
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A bio-orthogonal functionalization strategy for site-specific coupling of antibodies on vesicle surfaces after self-assembly†
Meiyu
Gai‡
 a,
Johanna
Simon‡
ab,
Ingo
Lieberwirth
a,
Volker
Mailänder
ab,
Svenja
Morsbach
*a and
Katharina
Landfester
a
a,
Johanna
Simon‡
ab,
Ingo
Lieberwirth
a,
Volker
Mailänder
ab,
Svenja
Morsbach
*a and
Katharina
Landfester
a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: morsbachs@mpip-mainz.mpg.de
bDepartment of Dermatology, University Medical Center of the Johannes Gutenberg-University Mainz, Langenbeckstrasse 1, 55131 Mainz, Germany
First published on 14th October 2019
Abstract
Attaching targeting ligands on the surface of self-assembled drug delivery systems is the key request for a controlled transport of the drug to a desired location. Most commonly, the amphiphilic molecules (block-copolymers, lipids etc.) are therefore pre-functionalized before the self-assembly takes place. However, this strategy cannot be applied, if it interferes with the self-assembly process, if the introduced functional groups react with loaded cargo or if natural carriers like extracellular vesicles should be functionalized. Here, we present the site-specific coupling of antibodies to the surface of amino group-terminated liposomes via bio-orthogonal copper-free click chemistry after liposome formation. The present primary amino groups were functionalized with a linker carrying a strained alkyne group for a bio-orthogonal strain-promoted alkyne–azide cycloaddition (SPAAC) reaction where Cu(I) as a catalyst can be avoided. Antibodies were site-specifically functionalized with azide moieties along the Fc region to avoid interference with the antigen binding sites. The liposome surface functionalization reaction was optimized by precisely analyzing the number of available functional groups (both amine and alkyne), which often represents a challenge for self-assembled systems. By finally confirming the successful antibody coupling, we provide a facile and robust functionalization strategy, which can be applied to a wide range of self-assembled systems and desired targeting antibodies maintaining physiological conditions throughout the procedure.
1. Introduction
Self-assembled systems of amphiphilic molecules are nowadays applied as drug delivery vehicles as they are biocompatible and can transport hydrophilic as well as hydrophobic cargos. Typically, such self-assembled systems can be categorized into the main groups of polymer-based structures, lipid-based structures and more recently introduced natural vesicles.1 Polymer-based self-assembled systems consist of amphiphilic block-copolymers, which typically form double-layers and assemble into polymersomes or micelles.2–4 Regarding lipid-based structures, liposomes, which are small artificial vesicles consisting of one or more phospholipid bilayers, were first introduced by Bangham et al. in the 1960s.5 They are still of high interest as their composition is similar to natural cellular membranes and as such have an extraordinary biocompatibility.6–9 Basically, there are four major types of liposomes: (1) MLVs, multilamellar vesicles with several lamellar lipid phase bilayers, (2) SUVs, small unilamellar vesicles with one lipid bilayer,10–14 (3) LUVs, large unilamellar vesicles, and (4) cochleate vesicles.15Compared to these conventional structures, “second-generation” vesicles are chemically modified to obtain specific properties such as a prolonged blood circulation time or targeting capability. More specifically, vesicles are functionalized with various targeting ligands such as antibodies, peptides, proteins or aptamers, with a variety of surface engineering techniques.14,16–18 These ligands are mostly chemically attached to the vesicles through interaction of reactive groups on the liposomes’ or polymersomes’ surface and specific groups present in the ligand. For this purpose, the pre-modification of various types of block-copolymers, lipids or cholesterol is often applied to introduce functional groups.19–24 One example is the commercially available DBCO modified liposome formulation (Immunosome®-DBCO). In this case, the functional DBCO groups were directly introduced via the phospholipid before the liposomes’ self-assembly. In a following reaction, azide containing peptides or proteins can be immobilized in the liposomes’ surface.
However, direct lipid or polymer functionalization might not be applicable when the self-assembly of the vesicle structure is disturbed by the chemical modification of one component. Similarly, pre-functionalization is not advisable when the functional groups should only be present on the outer vesicle surface to avoid reactions with sensitive cargos. Additionally, the functional moieties, which are introduced prior to self-assembly, might partially be buried in the hydrophobic environment of the formed membrane, as it was demonstrated for polymersomes.20 Ultimately, also for pre-assembled natural structures like extracellular vesicles25–27 a different functionalization strategy is required. Therefore, a robust way is needed to enable bio-orthogonal and highly specific functionalization of the vesicle surfaces after self-assembly. Additionally, the proposed functionalization strategy should allow for reliable surface characterization and precise reaction control, which is challenging for preformed self-assembled systems.
For a convenient and reliable coupling of two different functional moieties, click chemistry is widely used due to its unique reaction properties: the procedure is very efficient, mild and can be carried out in aqueous reaction conditions. Click chemistry is rapidly becoming a popular tool to functionalize the surface of biomacromolecules, including viruses, DNA, peptides, antibodies, liposomes, micelles, and nanoparticles with a wide variety of conjugates.28–30 There are two well established bio-orthogonal coupling approaches: (1) copper-catalyzed azide–alkyne cycloaddition (CuAAC) “click” reactions, which are attractive as the reaction kinetics are favorable and azide-containing reagents are widely available11,29,31,32 (2) the strain-promoted alkyne–azide cycloaddition (SPAAC) where the use of Cu(I) as a catalyst is avoided.33 This bio-orthogonal copper-free click chemistry offers a great advantage regarding the known deleterious effects associated with copper in cell-based studies and applications.34
In this work, we describe the formation of liposomes based on lipid thin-film hydration followed by extrusion and subsequent surface functionalization of lipid headgroups (Scheme 1). To enable chemical modification via the SPAAC approach, the lipid bilayer was composed out of cholesterol and two different phospholipids – 2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) and L-α-phosphatidylcholine (egg PC) – with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1. The formed liposomes were characterized thoroughly to determine the exact number of available surface primary amine groups (–NH2). Amino groups were further coupled with dibenzylcyclooctyne–PEG4–NHS ester to introduce a strained alkyne (DBCO group) to the liposomes’ surface under different reaction pH. Successful modification with DBCO groups was proven by an anthracene azide (Anth-N3) assay which was carried out for a precise DBCO group quantification as well as analysis of the stability in the aqueous solution. With this, we provide a universal functionalization strategy for any NH2-terminated nanocarrier. In addition, we give a detailed protocol on methods to monitor each functionalization step. Next to this, the antibody CD11c was site-specifically modified with azide groups by transferase and UDP-N-azidoacetylgalactosamine (UDP-GalNAz). Subsequently, the azide-functionalized antibody was coupled to the DBCO-functionalized liposomes via bio-orthogonal copper-free click chemistry (SPAAC). The successful coupling was confirmed by a flow cytometry based binding assay using fluorescently labeled secondary antibodies.
:1:1. The formed liposomes were characterized thoroughly to determine the exact number of available surface primary amine groups (–NH2). Amino groups were further coupled with dibenzylcyclooctyne–PEG4–NHS ester to introduce a strained alkyne (DBCO group) to the liposomes’ surface under different reaction pH. Successful modification with DBCO groups was proven by an anthracene azide (Anth-N3) assay which was carried out for a precise DBCO group quantification as well as analysis of the stability in the aqueous solution. With this, we provide a universal functionalization strategy for any NH2-terminated nanocarrier. In addition, we give a detailed protocol on methods to monitor each functionalization step. Next to this, the antibody CD11c was site-specifically modified with azide groups by transferase and UDP-N-azidoacetylgalactosamine (UDP-GalNAz). Subsequently, the azide-functionalized antibody was coupled to the DBCO-functionalized liposomes via bio-orthogonal copper-free click chemistry (SPAAC). The successful coupling was confirmed by a flow cytometry based binding assay using fluorescently labeled secondary antibodies.
