
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto-controlled one-pot strategy for the synthesis of asymmetric three-arm star polymers†
Yichuan
Zhang
 a,
Mark
Bradley
a and
Jin
Geng
*ab
a,
Mark
Bradley
a and
Jin
Geng
*ab
aEaSTCHEM School of Chemistry, University of Edinburgh, UK
bShenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen, P. R. China. E-mail: jin.geng@siat.ac.cn
First published on 25th July 2019
Abstract
A rapid photo-controlled one-pot strategy for the synthesis of asymmetric star polymers was developed using two different wavelengths of light to allow spatial and temporal control of the synthesis of the star polymers with controlled structures and narrow polydispersities. Several asymmetric star structures were successfully synthesised and the simultaneous photo-induced ATRP and RAFT polymerisation was demonstrated using this procedure.
Introduction
Block-copolymers with various architectures that include conventional linear forms as well as stars, combs and rings have attracted great interests in material science and biological sciences. Star-shape block-copolymers having either chemically different arms (also known as miktoarm stars) or arms with the same composition have attracted increasing attention over the past two decades owing to their distinct architectures and unique physical and chemical properties in both bulk and solution.1,2 For instance, the phase boundaries of star polymers are different from the linear analogues resulting in dissimilar self-assembly behaviours3,4 and viscosities.5 The high chain end functionality density gives the opportunity for high degrees of chemical modifications of the polymers,6 that contribute to potential applications such as nano/macro materials7,8 morphology modifiers,9 catalysis,10 emulsifiers11 and drug delivery carriers.12 Much effort has focused on the design and development of new star polymers, however complicated synthesis routes and the time consuming isolation processes resulting in many limitations.The ‘arm first’ approach is widely used in the synthesis of star polymers, which starts from an end functionalised linear polymer followed by a coupling reaction with a multifunctional reagent.6,13 In comparison to the ‘core first’ strategy, which utilise a multifunctional initiating motif (the core) upon which are grown multiple polymer chains,14 the ‘arm first’ approach avoids the tedious synthesis of complicated initiator cores, and therefore offers a more straightforward way of producing the target star polymers. However, it is still challenging to synthesise well defined asymmetric miktoarm star polymers because of the limited efficiency of core-arm coupling reaction and the complexity of isolation of intermediates/products from the macromolecular mixture.5
Reactions with high yield, minimal side product profiles and high reaction rates that form robust products are required for the coupling chemistry for the synthesis of star polymers. Active functionalities such as active esters,15 epoxides,16 isocyanates,17 chlorotriazines,18 maleimides,19 thiols20 and azides21 have been utilised for polymer–polymer and polymer–small molecule conjugations. In comparison to these conjugation reactions, strained alkene–tetrazine inverse electron-demand Diels–Alder (IEDDA) chemistry benefit from being catalyst-free, and offer high selectivity, rapid reaction rates and the generation of robust adducts.22–24
Pauses and terminations of polymerisations are often required to achieve a desired structure or molecular weight star and there is thus a growing interest in being able to controllably induce/initiate reactions by external stimuli, e.g. potential, light and mechanical forces.25 Light is one of the most favoured energy sources for the temporal and spatial control of reactions, because of the straightforward nature of the processes, the mild reaction conditions and minimised side reactions.26 One such example is photoinduced electron transfer (PET) polymerisation, which is able to precisely control the molecular weight and polydispersity of the synthesised polymers,27–29 while tolerating molecular oxygen and offering compatibility to many functional monomers.26,28
Herein we report new methods for the rapid synthesis of asymmetric miktoarm star polymers through a one-pot tandem strategy combining photo-controlled tetrazine formation chemistry, norbornene–tetrazine IEDDA chemistry and PET-atom transfer radical polymerisation (ATRP) with precise spatial and temporal control using different wavelength light. A bromine functionalised dihydrotetrazine ATRP initiator precursor was photo-oxidised aided by a photosensitiser to give a bromine functionalised tetrazine,30 which was subsequently coupled to a norbornene-terminated polyethylene glycol (PEG) resulting in a macro-ATRP initiator, and was polymerised with an acrylamide monomer via a metal free PET-ATRP process to give the desired asymmetric star polymer. The whole process was conducted at ambient temperature in presence of air without the need of any intermediate isolation steps.
