
Core-crosslinked worm-like micelles from polyether-based diblock terpolymers†
 ab
and
Felix H.
Schacher
*ab
ab
and
Felix H.
Schacher
*ab
Abstract
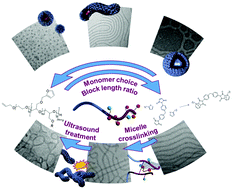
We herein report on the synthesis, purification and subsequent self-assembly of different polyether-based diblock terpolymers in aqueous solutions. While in all cases PEO was used as hydrophilic macroinitiator in anionic ring opening polymerization (AROP), the hydrophobic block was formed by different hydrophobic glycidyl ethers, thereby and by variation of the DP also influencing micellar morphologies in water as selective solvent. The application of sterically demanding, branched aliphatic glycidyl ethers (2-ethylhexyl glycidyl ether (EHGE), or hexyl, hexyl glyceryl glycidyl ether (HHGGE)) or aromatic glycidyl ethers (benzyl glycidyl ether (BGE) or naphthyl glycidyl ether (NGE)) as main comonomer for the hydrophobic block showed distinct influence on the morphologies formed by self-assembly in solution. In general, we observed a comparably broad window in the corresponding phase diagram for the formation of worm-like structures. Further, 10% of furfuryl glycidyl ether (FGE) was incorporated in the hydrophobic block of the diblock terpolymers to enable subsequent crosslinking of the micellar core. Hereby, thermally induced Diels–Alder reactions using a hydrophobic bismaleimide which was encapsulated in the micellar core prior to crosslinking could be successfully employed. We also carried out first experiments regarding ultrasound treatment of the crosslinked aggregates, revealing the formation of branched worm-like structures from previously well-defined filomicelles. All solution structures as well as the experiments on crosslinked worm-like micelles were investigated via light scattering (DLS) and transmission electron microscopy (cryo-TEM).
 Please wait while we load your content...
Please wait while we load your content...