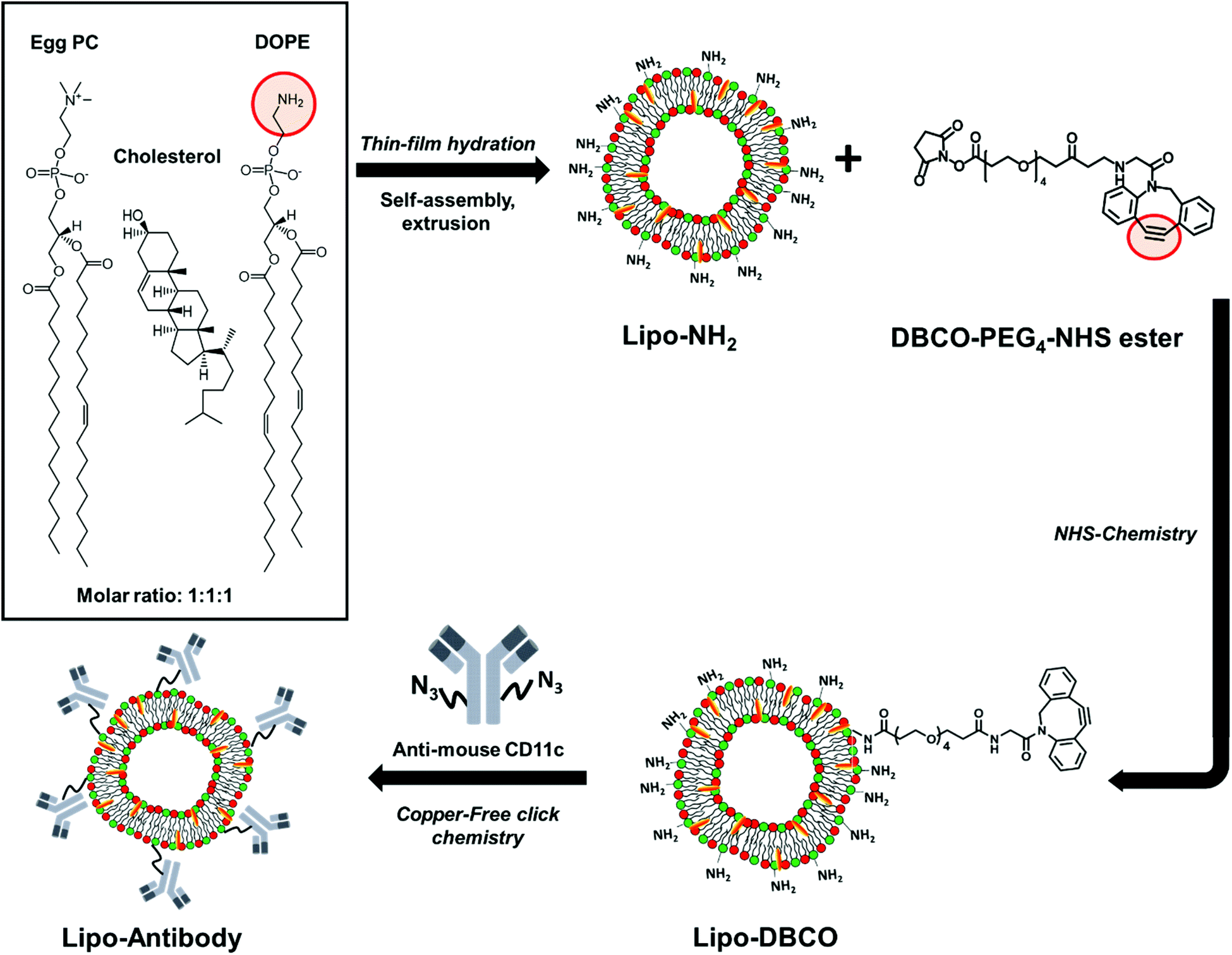 | ||
| Scheme 1 Schematic step-by-step illustration of liposome synthesis, surface functionalization and site-selective antibody attachment. | ||
2. Experimental
2.1. Materials
All chemicals and materials were used as received. 1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) with ≥99.0% purity (10 mg mL−1 in CHCl3), L-α-phosphatidylcholine (egg PC) with purity ≥99.0% (100 mg mL−1 in CHCl3), cholesterol (powder, BioReagent, ≥99%, Germany) and dimethylsulfoxide (DMSO) were purchased from Sigma Aldrich (>99% purity, Germany). The dye DiI Stain (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate) for labelling liposomes was purchased from Thermo Fisher (catalog number: D282, Germany). Dibenzylcyclooctyne (DBCO)–PEG4–NHS ester (cat. no. CLK-A134-100) was purchased from Jena Bioscience (purity: >95% (HPLC), Germany). Phosphate buffered saline (DPBS-buffer, –Mg2+, –Ca2+) was procured from Sigma-Aldrich (USA), while ethanol (99.5%) was acquired from Carl Roth, Germany. A MilliQ device (Merck Millipore, Germany) was used to obtain the demineralized water. Demineralized water was used for all experiments. The antibodies anti-mouse CD11c (clone N418) and the FITC-labeled goat anti-hamster (Armenian, clone Poly4055) was purchased from Biolegend (0.5 mg mL−1). The SiteClick™ Antibody Azido Modification Kit was obtained from Thermo Fisher (catalog number: S20026, America).2.2. Liposome preparation
:DOPE:Chol = 1:1:1 by film hydration followed by extrusion. Briefly, both lipids and cholesterol stock solutions in chloroform were prepared at a concentration of 10 mg mL−1. Then, 835 μL, 767 μL and 398 μL of the egg PC, DOPE and Chol solutions, respectively, were added into a 50 mL round-bottomed flask with an additional 2 mL of a mixture of chloroform with 1 vol% EtOH. The mixture was dried with a rotary evaporator at a reduced pressure of 450 mbar and 3 mbar each for half an hour at 42 °C. Subsequently, the flask was placed in a vacuum oven for 1 hour to remove organic solvent residues (images of the film formation procedure, see Fig. S1†).
2.3. Liposome characterization
2.4. Conjugation of DBCO groups to liposome surface via NHS ester reaction
Firstly, a dibenzylcyclooctyne (DBCO)–PEG4–NHS ester (Mw = 388.37 g mol−1) stock solution was prepared by dissolving 10 mg DBCO–PEG4–NHS ester in 250 μL of dry DMSO to obtain a final concentration of 40 mg mL−1. Secondly, in order to compare the DBCO conjugation efficiency, different reaction parameters were under consideration: (1) the molar ratio of added DBCO–PEG4–NHS ester to amine groups (–NH2) was varied to be 1:1, 3:1 and 6:1; (2) the reaction pH was varied from 7.1 to 7.6 to 8.2. To achieve the different coupling ratios, 126 μL, 63 μL or 21 μL of DBCO stock solution (concentration: 40 mg mL−1) were added into 500 μL of liposome sample (solid content: 0.35 wt%), separately. The different reaction pH was adjusted by addition of 0.5 M NaOH or 0.1 M HCl solution. All the NHS ester conjugation reactions were carried out by firstly vortexing for 20 s and then stirring (700 rpm) over night at room temperature. Afterwards, the liposomes were purified from excess functionalization reagent and organic solvent by centrifugation at 20000g and 4 °C for 1 h. The liposomes were then redispersed in PBS. This washing procedure was performed for a total of three times (see Fig. S2†).
2.5. Quantification of the DBCO groups on the liposomes’ surface
An anthracene-azide (Anth-N3) assay was performed to determine the DBCO conjugation efficiency. For the Anth-N3 stock solution (needs to be freshly prepared before the assay): 1.8 mg Anth-N3 yellow powder was added into 1.05 mL dry DMSO and covered with aluminum foil. The modified liposomes (solid content: 0.35–0.42 wt%, ∼3.5 mg mL−1) were dispersed in PBS buffer (0.1 M, pH = 7.4). The mixture solutions were vortexed for 20 s with maximum speed after addition of 5 different sample types into 1.5 ml Eppendorf tubes: (a–b) 17.3 μL Anth-N3/32.6 μL DMSO; (c–d) 25 μL liposome dispersion/17.3 μL Anth-N3/7.63 μL DMSO; (e) 25 μL liposome dispersion/25 μL DMSO. All the samples had the same total volume of 50 μL, were covered with aluminum foil and placed on the shaker over night at 25 °C. For the fluorescence intensity measurement by the plate reader (Ex: 370 nm, Em: 414 nm), all the mixtures were separated into two groups: 10 times dilution and 100 times dilution by DMSO. Namely, 10 μL of the samples were added into 90 μL DMSO and 1 μL of the samples added into 99 μL DMSO. All the measurement data were calculated according to a previously reported procedure.242.6. Site-specific, enzymatic modification of antibodies
Anti-mouse CD11c antibodies (clone N418, Biolegend, 0.5 mg mL−1) were modified site-specifically on the heavy chain Fc parts with an azide using the Site Click™ Antibody Azido Modification Kit from Thermo Fisher. In the first step, a buffer exchange was performed. Therefore, 250 μg of the anti-mouse CD11c antibody (500 μL, 0.5 mg mL−1) were loaded onto an antibody concentrator (supplemented with the kit) and centrifuged for 6 min at 5000g. The flow-through was discarded and 450 μL of antibody preparation buffer (supplemented with the kit) were added and centrifuged for 8 min at 5000g. For antibody recovery, the concentrator was inverted, placed into a fresh lo-bind Eppendorf tube (2 mL) and centrifuged (3 min, 1000g) to obtain a concentrated antibody solution (80 μL, 2 mg mL−1, 160 μg). Afterwards, galactosidase (supplemented with the kit, 10 μL) was added to the antibody solution and incubated overnight at 37 °C to remove the galactose residues on the carbohydrate domain at the Fc region. Further, the azide group was enzymatically attached to the carbohydrate modified-antibody. Therefore, 75 μL of H2O, 12.5 μL of 20× Tris buffer (pH = 7), 25 μL of buffer additive and 80 μL of GalT (Y289L) enzyme as well as the prepared carbohydrate modified antibody were added to a tube containing UDP-GalNAz (all components supplemented with the kit) and incubated overnight at 30 °C. To remove excess UDP-GalNAz, the azide-modified antibody was purified with a large antibody concentrator (supplemented with the kit). Therefore, the large antibody concentrator was pre-washed with 1 mL of 1× Tris (pH = 7) via centrifugation (10 min, 1200g). Afterwards, the azide-modified antibody was loaded onto the large antibody concentrator and 1.6 mL of 1× Tris (pH = 7) was added. The concentrator was centrifuged (6 min, 1200g) and the flow-through was discarded. This step was repeated three times. Finally, the concentrator was inverted, placed into the conical collection tube and centrifuged for 3 min at 1000g. The azide-modified antibody (∼40 μL, 2.6 mg mL−1) was recovered, transferred to a fresh lo-bind Eppendorf tube (1.5 mL) and stored at 4 °C.2.7. Liposome surface functionalization with antibody via copper-free click chemistry
167 μL DBCO-liposome (∼3 mg mL−1 assumption = 500 μg) and 6.7 μL of azide-modified anti-mouse CD11c-antibody (2.6 mg mL−1, 17.5 μg) were mixed and incubated overnight at room temperature. In order to remove unconjugated antibodies, the dispersion was centrifuged (20000g, 15 min, 4 °C) and washed with 500 μL of PBS. This step was repeated three times and in the end the liposome-antibody pellet was resuspended in 80 μL of PBS.