Results and discussion
The macro-initiator precursor, ω-norbornene-PEG IP1 was synthesised by coupling an amine functionalised PEG and a norbornene carboxylic acid (see ESI, Fig. S10†). The dihydrotetrazine precursor 1 was synthesised in two-steps in which the diaminopyridyl dihydrotetrazine S1 was synthesised from amino pyridinecarbonitrile according to the previous method30 with the α-bromoisobutyryl group introduced through an acid bromide–amine coupling chemistry (see ESI, Fig. S11†). The dihydrotetrazine was oxidised using i-pentyl nitrite, to give tetrazine 2 for comparison (see ESI, Fig. S11†).31In the one-pot system, reagents dihydrotetrazine 1 (2.1 mM), methylene blue (0.4 mM), 1,4-diazabicyclo[2.2.2]octane (DABCO) (34.0 mM), Eoisin Y (0.2 mM), an acrylamide monomer (1000 mM), and macro-initiator precursor IP1 (2.0 mM) were combined. The tetrazine formation reaction and the PET-ATRP were initiated independently using two different light sources (at 630 nm and 470 nm, respectively, Fig. 1).
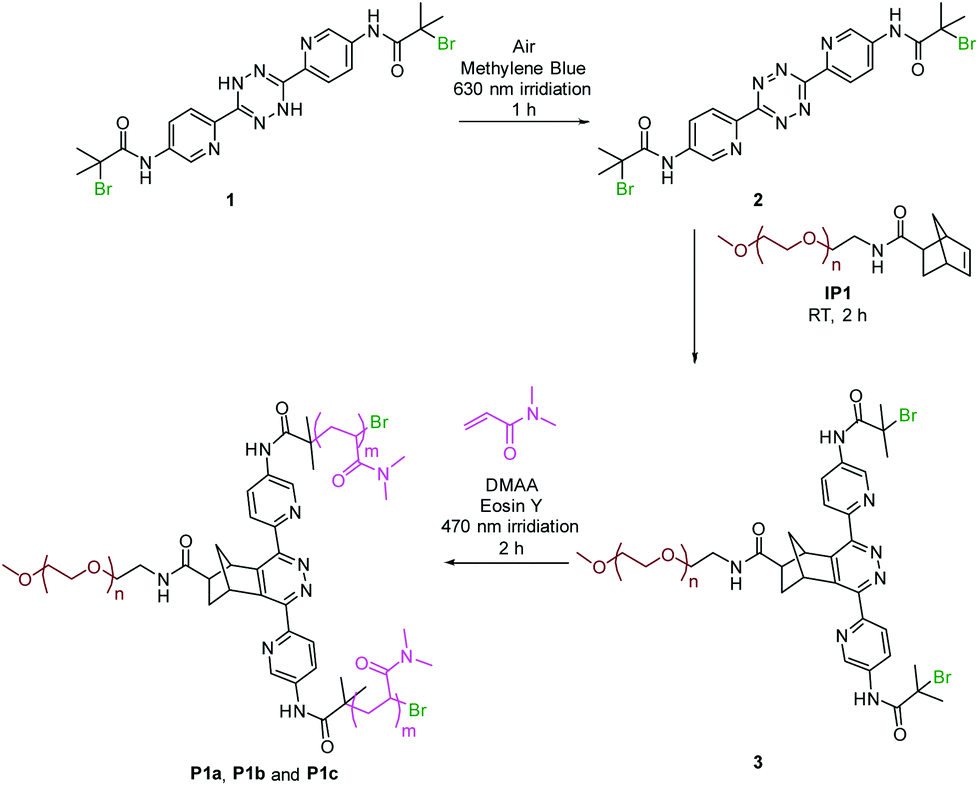 | ||
Fig. 1 One-pot synthesis of Star polymer P1a, P1b and P1c. Dihydrotetrazine 1 was oxidised to give tetrazine 2, which was subsequently coupled to the macro-initiator precursor IP1 to generate the macro-initiator 3. DMAA was then polymerised to give the asymmetric star polymers P1a, P1b and P1c ([DMAA]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[3] = 1000:1, 200:1 and 2000:1, respectively). :[3] = 1000:1, 200:1 and 2000:1, respectively). | ||
Methylene blue was used as a photosensitiser that catalysed the oxidation of the dihydrotetrazine with DABCO used as a singlet oxygen quencher. Eosin Y was used as the photo-catalyst for the polymerisation reaction due to its high efficiency while giving polymers with low polydispersities.32 In addition, during the polymerisation, DABCO also acted as an electron donor to accelerate the PET reaction.33
To evaluate this chemistry as a potentially “one-pot process” each step was initially optimised (see ESI, Table S1†). Thus the photo-catalysed oxidation of dihydrotetrazine and the subsequent norbornene–tetrazine IEDDA chemistry was investigated using 1H NMR and UV-vis spectroscopy (Fig. 2). Molecular oxygen was efficiently activated by methylene blue and 630 nm irradiation (4.0 mW cm−2) with oxidation of the dihydrotetrazine 1 to the corresponding tetrazine 2. The reaction was followed by both 1H NMR and UV-vis analysis (Fig. 2a and b) and the tetrazine 2 was identical to that generated using more conventional oxidation routes (see NMR data in ESI,† experimental). The non-specific oxidation of the dihydrotetrazine was minimised using DABCO, which has an agreement with previous studies (ESI, Fig. S6†).30
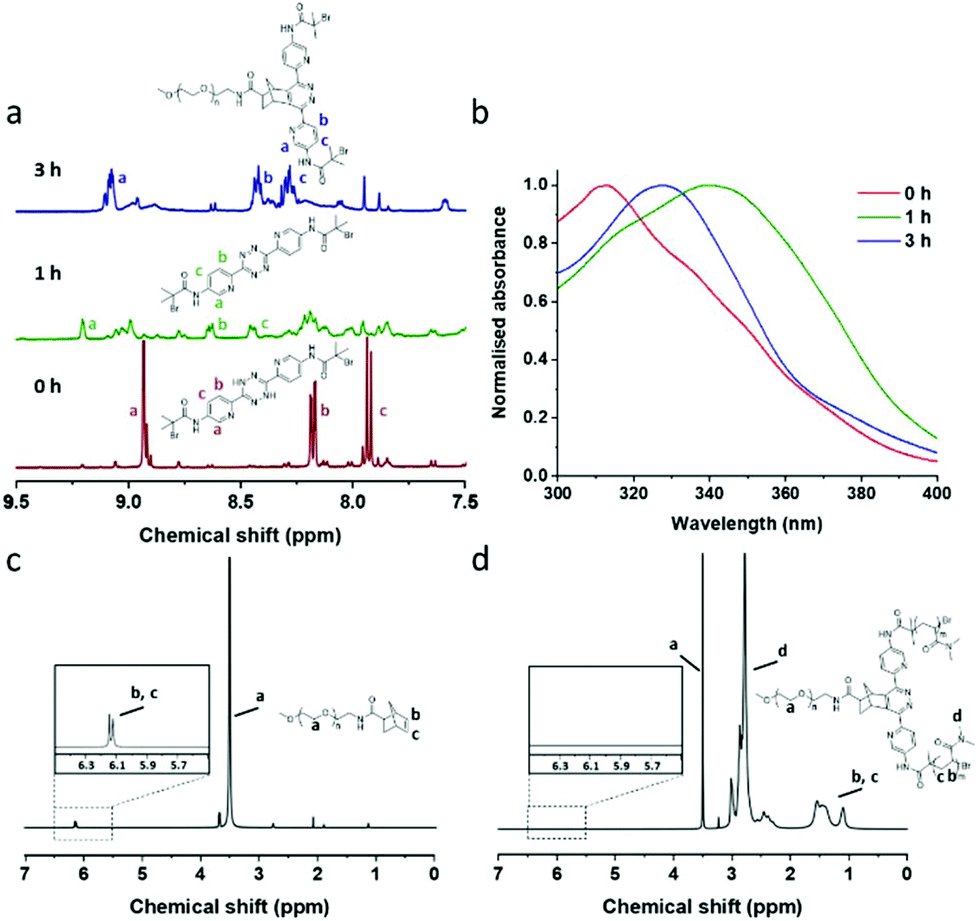 | ||
| Fig. 2 (a) 1H NMR spectra of the one-pot reaction mixture after 1 h (showing the aromatic region, from 7.5 ppm to 9.5 ppm, d6-DMSO). Red, t = 0 h, before oxidation (dihydrotetrazine 1); green, t = 1 h (generation of tetrazine 2 with 1 h irradiation at 630 nm); blue, t = 3 h (generating 3 by further incubation for 2 h). (b) UV-vis spectra of the reaction mixture. Red, t = 0 h; green, t = 1 h; blue, t = 3 h. (c) and (d) 1H NMR spectra (d6-DMSO) of macro-initiator precursor IP1 and star polymers P1b (isolated by dialysis, MWCO = 3500). | ||
The PET-ATRP progress of N,N-dimethylacrylamide (DMAA) on macro-ATRP initiator 3 was investigated using 1H NMR and GPC. Photoinduced polymerisation of DMAA was initiated by illumination at 470 nm (4.0 mW cm−2) to give asymmetric star polymers (see Fig. 1). We observed that longer irradiation times gave slightly higher conversions and higher molecular weight with polydispersities also increasing (Table 1), attributed to additional chain transfer.34 The resulting polymers, e.g.P1a (Đ = 1.48) and P1b (Đ = 1.47) showed moderate increases in molecular weight from the initiator precursor IP1 (Đ = 1.21), although longer polymers e.g.P1c (Đ = 1.93) gave significant increases.35,36 No polymerisation occurred in the absence of either Eosin Y or the ATRP initiator thus indicating that the photocatalyst Eosin Y did not directly generate free radicals, but undergoes a redox process with the bromine moiety on the initiator as previously reported.37
We observed that polymerisation was only initiated following consumption of the tetrazine 2 and reaction with the norbornenes, indicating that the tetrazine inhibited the polymerisation process. To confirm this, the commercially available tetrazine, 3,6-di-2-pyridyl-1,2,4,5-tetrazine was added to the reaction mixture consisting of the macro-initiator 3, DMAA, Eosin Y and DABCO. After illumination at 470 nm for 1 h no polymerisation was observed (ESI, Fig. S8†). However, after the full degradation of tetrazines, the polymerisation can be initiated and reached over 80% conversion within 2 h supporting the hypothesis that tetrazines inhibit PET polymerisation, thus providing a powerful switch-on process for controlling chemistry.
In order to further evaluate the one-pot strategy, two other initiator precursors (IP2 and IP3) were synthesised (ESI, Fig. S12 and S13†). Using the same procedure as described above, we synthesised three groups of star polymers P1a, P1b and P1c (A2-B); P2a, P2b and P2c (A2-B-A2); P3a, P2b and P3c (A-A′2) (Fig. 2 and 3). The photo-induced oxidation and the norbornene–tetrazine IEDDA reactions were complete within 3 h, while the monomer conversion of the polymerisations exceeded 80% after the 2 h photopolymerisation. The polydispersities of the star polymers were inherited from the macro-initiator precursors (IP1–IP3) and were nearly unchanged (Table 2).
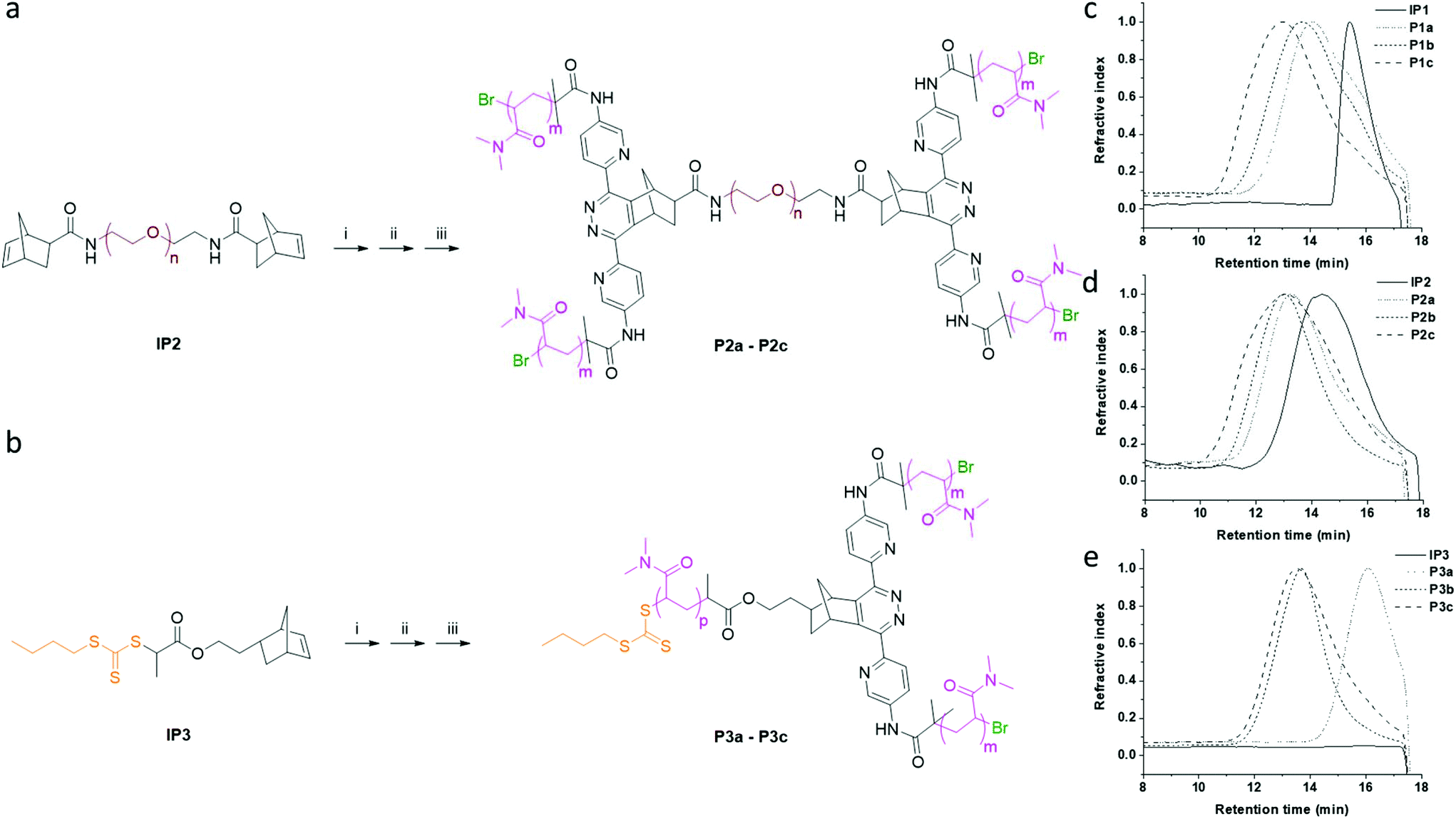 | ||
| Fig. 3 Synthesis of star polymers from different (macro) initiators. (a) and (b) Synthesis of the asymmetric star polymers P2a–P2c and P3a–P3c from initiator precursors IP2 and IP3: (i) 1, air, methylene blue, 630 nm irradiation, 1 h; (ii) RT, 2 h; (iii) DMAA, Eosin Y, 470 nm irradiation, 2 h. GPC traces of the initiator precursors: (c) IP1, (d) IP2 and (e) IP3 and their corresponding star polymers. | ||
| Polymer entry | Macro-initiator precursor | [DMAA]/[macro-initiator precursor] | Conversiona (%) |
M
nb (kDa) |
Đ |
|---|---|---|---|---|---|
| a Determined by 1H NMR. b Determined by GPC (eluting with DMF with 1% LiBr at 60 °C, calibrated with PMMA standards). | |||||
| P1a | IP1 | 200/1 | 90 | 47 | 1.48 |
| P1b | IP1 | 1000/1 | 84 | 173 | 1.47 |
| P1c | IP1 | 2000/1 | 85 | 323 | 1.93 |
| P2a | IP2 | 200/1 | 88 | 223 | 1.94 |
| P2b | IP2 | 1000/1 | 92 | 340 | 1.98 |
| P2c | IP2 | 2000/1 | 95 | 381 | 2.64 |
| P3a | IP3 | 200/1 | 99 | 24 | 1.28 |
| P3b | IP3 | 1000/1 | 99 | 164 | 1.26 |
| P3c | IP3 | 2000/1 | 99 | 208 | 1.59 |
After purification by dialysis (MWCO = 7 kDa), a small shoulder in GPC traces (15.8 min) in low mass region of the star polymers P1a and P1b was observed, which could due to the non-initiated macro-initiators (Fig. 3c).38 To get high quality star polymers, the mixture can be further purified by fractional precipitation39 or SEC column chromatography, although it was not conducted in the current study.
The norbornenyl functionalised trithiocarbonate RAFT chain transfer agent IP3 was synthesised (see ESI, Fig. S13†) in order to conduct simultaneous PET-ATRP and PET-RAFT polymerisations. As previous reported, the reactivity of ATRP and RAFT polymerisation are similar when carried out under the same reaction conditions, giving polymers with similar sizes and polydispersities.40 In this study, simultaneous initiation of both polymerisations resulted in A-A′2 structured star polymers (P3a, P3b and P3c) with the desired molecular weights and narrow dispersities (Table 2).
In general, to minimise the polydispersity and control the molecular weight of polymers, the conversions need to be precisely controlled.36 The photo-induced approach has been employed to allow a remote control of the polymerisation where the reaction can be switched “on” and “off” with and without illumination.37,41 In this study, we also demonstrated that the polymerisation can be conducted as “pause” and “resume” manners with the switches of light (Fig. 4). The polymerisation conversion increased steadily during this process, indicating a well-controlled polymerisation progress.
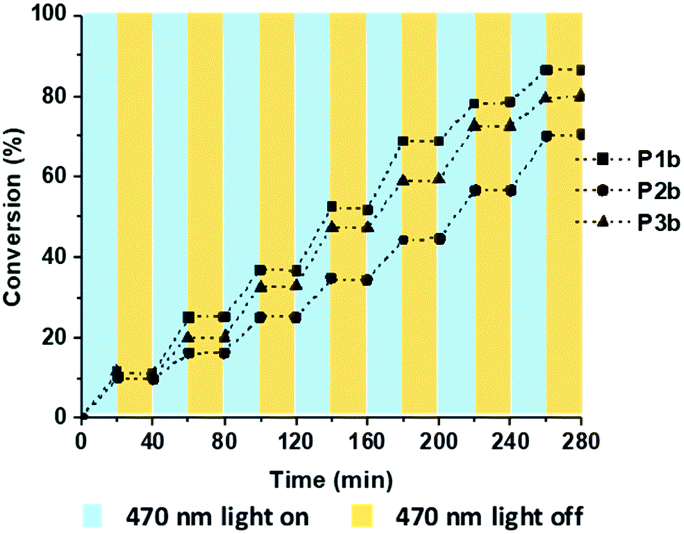 | ||
| Fig. 4 Plot of monomer conversion versus time. The polymer propagation was controlled by 470 nm light cycling (blue regions = on) and (yellow regions = off). Monomer conversions were quantified by 1H NMR. | ||
The compatibility of this approach was further evaluated using two additional monomers (methyl methacrylate and 2-(diethylamino)ethyl acrylate) which gave well-defined stars with 1H NMR showing identical resonances for different arm compositions and GPC showing narrow dispersities (see ESI, Fig. S9 and Table S2†).
Conclusions
A rapid, photo-controlled, asymmetric star polymer synthetic method was developed using the combination of tetrazine–norbornene chemistry and photo-induced polymerisation. This one-pot synthesis simplifies current asymmetric star polymer synthesis strategy, giving well-defined polymers within five hours. The use of two different wavelengths light allows each reaction to be controlled and initiated individually, allowing several well-defined asymmetric star polymers to be synthesised utilising this approach.The possibility of the synthesis of star polymers with different end functionalities via simultaneous PET-ATRP and PET-RAFT polymerisation were also demonstrated. The resulting terminated ω-bromine and the thiocarbonylthio could be further functionalised independently providing opportunities to generate heterogeneously end-functionalised star polymers.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank European Research Council (Advanced Grant ADREEM ERC-2013-340469) for funding.References
- N. Hadjichristidis, M. Pitsikalis, S. Pispas and H. Iatrou, Chem. Rev., 2001, 101(12), 3747–3792 CrossRef CAS PubMed.
- A. Blencowe, J. F. Tan, T. K. Goh and G. G. Qiao, Polymer, 2009, 50(1), 5–32 CrossRef CAS.
- S. Junnila, N. Houbenov, A. Karatzas, N. Hadjichristidis, A. Hirao, H. Iatrou and O. Ikkala, Macromolecules, 2012, 45(6), 2850–2856 CrossRef CAS.
- N. Hadjichristidis, H. Iatrou, S. Behal, J. Chludzinski, M. Disko, R. Garner, K. Liang, D. Lohse and S. Milner, Macromolecules, 1993, 26(21), 5812–5815 CrossRef CAS.
- T. Higashihara, S. Ito, S. Fukuta, T. Koganezawa, M. Ueda, T. Ishizone and A. Hirao, Macromolecules, 2014, 48(1), 245–255 CrossRef.
- X. Zhang, J. Xia and K. Matyjaszewski, Macromolecules, 2000, 33(7), 2340–2345 CrossRef CAS.
- X. Fan, K. Y. Win, Z. Hu, X. J. Loh and Z. Li, Macromol. Rapid Commun., 2018, 40(5), 1800217 CrossRef PubMed.
- S. Ito, R. Goseki, T. Ishizone and A. Hirao, Eur. Polym. J., 2013, 49(9), 2545–2566 CrossRef CAS.
- R. Phinjaroenphan, Y. Y. Kim, B. J. Ree, T. Isono, J. Lee, S. Rugmai, H. Kim, S. Maensiri, T. Kakuchi and T. Satoh, Macromolecules, 2015, 48(16), 5816–5833 CrossRef CAS.
- Y. Chi, S. T. Scroggins and J. M. Fréchet, J. Am. Chem. Soc., 2008, 130(20), 6322–6323 CrossRef CAS PubMed.
- Q. Qiu, G. Liu and Z. An, Chem. Commun., 2011, 47(47), 12685–12687 RSC.
- D. E. Discher and A. Eisenberg, Science, 2002, 297(5583), 967–973 CrossRef CAS PubMed.
- M. Morton, T. Helminiak, S. Gadkary and F. Bueche, J. Polym. Sci., 1962, 57(165), 471–482 CrossRef CAS.
- K. Matyjaszewski, P. J. Miller, J. Pyun, G. Kickelbick and S. Diamanti, Macromolecules, 1999, 32(20), 6526–6535 CrossRef CAS.
- A. Das and P. Theato, Chem. Rev., 2015, 116(3), 1434–1495 CrossRef PubMed.
- D. C. McLeod and N. V. Tsarevsky, J. Polym. Sci., Part A: Polym. Chem., 2016, 54(8), 1132–1144 CrossRef CAS.
- J. D. Flores, J. Shin, C. E. Hoyle and C. L. McCormick, Polym. Chem., 2010, 1(2), 213–220 RSC.
- C. A. Figg, T. Kubo and B. S. Sumerlin, ACS Macro Lett., 2015, 4(10), 1114–1118 CrossRef CAS.
- M. P. Robin, M. W. Jones, D. M. Haddleton and R. K. O'Reilly, ACS Macro Lett., 2011, 1(1), 222–226 CrossRef.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39(4), 1355–1387 RSC.
- V. Coessens, Y. Nakagawa and K. Matyjaszewski, Polym. Bull., 1998, 40(2–3), 135–142 CrossRef CAS.
- C. S. McKay and M. Finn, Chem. Biol., 2014, 21(9), 1075–1101 CrossRef CAS PubMed.
- K. Neumann, S. Jain, J. Geng and M. Bradley, Chem. Commun., 2016, 52(75), 11223–11226 RSC.
- S. Jain, K. Neumann, Y. Zhang, J. Geng and M. Bradley, Macromolecules, 2016, 49(15), 5438–5443 CrossRef CAS.
- F. A. Leibfarth, K. M. Mattson, B. P. Fors, H. A. Collins and C. J. Hawker, Angew. Chem., Int. Ed., 2013, 52(1), 199–210 CrossRef CAS PubMed.
- G. Li, W. Feng, N. Corrigan, C. Boyer, X. Wang and J. Xu, Polym. Chem., 2018, 9(20), 2733–2745 RSC.
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136(14), 5508–5519 CrossRef CAS PubMed.
- J. Wang, M. Rivero, A. Muñoz Bonilla, J. Sanchez-Marcos, W. Xue, G. Chen, W. Zhang and X. Zhu, ACS Macro Lett., 2016, 5, 1278–1282 CrossRef CAS.
- H. Ma, R. H. Davis and C. N. Bowman, Macromolecules, 2000, 33(2), 331–335 CrossRef CAS.
- H. Zhang, W. S. Trout, S. Liu, G. A. Andrade, D. A. Hudson, S. L. Scinto, K. T. Dicker, Y. Li, N. Lazouski and J. Rosenthal, J. Am. Chem. Soc., 2016, 138(18), 5978–5983 CrossRef CAS PubMed.
- J. Yuasa, A. Mitsui and T. Kawai, Chem. Commun., 2011, 47(20), 5807–5809 RSC.
- H. J. Avens and C. N. Bowman, J. Polym. Sci., Part A: Polym. Chem., 2009, 47(22), 6083–6094 CrossRef CAS PubMed.
- J. Xu, S. Shanmugam, H. T. Duong and C. Boyer, Polym. Chem., 2015, 6(31), 5615–5624 RSC.
- J. Qiu, T. Pintauer, S. G. Gaynor, K. Matyjaszewski, B. Charleux and J.-P. Vairon, Macromolecules, 2000, 33(20), 7310–7320 CrossRef CAS.
- K. Matyjaszewski, Curr. Opin. Solid State Mater. Sci., 1996, 1(6), 769–776 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101(9), 2921–2990 CrossRef CAS PubMed.
- X. Liu, L. Zhang, Z. Cheng and X. Zhu, Polym. Chem., 2016, 7(3), 689–700 RSC.
- R. Francis, B. Lepoittevin, D. Taton and Y. Gnanou, Macromolecules, 2002, 35(24), 9001–9008 CrossRef CAS.
- T. Higashihara, R. Faust, K. Inoue and A. Hirao, Macromolecules, 2008, 41(15), 5616–5625 CrossRef CAS.
- R. N. Kwak, K. Yungwan and K. Matyjaszewski, Macromolecules, 2008, 41(13), 4585–4596 CrossRef.
- N. D. Dolinski, Z. A. Page, E. H. Discekici, D. Meis, I. H. Lee, G. R. Jones, R. Whitfield, X. Pan, B. G. McCarthy and S. Shanmugam, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 268–273 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py00774a |
| This journal is © The Royal Society of Chemistry 2019 |