives.331H NMR analysis of the catalyst-free final reaction mixture of M1_Sx gave results along expected lines, i.e. no sulfur exchange reaction took place, while the TBD-catalyzed reaction delivered the targeted high sulfur content compound M1_Sx. The 1H NMR spectrum recorded after the purification of M1_Sx exhibited chemical shifts of the α-carbon protons as a function of sulfur content within the molecule (Fig. S13 in the ESI†). Explicitly, the multiplet in the region between 2.62 and 2.50 ppm associated with the hydrogen atom at the α-carbon adjacent to the disulfide bond was shifted to the following ranges: 3.03–2.93, 2.93–2.87 and 2.85–2.79 ppm, respectively, in such a way that each series of multiplets is assigned to the respective sulfur rank, hence nicely depicting the high sulfur content of the compound. In fact, this shift in the resonances of the disulfide bond in NMR analyses has been noted for polysulfides whose α-carbon protons exhibit multiplets, as in the current case.34,35 It can be postulated that these changes in chemical shifts result, in part, from the anisotropy or inductive effects of sulfur atoms, as reported previously.36 Notably, NMR analysis also revealed that the terminal methyl ester functional moiety remained intact and did not undergo any reaction, such as thioesterification as expected (refer to Fig. S13†). To shed further light on the process, an experiment was conducted under ambient atmosphere, which revealed that the presence of oxygen inhibits the process (data not disclosed). Furthermore, SEC traces in Fig. S14† revealed that the molar mass of M1 before and after the reaction (i.e.M1_Sx) varied significantly enough to be displayed as a new peak in the SEC traces. Although SEC and NMR spectroscopy evidenced the successful introduction of high sulfur content within M1, these data alone do not provide insight into the exact chemical composition of the model compound. Therefore, additional high resolution ESI-MS allowed assigning the incorporation of the high sulfur content, evidencing all possible fragments that can arise (Fig. S15 in the ESI†). Due to the shift of the molecular weight of the charged sodium cations of M1 by 31.972 m/z, it was apparent that the upper limit of sulfur content that can be additionally introduced via the TBD mediated sulfur exchange reaction between M1 and S8 is 6 sulfur atoms (m/zcalc = 873.3240, [M1_Sx=6 + Na]+). In other words, the maximum total sulfur content within the proof-of-concept molecule is 8 sulfur atoms per molecule. Importantly, this high sulfur content molecule is chemically stable and does not readily depolymerise back to M1 and S8, as it was bench stable for over a month under ambient conditions, in contrast to polymeric sulfur.
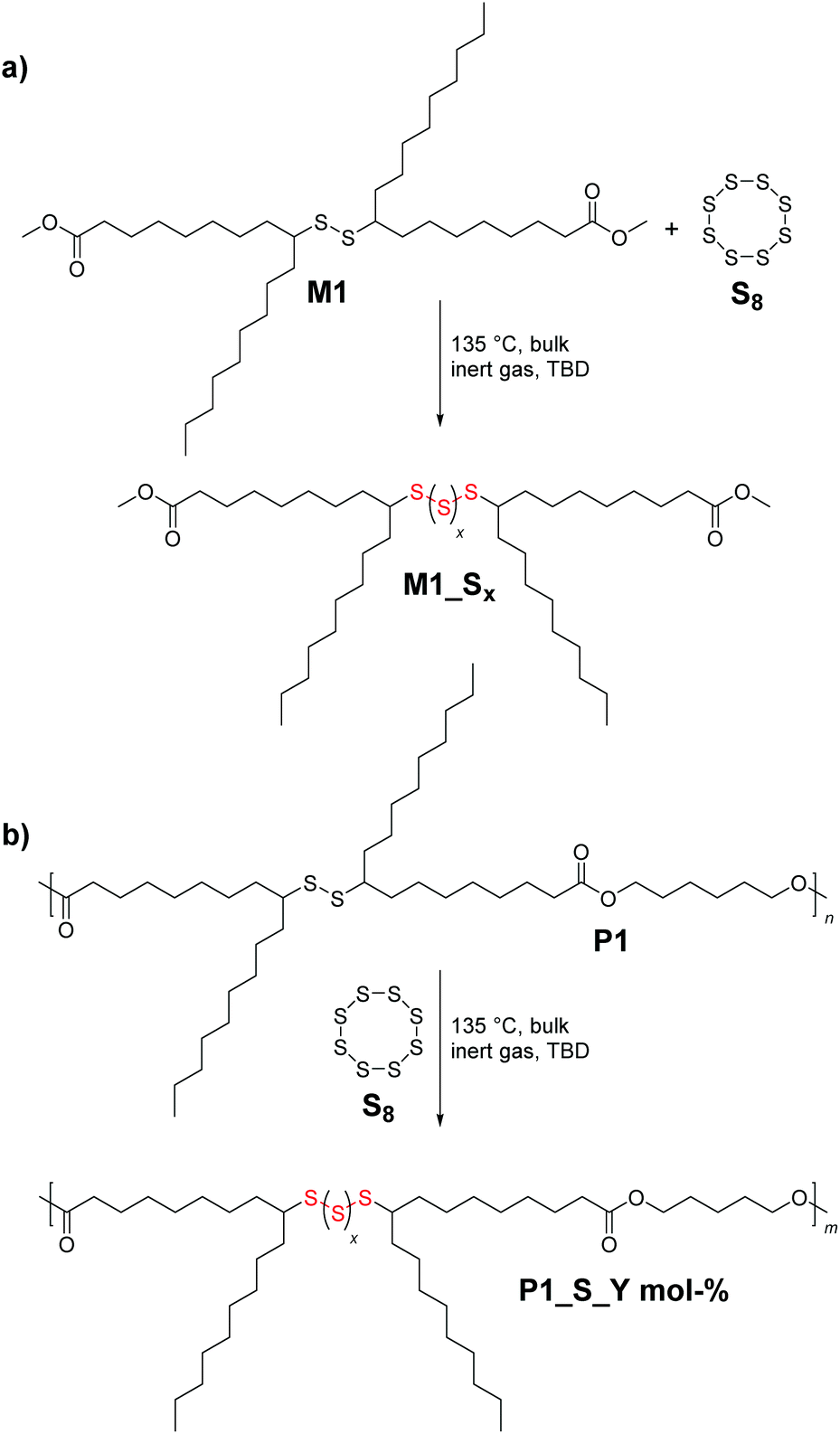 | ||
| Scheme 3 The organocatalytically (i.e. TBD) triggered sulfur exchange reaction between S8 and the secondary disulfide bonds of: (a) monomer M1 to synthesize a proof-of-concept molecule M1_Sx and (b) polymer P1 to deliver P1_S_Y mol%, respectively. “x” indicates the number of sulfur atoms introduced after the sulfur exchange reaction and “Y” stands for the loading of S8 (in mol%) during the reaction. | ||
Consequently, the final key step is to demonstrate that the custom designed polymer P1 decorated with secondary disulfide can also deliver high sulfur content polymers (P1_S_Y mol% in Scheme 3b, and Table S1 in the ESI†) with the aid of S8 by virtue of the sulfur exchange reaction (“Y” stands for the loading of S8 (in mol%) during the reaction). Initially, an equimolar mixture of P1 and elemental sulfur was reacted in the presence of 2.0 mol% TBD at 135 °C in a similar manner as it was performed for M1 (Table S1,† entry 1). Surprisingly, SEC traces of the reacted polymer P1_S_100.0 mol% (where 100.0 mol% indicates the equimolar loading of S8) after 1 h of reaction shifted to higher retention time revealing some degradation with decreasing apparent molar mass (see Fig. S16 in the ESI†). Indeed, it is conventionally considered that the sulfur content in the repeating unit of any polymeric structure is thermodynamically limited to a maximum of 9 sulfur atoms before the homopolymerization of elemental sulfur starts to compete with depolymerization, since the average bond dissociation energy of the –S–S– bond is expected to decrease to a certain value, resulting in an accelerated sulfur exchange reaction.37 Furthermore, it has also been reported that the reactivity of the thiolate anion decreases with increasing number of S atoms in the propagating chain because of negative charge stabilization, presumably via delocalization along the disulfide bond.38 To circumvent this problem, in situ incremental addition of elemental sulfur was postulated. Accordingly, under bulk conditions with a continuous flow of an inert gas, the first aliquot of S8 (5.0 mol%) was added to P1 and the reaction was performed for 1 h at 135 °C (Table S1,† entry 2). 1H NMR analysis of the crude reaction mixture showed chemical shifts of the α-carbon protons as a function of the sulfur content within the polymer, which was consistent with the results of the model reaction performed for M1 (refer Fig. S16 in the ESI†). Subsequently, additional aliquots of S8 (i.e. 5.0 mol%) were added at a time interval of 30 min to the reaction mixture (Table S1†). Characterization via1H and 13C NMR analyses of the crude reaction mixture after addition of 5.0, 25.0, 35.0 and 70.0 mol% aliquots of S8 revealed the progressive insertion of elemental sulfur into the secondary disulfide-repeating unit via the sulfur exchange reaction (Fig. S17 and S18†). On the one hand, the magnetic resonance pattern observed in the 1H NMR spectra of the crude reaction mixture after addition of 70.0 mol% S8 disclosed a similar pattern consistent with the proof-of-concept molecule (Fig. 3). In other words, the upper limit of the sulfur atoms that can be introduced via the TBD mediated sulfur exchange reaction between each repeating unit of P1 and S8 was also 6 sulfur atoms, resulting in a maximum of 8 sulfur atoms per repeating unit. Furthermore, comparative analysis of 13C NMR spectra (Fig. S18†) of the crude reaction mixtures after addition of 5.0, 25.0, 35.0 and 70.0 mol% aliquots of S8 to P1 indicated that the ester functional moiety remained intact (refer to 173.9 ppm and 64.2 ppm, respectively, at Fig. S18†), and the polymer did not undergo any depolymerization via thioesterification as one could have expected. On the other hand, SEC data clearly proved that the utmost amount attainable to polymer P1 is 70.0 mol% S8 (Fig. 2), resulting in a post-modified polymer with an apparent molar mass of 14600 g mol−1 and with a dispersity of Đ = 1.8. Above this value, for instance at a loading of 75.0 mol% S8, the depolymerization of elemental sulfur started to compete with sulfur insertion, hence resulting in the degradation of the final polymer (refer Fig. S19 and Table S1 in the ESI†). As previously mentioned, this result proved that the sulfur exchange reaction was accelerated at higher sulfur loadings per disulfide bond (i.e. x > 9 sulfur atoms per disulfide bond), and depolymerization ensued, while at x < 9 sulfur atoms per disulfide bond, due to the dynamic S–S present in the polymer backbone, it was not unexpected to observe marginal differences in the molar mass of the elemental sulfur treated polymer. The latter was also consistent with the earlier studies of Pyun39 and colleagues, who postulated that long chain polysulfides (with a high content of S–S bonds) are prone to thermally induced sulfur radical generation that is accompanied by fragmentation, and thus a decrease in the molar mass would be expected from the process.
Additional evidence for efficient sulfur insertion within the secondary disulfide bonds of P1 was provided by detailed UV, IR, DOSY and elemental analysis. Intriguingly, the increased sulfur content in polymer P1_S_70.0 mol% resulted in a higher molar absorbance at a given wavelength and more red-shifted spectra in comparison with P1. Indeed, an absorption maximum at 260–330 nm became pronounced when the sulfur content increased in comparison with M1 and P1, respectively (Fig. 5). The IR spectra of the polymer before and after sulfur insertion (Fig. 4) showed the intact CO stretching band of the ester functionality at 1736 cm−1. Moreover, additional valence vibrations were observable at 495–512 cm−1, associated with S–(S)x–S–. Furthermore, by evaluating the DOSY NMR measurements (Fig. S20 and S21 in the ESI†), particularly by applying the Stokes–Einstein equation (refer section B.2. in the ESI†), a hydrodynamic diameter of 6.5 nm for P1 in CDCl3 is determined. After sulfur insertion via the sulfur exchange reaction, the value increases by 90% to 11.9 nm for P1_S_70.0 mol%. The DOSY data were supported by the good agreement of the diffusion coefficients with each other (refer to Table S2†). Last but not least, elemental analysis confirmed the successful integration of sulfur atoms within the secondary disulfide bonds of polymer P1via the organocatalytic sulfur exchange reaction. The experimentally obtained values for P1_S_70.0 mol% (C: 56.30, H: 7.90, S: 28.20) compared to P1 (C: 70.20, H: 9.85, S: 9.72) were in good agreement, reaffirming the fact that the sulfur exchange of P1 with elemental sulfur (70.0 mol%) leads to the insertion of utmost six sulfur atoms in each –S–S– bond/repeating unit of the polyester derivatives.
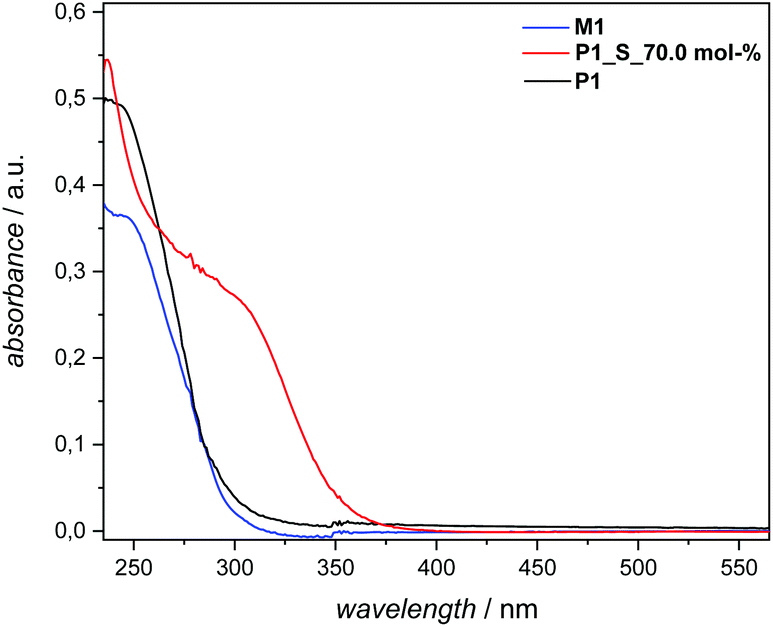 | ||
| Fig. 5 UV-Vis spectra of M1, P1 and P1_S_70.0 mol%. Sample concentrations were 1.23 × 10−5 mmol mL−1 (for M1) and 6.25 × 10−2 mg mL−1 (for P1 and P1_S_70.0 mol%.) in THF. Solvent cut-off wavelengths L0: 215 (λmax: 220 nm in THF). For details, refer to the main text and the ESI.† | ||
In summary, we have pioneered the synthesis of a sustainable bio-based aliphatic polyester derivative that bears secondary disulfide groups in each repeating unit. We evidenced that this specific functionality enabled the introduction of high sulfur content (up to 6 additional sulfur atoms per disulfide bond of each repeating unit within the polymer) by virtue of the organocatalytically (i.e. TBD) triggered sulfur exchange reaction with elemental sulfur. The successful synthesis of the monomer M1, polymer P1 and the sulfur-enriched polyester derivative P1_S_70.0 mol% was evidenced in depth via detailed analytical characterization by NMR, SEC, UV, IR, DOSY and elemental analysis. Hence, we believe that this powerful methodology could facilitate the synthesis of sustainable aliphatic polyesters with tailored functional properties (e.g. electrochemical, optical and chemical properties). Indeed, this approach may constitute a platform, as advanced yet simple, to implement these high sulfur content polymers in the formation of various materials with specified functions (e.g. self-organization, adaptive self-sorting, molecular motion, replication and transcription amongst others). For instance, these properties and functions may effectively be used for generating materials with potential applications ranging from biomedicine (such as protein biochips)40 to energy storage (e.g. Li–S batteries41 or IR imaging).42
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
M. E. D. acknowledges the UTN-DAAD (Universidad Tecnológica Nacional-Deutscher Akademischer Austauschdienst) for the scholarship. H. M. and P. T. acknowledge the continued support from the Karlsruhe Institute of Technology (KIT) in the context of the BIFTM programs of the Helmholtz Association. Professor M. A. R. Meier and N. Klaassen, respectively, are acknowledged for providing access to GC-MS, DSC, ATR-IR and elemental analysis.Notes and references
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef
.
- M. J. H. Worthington, R. L. Kucer and J. M. Chalker, Green Chem., 2017, 19, 2748–2761 RSC
- A. Llevot, P. K. Dannecker, M. von Czapiewski, L. C. Over, Z. Söyler and M. A. R. Meier, Chem. – Eur. J., 2016, 22, 11510–11521 CrossRef CAS
- Y. Q. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS
- Y. Xia and R. C. Larock, Green Chem., 2010, 12, 1893–1909 RSC
- K. F. Adekunle, Open J. Polym. Chem., 2015, 5, 34–40 CrossRef CAS
- H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Macromol. Rapid Commun., 2019, 40, 1800650 CrossRef
-
A. Gandini and T. M. Lacerda, Polymers from Plant Oils, in Thiol-ene and Thiol-yne Reactions for the Transformation of Oleochemicals into Monomers and Polymers, John Wiley & Sons Inc., Hoboken, NJ, 2018, pp. 109–134 Search PubMed
- J. G. Wagenfeld, K. Al-Ali, S. Almheiri, A. F. Slavens and N. Calvet, Waste Manage., 2019, 95, 78–89 CrossRef
- C. Song, Catal. Today, 2003, 86, 211–263 CrossRef CAS
- J. J. Griebel, R. S. Glass, K. Char and J. Pyun, Prog. Polym. Sci., 2016, 58, 90–125 CrossRef CAS
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, Á. Somogyi, P. Theato, M. E. Mackay, Y.-E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524 CrossRef CAS
- M. J. H. Worthington, R. L. Kucera and J. M. Chalker, Green Chem., 2017, 19, 2748–2276 RSC
- A. Hoefling, Y. J. Lee and P. Theato, Macromol. Chem. Phys., 2017, 218, 1600303 CrossRef
- D. J. Parker, H. A. Jones, S. Petcher, L. Cervini, J. M. Griffin, R. Akhtar and T. Hasell, J. Mater. Chem. A, 2017, 5, 11682–11692 RSC
- M. Arslan, B. Kiskan, E. C. Cengiz, R. Demir-Cakan and Y. Yagci, Eur. Polym. J., 2016, 80, 70–77 CrossRef CAS
- M. Worthington, R. Kucera, I. Albuquerque, C. Gibson, A. Sibley, A. Slattery, J. Campbell, S. Alboaiji, K. A. Muller, J. Young, N. Adamson, J. Gascooke, D. Jampaiah, Y. Sabri, S. Bhargava, S. J. Ippolito, D. Lewis, J. Quinton, A. Ellis, A. Johs, G. Bernardes and J. M. Chalker, Chem. – Eur. J., 2017, 64, 16219–16230 CrossRef
- M. P. Crockett, A. M. Evans, M. J. Worthington, I. S. Albuquerque, A. D. Slattery, C. T. Gibson, J. A. Campbell, D. A. Lewis, G. J. Bernardes and J. M. Chalker, Angew. Chem., Int. Ed., 2016, 55, 1714–1718 CrossRef CAS
- Y. Zhang, N. G. Pavlopoulos, T. S. Kleine, M. Karayilan, R. S. Glass, K. Char and J. Pyun, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 7–12 CrossRef CAS
- L. Montero de Espinosa and M. A. R. Meier, Eur. Polym. J., 2011, 47, 837–852 CrossRef CAS
- N. H. Koenig, G. S. Sasin and D. Swern, J. Org. Chem., 1958, 23, 1525–1530 CrossRef CAS
- M. Firdaus, M. A. R. Meier, U. Biermann and J. O. Metzger, Eur. J. Lipid Sci. Technol., 2014, 116, 31–36 CrossRef CAS
- I. R. Shaikh, J. Catal., 2014, 2014, 402860 Search PubMed
- https://www.bioplasticsmagazine.com , Rennovia enters piloting stage of its bio-based 1,6-hexanediol process, 27.07.2019.