2.8. Detection of antibodies on the liposomes’ surfaces via flow cytometry
The successful coupling of the anti-mouse CD11c antibodies on the liposomes’ surfaces was confirmed by our previously established flow cytometry protocol.36 Therefore, liposomes with DBCO or liposomes with attached antibodies (2 μL, 3 mg mL−1, 6 μg) were incubated with FITC-labeled goat anti-hamster (Armenian, clone Poly4055, Biolegend, 0.5 mg mL−1, 2 μL, 1 μg) for 30 min at room temperature in the dark. Afterwards, the mixture was filled up with PBS (1 mL). Flow cytometry measurements were performed on an Attune Nxt flow cytometer (Thermo Fisher Scientific) equipped with four lasers (405, 488, 561 and 637 nm) and 16 channels (VL1-4, BL1-4, YL1-4, RL1-4). Liposomes were displayed in a logarithmic scale in a dot plot (SSC vs. YL1). Further, the number of FITC-labeled liposomes was quantified in a histogram via the median fluorescent intensity (MFI) and amount of FITC positive liposomes (%).2.9. Nano differential scanning fluorimetry (nanoDSF)
Measurements were performed with the Prometheus NT.48 device from NanoTemper Technologies GmbH (Munich, Germany) using high sensitivity glass capillaries (NanoTemper). The excitation power was set to 50% for all experiments and a temperature ramp from 20 °C to 95 °C with a heat rate of 1 °C min−1 was used. The capillary was loaded with 10 μL of the antibody (0.5 mg mL−1). The fluorescence signal at a wavelength of 330 nm and 350 nm was recorded. For data analysis, the ratio of the 350 nm to the 330 nm channel was plotted against temperature. The melting point (TM) was determined from the plot of the first derivative of the fluorescence signal.2.10. SDS-PAGE
The antibody solution (3 μg in a total volume of 26 μL) was mixed with 4 μL of reducing agent and 10 μL of sample buffer. Samples were loaded onto a NuPAGE Novex 10% bis–tris gel and the gel was run for 1 h at 120 V. SeeBlue Plus2 Pre Stained Standard was used as marker. Protein bands were visualized with the SilverQuest Silver Staining Kit according to the manufactures’ instruction. All consumables were obtained from Thermo Fisher Scientific, Waltham, MA.2.11. Functionalization of azide-modified antibodies with DBCO–PEG5 ka
The azide-modified antibody (0.5 mg mL−1, 5 μL) was incubated with DBCO–PEG5 ka dissolved in DMSO (2.5 mg mL−1, 1 μL, Iris Biotech GmbH, Germany) overnight at room temperature. The resulting PEGylated antibody conjugate was further applied to SDS-PAGE.2.12. Functionalization of azide-modified antibodies with DIBO–PE
The azide-modified antibody (0.5 mg mL−1, 15 μL) was incubated with DIBO–PE (11.25 μL, supplemented with the Site Click™ Antibody Labeling Kit from Thermo Fisher) overnight at room temperature. The PE-conjugated antibody was purified according to the manufactures’ instruction.2.13. DC2.4 cell binding assay
Murine dendritic cells (DC2.4, Merck, Germany) were cultured in Iscove's Modified Dulbecco's Medium (IMDM), supplemented with 5% fetal bovine serum (FBS), 1% penicillin (100 U mL−1) and streptomycin (100 mg mL−1) as well as 1% 2-mercaptoethanol (100×), in an incubator with 37 °C and 5% CO2 humidity (CO2 Incubator C200, Labotect). For the cell binding assay, 100000 DC2.4 cells were incubated with the PE modified anti-mouse CD11c antibody (0.34 mg mL−1, 1 μL) for 30 min at 4 °C in a total volume of 100 μL PBS. For the blocking assay, DC2.4 cells were treated with unlabeled anti-mouse CD11 antibody (2 mg mL−1, 5 μL) for 30 min at 4 °C in a total volume of 100 μL PBS. All samples were filled up with 900 μL of PBS and run on an Attune NxT Flow cytometry. The cell population was selected with a FSC/SSC scatter plot, excluding cell debris populations. The gated events were evaluated by the fluorescent signal (YL1 channel for PE) expressed as median fluorescence intensity (MFI).
3. Results and discussion
3.1. Liposome preparation and characterization
The liposomes used for this study were produced by lipid dry-film hydration followed by extrusion with an original lipid concentration of 5 mg mL−1 (Fig. 1.A). This method is applicable to both lipids and block-copolymers for the formation of self-assembled vesicles. The final dispersion in PBS buffer yielded liposomes with very narrow size distribution and a hydrodynamic radius (Rh) of 97 ± 10 nm (Fig. 1.B, Fig. S3†). For the rehydration, also deionized water instead of PBS buffer can be used, which however changes the obtained final size slightly due to the different ionic strength (see Fig. S4†). In this case, PBS was chosen for conducting all further experiments to maintain a physiological sample environment. The zeta potential of the liposomes yielded a slightly negative surface charge of −16 ± 7 mV at pH 7 (zeta potential measurements see Fig. S5†). The final synthesized liposomes were obtained with a solid content of about 0.35–0.38 wt% (∼3.5–3.8 mg mL−1) after extrusion, which corresponds to a concentration of about 1.0 ± 0.2 × 1013 liposomes mL−1. The morphology of liposomes was then analyzed by cryo-TEM and the images demonstrated spherical nanosized unilamellar liposomes with a homogeneous size distribution (Fig. 1.C and D) and a membrane thickness of 4.0 ± 0.5 nm. Additionally, the prepared liposomes were characterized with regards to their number of accessible functional amine groups on the surface, which is extremely important to yield defined conditions for the subsequent coupling reactions. According to the fluorescamine assay (reaction scheme see Fig. S6†), the synthesized egg PC/DOPE/Chol liposomes contained (1.68 ± 0.01) × 1018 NH2 groups mL−1 corresponding to (1.6 ± 0.3) × 105 NH2 groups per liposome.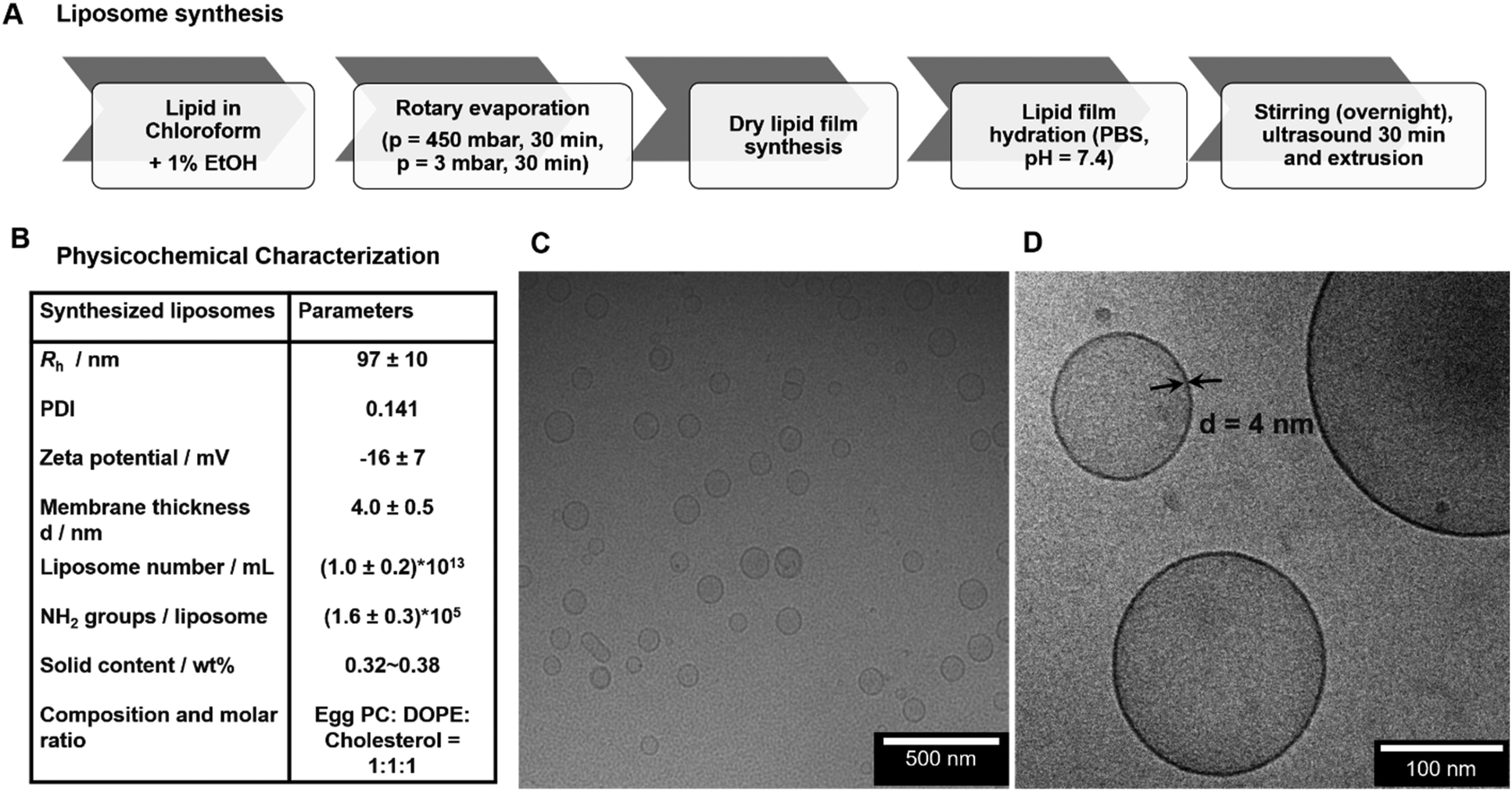 | ||
| Fig. 1 (A) Schematic overview of liposome preparation. (B) Physicochemical characterization of synthesized liposomes. C, (D) cryo-TEM images of liposomes. From images with high magnification (D) the membrane thickness of the liposomes d was determined. | ||
3.2. Conjugation of DBCO groups to liposomes via NHS ester reaction
The DBCO–PEG4–NHS ester was selected for liposome surface modification to introduce active alkyne groups, which react specifically and efficiently with a primary amine (–NH2) at pH 7.0–8.5 to form a covalent bond (see Fig. 2A). This is especially desired since amine groups can also easily be introduced into e.g. polymersomes and are present on other natural carrier systems such as extracellular vesicles, where they mostly appear in the form of membrane proteins. Like this, the functionalization strategy could directly be adapted for all kinds of self-assembled structures. The hydrophilic polyethylene glycol (PEG) spacer arm imparts water solubility and provides a flexible connection that minimizes steric hindrance involved with ligation to complementary azide-containing molecules. This spacer can further be varied in length to later provide optimal accessibility of the coupled compound.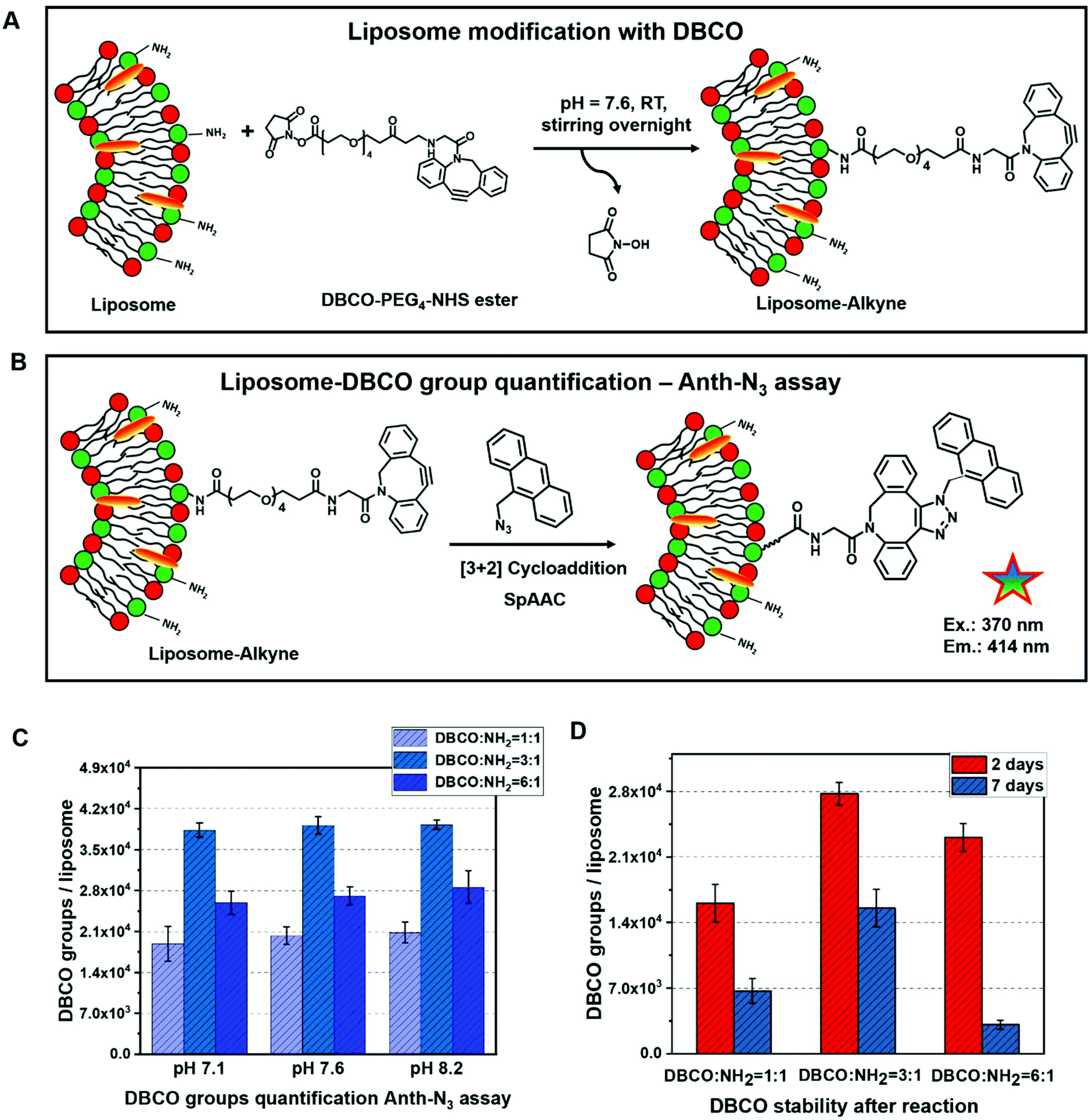 | ||
| Fig. 2 Quantification of DBCO groups on the liposomes’ surface. Schematic illustration of (A) the functionalization procedure with a DBCO–PEG4–NHS ester. (B) The anthracene-azide assay: the coupling of anthracene provides fluorescence emission proportional to the number of DBCO groups. (C) Anthracene-azide assay quantification results for different reaction conditions (varied DBCO/NH2 group ratio and different reaction pH). (D) DBCO group quantification after different storage times. | ||
At this point, it is worth to point out that the liposomes (including all other self-assembled structures comprised of amphiphilic molecules) are sensitive towards organic solvents, which can cause liposome aggregation. Thus, the DBCO–PEG4–NHS ester was dissolved in DMSO in a very high concentration (40 μg μL−1) so that only a minimal addition of organic solvent DMSO was required to carry out the functionalization reaction successfully. With this low amount of organic solvent, no aggregation of liposomes was observed.
The reaction of NHS esters with amines is strongly pH-dependent: on one hand at a low pH, the amino groups on the liposomes’ surface will be protonated and therefore the reactivity is decreased. On the other hand, if the reaction is performed at higher pH, hydrolysis of the NHS ester will be very fast and thus the functionalization yield of liposomes will be reduced. Therefore, optimization of the pH value for this reaction is crucial. In our work, different reaction pH values (7.1, 7.6 and 8.2) as well as different molar ratios of DBCO/NH2 groups (1:1, 3:1 and 6:1) were compared carefully with regards to the modification efficiency.
3.3. Quantification of DBCO groups via an anthracene-azide (Anth-N3) assay
For further antibody click reaction, it was important to determine the total number of DBCO groups that could participate in a further click reaction. Therefore, a click reaction of Anth-N3 (9-(azidomethyl) anthracene) to the Lipo-DBCO samples was performed for the quantification of the surface alkyne groups. This procedure was described in literature by Baier et al.31 The Anth-N3 click reaction mechanism is shown in Fig. 2.B. Anth-N3 itself is already fluorescent (I0, in eqn (1)) (Ex: 370 nm, Em: 414 nm). However, after the click reaction with the Lipo-DBCO the quantum yield increases by a factor of 48 resulting in an increase of the overall fluorescence intensity (I, in eqn (1)) due to a photoinduced electron transfer (PET) effect. A 48-fold increased quantum yield was used for our calculation for the following reason: the quantum yield of the azide is 0.02 and the newly formed triazole has a quantum yield of 0.95–0.97 based on literature reports.37 Therefore, the ratio and increase of the quantum yield is 0.96/0.02 = 48. The amount of DBCO groups on the liposome surface could further be quantitatively calculated viaeqn (1) and (2). In the equations, Ω represents the fluorescent enhancement and n(react-Azido) the total amount of reacted azide groups, which can be calculated. The obtained values are as shown in Fig. 2C.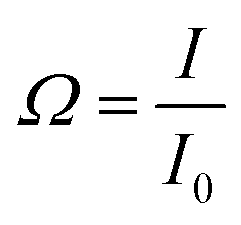 | (1) |
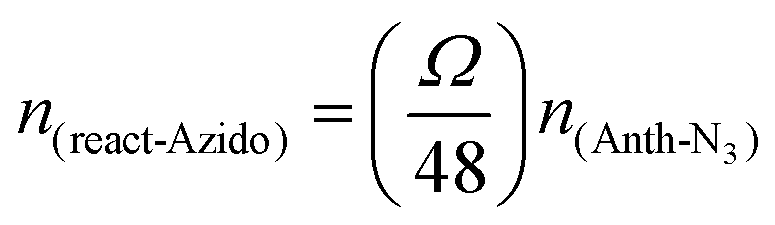 | (2) |
In the Anth-N3 assay, the quantitative determination38–40 of DBCO groups on the liposomes’ surface was performed for the three different DBCO/NH2 group ratios 1:1, 3:1 and 6:1 with three different reaction pH values (7.1, 7.6 and 8.2) (as shown in Fig. 2C). From the measured fluorescence, the number of DBCO groups per liposome were calculated. Additionally, the DBCO conjugation stability after the reaction with the liposomes was determined (Fig. 2C and D). We found that the highest modification efficiency is observed for a 3-fold molar ratio of DBCO to liposomes for the same reaction pH value. In this case, the maximum conjugation efficiency is ∼20%. In addition, the conjugation efficiency increased slightly along with the increasing reaction pH for all reaction molar ratios between DBCO/NH2. Next to this, the stability of the DBCO groups after the liposome modification was investigated (Fig. 2D). The value of DBCO groups per liposomes decreased by more than 50% after 7 days compared to the fresh samples. Moreover, some floccules started to appear within the Lipo-DBCO suspension, especially for the molar ratio DBCO/NH2 = 6:1 after 7 days. This means that further coupling by click chemistry ideally should be performed within the same day. Since during this reaction optimization step it was found that the ratio of DBCO/NH2 = 6:1 did not result in further increase of the reaction yield, but rather in a significant decrease, subsequent coupling of the chosen antibody was only performed for the 1:1 and 3:1 ratio conditions, which were coupled at a reaction pH of 8.2.
3.4. Antibody functionalization and coupling to liposomes via copper-free click reaction
Most widely in literature, antibodies are functionalized via NHS-chemistry.41–43 In this case, the functional group is attached on the lysine residues or the N-termini of the antibodies. This functionalization strategy is highly unspecific as lysine residues are distributed over the whole antibody sequence including the antigen-binding site (Fab region).44 Next to this, the two N-termini of the antibody are also located within the Fab region. Therefore, introducing functional groups along those groups can cause a loss of the antibody functionality and decreased cellular recognition.36 Therefore, we chose a site-selective antibody functionalization strategy to overcome the loss of binding specificity and antibody functionality. Monoclonal antibodies have two conserved N-glycosylation sites on the heavy chain of the fragment crystallizable region (Fc-part).45 It is known that N-glycosylation occurs at the amino acid asparagine (Asn) in the position Asn-297.46 Therefore, here we used a functionalization strategy, which allows the site-selective attachment of a maximum of four azide groups (two per heavy chain) at the Asn-297 position to the CD11c antibody. It is a two-step protocol, where in the first step galactose is enzymatically removed from the sugar-tree and an N-acetylglucosamine residue is exposed (Fig. 3.A). In a second step, an azide-labeled sugar derivate (GalNAz, N-azidoacetylgalactosamine-tetraacylated) is enzymatically attached via the galactose-specific transferase (β1,4-galactosyltranferase mutant Y289L-Gal-T1) to the glycans of the antibody (Fig. 3.B). This method was first developed for the site-selective radiolabeling of antibodies.47 In this case, the azide-modified antibody was conjugated with a desferrioxamine-modified dibenzocyclooctyne (DIBO) in a copper-free click reaction and 89Zr was further added for radiolabeling. The same antibody functionalization strategy was also applied for engineering site-specific antibody–drug-conjugates.48,49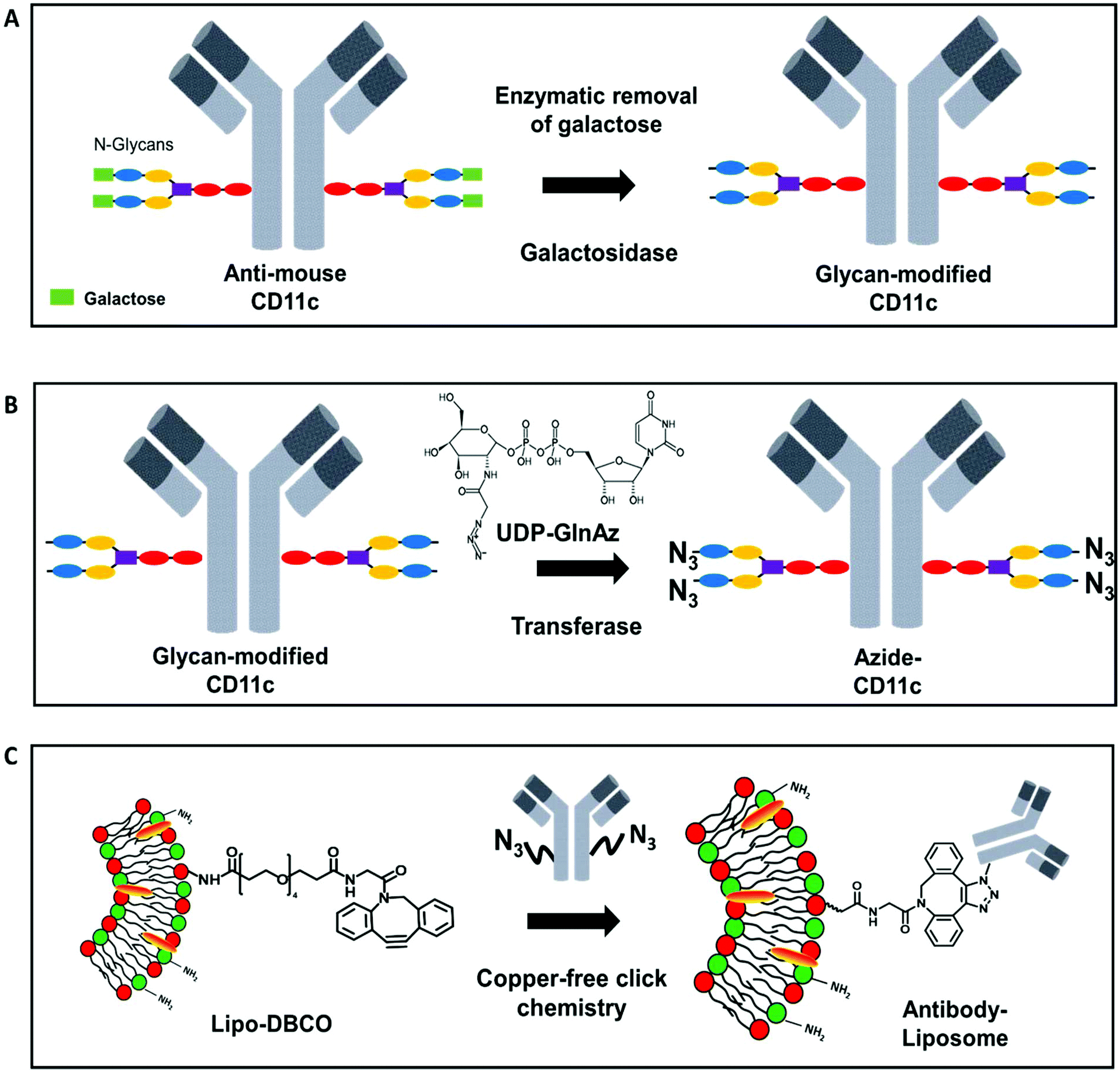 | ||
| Fig. 3 Antibody functionalization and attachment to surface-modified liposomes. (A) Enzymatic removal of galactose from the Fc-part of the CD11c-antibody with galactosidase. (B) Site-specific, enzymatic attachment of an azide functionality using UDP-N-azidoacetylgalactosamine (UDP-GalNAz) to the sugar residues on the Fc-part of the CD11c-antibody (C) bio-orthogonal copper-free click reaction to attach the azide-modified antibody on the DBCO-modified liposome surface. | ||
To investigate the structural and functional integrity of the azide-modified antibody compared to the native antibody, nanoDSF measurements, SDS-PAGE (Fig. 4) and a cell binding assay (Fig. S7†) were performed. nanoDSF is a method to determine protein stability based on the intrinsic fluorescence intensity of a protein upon heating. The characteristic melting temperature (TM) is defined at the point, where 50% of the protein is unfolded. For the native anti-mouse CD11c antibody (blue) and the azide-modified antibody (orange), the thermal behavior determined by nanoDSF was highly comparable (Fig. 4A). Next to this, the melting temperature before and after functionalization was about 67 °C, which is in accordance with literature reports for other antibodies.50 This indicates that the azide modification of the antibody had no major impact on the structural properties of the antibody.
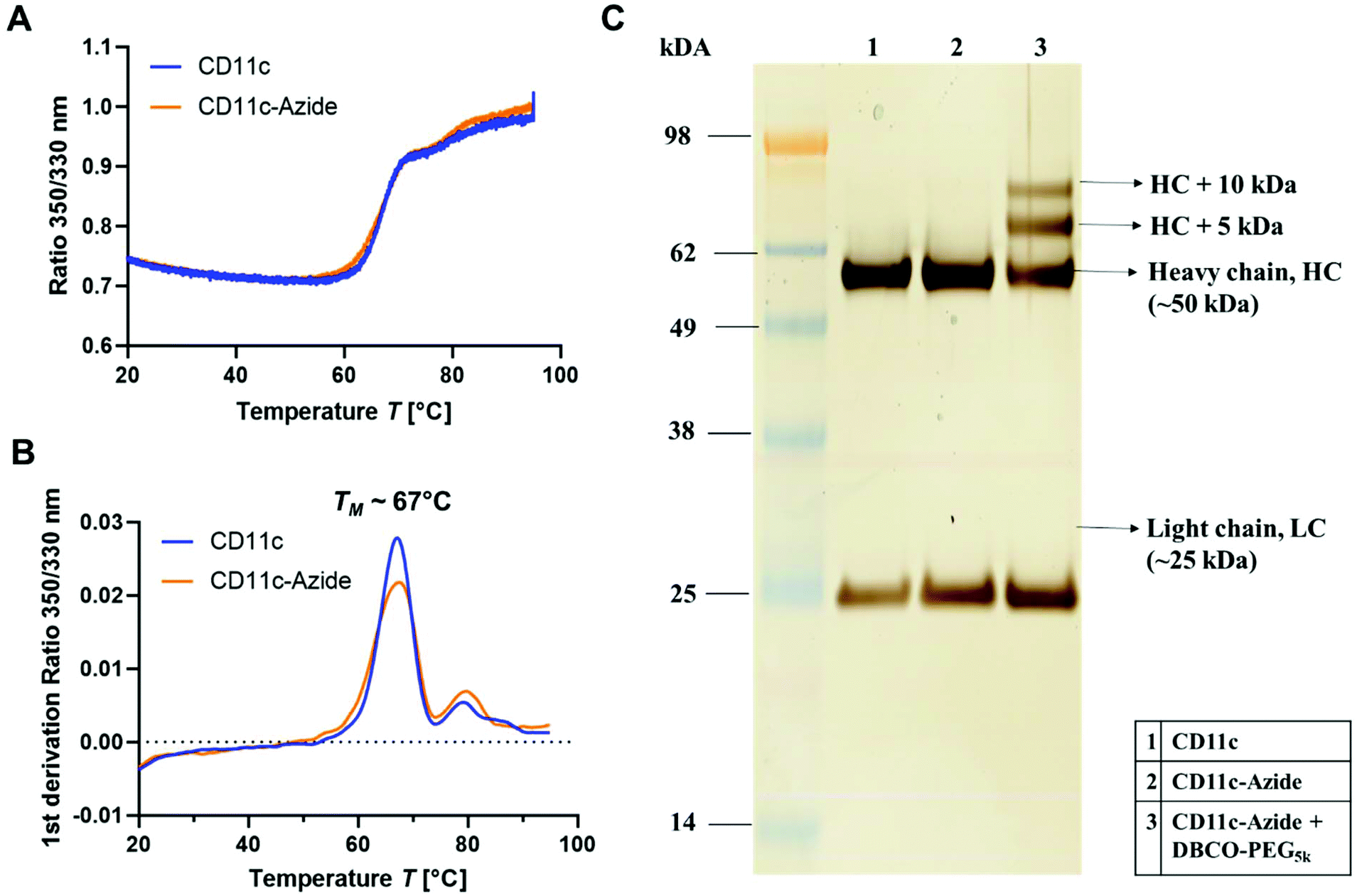 | ||
| Fig. 4 Characterization of the azide-modified antibody. (A) and (B) nanoDSF measurements of the unmodified and azide-modified anti-mouse CD11c antibody (10 μL, ∼0.5 mg mL−1). The antibody was heated up (1 °C min−1) from 20 °C to 95 °C and the intrinsic fluorescence intensity at 330 nm and 350 nm was recorded. The ratio of the fluorescence intensity (350/330 nm, A) and the first derivation (B) is plotted. The melting temperature TM is determined from the maximum of the 1st derivation of the ratio. (C) SDS-PAGE of unmodified (1), azide modified CD11c (2) and PEGylated CD11c antibody (3). Proteins (3 μg) are reduced, applied to an SDS-PAGE and protein bands are visualized by Silver Staining. | ||
Next to this, the unmodified (1) and modified (2) antibody was applied to SDS-PAGE (Fig. 4C) to investigate molecular weight differences and possible degradation products after antibody functionalization. We could observe no significant difference for the azide-modified antibody compared to the unmodified one. To verify the successful azide functionalization of the antibody, a model reaction with a DBCO-PEG-linker (5 kDa) was carried out and the resulting conjugate was applied to SDS-PAGE. In lane 3 (Fig. 4C) there are two additional protein bands visualized, which correspond to the heavy chain (HC) fragment of the antibody (50 kDa) modified with one DBCO–PEG5 kDa (+5 kDa) and thus one azide group or an HC fragment conjugated with two DBCO–PEG5 kDa (+10 kDa) and thus two azide groups per HC fragment. This means for the full antibody (having two HC fragments) the labeling efficiency is between 2–4 azides per antibody. This is also in agreement with literature reports, stating a labeling degree of 2.7 ± 0.2 GalNAz residues per antibody.47
To determine the functional binding properties of the modified antibody, a DIBO-modified fluorescent dye (PE) was incubated with the azide-modified anti-mouse CD11c antibody. Murine dendritic cells (DC2.4), which express CD11c antigen, were incubated with the PE-modified anti-mouse CD11c antibody for 30 min at 4 °C (Fig. S7†). The PE-modified anti-mouse CD11c antibody strongly bound to the DC2.4 cells. To confirm the antibody specificity, cells were pre-treated with free unlabeled anti-mouse CD11c antibody (blocking assay). In this case, the PE-modified anti-mouse CD11c antibody was not able to bind to the DC2.4 cells proving the receptor specificity of the PE-modified anti-mouse CD11c antibody.
In a last step, the azide-modified anti-mouse CD11c antibodies were incubated with the DBCO-functionalized liposomes overnight at room temperature (Fig. 3C). Via the bio-orthogonal copper-free click reaction (SPAAC), it was possible to covalently immobilize the azide-modified antibodies on the DBCO-functionalized liposomes’ surface.
To prove the successful coupling, flow cytometry measurements were performed using our previously established protocol.36 Here, we chose a fluorescently-labeled secondary antibody (FITC-anti hamster antibody), which specifically binds to the immobilized anti-mouse CD11c antibodies (clone N418, host species Armenian Hamster) on the liposomes’ surface by recognition of the Fc-region (Fig. 5A). As a control, liposomes functionalized with DBCO only were incubated with the FITC-labeled secondary antibody.
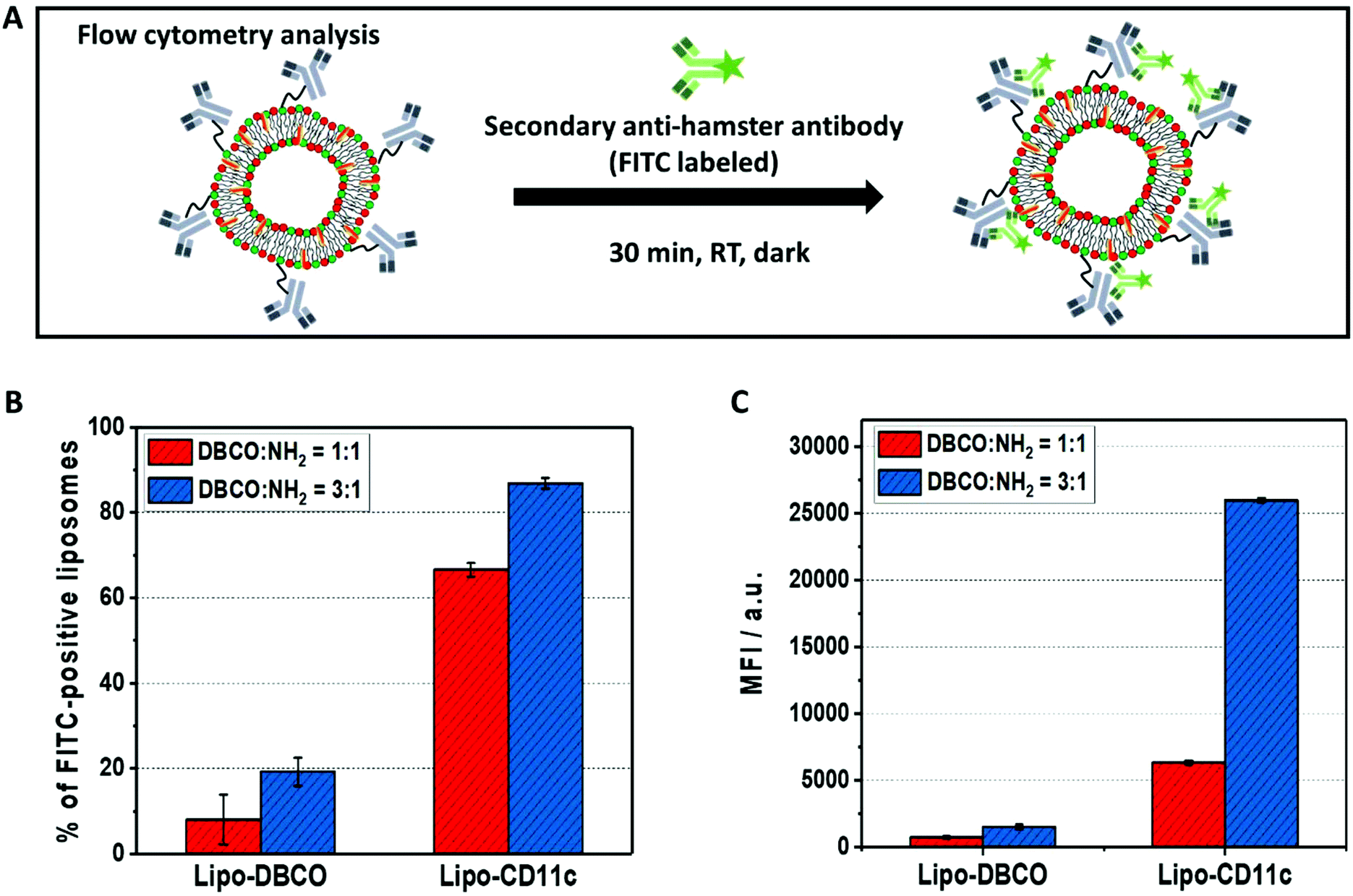 | ||
| Fig. 5 Verification of antibody presence on the liposomes’ surface. (A) Schematic illustration of the flow cytometry measurements for the antibody quantification on the liposome surface. (B) and (C) Flow cytometry measurements for liposomes functionalized with anti-mouse-CD11c antibodies. A secondary labeled FITC-anti-hamster antibody was used to detect the antibodies on the liposome surface. Different reaction conditions (ratio between DBCO/NH2 groups 1:1 or 3:1) were investigated. The number of FITC-positive liposomes in % (B) or the median fluorescence intensity (MFI) is shown (C). | ||
As a result of the flow cytometry analysis, we detected a high amount of FITC-fluorescence positive liposomes for Lipo-CD11c (∼60% to 80%, Fig. 5B). In contrast, there was a very low unspecific binding of the FITC-labeled secondary antibody to the Lipo-DBCO samples (<20%) as expected without any attached proteins being present. Along with the quantification of the DBCO groups on the liposomes’ surface (Fig. 2B), a higher amount of antibodies was detected for liposomes modified with the DBCO/NH2 = 3:1 ratio (Fig. 5C). This means that, indeed, the number of attached antibodies was also proportional to the number of available DBCO groups on the liposomes and the proposed coupling strategy provides precise control over the obtained liposomal surface. It has to be noted, however, that the exact number of antibodies per liposome cannot be deducted from the fluorescence intensity, as the unspecific binding also has to be taken into account. The theoretical maximum number of antibodies per liposome was calculated to be 42 (see Table S1†).
Finally, the liposome–antibody conjugates were again characterized with regards to their physico-chemical properties as shown in Fig. 6. The size and surface charge of the non-functionalized as well as the functionalized liposomes was compared for both coupling ratios DBCO/NH2 1:1 and 3:1.
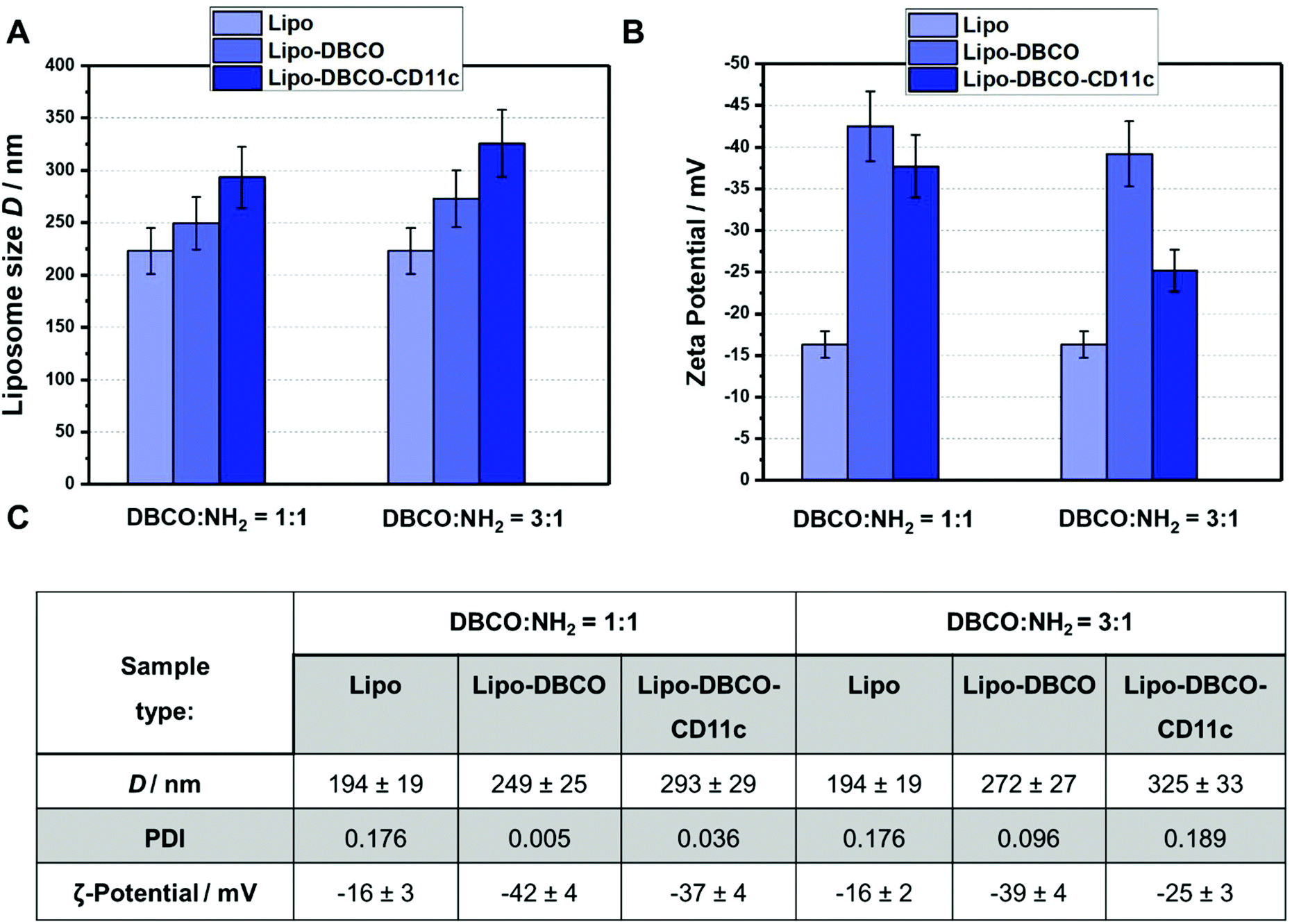 | ||
| Fig. 6 Physicochemical characterization of liposomes before and after functionalization with DBCO/antibodies. (A) Size of liposomes before/after DBCO and antibody attachment using 1:1 and 3:1 DBCO/NH2 ratios. (B) Surface charge before and after liposome functionalization. (C) Overview of the physicochemical properties of the liposomes for the different reaction conditions. | ||
As indicated in Fig. 6.A and C, the size of the liposomes increased step-wise after functionalization. Coupling of the DBCO groups and the antibody CD11c slightly enlarged the liposomes hydrodynamic diameter from 194 nm up to 293 and 325 nm (1:1 and 3:1 DBCO/NH2 groups, respectively). There are mainly two reasons: (1) for the DBCO conjugation/antibody click reaction, the DBCO-liposomes and the liposome-antibody conjugates have to be purified from free excess DBCO–PEG4–NHS ester and further free unreacted antibodies by centrifuging 3 times at 20000g. During these washing steps, smaller liposomes could also be removed. (2) Long chain molecule PEG and DBCO groups as well as the attached antibodies result in larger overall liposomes. Furthermore, we observed that the zeta potential of the functionalized liposomes was more negative compared to the non-functionalized surface. After attachment of the CD11c antibodies, the zeta potential approached a value of −25 mV (for the 3:1 DBCO/NH2 ratio), which is typical for nanocarrier surfaces covered with proteins.
Finally, with the performed characterization approaches, we are confident that we have presented a facile functionalization approach for bio-orthogonal and site-specific attachment of antibodies on a liposome surface. By making use of specific quantification techniques, the available surface functional groups can precisely be analyzed so that the click chemistry reaction stoichiometry can be adjusted to yield an optimal efficiency. With the site-specific antibody modification, additionally it can be assured that the final conjugation product carries recognizable antibody units, which is of importance for further application. The availability of the coupled antibody could even be increased if necessary upon introduction of a longer PEG linker, so that e.g. the adsorption of proteins would not cover the targeting structures.
4. Conclusion
In this study, we have shown the synthesis of amine group-terminated liposomes via lipid dry film rehydration followed by extrusion. In contrast to common vesicle functionalization approaches, which rely on pre-modification of lipids or block-copolymers, we introduced further functional groups and performed antibody coupling via bio-orthogonal copper-free click chemistry after liposome self-assembly. For precise reaction control and tunability, a fluorescamine assay (FA assay) and an anthracen-azide (Anth-N3) assay were utilized to determine the number of present NH2 groups as well as DBCO groups after functionalization. The antibody, which was introduced as a possible targeting structure, was modified site specifically at the Fc fragment and coupled to the liposomes using the strain-promoted alkyne–azide click (SPAAC) reaction. This surface functionalization procedure offers the advantage to be transferred to all self-assembled systems bearing primary surface amino groups without damaging the assembled structure because no large amounts of organic solvent are required and the carriers can be kept in physiological conditions. Amino groups can easily be introduced as end groups of block-copolymers and are already present on natural structures such as extracellular vesicles (in the form of membrane proteins). Additionally, this type of antibody coupling is applicable for all antibody types without the disadvantage of potentially covering or modifying the antigen binding sites. If needed, the introduced PEG spacer can also be varied in length or type of building block to provide additional characteristics like enhanced stealth properties or better antibody accessibility. In summary, this versatile vesicle surface functionalization strategy is applicable for a wide range of drug delivery systems as well as straightforward and robust in terms of handling.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement No. 801338 (Ves4Us project). Open Access funding provided by the Max Planck Society.References
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Liposomes and polymersomes: a comparative review towards cell mimicking, Chem. Soc. Rev., 2018, 47(23), 8572–8610 RSC.
- J. Braun, N. Bruns, T. Pfohl and W. Meier, Phase Behavior of Vesicle-Forming Block Copolymers in Aqueous Solutions, Langmuir, 2007, 23(14), 7484–7490 CrossRef.
- D. E. Discher and A. Eisenberg, Polymer vesicles, Science, 2002, 297(5583), 967–973 CrossRef CAS PubMed.
- R. P. Brinkhuis, F. P. J. T. Rutjes and J. C. M. van Hest, Polymeric vesicles in biomedical applications, Polym. Chem., 2011, 2(7), 1449–1462 RSC.
- A. D. Bangham and R. W. Horne, Negative staining of phospholipids and their structural modification by surface-active agents as observed in the electron microscope, J. Mol. Biol., 1964, 8(5), 660–668 CrossRef CAS.
- J. Li, X. Wang, T. Zhang, C. Wang, Z. Huang, X. Luo and Y. Deng, A review on phospholipids and their main applications in drug delivery systems, Asian J. Pharm. Sci., 2015, 10(2), 81–98 CrossRef.
- G. Caracciolo, Clinically approved liposomal nanomedicines: lessons learned from the biomolecular corona, Nanoscale, 2018, 10(9), 4167–4172 RSC.
- D. G. Villalva, L. Giansanti, A. Mauceri, F. Ceccacci and G. Mancini, Influence of the state of phase of lipid bilayer on the exposure of glucose residues on the surface of liposomes, Colloids Surf., B, 2017, 159, 557–563 CrossRef CAS.
- N. Monteiro, A. Martins, L. Reis Rui and M. Neves Nuno, Liposomes in tissue engineering and regenerative medicine, J. R. Soc., Interface, 2014, 11(101), 20140459 CrossRef.
- Y.-C. Kuo, I. Y. Chen and R. Rajesh, Use of functionalized liposomes loaded with antioxidants to permeate the blood–brain barrier and inhibit β-amyloid-induced neurodegeneration in the brain, J. Taiwan Inst. Chem. Eng., 2018, 87, 1–14 CrossRef CAS.
- S. Cavalli, A. R. Tipton, M. Overhand and A. Kros, The chemical modification of liposome surfaces via a copper-mediated [3 + 2] azide–alkyne cycloaddition monitored by a colorimetric assay, Chem. Commun., 2006,(30), 3193–3195 RSC.
- C. Saraiva, C. Praça, R. Ferreira, T. Santos, L. Ferreira and L. Bernardino, Nanoparticle-mediated brain drug delivery: Overcoming blood–brain barrier to treat neurodegenerative diseases, J. Controlled Release, 2016, 235, 34–47 CrossRef CAS.
- J. Kreuter, Nanoparticulate systems for brain delivery of drugs, Adv. Drug Delivery Rev., 2001, 47(1), 65–81 CrossRef CAS.
- A. Akbarzadeh, R. Rezaei-Sadabady, S. Davaran, S. W. Joo, N. Zarghami, Y. Hanifehpour, M. Samiei, M. Kouhi and K. Nejati-Koshki, Liposome: classification, preparation, and applications, Nanoscale Res. Lett., 2013, 8(1), 102 CrossRef.
- B. S. Pattni, V. V. Chupin and V. P. Torchilin, New Developments in Liposomal Drug Delivery, Chem. Rev., 2015, 115(19), 10938–10966 CrossRef CAS.
- M. K. Riaz, M. A. Riaz, X. Zhang, C. Lin, K. H. Wong, X. Chen, G. Zhang, A. Lu and Z. Yang, Surface Functionalization and Targeting Strategies of Liposomes in Solid Tumor Therapy: A Review, Int. J. Mol. Sci., 2018, 19(1), 195 CrossRef.
- P. V. Pawar, S. V. Gohil, J. P. Jain and N. Kumar, Functionalized polymersomes for biomedical applications, Polym. Chem., 2013, 4(11), 3160–3176 RSC.
- S. Egli, H. Schlaad, N. Bruns and W. Meier, Functionalization of Block Copolymer Vesicle Surfaces, Polymers, 2011, 3(1), 252–280 CrossRef CAS.
- C. Weber, M. Voigt, J. Simon, A.-K. Danner, H. Frey, V. Mailänder, M. Helm, S. Morsbach and K. Landfester, Functionalization of Liposomes with Hydrophilic Polymers Results in Macrophage Uptake Independent of the Protein Corona, Biomacromolecules, 2019, 20(8), 2989–2999 CrossRef CAS.
- S. A. Meeuwissen, M. F. Debets and J. C. M. van Hest, Copper-free click chemistry on polymersomes: pre- vs. post-self-assembly functionalisation, Polym. Chem., 2012, 3(7), 1783–1795 RSC.
- P. Vabbilisetty and X.-L. Sun, Liposome surface functionalization based on different anchoring lipids via Staudinger ligation, Org. Biomol. Chem., 2014, 12(8), 1237–1244 RSC.
- Y. Ma, H. Zhang, V. Gruzdys and X.-L. Sun, Azide-Reactive Liposome for Chemoselective and Biocompatible Liposomal Surface Functionalization and Glyco-Liposomal Microarray Fabrication, Langmuir, 2011, 27(21), 13097–13103 CrossRef CAS.
- S. J. Rijpkema, B. J. Toebes, M. N. Maas, N. R. M. de Kler and D. A. Wilson, Designing Molecular Building Blocks for Functional Polymersomes, Isr. J. Chem., 2019, 59, 1–18 CrossRef.
- S. F. M. van Dongen, M. Nallani, S. Schoffelen, J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, A Block Copolymer for Functionalisation of Polymersome Surfaces, Macromol. Rapid Commun., 2008, 29(4), 321–325 CrossRef CAS.
- R. C. Lai, R. W. Y. Yeo, K. H. Tan and S. K. Lim, Exosomes for drug delivery—a novel application for the mesenchymal stem cell, Biotechnol. Adv., 2013, 31(5), 543–551 CrossRef CAS.
- S. Stremersch, R. E. Vandenbroucke, E. Van Wonterghem, A. Hendrix, S. C. De Smedt and K. Raemdonck, Comparing exosome-like vesicles with liposomes for the functional cellular delivery of small RNAs, J. Controlled Release, 2016, 232, 51–61 CrossRef CAS.
- J. G. van den Boorn, J. Daßler, C. Coch, M. Schlee and G. Hartmann, Exosomes as nucleic acid nanocarriers, Adv. Drug Delivery Rev., 2013, 65(3), 331–335 CrossRef CAS.
- M. A. Bruckman, G. Kaur, L. A. Lee, F. Xie, J. Sepulveda, R. Breitenkamp, X. Zhang, M. Joralemon, T. P. Russell, T. Emrick and Q. Wang, Surface Modification of Tobacco Mosaic Virus with “Click” Chemistry, ChemBioChem, 2008, 9(4), 519–523 CrossRef CAS.
- R. K. O'Reilly, M. J. Joralemon, K. L. Wooley and C. J. Hawker, Functionalization of Micelles and Shell Cross-linked Nanoparticles Using Click Chemistry, Chem. Mater., 2005, 17(24), 5976–5988 CrossRef.
- J. Gierlich, G. A. Burley, P. M. E. Gramlich, D. M. Hammond and T. Carell, Click Chemistry as a Reliable Method for the High-Density Postsynthetic Functionalization of Alkyne-Modified DNA, Org. Lett., 2006, 8(17), 3639–3642 CrossRef CAS.
- G. Baier, J. M. Siebert, K. Landfester and A. Musyanovych, Surface Click Reactions on Polymeric Nanocapsules for Versatile Functionalization, Macromolecules, 2012, 45(8), 3419–3427 CrossRef CAS.
- E. M. Alexandrino, P. Buchold, M. Wagner, A. Fuchs, A. Kreyes, C. K. Weiss, K. Landfester and F. R. Wurm, A molecular “screw-clamp”: accelerating click reactions in miniemulsions, Chem. Commun., 2014, 50(72), 10495–10498 RSC.
- C. Ornelas, J. Broichhagen and M. Weck, Strain-Promoted Alkyne Azide Cycloaddition for the Functionalization of Poly(amide)-Based Dendrons and Dendrimers, J. Am. Chem. Soc., 2010, 132(11), 3923–3931 CrossRef CAS.
- E. Oude Blenke, G. Klaasse, H. Merten, A. Plückthun, E. Mastrobattista and N. I. Martin, Liposome functionalization with copper-free “click chemistry”, J. Controlled Release, 2015, 202, 14–20 CrossRef CAS.
- S. W. Provencher, Contin - a General-Purpose Constrained Regularization Program for Inverting Noisy Linear Algebraic and Integral-Equations, Comput. Phys. Commun., 1982, 27(3), 229–242 CrossRef.
- M. Tonigold, J. Simon, D. Estupiñán, M. Kokkinopoulou, J. Reinholz, U. Kintzel, A. Kaltbeitzel, P. Renz, M. P. Domogalla, K. Steinbrink, I. Lieberwirth, D. Crespy, K. Landfester and V. Mailänder, Pre-adsorption of antibodies enables targeting of nanocarriers despite a biomolecular corona, Nat. Nanotechnol., 2018, 13(9), 862–869 CrossRef CAS.
- F. Xie, K. Sivakumar, Q. Zeng, M. A. Bruckman, B. Hodges and Q. Wang, A fluorogenic ‘click'reaction of azidoanthracene derivatives, Tetrahedron, 2008, 64(13), 2906–2914 CrossRef CAS.
- I. S. Marks, J. S. Kang, B. T. Jones, K. J. Landmark, A. J. Cleland and T. A. Taton, Strain-promoted “click” chemistry for terminal labeling of DNA, Bioconjugate Chem., 2011, 22(7), 1259–1263 CrossRef CAS.
- P. Van Delft, N. J. Meeuwenoord, S. Hoogendoorn, J. Dinkelaar, H. S. Overkleeft, G. A. van der Marel and D. V. Filippov, Synthesis of oligoribonucleic acid conjugates using a cyclooctyne phosphoramidite, Org. Lett., 2010, 12(23), 5486–5489 CrossRef CAS.
- A. Jawalekar, S. Malik, J. Verkade, B. Gibson, N. Barta, J. Hodges, A. Rowan and F. van Delft, Oligonucleotide tagging for copper-free click conjugation, Molecules, 2013, 18(7), 7346–7363 CrossRef CAS PubMed.
- H. Gong, I. Holcomb, A. Ooi, X. Wang, D. Majonis, M. A. Unger and R. Ramakrishnan, Simple Method To Prepare Oligonucleotide-Conjugated Antibodies and Its Application in Multiplex Protein Detection in Single Cells, Bioconjugate Chem., 2016, 27(1), 217–225 CrossRef CAS.
- J. A. G. L. van Buggenum, J. P. Gerlach, S. Eising, L. Schoonen, R. A. P. M. van Eijl, S. E. J. Tanis, M. Hogeweg, N. C. Hubner, J. C. van Hest, K. M. Bonger and K. W. Mulder, A covalent and cleavable antibody-DNA conjugation strategy for sensitive protein detection via immuno-PCR, Sci. Rep., 2016, 6, 22675 CrossRef CAS.
- S. L. Filbrun, A. B. Filbrun, F. L. Lovato, S. H. Oh, E. A. Driskell and J. D. Driskell, Chemical modification of antibodies enables the formation of stable antibody–gold nanoparticle conjugates for biosensing, Analyst, 2017, 142(23), 4456–4467 RSC.
- H. Yao, F. Jiang, A. Lu and G. Zhang, Methods to Design and Synthesize Antibody-Drug Conjugates (ADCs), Int. J. Mol. Sci., 2016, 17(2), 194 CrossRef.
- F. Cymer, H. Beck, A. Rohde and D. Reusch, Therapeutic monoclonal antibody N-glycosylation – Structure, function and therapeutic potential, Biologicals, 2018, 52, 1–11 CrossRef CAS.
- M. Kiyoshi, K. Tsumoto, A. Ishii-Watabe and J. M. M. Caaveiro, Glycosylation of IgG-Fc: a molecular perspective, Int. Immunol., 2017, 29(7), 311–317 CrossRef CAS.
- B. M. Zeglis, C. B. Davis, R. Aggeler, H. C. Kang, A. Chen, B. J. Agnew and J. S. Lewis, Enzyme-Mediated Methodology for the Site-Specific Radiolabeling of Antibodies Based on Catalyst-Free Click Chemistry, Bioconjugate Chem., 2013, 24(6), 1057–1067 CrossRef CAS PubMed.
- P. Thompson, E. Ezeadi, I. Hutchinson, R. Fleming, B. Bezabeh, J. Lin, S. Mao, C. Chen, L. Masterson, H. Zhong, D. Toader, P. Howard, H. Wu, C. Gao and N. Dimasi, Straightforward Glycoengineering Approach to Site-Specific Antibody–Pyrrolobenzodiazepine Conjugates, ACS Med. Chem. Lett., 2016, 7(11), 1005–1008 CrossRef CAS.
- X. Cai and K. D. Janda, A chemoenzymatic approach toward the preparation of site-specific antibody–drug conjugates, Tetrahedron Lett., 2015, 56(23), 3172–3175 CrossRef CAS.
- M. Tonigold, J. Simon, D. Estupiñán, M. Kokkinopoulou, J. Reinholz, U. Kintzel, A. Kaltbeitzel, P. Renz, M. P. Domogalla, K. Steinbrink, I. Lieberwirth, D. Crespy, K. Landfester and V. Mailänder, Pre-adsorption of antibodies enables targeting of nanocarriers despite a biomolecular corona, Nat. Nanotechnol., 2018, 1 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py01136f |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |