
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlled synthesis of methacrylate and acrylate diblock copolymers via end-capping using CCTP and FRP†
Georgios
Patias
 ,
Alan M.
Wemyss
,
Spyridon
Efstathiou
,
James S.
Town
,
Christophe J.
Atkins
,
Ataulla
Shegiwal
,
Richard
Whitfield
and
David M.
Haddleton
*
,
Alan M.
Wemyss
,
Spyridon
Efstathiou
,
James S.
Town
,
Christophe J.
Atkins
,
Ataulla
Shegiwal
,
Richard
Whitfield
and
David M.
Haddleton
*
University of Warwick, Department of Chemistry, Gibbet Hill, CV4 7AL, Coventry, UK. E-mail: d.m.haddleton@warwick.ac.uk
First published on 13th November 2019
Abstract
Scalable methods of producing functional materials is of importance. Herein, we report the use of free radical copolymerisation of ω-unsaturated methacrylic macromonomers, as derived from catalytic chain transfer polymerisation (CCTP), with acrylic monomers in solution leading to block copolymers by varying the nature of the macromonomer. We demonstrate the effect that varying the molecular weight and ester side chain length of macromonomer from CCTP, has on the polymerisation leading to either graft or diblock copolymers. DOSY NMR, MALDI-ToF/ToF MS has allowed for the exploration of the extent to which varying the concentration of macromonomer has on the products formed. Importantly, during the successful synthesis of acrylic and methacrylic diblock copolymers with broad dispersities of each block, full consumption of the macromonomers was observed.
Introduction
Low molecular weight methacrylic polymers/oligomers bearing propenyl ω-end groups have been used as precursors in the synthesis of a range of materials, such as functionalised branched copolymers, amphiphilic dispersants, or multi-block copolymers.1–8 Copolymerisation with acrylates can be challenging and has been the subject of a number of previous investigations.9–13 Propagating radicals, as produced from conventional free radical polymerisation, from both of these monomer types have been shown to readily add to the vinyl end groups of these oligomers, and following addition, the relative rates of monomer propagation (kp) and beta-scission/fragmentation of the radical adduct (kβ) largely determine the composition of the products.14–19An efficacious method to produce low molecular weight oligomers containing functional unsaturated end groups which has been exploited industrially for over 20 years, is cobalt(II) mediated catalytic chain transfer polymerisation (CCTP).20–27 The CoII catalysts used in CCTP are highly efficient in chain transfer from propagating polymer to cobalt(II) leading to CoIII–H enabling for control over the molecular weight of the polymers produced at very low catalyst concentrations (ppm). CCTP of methacrylates leads to the abstraction of a hydrogen atom from the α-methyl group relative to the radical centre resulting in a vinyl terminated chain and an unstable CoIII–H complex that reinitiates polymerisation through hydrogen transfer to monomer, regenerating the CoII catalyst. This is a highly efficient process with observed chain transfer constants over four orders of magnitude higher than thiols at up to Cs = 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. Through this process, macromonomers are produced with α-hydrogen and ω-vinyl groups (mechanism ESI Fig. 1†).
000. Through this process, macromonomers are produced with α-hydrogen and ω-vinyl groups (mechanism ESI Fig. 1†).
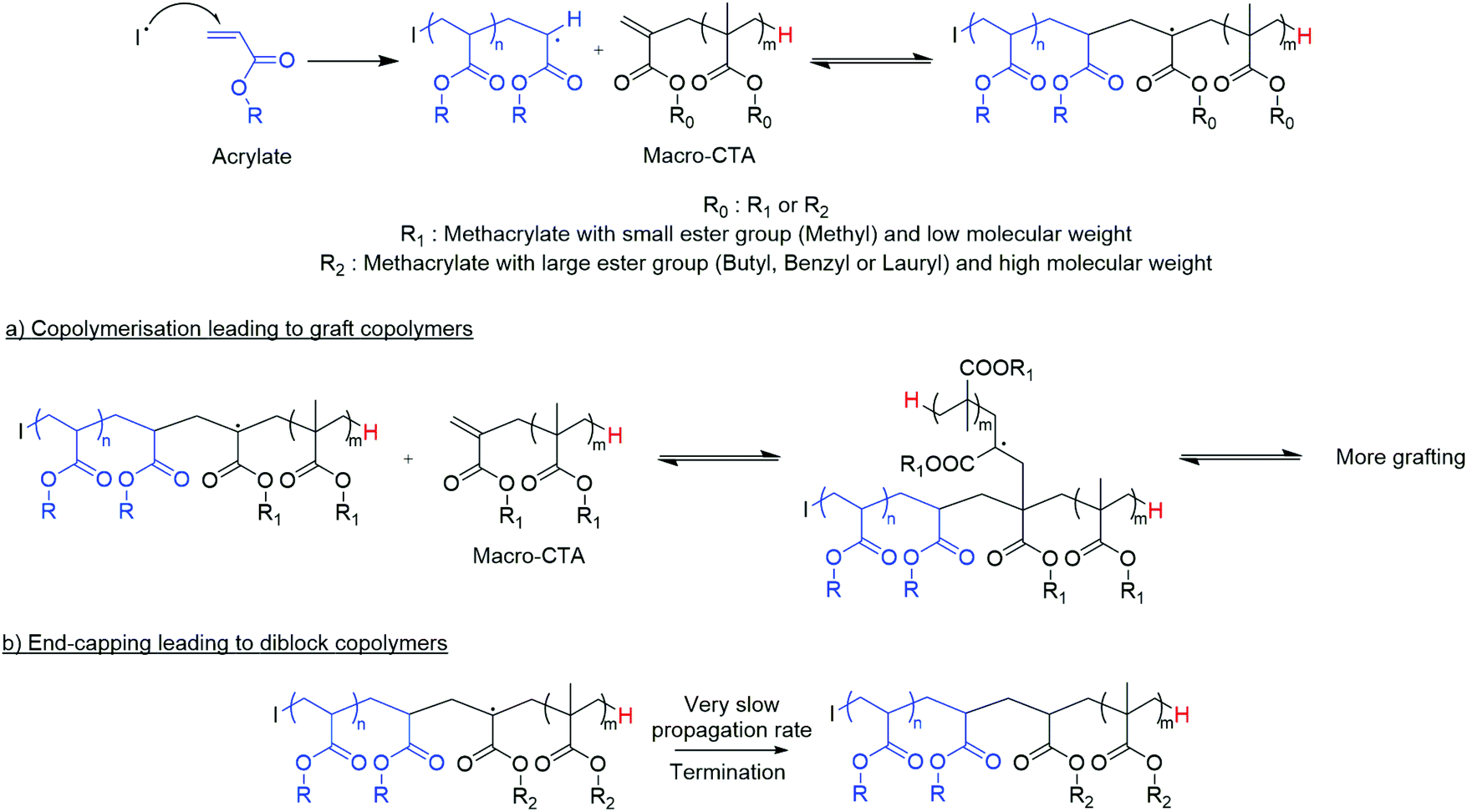
We have previously investigated the copolymerisation of methacrylic macromonomers from CCTP with methacrylates.3,12,28,29 Following the addition of a propagating polymeric methacrylic radical to the vinyl functionality of these macromonomers, the relative rates of propagation and β-scission (kp/kβ) is low, hence β-scission (fragmentation) is favoured. This addition–fragmentation chain transfer (AFCT) process produces a propagating radical of the original macromonomer and a new methacrylic macromonomer that is also capable of undergoing an AFCT reaction.9,12,30 In this way, block copolymers can be produced in a process similar to conventional sulphur containing RAFT agents, mediated by dithioester or trithiocarbonate chain transfer agents (CTAs).
In contrast, acrylic monomers generally have higher propagation rate constants than methacrylates. For example, the kp of methyl methacrylate has been measured as 323 L mol−1 s−1, whereas the kp of methyl acrylate is nearly fifty times higher at 15600 L mol−1 s−1.31,32 Based upon this, the copolymerisation behaviour of acrylate monomers and these CCTP methacrylic macromonomers is expected to be significantly different, with a number of reports in the literature describing the formation of graft copolymers.33–35 However, studies from Yamada and co-workers show that the copolymerisation of acrylates can be much more complicated than suggested in previous studies.13,36–38
Herein, we give new insight into the effective copolymerisation of two types of monomers, methacrylates and acrylates, Scheme 1. A better mechanistic understanding of the copolymerisation of methacrylic macromonomers with methacrylates is reported utilising MALDI-ToF/ToF MS. Moreover, the effect of the chain length of methacrylic macromonomers from CCTP into the free radical polymerisation of acrylates is investigated. We have also explored the extent to which the nature of the macromonomer changes the nature of the final product. Grafted polymers were obtained when methyl methacrylate oligomers were present in the methyl acrylate polymerisation, whereas end-capped diblock copolymers of acrylates with pLMA, pBMA, pBzMA were synthesized under similar conditions. Notably, the 1H NMR spectroscopy showed full consumption of the macromonomer and successful synthesis of diblock copolymers with various molecular weights, albeit macromonomers with broad dispersities were used in their free radical copolymerisation with acrylates. This leads to block and graft copolymers without the requirement of transition metal catalysts, sulphur containing RAFT agents or other expensive and often troublesome reagents. This work is focussed on free radical polymerisation which is widely used and accepted.
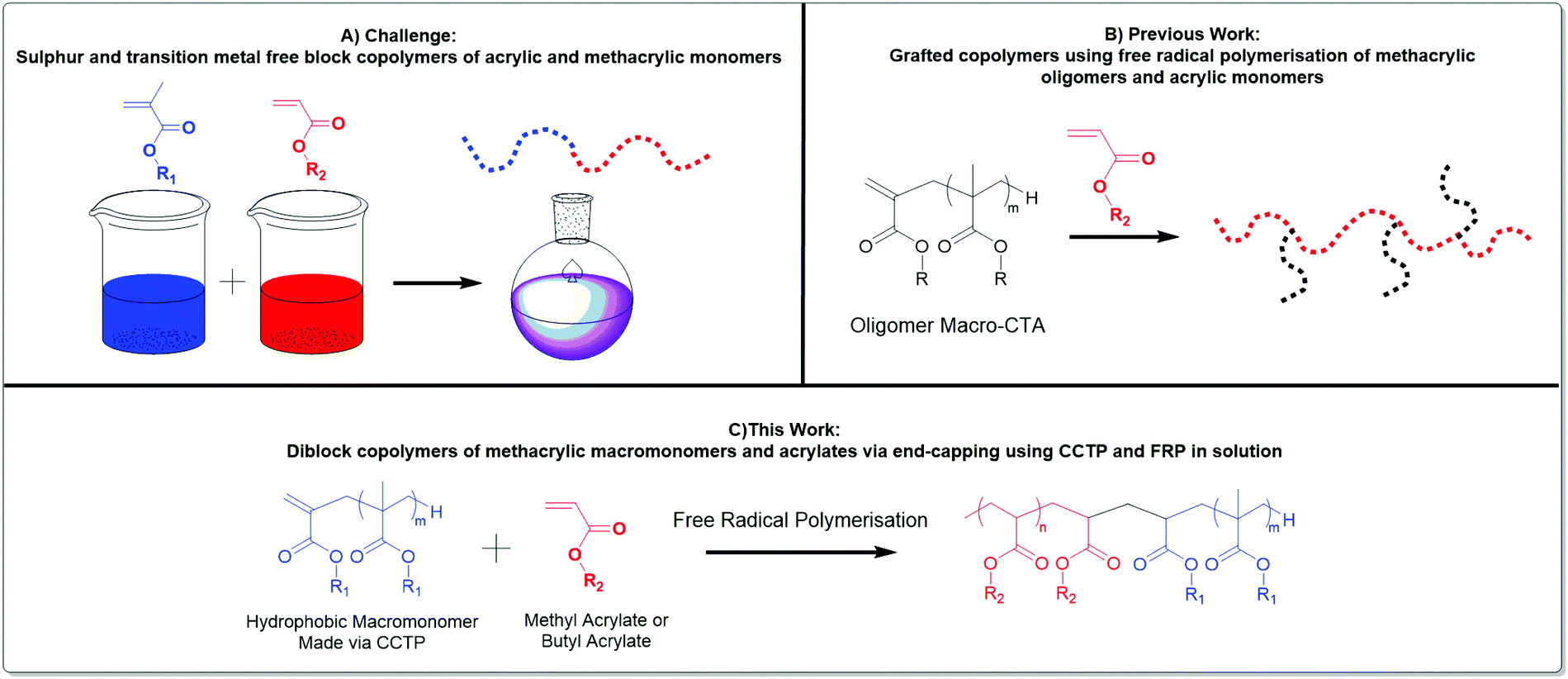 | ||
| Scheme 1 A) Target is to synthesise AB block copolymers using conventional free radical polymerisation. (B) Oligomers containing terminal vinyl functionality might be expected to copolymerise with acrylates to give graft copolymers. (C) This work demonstrates the formation of AB block copolymers by use of macroCTA agents prepared from catalytic chain transfer polymerisation. | ||
Experimental section
Materials and methods
General procedure for the preparation of methacrylic macromonomers via catalytic chain transfer polymerisation
For a CCTP in solution, 0.09 mg bis[(difluoroboryl)dimethyl glyoximato]cobalt(II) (CoBF, 5 ppm relative to monomer) for MMA or bis[(difluoroboryl)dimethyl phenyl-glyoximato]cobalt(II) (Co(MePh)BF) for BMA, BzMA and LMA and a stirring bar were charged into a 100 mL round bottom flask. Nitrogen was purged into the flask for 1 minute (ESI Fig. 4b†). Subsequently, 10 ml of methyl methacrylate (MMA) previously deoxygenated for 30 minutes (ESI Fig. 4a†), were added to the flask via a deoxygenated syringe. The mixture was stirred under a nitrogen atmosphere until the dissolution of the catalyst. Meanwhile, 107 mg dimethyl 2,2′-azobis(2-methyl propionate) (V601, 1 mol% relative to monomer) were dissolved in 10 ml toluene (1/1 v/v to monomer) and the solution was charged into a 50 mL round bottom flask and bubbled with nitrogen for 30 min. Subsequently, the 100 mL flask with the monomer and catalyst was heated to 75 °C under an inert atmosphere. When the temperature of the catalyst solution reached 75 °C, the initiator solution added. The reaction was allowed to continue for 6 hours with continuous stirring.The number average molecular weight of macromonomers was calculated from 1H NMR spectra by integrating the vinyl resonances (6.17 and 5.44 ppm) against the methoxy peak (3.57 ppm). 1H NMR (400 MHz, CDCl3): δ 6.17 (s, 1H), 5.44 (s, 1H), 3.57 (s, 3H, broad), 2.00–1.80 (s, 2H, broad), 1.20 (s, 3H, mm), 1.01 (s, 3H, mr), 0.82 (s, 3H, rr). The dispersity and molecular weights of the final products was determined by Gel Permeation Chromatography (GPC), Mn = 1400 g mol−1, Đ = 1.76.
General procedure for the copolymerisation of methyl methacrylate tetramer with deuterated methyl methacrylate (MMAd8)14
For the copolymerisation in solution, MMA tetramer (1 g), butanone (4 ml) and a stirring bar were charged into a 50 mL round bottom flask. Nitrogen was purged in the flask for 30 min. Subsequently, the MMAd8 (2 g) and a solution of 2,2′-azobis(2-methylpropionitrile) (AIBN, 1 mol% relative to monomer) in butanone, both previously deoxygenated for 15 min, were added. A deoxygenated stock solution of CoBF in butanone was also prepared. When CoBF was used in the experiment, the appropriate amount (40 ppm relative to monomer) of the stock solution was added in the round bottom flask. The reaction was heated at 60 °C, under a nitrogen atmosphere and stirred for 24 hours.General procedure for the copolymerisation of methacrylic macromonomers with acrylic monomers in solution
For a standard copolymerisation in solution at 20% solids content, toluene, macromonomer and a stirring bar were charged into a 50 mL round bottom flask and the flask was purged with nitrogen for 30 min. Subsequently, the second monomer and a solution of tert-butyl peroxy-2-ethylhexanoate (Trigonox-21s, 0.5 mol% of the second monomer) in toluene (same volume with the second monomer) were added, previously deoxygenated for 30 min. The reaction was heated under nitrogen for 18 hours. The number average molecular weight of each block was calculated by analysing the 1H NMR and SEC spectra.Results and discussion
Synthesis of methacrylic macromonomers via catalytic chain transfer polymerisation
Macromonomers of poly(methyl methacrylate) (pMMA), poly(butyl methacrylate) (pBMA), poly(benzyl methacrylate) (pBzMA) and poly(lauryl methacrylate) (pLMA) were synthesized using CCTP, with targeted molecular weights, ranging from 200 to 16700 g mol−1. Two different catalysts were selected according to their solubility in the monomer. In the polymerisation of MMA, CoBF was used, whereas the more hydrophobic Co(MePh)BF was used for the polymerisation of BMA, BzMA and LMA (Table 1). In all reactions, 1 mol% of initiator to monomer and 1/1 v/v solvent to monomer were used. Moreover, the amount of catalyst was varied depending on the targeted molecular weight of the polymer desired.
| Monomer | Catalyst | Number average degree of polymerisation (DPn) | ||
|---|---|---|---|---|
| MMA | CoBF | 2 | 4 | 14 |
| BMA | Co(MePh)BF | 24 | ||
| BzMA | Co(MePh)BF | 47 | ||
| LMA | Co(MePh)BF | 12 | 18 | |
The products were characterized by 1H NMR and MALDI-ToF MS. Their NMR spectra were used to determine the DPn, monitor conversion and confirm the presence of vinyl peaks from the propenyl end group, for instance for pLMA, at 6.19 and 5.46 ppm (ESI Fig. 2a†). MALDI-ToF was used to further demonstrate end group fidelity, which confirmed that the macromonomers have two distinct and characteristic terminal groups, a hydrogen atom at the α-chain end and a vinyl group at their ω-terminal.
It is noted that the lauryl methacrylate was used as purchased and is actually a mixture of C12 and C14 methacrylates (77% and 23%), presumably arising from the alkyl chain being derived from the hydrolysis of natural oils prior to transesterification with MMA. This mixture was considered to be acceptable for the purposes of this study and although difficult to see by NMR is clear from GC of the monomer and MALDI-ToF MS of the products, as shown in ESI Fig. 2b† and Fig. 1 respectively.
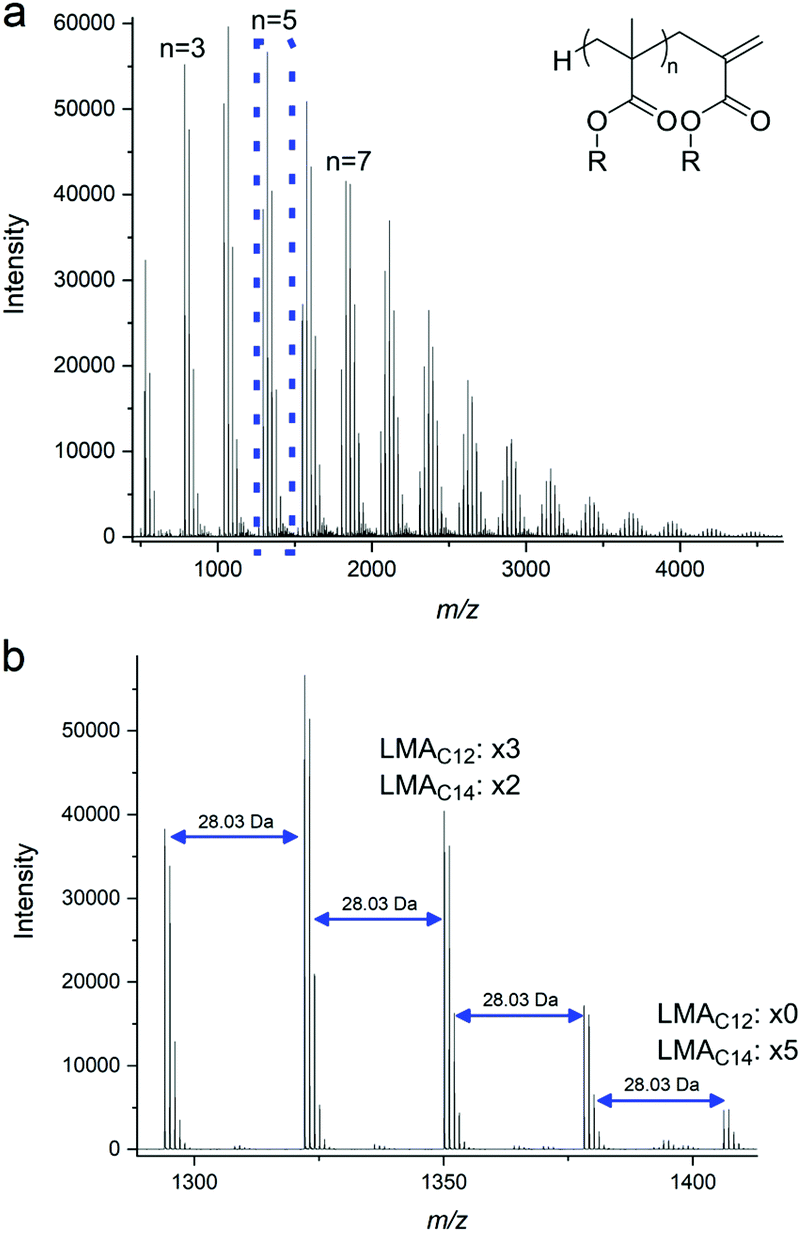 | ||
| Fig. 1 (a) MALDI-ToF spectrum of the pLMA6 macromonomer as synthesized via CCTP. The distribution of the peaks is partly due to the existence of the two different monomers. (b) Expansion of the MALDI-ToF spectrum (a) in the 1250–1450 m/z range, showing the mass difference of 28.03 m/z due to the occurrence of the C14 together with the C12 monomer. | ||
Size-exclusion chromatography (SEC) was used for the characterization of all products. Typical examples are shown for the synthesis of pLMA (ESI Fig. 3†) as prepared with Co(MePh)BF as a catalyst. Molecular weight data from GPC is given in ESI Table 1.†
Increasing the concentration of the cobalt catalyst in the reaction results in a lowering of the molecular weight of the product as in conventional chain transfer.21
The effective chain transfer activity (CEs) of the catalysts, measured at high conversions, used in the CCTP of pLMA was measured using the Mayo equation (eqn (1)), where DPn is the number average degree of polymerisation in the presence of the CTA, DP0n the number average degree of polymerisation in the absence of the CTA, [S] the concentration of CTA and [M] the concentration of the monomer.
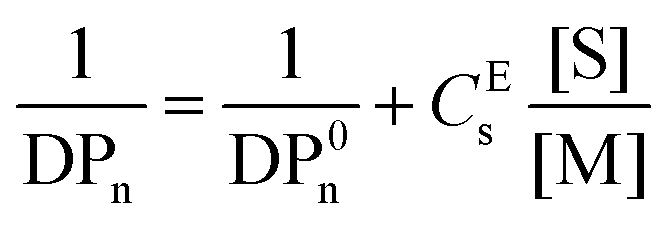 | (1) |
The activity of the relatively hydrophobic catalyst Co(MePh)BF for the polymerisation of LMA gave CEs = 1700. Note that this is lower than the CEs of CoBF for the polymerisation of MMA, which is reported as ≈30000.21 Thus, when the targeted Mn of pLMA was approximately 2000, 40 ppm of Co(MePh)BF was used, whereas the same ratio of CoBF is required only when a mixture of MMA dimer and trimer is targeted.
Copolymerisation of methacrylic macromonomers with MMAd8
In an earlier report, we reported on the mechanism of the addition fragmentation by addition of MMA tetramer (MMA4 as produced by CCTP of MMA followed by isolation by distillation) to further polymerisation reactions of BMA.14 Using MALDI-ToF, we showed that following the addition of a methacrylic radical to MMA4, the only detectable reaction pathway was β-scission (fragmentation) of the radical adduct, with no chain transfer to cobalt detected or copolymerisation. Herein, we repeated these reactions, substituting BMA with fully deuterated MMA (MMAd8) given advances in analytical techniques since the original studies to reaffirm this in relation to the present study. As it is always preferable to remove oxygen from a free radical polymerisation prior to the reaction we monitoring this with an oxygen concentration probe (FireStingGO2 optical probe utilising quenching of oxygen in the NIR), we found that the time taken to deoxygenate reactants varied from 1 to 30 minutes, depending on their physical state (ESI Fig. 4†).The molecular weight of MMAd8 = 108.15 g mol−1 which allows for differentiation from the MMA macromonomer in MALDI-ToF MS. Four products are observed in the MALDI-ToF MS spectra (Fig. 2a and b), in the presence of CoBF, (Fig. 2b). In order to achieve more detailed information of the products MALDI-ToF/ToF was also used, Fig. 2c and d. This technique provides the fragmentation patterns of the polymers, allowing for the exploration of the arrangement of the monomeric units in the products. Thus, analysing the peak at m/z = 1319.857 (d in Fig. 2b) using MALDI-ToF/ToF, Fig. 2d, the highest magnitude signal originates from products of the general formula D-(MMAd8)x, which would be the product expected from the CCTP of MMAd8 in the absence of MMA4. However, smaller peaks are also apparent from products that contain 1 MMA, 4 MMA and 3 MMA units with an absence of products containing 2 MMA monomers. These are derived from MMA3(MMAd8)xMMA1, MMA3(MMAd8)x and H(MMAd8)xMMA1 which arise from fragmentation and reinitiation. This supports the observed results from our previously published work, that β-scission is the predominant reaction pathway of the macromonomer radical adduct under these reaction conditions, Fig. 2 and ESI Table 2.†18
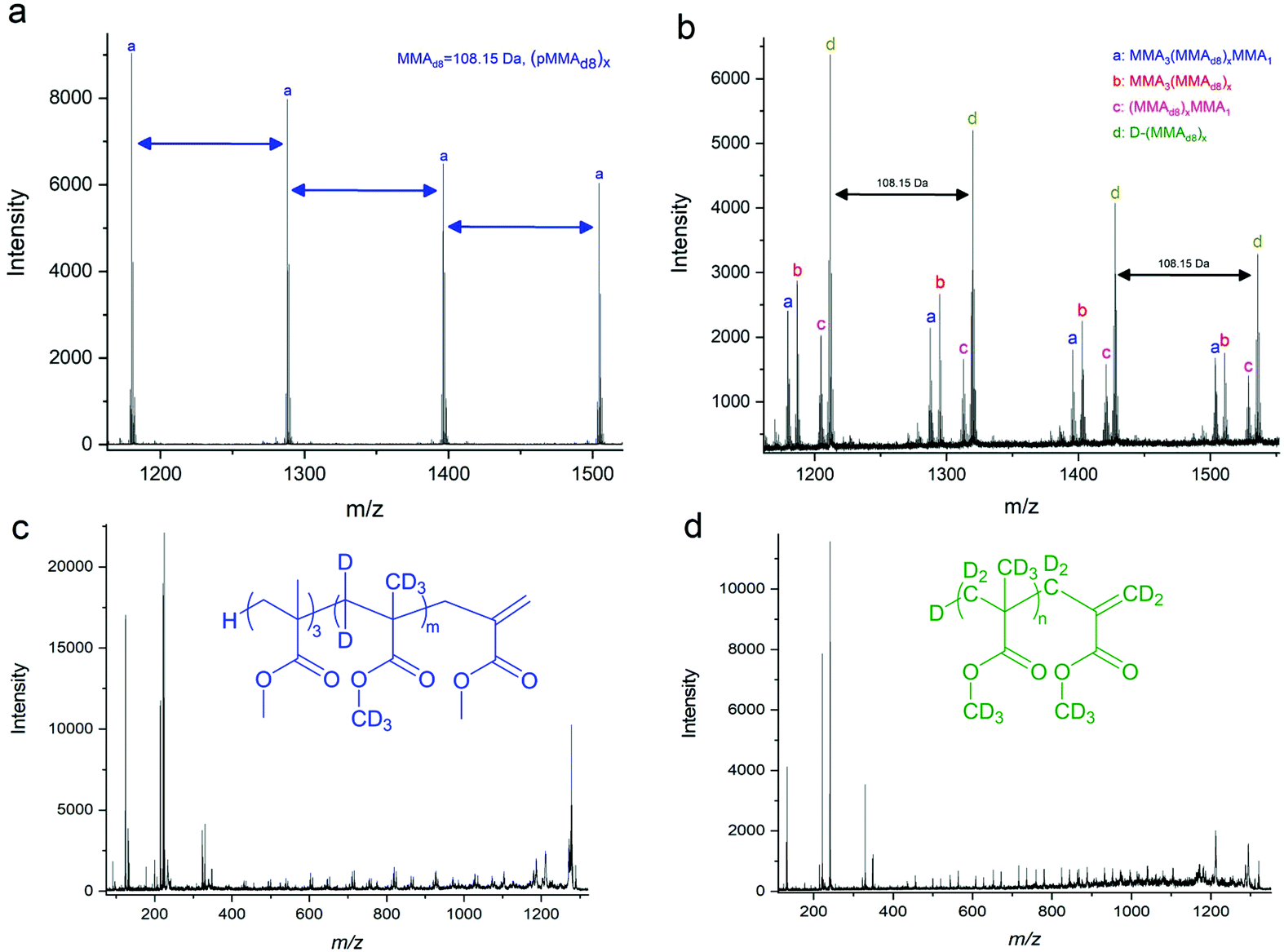 | ||
| Fig. 2 (a) Expansion of the full MALDI-ToF spectrum, ESI Fig. 6a,† of the copolymerisation of pMMA4 with MMAd8 in the 1150–1550 m/z range. (b) Expansion of the full MALDI-ToF spectrum, ESI Fig. 6b,† of the copolymerisation of pMMA4 with MMAd8 in the presence of CoBF in the 1150–1550 m/z range. (c) MALDI-ToF/ToF of peak “a” in Fig. 2b showing the backbone cleavages. (d) MALDI-ToF/ToF of peak “d” in Fig. 2b showing the backbone cleavages. | ||
Copolymerisation of methacrylic macromonomers with acrylates
Following from the copolymerisation with methacrylates, we investigated the effect of polymerisation of acrylic monomers in the presence these macromonomers noting acrylates have a higher propagation rate constant than corresponding methacrylates, kp. Initially, MA was used for copolymerisation with MMA dimer. The molar ratio of [MMA2] to [MA] was varied between 0 and 4.3 mol% which had a marked effect on the molecular weight of the final product, Table 2 and Fig. 3a. MALDI-ToF MS was used to analyse the products and the results confirmed that the copolymerisation of a MMA dimer with methyl acrylate leads to graft copolymers, Fig. 3d.33,34,36 According to Fig. 4a, grafted copolymers of pMA with 2, 3 and 4 units of MMA dimer are produced during the copolymerisation (ESI Table 3†). Furthermore, pMA polymers with up to six MMA dimers per molecule were calculated in higher molecular weights of the same spectrum.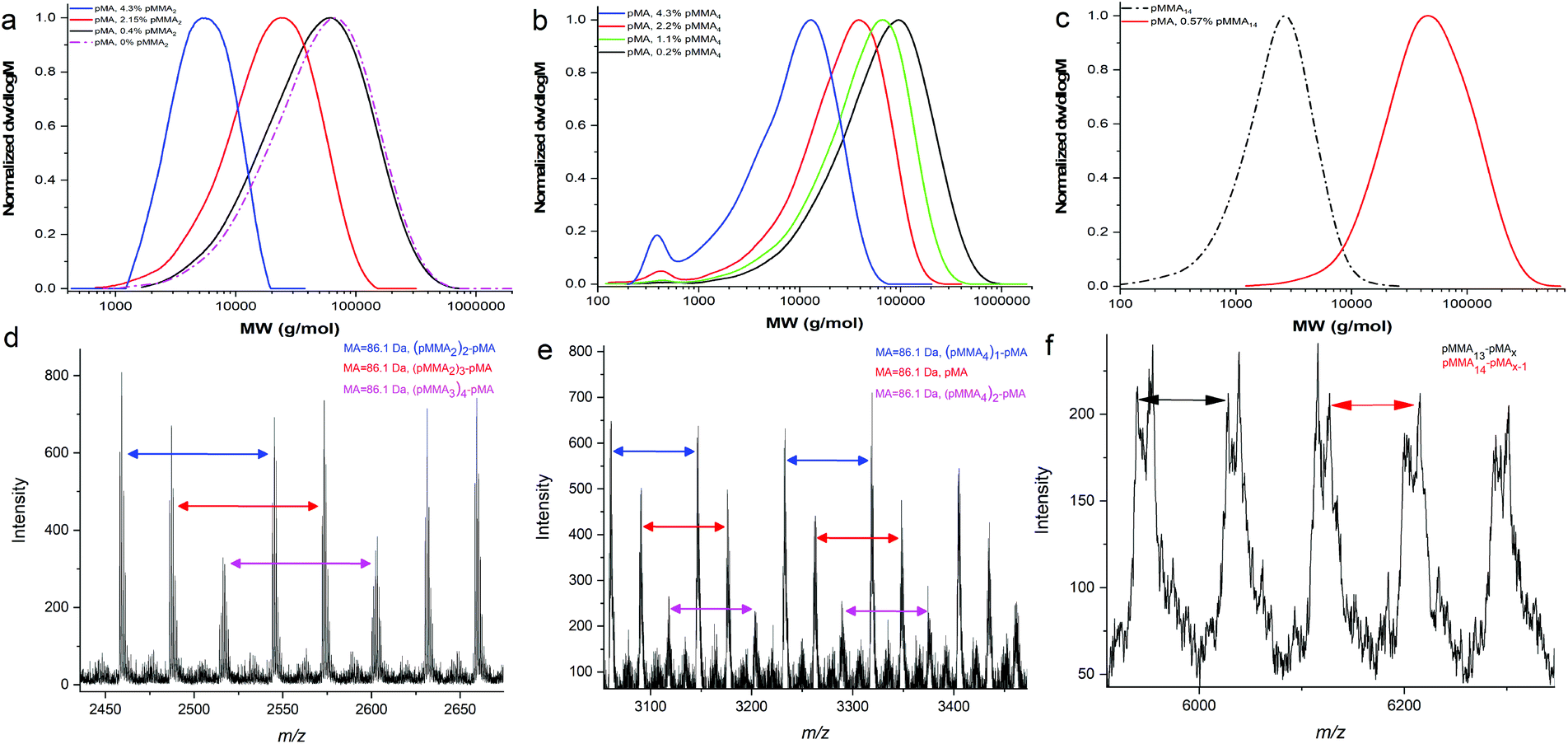 | ||
| Fig. 3 (a) GPC results of the pMA homopolymer and the copolymerisation of MA with different amount of pMMA2. (b) GPC results of the copolymerisation of MA with different amount of pMMA4. (c) GPC results of the copolymerisation of MA with different amount of pMMA14. (d) Expansion of the full MALDI-ToF spectrum of the copolymerisation of pMA with MMA2 (sample 4) in the 2430–2700 m/z range. (e) Expansion of the full MALDI-ToF spectrum of the copolymerisation pMA with MMA4 (sample 9) in the 3050–3470 m/z range. (f) Expansion of the MALDI-ToF spectrum of the copolymerisation of pMA with pMMA14 in the 5900–6350 m/z range. | ||
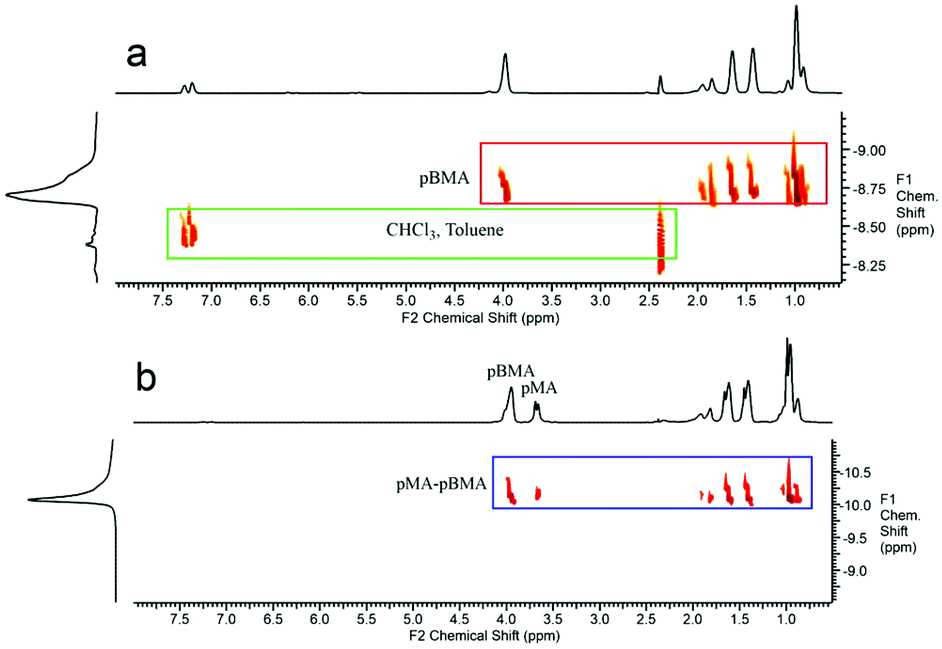 | ||
| Fig. 4 (a) DOSY NMR of pBMA macromonomer. The peaks of the macromonomer and solvents have different diffusion constants. (b) DOSY NMR of the free radical polymerisation of MA in the presence of 3.3 mol% pBMA macromonomer. The diffusion constant of the pure final product is different from that of the macromonomer. | ||
| Sample | Monomer | pMMAx | pMMAx to MA (mol%) | M n (g mol−1) | Đ |
|---|---|---|---|---|---|
| 1 | MA | — | — | 25600 |
2.91 |
| 2 | MA | pMMA2 | 0.4 | 23400 |
3.21 |
| 3 | MA | pMMA2 | 2.1 | 12600 |
2.11 |
| 4 | MA | pMMA2 | 4.3 | 3600 | 1.60 |
| 5 | MA | — | — | 37500 |
2.83 |
| 6 | MA | pMMA4 | 0.2 | 33800 |
3.01 |
| 7 | MA | pMMA4 | 1.1 | 23300 |
2.71 |
| 8 | MA | pMMA4 | 2.2 | 16400 |
2.34 |
| 9 | MA | pMMA4 | 4.3 | 5100 | 2.39 |
| 10 | MA | pMMA14 | 0.6 | 30100 |
2.12 |
Distilled MMA4 macromonomer was studied in the copolymerisation with MA and the results compared with those obtained when using the equivalent MMA dimer, Fig. 3b and e. Interestingly, the chain length of the macromonomer affects the graft amount incorporated into the final product. The grafting density of MMA4 is limited to two per polymer chain, and the amounts of the copolymer pMAx-b-(MMA4)1 and pMAx homopolymer were detected in larger amounts relative to the graft copolymer.
Subsequently, the copolymerisation of pMMA14 with MA was carried out, Fig. 3c. The main peaks of the MALDI-ToF spectra indicate the formation of an end-capped diblock copolymer pMMA-b-pMA arising from a chain stopping reaction whereby a propagating PMA chain adds a PMMA macromonomer with subsequent termination occuring in preference to propagation. However, due to the small difference in the masses of the monomers it is not 100% certain if there is also graft copolymer in the product, Fig. 3f.
In order to further investigate this copolymerisation reaction, a pBMA CCTP macromonomer was copolymerised with MA. The larger side group alkyl chain and the higher molecular weight of this macromonomer were chosen so as to help gain a better insight into this system. Methyl acrylate was polymerised in the presence of 0.26 and 3.3 mol% of pBMA, under similar reaction conditions as described above, and a shift to higher molecular weight for the product observed (ESI Fig. 7 and Table 4†). The DOSY NMR spectra of the macromonomer, Fig. 4a, when compared to the final product of the copolymerisation with MA, Fig. 4b, show distinct differences in the diffusion constants of the pBMA before and after copolymerisation. Moreover, all of the protons of the final product are aligned on the same region of the spectrum (same diffusion rate), indicating that they are part of the same molecule.
In order to further investigate the effect of the nature of the macromonomer, methyl acrylate (MA) was copolymerised with both poly(benzyl methacrylate) and poly(lauryl methacrylate) macromonomers, Fig. 5. Similarly, to previous results, a decrease of the Mn of the products with increasing macromonomer concentration is observed (Table 3 and ESI Table 5†). However, interestingly, from MALDI-ToF analysis, only end-capped diblock copolymers were obtained, Fig. 5 and ESI Fig. 8.† The 77% to 23% ratio of the isomers of LMA is observed in the MALDI-ToF spectra of the diblock copolymer pLMA-b-pMA. The main peaks refer to end-capped pLMAC12 macromonomer, as it is in the higher percentage. The 28 and 56 m/z difference is due to block copolymers with a different number of the monomeric units, C12 or C14 LMA monomers, in the final product.
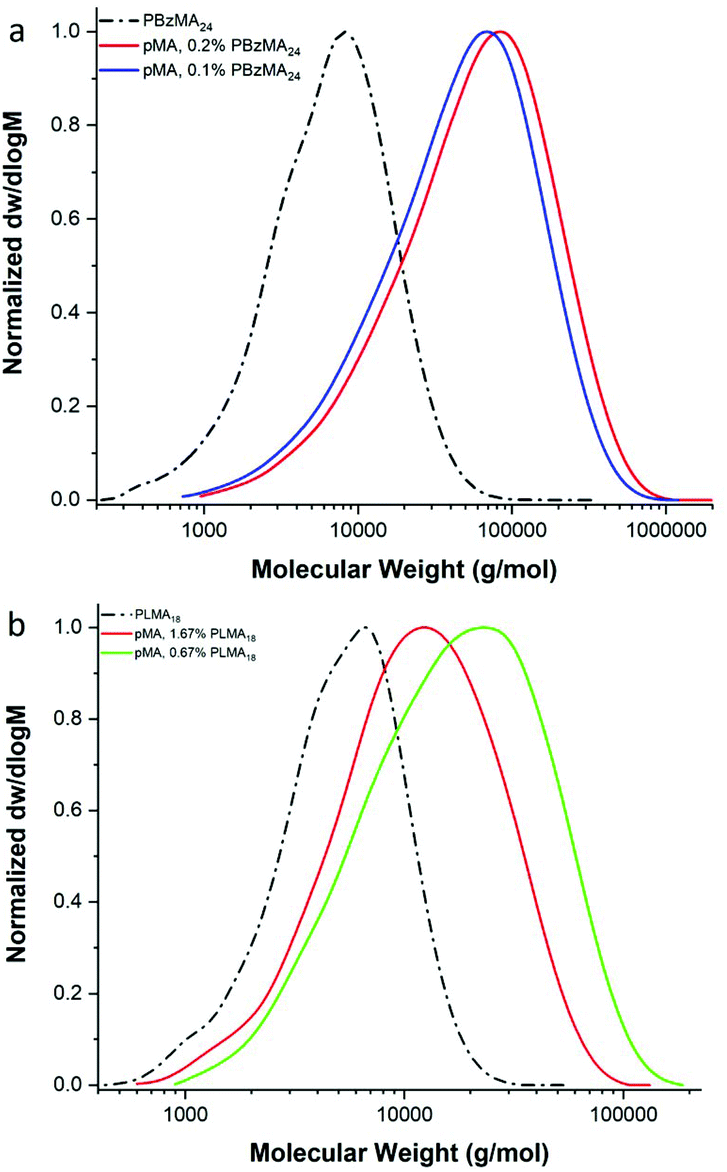 | ||
| Fig. 5 GPC results of the free radical polymerisation of (a) MA with 0.1 mol% and 0.2 mol% pBzMA24 and (b) MA with 0.67 mol% and 1.67 mol% pLMA18. | ||
| Sample | Monomer | pLMA18 | pLMA18 to MA (mol%) | M n (g mol−1) | Đ |
|---|---|---|---|---|---|
| 1 | MA | — | — | 25600 |
2.91 |
| 11 | MA | pLMA18 | 0.17 | 24500 |
3.93 |
| 12 | MA | pLMA18 | 0.52 | 20000 |
3.76 |
| 13 | MA | pLMA18 | 0.67 | 10700 |
2.27 |
| 14 | MA | pLMA18 | 1.67 | 7600 | 2.05 |
| 15 | MA | pLMA18 | 10 | 5500 | 1.72 |
Finally, the methacrylic macromonomer pLMA was added to the free radical polymerisation of the more hydrophobic acrylate, butyl acrylate (BA). Similarly, the macromonomer acts as terminating species via an end-capping event as opposed to a monomer, leading to the block copolymer pLMA-b-pBA as opposed to graft copolymers, ESI Table 6 and Fig. 10.†
In the case of the pLMA with higher steric hindrance, only block copolymers result via this end-capping by the macromonomer to the macroradical pMA (Fig. 6). This suggests that during the copolymerisation of a methacrylic macromonomer from CCTP and acrylic monomers, an increase of the molecular weight of the macromonomer, and also of the ester side chain length, reduces the likelihood of obtaining graft copolymers (Scheme 2). On the contrary, if the steric hindrance of the macromonomer is high, it results in an end-capping reaction of the propagating polyacrylate radical and diblock copolymers are the only product.
 | ||
| Scheme 2 Schematic presentation of the proposed routes of the free radical polymerisation of acrylates in the present of methacrylic macromonomers leading to graft or diblock copolymers. | ||
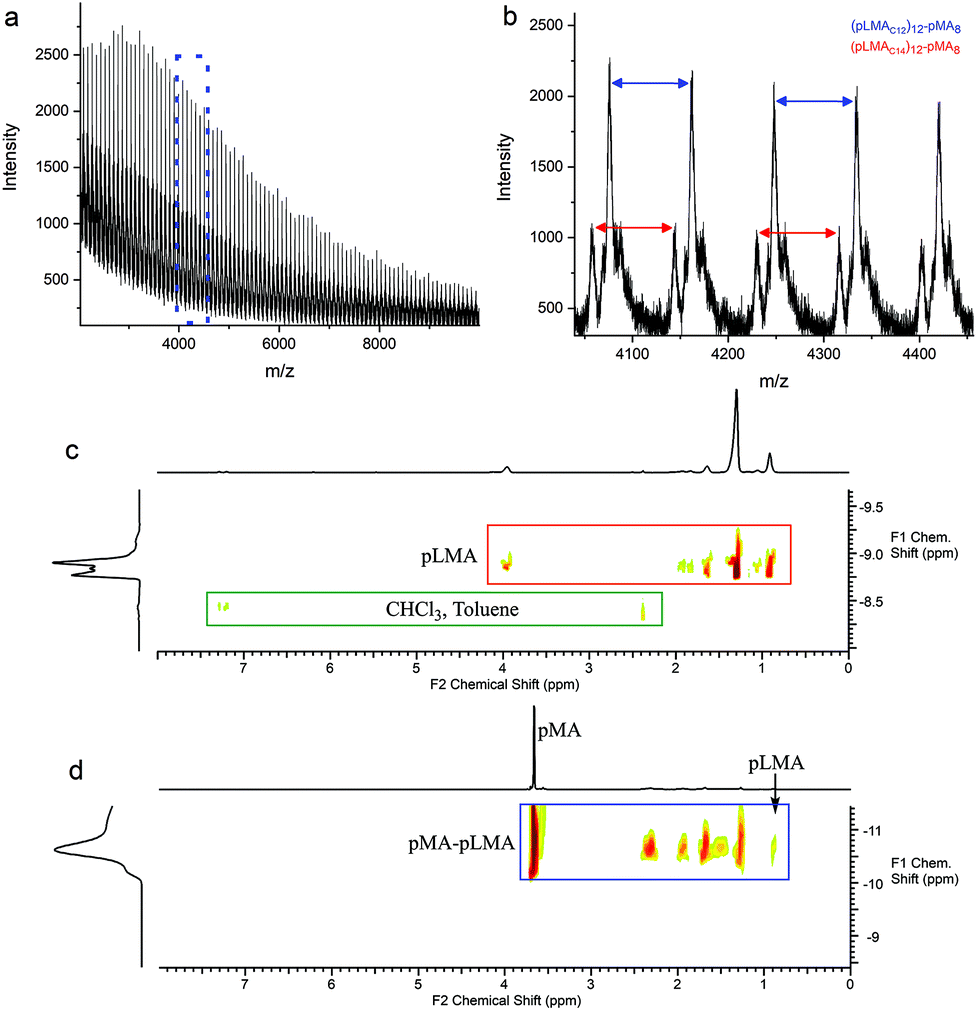 | ||
| Fig. 6 (a) Full MALDI-ToF spectra of the copolymerisation of MA with pLMA macromonomer, sample 11, showing the diblock copolymer formation via end-capping. (b) Expansion of the MALDI-ToF (a) of the in the 4050–4450 m/z range. (c) DOSY NMR of pLMA macromonomer. The peaks of the macromonomer and solvents have different diffusion constants. (d) DOSY NMR of the free radical polymerisation of MA in the presence of 0.17 mol% pLMA macromonomer, sample 11. The diffusion constant of the pure final product is different from that of the macromonomer. | ||
Thus diblock copolymers of methacrylic and acrylic monomers can be synthesised using the appropriate methacrylic macromonomer as derived from CCTP. Further studies have already been conducted in order to optimize the conditions in solution, as this is a highly useful and scalable technique to combine the properties of different monomers, in relatively facile synthetic conditions, applicable to scale-up and application.
Conclusions
In summary, we present a reproducible, easy to use and scalable method of producing block copolymers of different types of methacrylate/acrylate monomers with relatively broad dispersities of each block. Moreover, the significant effect of the size of CCTP pMMA macromonomers in the grafting density during their copolymerisation with methyl acrylate was demonstrated. Methacrylic macromonomers with larger side ester chains were shown to end-cap the acrylic macroradical leading to AB diblock copolymers. During the diblock formation, no by products were produced, although the high polydispersity of each block, making this procedure versatile for the synthesis of materials without the presence of sulphur substances or transition metals.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Polymer Characterisation RTP and Dr Daniel Lester (University of Warwick) for providing use of SEC equipment. We thank Lubrizol (GP, CJA), Unilever (SE), DSM (AS) and Syngenta (RIW) as well as the University of Warwick and the EPSRC Centre for Doctoral Training in Molecular Analytical Science, grant number EP/L015307/1 with additional funding granted by Syngenta and AstraZeneca (JT) for studentship funding.The raw data for all information presented in this paper can be accessed from The University of Warwick repository at https://wrap.warwick.ac.uk/127874.
Notes and references
- D. J. Krasznai, T. F. L. McKenna, M. F. Cunningham, P. Champagne and N. M. B. Smeets, Polym. Chem., 2012, 3, 992–1001 RSC.
- C. J. Atkins, G. Patias, J. S. Town, A. M. Wemyss, A. M. Eissa, A. Shegiwal and D. M. Haddleton, Polym. Chem., 2019, 10, 646–655 RSC.
- G. Nurumbetov, N. Engelis, J. Godfrey, R. Hand, A. Anastasaki, A. Simula, V. Nikolaou and D. M. Haddleton, Polym. Chem., 2017, 8, 1084–1094 RSC.
- D. M. Haddleton, E. Depaquis, E. J. Kelly, D. Kukulj, S. R. Morsley, S. A. F. Bon, M. D. Eason and A. G. Steward, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2378–2384 CrossRef CAS.
- A. Anastasaki, C. Waldron, V. Nikolaou, P. Wilson, R. McHale, T. Smith and D. M. Haddleton, Polym. Chem., 2013, 4, 4113–4119 RSC.
- A. H. Soeriyadi, G. Z. Li, S. Slavin, M. W. Jones, C. M. Amos, C. R. Becer, M. R. Whittaker, D. M. Haddleton, C. Boyer and T. P. Davis, Polym. Chem., 2011, 2, 815–822 RSC.
- N. M. B. Smeets, Eur. Polym. J., 2013, 49, 2528–2544 CrossRef CAS.
- L. Nurmi, J. Lindqvist, R. Randev, J. Syrett and D. M. Haddleton, Chem. Commun., 2009, 2727–2729 RSC.
- J. Krstina, G. Moad, E. Rizzardo, C. L. Winzor, C. T. Berge and M. Fryd, Macromolecules, 1995, 28, 5381–5385 CrossRef CAS.
- C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1996, 29, 7717–7726 CrossRef CAS.
- D. G. Hawthorne, R. L. Laslett, E. Rizzardo and D. H. Solomon, J. Macromol. Sci., Part A, 1986, 23, 839–852 CrossRef.
- N. G. Engelis, A. Anastasaki, G. Nurumbetov, N. P. Truong, V. Nikolaou, A. Shegiwal, M. R. Whittaker, T. P. Davis and D. M. Haddleton, Nat. Chem., 2017, 9, 171–178 CrossRef CAS PubMed.
- B. Yamada, P. Zetterlund and E. Sato, Utility of propenyl groups in free radical polymerization: Effects of steric hindrance on formation and reaction behavior as versatile intermediates, 2006 Search PubMed.
- D. M. Haddleton, D. R. Maloney and K. G. Suddaby, Macromolecules, 1996, 29, 481–483 CrossRef CAS.
- D. M. Haddleton, D. R. Maloney, K. G. Suddaby Adam Clarke and S. N. Richards, Polymer, 1997, 38, 6207–6217 CrossRef CAS.
- T. Davis, D. Kukulj, D. M. Haddleton and D. R. Maloney, Cobalt-mediated free-radical polymerization of acrylic monomers, 1995 Search PubMed.
- D. M. Haddleton, D. R. Maloney, K. G. Suddaby, A. V. G. Muir and S. N. Richards, Macromol. Symp., 1996, 111, 37–46 CrossRef CAS.
- D. M. Haddleton, D. R. Maloney and K. G. Suddaby, Macromolecules, 1996, 29, 481–483 CrossRef CAS.
- D. M. Haddleton, M. C. Crossman, K. H. Hunt, C. Topping, C. Waterson and K. G. Suddaby, Macromolecules, 1997, 30, 3992–3998 CrossRef CAS.
- A. Gridnev, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1753–1766 CrossRef CAS.
- J. P. A. Heuts and N. M. B. Smeets, Polym. Chem., 2011, 2, 2407–2423 RSC.
- S. Slavin, E. Khoshdel and D. M. Haddleton, Polym. Chem., 2012, 3, 1461–1466 RSC.
- Q. Zhang, S. Slavin, M. W. Jones, A. J. Haddleton and D. M. Haddleton, Polym. Chem., 2012, 3, 1016–1023 RSC.
- D. M. Haddleton, E. Depaquis, E. J. Kelly, D. Kukulj, S. R. Morsley, S. A. F. Bon, M. D. Eason and A. G. Steward, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2378–2384 CrossRef CAS.
- K. G. Suddaby, D. M. Haddleton, J. J. Hastings, S. N. Richards and J. P. O'Donnell, Macromolecules, 1996, 29, 8083–8091 CrossRef CAS.
- I. Schreur-Piet and J. P. A. Heuts, Polym. Chem., 2017, 8, 6654–6664 RSC.
- G. E. Roberts, T. P. Davis, J. P. A. Heuts and G. E. Ball, Macromolecules, 2002, 35, 9954–9963 CrossRef CAS.
- N. G. Engelis, A. Anastasaki, R. Whitfield, G. R. Jones, E. Liarou, V. Nikolaou, G. Nurumbetov and D. M. Haddleton, Macromolecules, 2018, 51, 336–342 CrossRef CAS.
- A. Shegiwal, A. M. Wemyss, M. A. J. Schellekens, J. de Bont, J. Town, E. Liarou, G. Patias, C. J. Atkins and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, E1–E9 CrossRef.
- L. Hutson, J. Krstina, C. L. Moad, G. Moad, G. R. Morrow, A. Postma, E. Rizzardo and S. H. Thang, Macromolecules, 2004, 37, 4441–4452 CrossRef CAS.
- S. Beuermann, M. Buback, T. P. Davis, R. G. Gilbert, R. A. Hutchinson, O. F. Olaj, G. T. Russell, J. Schweer and A. M. van Herk, Macromol. Chem. Phys., 1997, 198, 1545–1560 CrossRef CAS.
- M. Buback and C. H. Kurz, Macromol. Chem. Phys., 1998, 199, 2301–2310 CrossRef CAS.
- P. Cacioli, D. G. Hawthorne, R. L. Laslett, E. Rizzardo and D. H. Solomon, J. Macromol. Sci., Part A, 1986, 23, 839–852 CrossRef.
- K. J. Abbey, D. L. Trumbo, G. M. Carlson, M. J. Masola and R. A. Zander, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 3417–3424 CrossRef CAS.
- L. M. Muratore, K. Steinhoff and T. P. Davis, J. Mater. Chem., 1999, 9, 1687–1691 RSC.
- B. Yamada, F. Oku and T. Harada, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 645–654 CrossRef CAS.
- B. Yamada and S. Kobatake, Prog. Polym. Sci., 1994, 19, 1089–1131 CrossRef CAS.
- D. Colombani and P. Chaumont, Prog. Polym. Sci., 1996, 21, 439–503 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py01133a |
| This journal is © The Royal Society of Chemistry 2019 |