- S. Nevejans, N. Ballard, J. I. Miranda, B. Reck and J. M. Asua, Phys. Chem. Chem. Phys., 2016, 18, 27577–27583 RSC
- U. Schuchardta, R. Serchelia and R. M. Vargas, J. Braz. Chem. Soc., 1998, 9, 199 Search PubMed
- H. Mutlu and M. A. R. Meier, Macromol. Chem. Phys., 2009, 210, 1019–1025 CrossRef CAS
- O. Türünç and M. A. R. Meier, Macromol. Rapid Commun., 2010, 31, 1822–1826 CrossRef
- H. Mutlu, J. Ruiz, S. C. Solleder and M. A. R. Meier, Green Chem., 2012, 14, 1728–1735 RSC
- H. Mutlu, R. Hofsass, R. E. Montenegro and M. A. R. Meier, RSC Adv., 2013, 3, 4927–4934 RSC
- A. Bossion, K. V. Heifferon, L. Meabe, N. Zivic, D. Taton, J. L. Hedrick, T. E. Long and H. Sardon, Prog. Polym. Sci., 2019, 90, 164–210 CrossRef CAS
- K. Kaupmees, A. Trummal and I. Leito, Croat. Chem. Acta, 2014, 87, 385–395 CrossRef
- D. Nyoni, K. A. Lobb, P. T. Kaye and M. R. Cairab, ARKIVOC, 2012, 2012, 245–252 Search PubMed
- F. Freeman and C. S. Lee, Magn. Reson. Chem., 1988, 26, 813–816 CrossRef CAS
- A. Duda and S. Penczek, Makromol. Chem., 1980, 181, 995–1001 CrossRef CAS
- D. J. Martin and R. H. Pearce, Anal. Chem., 1966, 38, 1604–1605 CrossRef CAS
- R. Steudel, Angew. Chem., Int. Ed. Engl., 1975, 14, 655–720 CrossRef
- A. Duda and S. Penczek, Macromolecules, 1982, 15, 36–40 CrossRef CAS
- Y. Zhang, K. M. Konopka, R. S. Glass, K. Char and J. Pyun, Polym. Chem., 2017, 8, 5167–5173 RSC
- F. Rusmini, Z. Zhong and J. Feijen, Biomacromolecules, 2007, 8, 1775–1789 CrossRef CAS PubMed
- Z. Chen, J. Droste, G. Zhai, J. Zhu, J. Yang, M. R. Hansen and X. Zhuang, Chem. Commun., 2019, 55, 9047–9050 RSC
- D. A. Boyd, V. Q. Nguyen, C. C. McClain, F. H. Kung, C. C. Baker, J. D. Myers, M. P. Hunt, W. Kim and J. S. Sanghera, ACS Macro Lett., 2019, 8, 113–116 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py01152h |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |