
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Defining side chain successions in anthracene-based poly(arylene ethynylene)-alt-poly(phenylene vinylene)s: probing structure–property relationships†
Christoph
Ulbricht
 *ab,
Nassima
Bouguerra
bc,
Samuel
Inack Ngi
b,
Oliver
Brüggemann
d and
Daniel A. M.
Egbe
*ab
*ab,
Nassima
Bouguerra
bc,
Samuel
Inack Ngi
b,
Oliver
Brüggemann
d and
Daniel A. M.
Egbe
*ab
aInstitute of Polymeric Materials and Testing, Johannes Kepler University Linz, Altenberger Straße 69, 4040 Linz, Austria. E-mail: christoph.ulbricht@jku.at; daniel_ayuk_mbi.egbe@jku.at
bLinz Institute for Organic Solar Cells, Physical Chemistry, Johannes Kepler University Linz, Altenberger Straße 69, 4040 Linz, Austria
cDepartment of Chemical Engineering, Electrochemistry, Corrosion and Energetic Valorization Laboratory, A. MIRA University, Targa Ouzemmour, 06000 Bejaia, Algeria
dInstitute of Polymer Chemistry, Johannes Kepler University Linz, Altenberger Straße 69, 4040 Linz, Austria
First published on 18th September 2019
Abstract
By optimizing and adapting the synthesis of double dissymmetric functionalized phenylene building blocks, a library of nine anthracene-containing poly(arylene ethynylene)-alt-poly(phenylene vinylene)s (AnE-PVs) with diverse successions of octyloxy and 2-ethylhexyloxy side chains was realized. Despite the structural resemblance of the regioisomeric comonomers and the polymers, their 1H NMR spectra were found to feature perceptible side chain configuration-specific patterns. The differentiation of the photophysical characteristics of the polymers induced by the various defined, semi-defined and random side chain sequences has been thoroughly investigated by UV-vis absorption and photoluminescence spectroscopy in solution and in thin film. The gained insights allowed the specification of the notions on the complex structure–property relationships in AnE-PV systems. In particular, the side chain assignment in direct proximity to the anthracene moieties distinctly affects the optical properties. This systematic study presents an illustrative example for the versatile but also intricate capabilities of side chain engineering.
Introduction
For decades, research on organic semiconductors, small molecules and polymers,1 and their optoelectronic features has prospered due to the vast possibilities of structural design and the promising aspects for a broad spectrum of applications. Catalysis,2 sensing3,4 and organic electronic applications such as in conductors,5 batteries,6 organic field effect transistors (OFETs),7–9 organic light emitting devices (OLEDs)10,11 and solar cells,12–14 are highly attractive. The optoelectronic characteristics arising from the layout of the conjugated system can be modified by the introduction of substituents at the periphery or metal centers at coordination sites. In addition, the impact of side chains can substantially exceed the “mere” facilitation of solubility and processability. Depending on their structure, number and positioning, side chains can affect the alignment of a conjugated scaffold (torsion/planarization), the packing (π–π stacking, etc.), the orientation and the morphology in neat and in blended layers.15–17 Absorption and emission as well as electronic and charge transport characteristics can be manipulated significantly.8,18 As a consequence side chain engineering can serve as a highly potent tool to modify materials for optimized device performance.19–23Poly(arylene vinylene)s (PAVs) were the first group of conjugated polymers for which electroluminescence was reported, and are intensively investigated.24 Poly(arylene ethynylene)s (PAEs) are more rigid structures with commonly enhanced photostability and high emission quantum yield in solution.25 The assembly of arylene, vinylene and ethynylene elements gave rise to hybrid systems, poly(arylene ethynylene)-alt-poly(arylene vinylene)s (PAE-PAVs),15,26,27 with appealing characteristics, such as good solid state fluorescence quantum efficiency and low turn-on voltages in OLED applications.10 The most common synthetic approach, involving Sonogashira cross-couplings as well as the Honor–Wadsworth–Emmons (HWE) reaction (Scheme 1), provides high modularity. This enables the realization of versatile PAE–PAV layouts, and allows for the implementation and systematic investigation of diverse side chain configurations.28–33 The anthracene-containing polymer AnE-PVstat exhibits the best solar cell performance for a PPV-type material so far. Due to the balanced effects of linear octyloxy, permitting strong π–π stacking, and branched 2-ethylhexyloxy chains, stimulating an amorphous film morphology, good power conversion efficiencies of up to nearly 5% could be achieved.34–36 Although the majority of PAE–PAVs possess identical alkoxy side chains at individual phenylene units, a few with mixed configuration, methoxy/2-ethylhexlyoxy or octyloxy/octadecyloxy, were prepared as well.18,31,37–39
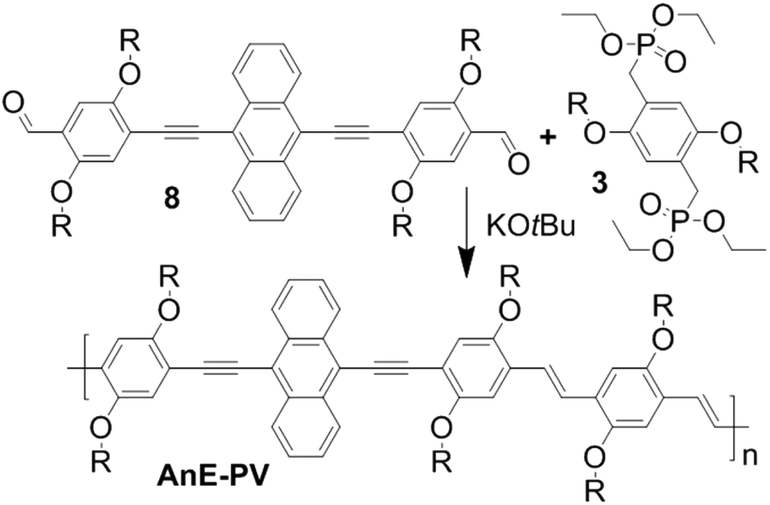 | ||
| Scheme 1 Synthesis of AnE-PVs via HWE polycondensation of dialdehydes 8 and bisphosphonates 3. | ||
To further elaborate the insights on side chain-induced structure–properties relationships the present article reports on the synthesis and investigation of anthracene-containing poly(arylene ethynylene)-alt-poly(phenylene vinylene)s (AnE-PVs) bearing various combinations and sequences of isomeric octyloxy and 2-ethylhexyloxy side chains. The crucial step for the structural layout lies in the preparation of double dissymmetric functionalized phenylene building blocks (Schemes 2 and 3). Equipping a phenylene unit with two different opposing side chains and with two different opposing reactive groups provides two regioisomer options. Such phenylene units were synthesized in mixture as well as selectively using three different pathways, and subsequently utilized in the preparation of anthracene-containing dialdehydes. HWE reactions with three bisphosphonate derivatives yielded nine AnE-PVs with fully random to highly specific octyloxy/2-ethylhexyloxy side chain successions (Table 1). NMR as well as UV-vis absorption and photoluminescence investigations revealed the distinct impact of the various side chain arrangements.
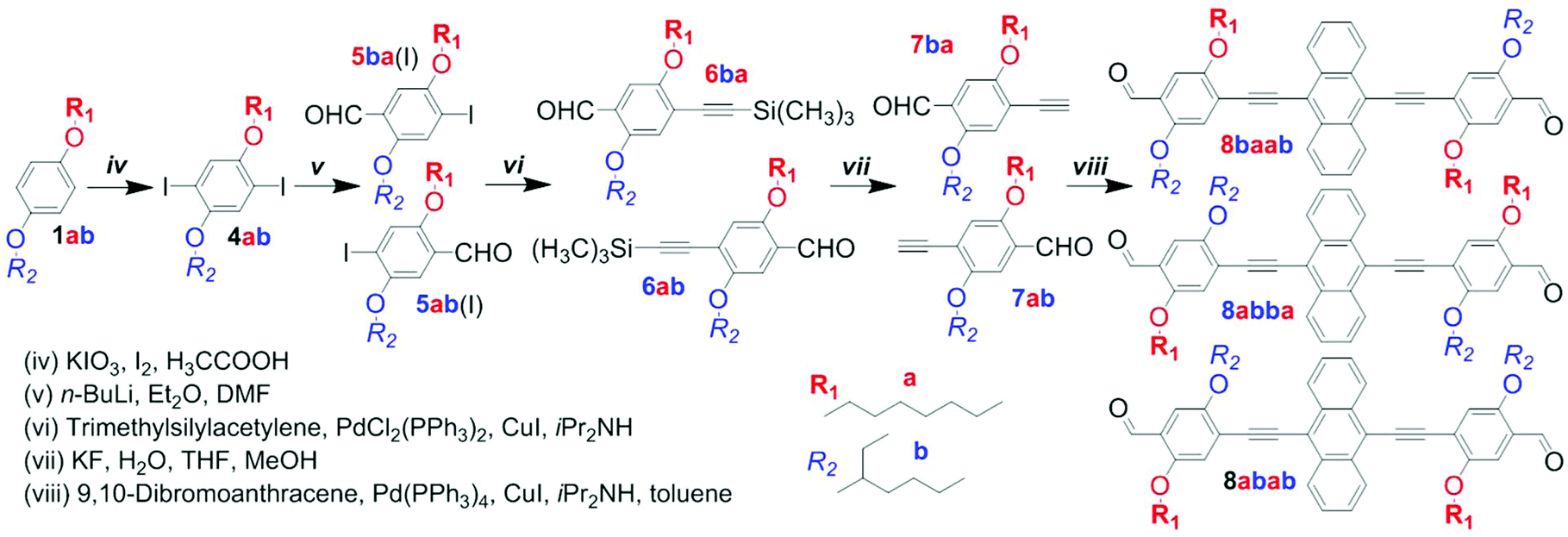 | ||
| Scheme 2 Synthesis of the dialdehyde comonomer mixture 8a/ba/b (8baab/8abba/8abab) by a non-selective approach. | ||
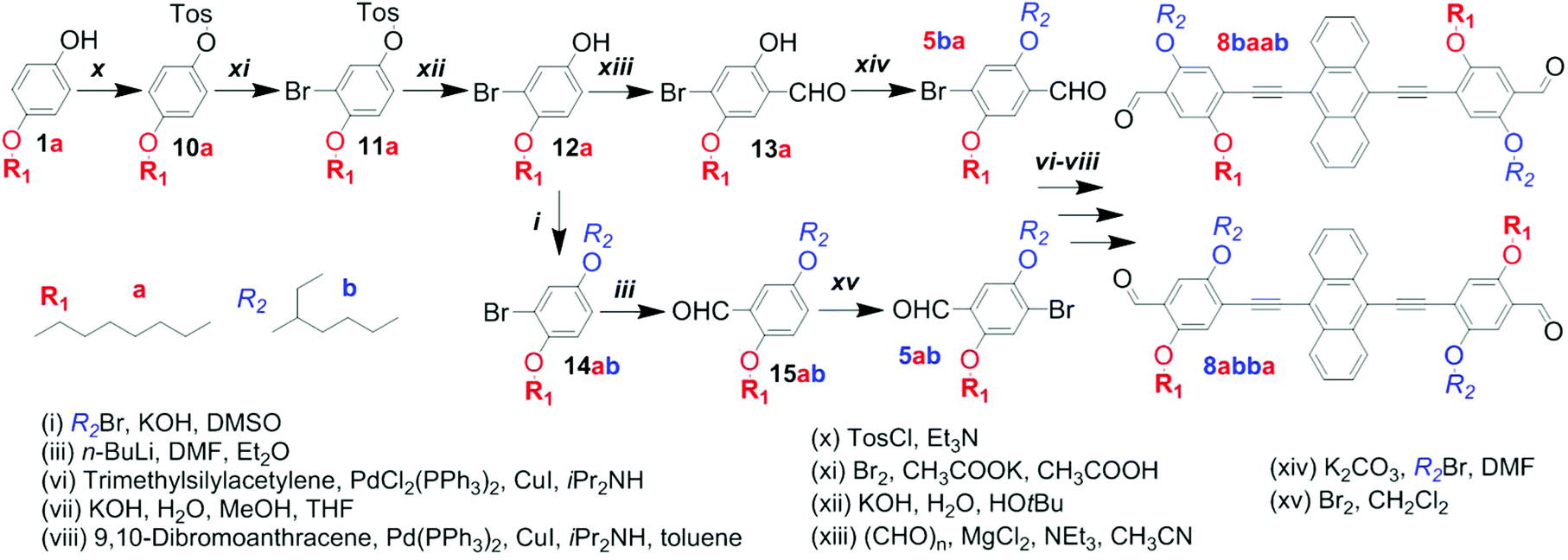 | ||
| Scheme 3 Selective syntheses of the isomeric dialdehydes 8baab and 8abba. | ||
| Depending on the used bisphosphonate (top row) and dialdehyde batch (left column) polymers with various side chain configurations – defined successions (orange cells); alternating, defined and ill-defined elements (green cells); and a highly random sequence (blue cell) – were obtained. |
|---|
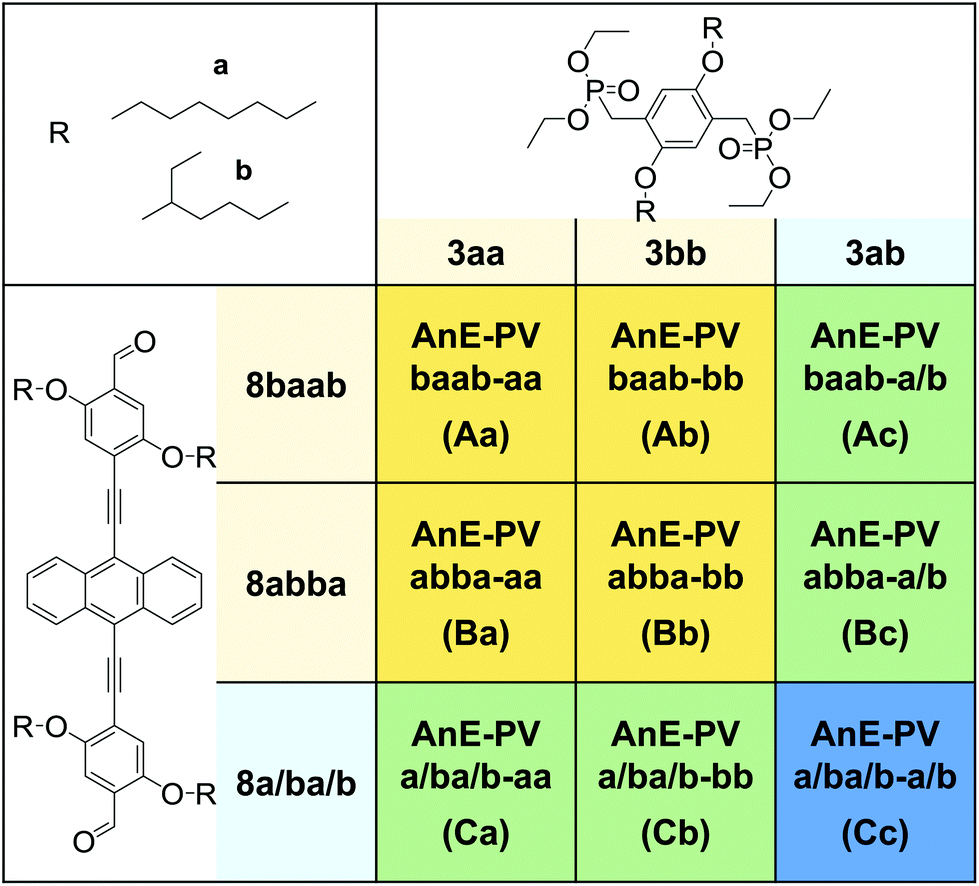
|
Results and discussion
Synthesis
The preparation of anthracene-containing poly(arylene ethynylene)-alt-poly(phenylene vinylene)s (AnE-PVs) by the Horner–Wadsworth–Emmons (HWE) reaction (Scheme 1) is highly effective.28,34,39 Syntheses of the bisphosphonate and dialdehyde comonomers with phenylene elements bearing symmetric or dissymmetric configurations of octyloxy (a) and 2-ethylhexyloxy (b) side chains are illustrated in Scheme S1† and Schemes 2 and 3.The three 2,5-dialkoxy-p-xylylene bis(diethylphosphonate)s equipped with two octyloxy (3aa), two 2-ethylhexyloxy (3bb) or both side chains (3ab) were prepared from hydroquinone and p-octyloxyphenol, respectively.31,40 Sequences of Williamson etherification, bromomethylation and the Michaelis–Arbuzov reaction provided the bisphosphonates via the respective intermediates (1aa, 1bb, 1ab and 2aa, 2bb, 2ab; Scheme S1†).
Our first attempt to synthesize an anthracene-containing dialdehyde comonomer with mixed side chain configuration followed a multi-step procedure well-established for the preparation of analogous dialdehydes with all-identical side chains (Scheme 2).28,34,39 Subjecting the dialkoxy derivative 1ab to an iodination reaction produced the diiodo compound 4ab in good yields. Through sequential addition of n-butyl lithium and dimethylformamide in defined amounts, one of the iodine functionalities is targeted for substitution with a formyl group. In a previous work, A. Wild et al. have demonstrated that the analogous compound 5-(2-ethylhexyloxy)-4-iodo-2-methoxy-benzaldehyde could be obtained with high selectivity if the reaction was performed at low temperature (−78 °C).37 For our system, however, regioselectivity is less pronounced, as the reaction repeatedly yielded mixtures of both possible monoaldehydes 5ab(I) and 5ba(I), which could not be separated by standard column chromatography techniques. Using Sonogashira cross-coupling the remaining iodo group was substituted with trimethylsilylethynylene. After desilylation, the obtained mixture of alkynes (7ab and 7ba) was reacted with 9,10-dibromoanthracene in a second Sonogashira reaction to yield a mixture of dialdehydes 8a/ba/b (8abba, 8baab and 8abab; Schemes 2 and S2†). Analogous to the preceding intermediates (5a/b, 6a/b, 7a/b), a separation of the regioisomers could not be accomplished. While all three dialdehydes have a plane of symmetry within their planar backbone, 8abba and 8baab also possess an inversion center (point group C2h) or another mirror plane (point group C2v), depending on the alignment of their subunits (Scheme S3†). The dialdehyde 8abab and the bisphosphonate 3ab, on the other hand, merely belong to point group Cs.
In order to prepare the phenylene building blocks 5ab and 5ba, and eventually the dialdehydes 8abba and 8baab, in a selective manner, the synthetic pathway needed to be altered. A report by X. Zhu et al. on the synthesis of phenylene–vinylene oligomers with regioregular side chain arrangements provided a promising approach.41 Employing, adapting and optimizing the described synthetic elements enabled the selective preparation of both isomers, 5ba and 5ab, from the same starting material, 4-octyloxyphenol (1a; Scheme 3). The regio-specific functionalization of aryl derivatives requires a sufficient electronic differentiation of the ring positions.41,42
The reaction of 1a with 4-toluenesulfonyl chloride provided the intermediate 10a. The tosyl group reduces the electron density at the ortho-positions significantly, leading to a strong deactivation for electrophilic attacks. The subsequent bromination addressed one of the electron-rich positions next to the octyloxy chain, yielding compound 11a.
Hydrolytic cleavage of the tosyl group provided the brominated p-alkoxyphenol 12a, which constitutes the “branching point” in the here-described syntheses of the two regioisomeric building blocks 5ba and 5ab. The synthesis of 5ba involved the selective formylation in the ortho-position to the hydroxy group and the etherification with 2-ethylhexyl bromide.
The protocol of X. Zhu et al. relies on two additional steps – (1) protection of the aldehyde function by condensation with neopentyl glycol, prior to etherification, and (2) hydrolysis of the acetal, afterwards (Scheme S4†). By adapting this protocol, the synthesis could be crucially improved and simplified. Using potassium carbonate instead of the hydroxide, the etherification could be performed directly in the presence of the aldehyde functionality. For the synthesis of 5ab a new route utilizing the intermediate 12a was developed. Instead of starting the original sequence with the corresponding substrate, 4-(2-ethylhexyloxy)phenol, all over again, only three steps – etherification, formylation and bromination – were needed to obtain 5ab selectively.43,44 Pursuing the synthesis (Sonogashira reactions and intermediate desilylation) with the individual regioisomers 5ba and 5ab eventually led to the selective formation of 8baab and 8abba.
Each of the three dialdehyde batches, the mixture 8a/ba/b as well as the pure regioisomers 8baab and 8abba, were subjected to HWE reactions with each of the bisphosphonate derivatives, 3aa, 3bb and 3ab. Nine AnE-PVs with various side chain sequences were prepared (Table 1). Combinations of a symmetric dialdehyde (8abba or 8baab) and a symmetric bisphosphonate (3aa or 3bb) yielded polymers with highly defined side chain successions, identical for each repeating unit – AnE-PVbaab-aa (Aa), AnE-PVbaab-bb (Ab), AnE-PVabba-aa (Ba) and AnE-PVabba-bb (Bb). Substituting one of the two comonomers with a dissymmetric specimen, such as 8abab or 3ab, or a mixture, such as 8a/ba/b, produced polymeric materials with less defined side chain configurations. This reduction in sequence definition originates from the two distinguishable orientations in which a dissymmetric comonomer can be attached (Scheme S5†), or, in the case of a comonomer mixture, from random incorporation of the various species. The resulting polymers consist of alternating comonomer-derived segments with defined and non-defined side chain arrangements – AnE-PVbaab-a/b (Ac), AnE-PVabba-a/b (Bc), AnE-PVa/ba/b-aa (Ca) and AnE-PVa/ba/b-bb (Cb). Using the dissymmetric bisphosphonate 3ab together with the dialdehyde mixture 8a/ba/b produced a polymer, AnE-PVa/ba/b-a/b (Cc), with a highly random side chain configuration.
While the majority of the synthesized polymers exhibited good solubility in common organic solvents such as chloroform, tetrahydrofuran, etc., the polymers AnE-PVbaab-aa (Aa) and AnE-PVbaab-a/b (Ac) appeared slightly reluctant. Gentle heating was required in order to dissolve elevated amounts of AnE-PVbaab-a/b or in particular of AnE-PVbaab-aa in aromatic solvents such as toluene, chlorobenzene and 1,2,4-trichlorobenzene.
All synthesized materials were analyzed by NMR spectroscopy. The characteristics of the new polymers were investigated by size exclusion chromatography (SEC), UV-vis absorption and photoluminescence spectroscopy. The detailed synthesis descriptions and analysis data can be found in the ESI.†
Structural analysis
Structural and purity affirmation for all intermediates, comonomers and polymers was acquired by NMR spectroscopy in deuterated chloroform. The results exhibit many similarities to findings on related structures in preceding studies.18,31,34,39,41 While the presence of the linear octyloxy and branched 2-ethylhexyloxy side chain is clearly reflected in the 1H NMR spectra by several specific signal sets, the differentiation between the spectral outputs for the respective double dissymmetric regioisomeric phenylene derivatives, such as 7ba and 7ab (Fig. S1†), is subtle. The spectra of the dialdehyde comonomers and of the AnE-PV polymer series, respectively, exhibit overall high resemblance with each other. However, some clear distinctions become evident at close inspection. With the attachment of 7ba or 7ab to a central anthracene unit the difference in the side chain configuration is more visible. Fig. 1 displays select 1H NMR spectral details of the three dialdehyde batches, 8baab, 8abba and 8a/ba/b. Full range depictions can be found in the ESI (Fig. S2†).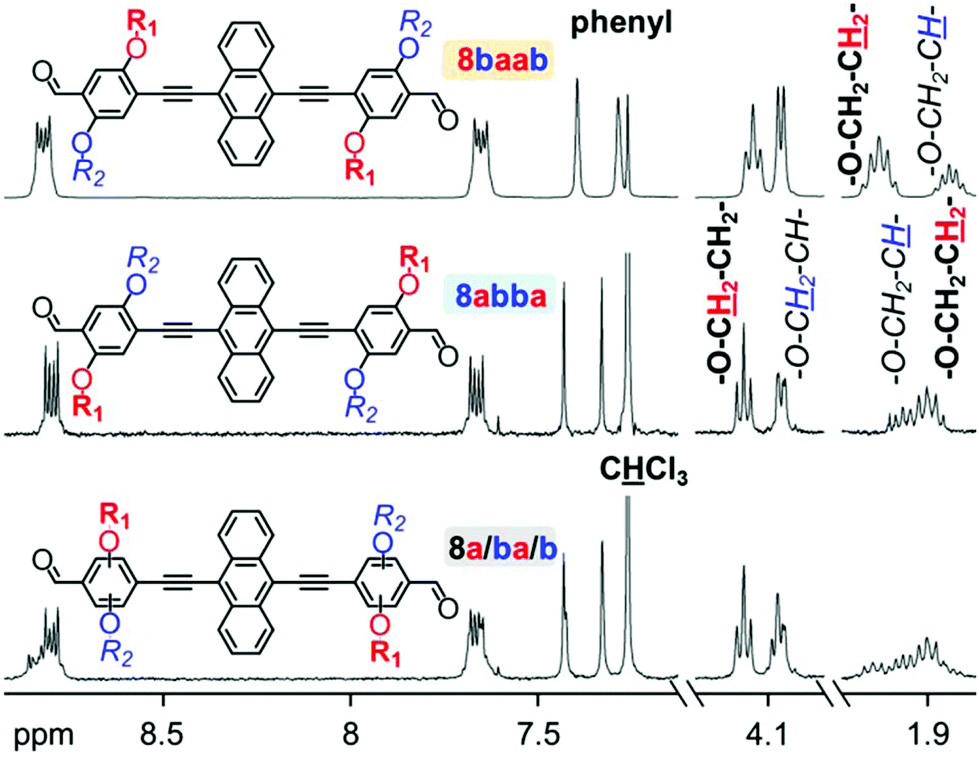 | ||
| Fig. 1 Details of 1H NMR (300 MHz, CDCl3) spectra recorded from the dialdehyde batches 8a/ba/b, 8abba and 8baab. | ||
In particular the signal sets originating from the protons at the second carbon of the alkoxy chains (–O–CH2–C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) x–) allow for an unambiguous distinction between the two regioisomers 8baab and 8abba. While octyloxy chains exhibit two protons at this position (–O–CH2–C2–), only one proton resides at the branching point of 2-ethylhexyloxy chains (–O–CH2–C–). In the spectrum of 8baab, the octyloxy signal (–O–CH2–C2–; 2.04 ppm) is significantly more downfield-shifted than the less intense 2-ethylhexyloxy peaks (–O–CH2–C–, 1.84 ppm). Even though the corresponding multiplets in the spectrum of 8abba are not completely separated, the inversion in their succession is evident and reflects the repositioning of the side chains. The 1H NMR spectrum of the dialdehyde batch 8a/ba/b exhibits clear evidence for the presence of various dialdehyde species, as it displays features with high resemblance to an overlap of signals from both regioisomers, 8baab and 8abba.
x–) allow for an unambiguous distinction between the two regioisomers 8baab and 8abba. While octyloxy chains exhibit two protons at this position (–O–CH2–C2–), only one proton resides at the branching point of 2-ethylhexyloxy chains (–O–CH2–C–). In the spectrum of 8baab, the octyloxy signal (–O–CH2–C2–; 2.04 ppm) is significantly more downfield-shifted than the less intense 2-ethylhexyloxy peaks (–O–CH2–C–, 1.84 ppm). Even though the corresponding multiplets in the spectrum of 8abba are not completely separated, the inversion in their succession is evident and reflects the repositioning of the side chains. The 1H NMR spectrum of the dialdehyde batch 8a/ba/b exhibits clear evidence for the presence of various dialdehyde species, as it displays features with high resemblance to an overlap of signals from both regioisomers, 8baab and 8abba.
The NMR characteristics of the HWE products reveal the transformation of the dialdehyde and bisphosphonate comonomers (Fig. S3†). While the characteristic comonomer signals of the formyl and phosphonate groups vanished from the polymer spectrum, new signals accounting for the vinylene linkages have emerged in the aromatic/olefinic region. In addition, a strong signal broadening and loss in resolution can be observed. Despite some shifts, most signal patterns are rather consistent with their counterparts in the comonomer spectra. Even most of the side chain configuration-specific features are well-preserved. 1H NMR spectra depictions of all synthesized polymers, in full range as well as selected magnifications, can be found in the ESI (Fig. S4 and S5†). Commonly the HWE olefination reaction produces vinylene linkages with a high trans(E)-selectivity. While this turns out to be valid for the polymers Aa, Ab and Ac, the 1H NMR spectra of the other polymers (Ba, Bb, Bc, Ca, Cb and Cc) display some weak signals at about 6.8 and 3.6 ppm, indicating a small percentage of cis(Z)-vinylene linkages.45,46 This suggests that the side chain configuration of the dialdehydes might affect the vinylene linkage formation during HWE reactions in a detectable manner. Here, the presence of an octyloxy chain instead of a 2-ethylhexyloxy chain right next to the formyl group slightly increases the probability of cis-vinylene linkage formation. More details and discussions of NMR features as well as an attempt to correlate the findings with the side chain sequences of the polymers can be found in the ESI.†
For further characterization of the new polymers, high temperature size exclusion chromatography (HT-SEC) measurements were performed. A list of the results and the SEC plots are depicted in the ESI (Table S1, Fig. S6†). The polymer characteristics, namely the number average molecular mass (Mn), the mass average molecular mass (Mw) and the dispersity (Đ = Mw/Mn), of the nine polymers do not show very large variations. The Mn and Mw results range from 7200 (Aa) to 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 g mol−1 (Cb) and from 17400 (Bb) to 34700 g mol−1 (Cb), respectively. Correlation with the molar mass of the repeating unit, M(C88H120O6) = 1273.92 g mol−1, suggests degree of polymerization (DP) values between roughly 6 and 11. Almost all polymers exhibit dispersity (Đ) values between 2.3 (Ab) and 2.64 (Cc). Only AnE-PVbaab-aa (Aa) exhibits a rather high dispersity of 4.46. As pointed out previously, AnE-PVbaab-a/b (Ac) and especially AnE-PVbaab-aa (Aa) exhibit a discernibly lower solubility in aromatic solvents than AnE-PVbaab-bb (Ab) and the rest of the polymers. It is evident that the reduction in solubility does not originate from a higher molar mass, as it could be suspected. A correlation with the specific side chain configurations at the alternating segments is apparent. The polymers produced with the dialdehyde 8abba, in which the 2-ethylhexyloxy side chains are positioned in direct proximity to the anthracene unit, exhibit good solubility, including the ones with a comparable high molar mass. The polymers prepared from 8baab, on the other hand, exhibit tendencies of reduced solubility depending on the side chains at the bisphosphonate-derived segments. The switching from octyloxy to 2-ethylhexyloxy side chains leads to a gradual increase in solubility, AnE-PVbaab-aa (Aa) < AnE-PVbaab-a/b (Ac) < AnE-PVbaab-bb (Ab). These observations are clear indications for varying aggregation tendencies directly induced by the side chain successions and combinations, and probably also slightly affected by the various small contents of cis-vinylene linkages.
500 g mol−1 (Cb) and from 17400 (Bb) to 34700 g mol−1 (Cb), respectively. Correlation with the molar mass of the repeating unit, M(C88H120O6) = 1273.92 g mol−1, suggests degree of polymerization (DP) values between roughly 6 and 11. Almost all polymers exhibit dispersity (Đ) values between 2.3 (Ab) and 2.64 (Cc). Only AnE-PVbaab-aa (Aa) exhibits a rather high dispersity of 4.46. As pointed out previously, AnE-PVbaab-a/b (Ac) and especially AnE-PVbaab-aa (Aa) exhibit a discernibly lower solubility in aromatic solvents than AnE-PVbaab-bb (Ab) and the rest of the polymers. It is evident that the reduction in solubility does not originate from a higher molar mass, as it could be suspected. A correlation with the specific side chain configurations at the alternating segments is apparent. The polymers produced with the dialdehyde 8abba, in which the 2-ethylhexyloxy side chains are positioned in direct proximity to the anthracene unit, exhibit good solubility, including the ones with a comparable high molar mass. The polymers prepared from 8baab, on the other hand, exhibit tendencies of reduced solubility depending on the side chains at the bisphosphonate-derived segments. The switching from octyloxy to 2-ethylhexyloxy side chains leads to a gradual increase in solubility, AnE-PVbaab-aa (Aa) < AnE-PVbaab-a/b (Ac) < AnE-PVbaab-bb (Ab). These observations are clear indications for varying aggregation tendencies directly induced by the side chain successions and combinations, and probably also slightly affected by the various small contents of cis-vinylene linkages.
Photophysical properties
The spectroscopic properties of all nine polymers have been studied in dilute solution – in aromatic (chlorobenzene) and non-aromatic (chloroform) solvents – as well as in thin solid film, cast from chlorobenzene and chloroform solutions, respectively. The recorded UV-vis absorption and photoluminescence (PL) spectra are depicted in Figs. 2 and 3. A summary of the photophysical data, acquired from chlorobenzene solutions and chlorobenzene-cast films, is presented in Table 2, namely the absorption and the photoluminescence maximum (λabs,max and λPL,max) as well as the optical band gap energy (Eoptg). An extended list, which also includes the data for the chloroform solutions and the films cast from chloroform solutions, can be found in the ESI (Table S2†), together with additional arrays of the UV-vis and PL spectra (Fig. S7–S12†). These depictions allow for a systematic and detailed comparison of the optical variations. As the polymer characteristics (Mn, Mw, Đ) of all nine polymers are in an overall similar range, differences in the optical responses are not expected to be significantly linked to the “slight” variations within these parameters.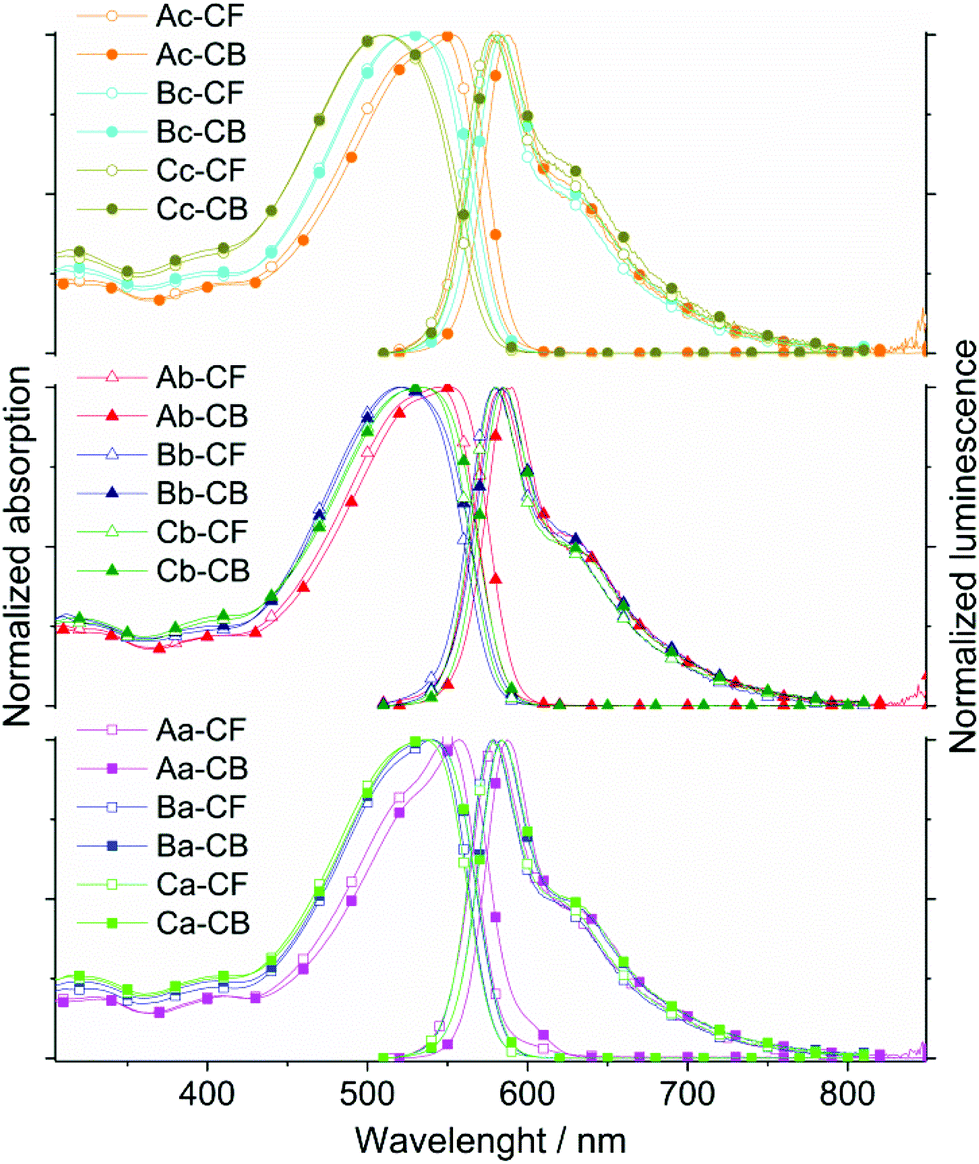 | ||
| Fig. 2 Normalized UV-vis and PL spectra of the polymers AnE-PVbaab/abba/a/ba/b-aa (Aa, Ba, and Ca; bottom), AnE-PVbaab/abba/a/ba/b-bb (Ab, Bb, and Cb; middle) and AnE-PVbaab/abba/a/ba/b-a/b (Ac, Bc, and Cc; top), in chloroform (CF, open symbols) and in chlorobenzene (CB, closed symbols) solution, respectively. | ||
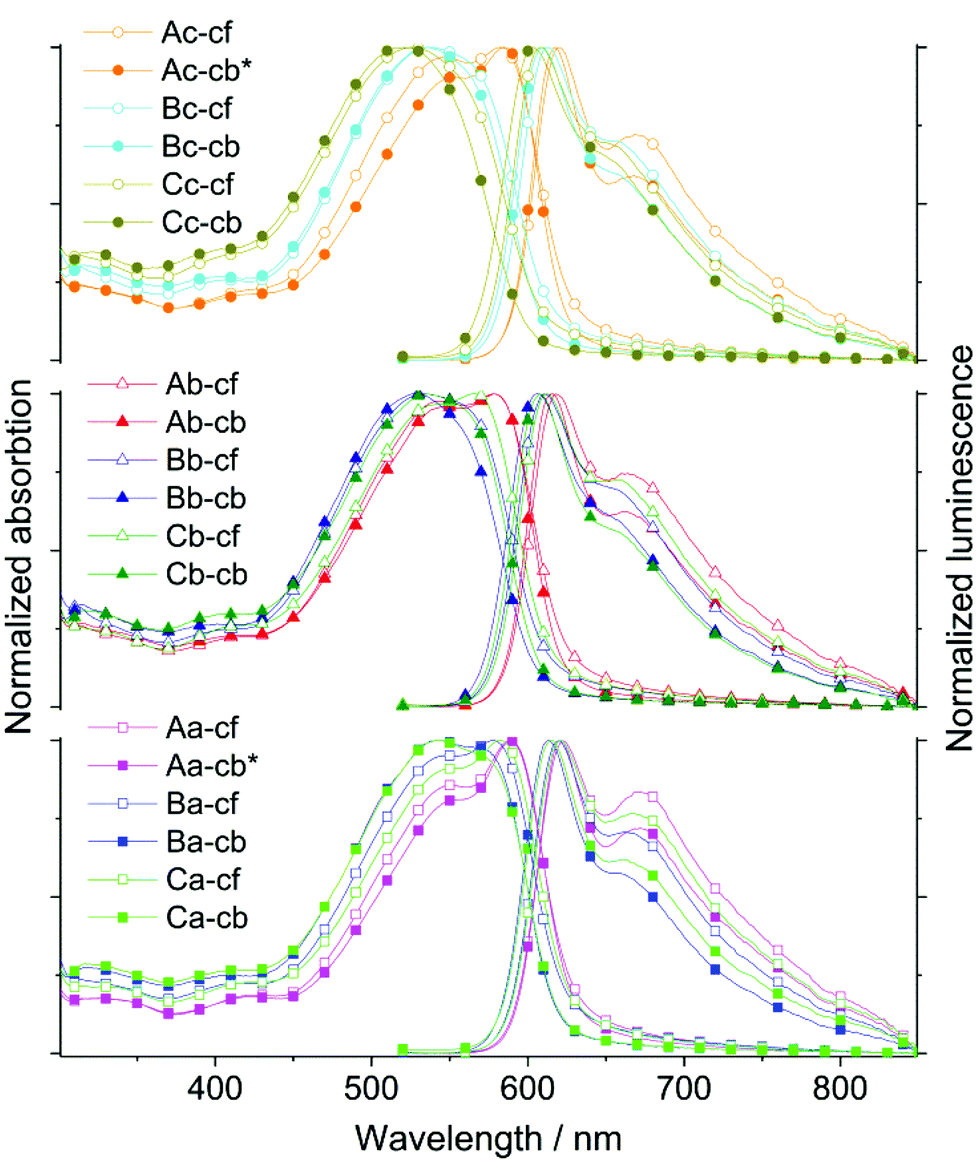 | ||
| Fig. 3 Normalized UV-vis and PL spectra of the polymers AnE-PVbaab/abba/a/ba/b-aa (Aa, Ba, and Ca; bottom), AnE-PVbaab/abba/a/ba/b-bb (Ab, Bb, and Cb; middle) and AnE-PVbaab/abba/a/ba/b-a/b (Ac, Bc, and Cc; top) cast into thin films from chloroform (cf, open symbols) and chlorobenzene solutions (cb, closed symbols), respectively. The asterisk indicates the processing by blade-coating instead of spin-coating. | ||
| AnE-PV | λ abs,max (nm) | λ PL,max (nm) |
E
optg
(eV) |
|
|---|---|---|---|---|
| a Excitation at 500 nm. b Optical band gap energy calculated using 1240/λ10%abs,max [eV], λ10%abs,max – wavelength at 10% of the absorption maximum intensity (at the onset side of the spectrum). c Film cast using a doctor blade. Keys: s – dilute solution, f – thin film; λabs,max – wavelength of the absorption maximum; and λPL,max – wavelength of the photoluminescence maximum. Additional values are given in parentheses (local maximum) and square brackets [shoulder]. | ||||
| baab-aa (Aa) | s | [530] 557 [602] | 587 [634] | 2.05 |
| fc | [554] 589 | 622 (672) | 1.93 | |
| baab-bb (Ab) | s | [529] 552 | 589 [634] | 2.09 |
| f | (547) 578 | 616 (660) | 1.97 | |
| baab-a/b (Ac) | s | [530] 553 | 588 [633] | 2.05 |
| fc | [554] 585 | 618 (668) | 1.97 | |
| abba-aa (Ba) | s | [521] 539 | 584 [630] | 2.12 |
| f | 544 [569] | 614 (656) | 1.99 | |
| abba-bb (Bb) | s | 522 | 584 [635] | 2.13 |
| f | 526 | 606 [655] | 2.04 | |
| abba-a/b (Bc) | s | 526 | 584 [632] | 2.13 |
| f | 536 | 610 [656] | 2.02 | |
| a/ba/b-aa (Ca) | s | 537 | 584 [633] | 2.12 |
| f | 541 [573] | 616 (660) | 1.99 | |
| a/ba/b-bb (Cb) | s | 533 | 585 [633] | 2.12 |
| f | 536 | 608 [658] | 2.01 | |
| a/ba/b-a/b (Cc) | s | 511 | 580 [630] | 2.15 |
| f | 520 | 604 [654] | 2.06 | |
The photophysical characterization of the new polymers reveal conspicuous resemblances to previously investigated AnE-PV polymers with different successions of linear and/or branched side chains.34,35,39 The polymers presented in these studies were prepared from dialdehydes and bisphosphonates bearing four and two identical side chains, respectively, or a mixed methoxy/2-ethylhexyloxy configuration. Already in solution, the absorption spectra of the new AnE-PV polymers are distinguishable at close inspection, with the side chain layout at the dialdehyde- and bisphosphonate-derived phenylene elements perceptively affecting the absorption response (Fig. 2, S7 and S9;†Tables 2 and S2†).
The dissolution in chloroform or in chlorobenzene barely impacts the absorption spectra of the majority of the new polymers. Solely for the polymers with octyloxy side chains in direct proximity to the anthracene unit, AnE-PVbaab-aa,bb,a/b (Aa, Ab, and Ac), a significant bathochromic shift by 7 to 8 nm is observed upon exchanging chloroform with chlorobenzene. For the remaining polymers, with 2-ethylhexyloxy chains in direct proximity to the anthracene moiety, AnE-PVabba-aa,bb,a/b (Ba, Bb, and Bc), or with more random combinations of side chains, AnE-PVa/ba/b-aa,bb,a/b (Ca, Cb, and Cc), the exchange of solvent induces only a very small signal broadening and/or shift (Δλabs,max ∼ 2 nm).
The main characteristic of all spectra in solution is an intense absorption band arising between 425 and 600 nm. With maxima at 550, 546 and 544 nm, respectively, and a distinct shoulder at higher energy (ca. 524 nm), the polymers Aa, Ac and Ab exhibit the most bathochromic absorption in chloroform solution among all nine polymers. The absorption bands of Ba, Bc and Bb peak at 541, 528 and 520 nm, respectively, and an additional shoulder (518 nm) was observed only for Ba. The slight blue shift in the absorption of the polymer sets Aa, Ac, and Ab and Ba, Bc, and Bb correlates with the incremental substitution of octyloxy chains (a) with 2-ethylhexyloxy chains (b) at the bisphosphonate-derived segments. In the third set of polymers Ca, Cb, and Cc, the specimen with the most random side chain sequences (Cc) exhibits by far the most hypsochromic absorption of all nine polymers, with a maximum at 509 nm. The absorption spectra of the other two polymers, Ca and Cb, strongly overlap revealing a maximum at about 535 nm (in CHCl3). Within the new series of polymers, only AnE-PVbaab-aa (Aa) dissolved in chlorobenzene exhibits a distinct low energy shoulder in the absorption spectrum at about 602 nm. In chloroform solution, this feature of Aa is strongly reduced. Such a shoulder can be ascribed to aggregation in solution and has been described for AnE-PV derivatives with solely linear side chains at the alternating conjugated segments.39 Commonly the exchange of linear with branched side chains considerably inhibits aggregation tendencies.
A comprehensive comparison of the absorption spectra of the new polymers with the spectra of AnE-PVs from previous studies in chloroform39 and chlorobenzene solution,34 respectively, shows significant correlations (Table S3†). The absorption spectra recorded for solutions of AnE-PVs bearing solely linear side chains at the dialdehyde-derived segments exhibit high resemblance to the spectra of the new polymers Aa, Ab and Ac (AnE-PVbaab-aa, bb, a/b), which feature octyloxy chains in direct proximity, and branched 2-ethylhexyloxy chains in the second-closest positions to the anthracene units. Similar correlations are observed between the absorption spectra of AnE-PVs with solely branched 2-ethylhexyloxy side chains at the dialdehyde-derived segments, and the new polymers Bb and Bc (AnE-PVabba-bb, a/b) with 2-ethylhexyloxy chains right next to anthracene, and octyloxy side chains in the following positions. The steric demand of the side chains in direct proximity to the anthracene unit obviously has the most dominant impact on the differentiation of the absorption properties of AnE-PVs in solution. The exchange of linear side chains with bulky branched side chains here apparently induces twisting in the conjugated backbone. The effect of the side chains in the following positions, on the other hand, is much less pronounced. The optical characteristics of the polymer Ba (AnE-PVabba-aa) seem not to be in good accordance with the suggested structure–property correlations. In comparison to Ca, which possesses some dialdehyde-derived segments with octyloxy side chains in direct proximity to anthracene, the absorption of the chloroform solution of Ba is slightly bathochromic-shifted. This is likely to be attributed to the extended sections with solely linear octyloxy chains occurring in a highly regular succession. The two 2-ethylhexyloxy chains right next to the anthracene center alternate with four octyloxy side chains in the following section. Due to the seemingly peculiar absorption characteristics of the polymers Ba and Cc, only the optical responses of the polymer series Ab, Cb, and Bb (2-ehtylhexyloxy/octyloxy = 4/2) completely fulfill the expected trend – increasing the percentage of 2-ethylhexyloxy side chains in close proximity to anthracene units induces a gradual hypsochromic shift in the absorption.
The current findings also allow for a more specific interpretation of previous results obtained for an AnE-PV derivative with a mixed 2-ethylhexyloxy/methoxy side chain configuration.39 Also in this case the sterically less demanding “linear” methoxy side chains occupy the positions closest to the anthracene moiety, while the bulky 2-ethylhexyloxy side chains at the following positions face away from the central anthracene moiety of the dialdehyde-derived segments, which caused the observed differences in the absorption features in chloroform solution (Table S3†).
Optical band gap energy (Eoptg) calculations were performed using the wavelength value at 10% maximum intensity of the onset side of the respective absorption spectrum (λ10%abs,max) in the equation Eoptg = 1240/λ10%abs,max [eV].47 All results were found within narrow bounds – 2.11 (Aa) to 2.16 eV (Cc) for chloroform solutions, and slightly lower in chlorobenzene, 2.05 (Aa) to 2.15 eV (Cc). These numbers correlate well with the results of previously reported AnE-PVs.34,39
For the photoluminescence investigations, all samples were excited at 500 nm. The emissions for all nine polymers dissolved in chloroform or in chlorobenzene exhibit very similar characteristics (Fig. 2, S7 and S11†; Tables 2 and S2†). Also a close resemblance with the emissive behavior of other AnE-PVs is evident (Table S3†).34,39 The solvent switch from chloroform to chlorobenzene leads to slight bathochromic shifts, of up to 8 nm. The main emission band, which is ascribed to the transition from the first excited state (S10) to the ground state (S00), arises between 575 and 587 nm. It is followed by a shoulder at around 627 to 638 nm, indicating the transition from the first excited state (S10) to the first vibronic state of the electronic ground state (S01). The photoluminescence maximum (λPL,max) ranges between 578 (Bc, Cc) and 584 nm (Ab) in chloroform, and between 580 (Cc) and 589 nm (Ab) in the chlorobenzene solution.
Spin-coating (Ab, Ba, Bb, Bc, Ca, Cb, and Cc) and blade-coating (Aa and Ac) were applied to prepare thin polymer films from chlorobenzene solutions on glass. All films from chloroform solutions were fabricated by spin-coating. The films were used to conduct photophysical investigations in solid state (Fig. 3, S8, S10 and S12;†Tables 2 and S2†).
Due to the enhanced planarization and organization in films (e.g. intermolecular interactions such as π–π stacking), the optical response of all nine polymers is substantially bathochromic-shifted as well as broadened, and specific features are enhanced and more resolved in comparison to the spectra recorded in solution. Many of the trends and peculiarities which were observed in solution subsist in the AnE-PV thin films. The dominant effect of the side chains in direct proximity to the anthracene unit on the differentiation of the optical response is once again evident. The impact of the side chains at the other positions, especially at the bisphosphonate-derived segments, appears quite significant, too. The correlations with AnE-PVs in previous studies are often remarkable (Table S3†).34,39 The applied casting solvent, chloroform or chlorobenzene, influences, in part considerably, the spectral characteristics of the obtained films. In most cases, the use of chloroform leads to an amplification of low energy features in the spectral response.
Among the thin films cast from chlorobenzene, the polymer set Aa, Ac, and Ab exhibits the most bathochromically shifted and well-resolved absorption. Within this set, the absorption maximum shifts from 589 (Aa) to 585 (Ac) and to 578 nm (Ab), and the proportion of the high energy shoulder at about 550 nm increases to form a local maximum in the spectrum of Ab. These observations correlate with the incremental substitution of octyloxy by 2-ethylhexyloxy side chains at the bisphosphonate-derived segments, Aa(0) < Ac(1) < Ab(2). Moreover, only for this polymer set the switching of the casting solvent from chlorobenzene to chloroform leads to slight hypsochromic shifts as well as an apparent increase in the high-energy shoulder/maximum in the absorption spectra.
The π–π* transition of polymers Bc and Bb in films cast from chlorobenzene is structureless with their maxima arising at 536 and 526 nm, respectively. The absorption of the chloroform-cast films peaks at 536 (Bc) and 533 nm (Bb), respectively. For the polymer with the higher octyloxy side chain proportion (Bc), a very weak low-energy shoulder presents itself at about 558 nm. Similar to the results in solution, the absorption characteristics of Ba in film stand out within the polymer set Ba, Bc, and Bb. In addition to the maximum at 544 nm, the absorption spectrum of the chlorobenzene-cast Ba film also exhibits a distinct shoulder at about 569 nm. Changing the casting solvent to chloroform leads to bathochromic shifts and changes the ratio of the two main absorption features significantly – the former maximum is “reduced” to a shoulder located at about 548 nm, while the low-energy shoulder is “transformed” into the new maximum at about 577 nm. Cast from chloroform solution, the polymer films of Ba, Ab and Ac exhibit a rather similar normalized absorption. Analogous to the results obtained in solution, the optical response of Cc in thin film seems to be affected by its highly randomized side chain configuration. The films of Cc exhibit the most hypsochromic absorption among all nine polymers – λabs,max = 520 (chlorobenzene-cast) and 525 nm (chloroform-cast) – with a structureless broad π–π* transition band. Similar to the observations in solution, the optical responses of Ca and Cb in thin film arise between the spectral outputs of Bb and Bc and Ab, Ac, and Aa. Chlorobenzene-cast films of Ca and Cb exhibit absorption maxima at 541 and 536 nm, respectively. While the main spectral transition of Cb is rather structureless and bears similarity to the absorption spectrum of a chlorobenzene-cast Bc film, Ca exhibits a more structured spectrum with a low-energy shoulder at 573 nm and a rather close resemblance to the absorption features of Ba films cast from chlorobenzene. Switching the casting solvent from chlorobenzene to chloroform leads to bathochromic shifting and significant changes in the ratio of the main absorption features. Both chloroform-cast films, Ca and Cb, exhibit “new” absorption maxima at 583 and 565 nm, respectively, as well as “new” high-energy shoulders at 548 and 538 nm, respectively.
Also in film, all PL spectra were recorded upon excitation at 500 nm. Despite the differences in the absorption characteristics, all emission spectra basically exhibit the same two main components, a high intensity peak at about 615 and a low-energy tailing shoulder or local maximum at around 663 nm (Fig. 3, S8 and S12,† and Tables 2 and S2†). The films of Cc show the most hypsochromic emissions with maxima at 604 (chlorobenzene-cast film) and 608 nm (chloroform-cast film), respectively, as well as a shoulder at about 654 nm. The films of Aa, cast from chloroform or chlorobenzene, exhibit the most bathochromic emissions with the main maximum at 622 and a local maximum at 672 nm. While the switch in casting solvent often leads to only small shifts, the impact on the ratio between PL maximum and low-energy feature (shoulder or local maximum) is commonly quite significant. Casting from chloroform seemingly provides a general “boost” for the low-energy component. The optical band gap (Egopt) values are smaller than in solution, which originates from the enhanced planarization and intermolecular interaction.
Conclusions
The optimized and customized selective syntheses of double dissymmetric functionalized phenylene building blocks offer great potential for the realization of elaborate side chain configurations within diverse conjugated systems. Formyl-phenylene elements with octyloxy (a) and 2-ethylhexyloxy (b) side chains, in both regioisomeric configurations, were prepared as a mixture and in isomer-pure forms. Using the three preparations in the synthesis of bis(4-formyl-phenylethynyl)anthracene derivatives yielded, in turn, a mixture and pure regioisomers with specific side chain configurations (baab and abba), respectively. Nine anthracene-containing poly(arylene ethynylene)-alt-poly(phenylene vinylene)s with individual side chain sequences – highly defined (e.g.AnE-PVbaab-aa), partly defined (e.g.AnE-PVabba-a/b) and random (AnE-PVa/ba/b-a/b) – were produced and investigated spectroscopically. Despite the resemblance among the new materials, detailed analysis of the 1H NMR data sets revealed side chain configuration-specific features. The comprehensive spectral UV-vis absorption and PL characteristics of the numerous polymer sample sets, in solution and in film, allowed the verification of side chain combination- and succession-specific structure–property relationships. The correlation with data of AnE-PV specimens from previous studies enabled further elaboration of side chain-induced effects. The current study clarifies that in particular the steric demand of the side chains in direct proximity to the anthracene unit has the most dominant impact on the differentiation of the optical properties of AnE-PVs. While bulky branched 2-ethylhexyloxy chains at these anthracene-facing positions are capable of inducing twisting in the conjugated backbone and strongly inhibit aggregation tendencies, the impact of the side chains at the following positions is often less substantial, but by far not negligible. The investigated examples illustrate the extensive possibilities to fine-tune material characteristics, such as photophysical response, by elaborate side chain engineering.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the support of Professor N. S. Sariciftci for providing the experimental facilities of the Linz Institute for Organic Solar Cells (LIOS) for this study. Professor C. Paulik and the Institute for Chemical Technology of Organic Materials (CTO) are highly acknowledged for the support to perform and evaluate HT-SEC measurements. Gunnar Spiegel (CTO) and Herwig Heilbrunner (LIOS) are thanked for support and discussions. The FWF is gratefully thanked for the funding through the grant no. I 1703-N20.References
- X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832–1908 CrossRef CAS.
- G. Zhang, Z.-A. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 CrossRef PubMed.
- U. H. F. Bunz, K. Seehafer, M. Bender and M. Porz, Chem. Soc. Rev., 2015, 44, 4322–4336 RSC.
- Y. H. Lee, M. Jang, M. Y. Lee, O. Y. Kweon and J. H. Oh, Chem, 2017, 3, 724–763 CAS.
- D. Farka, H. Coskun, J. Gasiorowski, C. Cobet, K. Hingerl, L. M. Uiberlacker, S. Hild, T. Greunz, D. Stifter, N. S. Sariciftci, R. Menon, W. Schoefberger, C. C. Mardare, A. W. Hassel, C. Schwarzinger, M. C. Scharber and P. Stadler, Adv. Electron. Mater., 2017, 3, 1700050 CrossRef.
- S. Muench, A. Wild, C. Friebe, B. Häupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS.
- J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724–6746 CrossRef CAS.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS PubMed.
- S. Riera-Galindo, F. Leonardi, R. Pfattner and M. Mas-Torrent, Adv. Mater. Technol., 2019, 1900104 CrossRef.
- M. S. White, M. Kaltenbrunner, E. D. Glowacki, K. Gutnichen-ko, G. Kettlgruber, I. Graz, S. Aazou, C. Ulbricht, D. A. M. Egbe, M. C. Miron, Z. Major, M. C. Scharber, T. Sekitani, T. Someya, S. Bauer and N. S. Sariciftci, Nat. Photonics, 2013, 7, 811–816 CrossRef CAS.
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS.
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS.
- Y. Cui, H. Yao, J. Zhang, T. Zhang, Y. Wang, L. Hong, K. Xian, B. Xu, S. Zhang, J. Peng, Z. Wei, F. Gao and J. Hou, Nat. Commun., 2019, 10, 2515 CrossRef PubMed.
- D. A. M. Egbe, B. Carbonnier, E. Birckner and U.-W. Grummt, Prog. Polym. Sci., 2009, 34, 1023–1067 CrossRef CAS.
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS.
- Z.-G. Zhang and Y. Li, Sci. China: Chem., 2015, 58, 192–209 CrossRef CAS.
- Ö. Usluer, C. Kästner, M. Abbas, C. Ulbricht, V. Cimrova, A. Wild, E. Birckner, E. Tekin, N. S. Sariciftci, H. Hoppe, S. Rathgeber and D. A. M. Egbe, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3425–3436 CrossRef.
- M. Jung, Y. Yoon, J. H. Park, W. Cha, A. Kim, J. Kang, S. Gautam, D. Seo, J. H. Cho, H. Kim, J. Y. Choi, K. H. Chae, K. Kwak, H. J. Son, M. J. Ko, H. Kim, D.-K. Lee, J. Y. Kim, D. H. Choi and B. S. Kim, ACS Nano, 2014, 8, 5988–6003 CrossRef CAS PubMed.
- T. D. Leitner, A. Vogt, D. Popović, E. Mena-Osteritz, K. Walzer, M. Pfeiffer and P. Bäuerle, Mater. Chem. Front., 2018, 2, 959–968 RSC.
- T. Liu, X. Pan, X. Meng, Y. Liu, D. Wie, W. Ma, L. Huo, X. Sun, T. H. Lee, M. Huang, H. Choi, J. Y. Kim, W. C. H. Choy and Y. Sun, Adv. Mater., 2017, 29, 1604251 CrossRef PubMed.
- X. Liu, C. Zhang, C. Duan, M. Li, Z. Hu, J. Wang, F. Liu, N. Li, C. J. Brabec, R. A. J. Janssen, G. C. Bazan, F. Huang and Y. Cao, J. Am. Chem. Soc., 2018, 140, 8934–8943 CrossRef CAS PubMed.
- D. Joly, M. Godfroy, L. Pellejà, Y. Kervella, P. Maldivi, S. Narbey, F. Oswald, E. Palomares and R. Demadrille, J. Mater. Chem. A, 2017, 5, 6122–6130 RSC.
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed.
- U. H. F. Bunz, Macromol. Rapid Commun., 2009, 30, 772–805 CrossRef CAS PubMed.
- G. Brizius, G. N. Pschirer, W. Steffen, K. Stitzer, H. C. zur Loye and U. H. F. Bunz, J. Am. Chem. Soc., 2000, 122, 12435–12440 CrossRef CAS.
- D. A. M. Egbe, H. Tillmann, E. Birckner and E. Klemm, Macromol. Chem. Phys., 2001, 202, 2712–2726 CrossRef CAS.
- D. A. M. Egbe, H. Neugebauer and N. S. Sariciftci, J. Mater. Chem., 2011, 21, 1338–1349 RSC.
- D. A. M. Egbe, C. Ulbricht, T. Orgis, B. Carbonnier, T. Kietzke, M. Peip, M. Metzner, M. Gericke, E. Birckner, T. Pakula, D. Neher and U.-W. Grummt, Chem. Mater., 2005, 17, 6022–6032 CrossRef CAS.
- E. Tekin, D. A. M. Egbe, J. M. Kranenburg, C. Ulbricht, S. Rathgeber, E. Birckner, N. Rehmann, K. Meerholz and U. S. Schubert, Chem. Mater., 2008, 20, 2727–2735 CrossRef CAS.
- N. Bouguerra, A. Růžička, C. Ulbricht, C. Enengl, S. Enengl, V. Pokorná, D. Výprachtický, E. Tordin, R. Aitout, V. Cimrová and D. A. M. Egbe, Macromolecules, 2016, 49, 455–464 CrossRef CAS PubMed.
- S. Boudiba, A. Růžička, C. Ulbricht, S. Enengl, C. Enengl, J. Gasiorowski, C. Yumusak, V. Pokorná, D. Výprachtický, K. Hingerl, D. R. T. Zahn, F. Tinti, N. Camaioni, S. Bouguessa, A. Gouasmia, V. Cimrová and D. A. M. Egbe, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 129–143 CrossRef CAS.
- G. Adam, A. Pivrikas, A. M. Ramil, S. Tadesse, T. Yohannes, N. S. Sariciftci and D. A. M. Egbe, J. Mater. Chem., 2011, 21, 2594–2600 RSC.
- D. A. M. Egbe, G. Adam, A. Pivrikas, A. M. Ramil, E. Birckner, V. Cimrova, H. Hoppe and N. S. Sariciftci, J. Mater. Chem., 2010, 20, 9726–9734 RSC.
- S. Rathgeber, D. B. de Toledo, E. Birckner, H. Hoppe and D. A. M. Egbe, Macromolecules, 2010, 43, 306–315 CrossRef CAS.
- C. Kästner, C. Ulbricht, D. A. M. Egbe and H. Hoppe, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1562–1566 CrossRef.
- A. Wild, D. A. M. Egbe, E. Birckner, V. Cimrová, R. Baumann, U.-W. Grummt and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2243–2261 CrossRef CAS.
- S. Günes, A. Wild, E. Cevik, A. Pivrikas, U. S. Schubert and D. A. M. Egbe, Sol. Energy Mater. Sol. Cells, 2010, 94, 484–491 CrossRef.
- D. A. M. Egbe, S. Türk, S. Rathgeber, F. Kühnlenz, R. Jadhav, A. Wild, E. Birckner, G. Adam, A. Pivrikas, V. Cimrová, G. Knör, N. S. Sariciftci and H. Hoppe, Macromolecules, 2010, 43, 1261–1269 CrossRef CAS.
- C. Weder and M. S. Wrighton, Macromolecules, 1996, 29, 5157–5165 CrossRef CAS.
- X. Zhu and K. N. Plunkett, J. Org. Chem., 2014, 79, 7093–7102 CrossRef CAS.
- R. Nambiar, K. B. Woody, J. D. Ochocki, G. L. Brizius and D. M. Collard, Macromolecules, 2009, 42, 43–51 CrossRef CAS.
- R. W. Wagner, J. S. Lindsey, I. Turowka-Tyrk and W. R. Scheidt, Tetrahedron, 1994, 50, 11097–11112 CrossRef CAS.
- A. Brun and G. Etemad-Moghadam, Synthesis, 2002, 1385–1390 CrossRef CAS.
- H. Katayama, M. Nagao, T. Nishimura, Y. Matsui, K. Umeda, K. Akamatsu, T. Tsuruoka, H. Nawafune and F. Ozawa, J. Am. Chem. Soc., 2005, 127, 4350–4353 CrossRef CAS.
- F. Wang, F. He, Z. O. Xie, Y. P. Li, M. Hanif, M. Li and Y. Ma, Macromol. Chem. Phys., 2008, 209, 1381–1388 CrossRef CAS.
- D. A. M. Egbe, B. Cornelia, J. Nowotny, W. Günther and E. Klemm, Macromolecules, 2003, 36, 5459–5469 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental information including details about the employed materials, conducted syntheses, applied analytical methods and obtained results. Additional depictions of UV-vis and PL spectra. Additional synthesis schemes and structural images. See DOI: 10.1039/c9py01030k |
| This journal is © The Royal Society of Chemistry 2019 |