
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDegradable poly(ethylene oxide) through metal-free copolymerization of ethylene oxide with L-lactide†
Jobi Kodiyan
Varghese
a,
Nikos
Hadjichristidis
 b,
Yves
Gnanou
*a and
Xiaoshuang
Feng
*a
b,
Yves
Gnanou
*a and
Xiaoshuang
Feng
*a
aPhysical Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955, Kingdom of Saudi Arabia. E-mail: yves.gnanou@kaust.edu.sa; fxs101@gmail.com
bKAUST Catalysis Center, Physical Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955, Kingdom of Saudi Arabia
First published on 31st May 2019
Abstract
A simple and convenient method for the preparation of degradable poly(ethylene oxide) (PEO) is presented in this work. Through metal-free copolymerization of ethylene oxide with L-Lactide (LLA), very low content of LLA could be randomly incorporated into the backbone of PEO in the presence of triethylborane. In the presence of the latter Lewis acid, the reactivity of LLA could be curtailed, and transesterification reactions suppressed. The copolymerization of EO with LLA resulted in P(EO-co-LLA) samples of low to moderate content in ester units, and of controlled molar mass and low dispersity value. Reactivity ratios were determined using the non-terminal model (Beckingham-Sanoja-Lynd BSL equation) and the terminal model (Meyer–Lowry ML equation), respectively. The NMR characterization of the copolymer samples conbined with the kinetic treatment shows that non-terminal model describes adequately the copolymerization of EO with LLA. The resulting copolymers were further studied by differential scanning calorimetry (DSC); hydrolysis experiments were carried out to show the degradability of the prepared PEO samples containing a few percentage of ester units.
Introduction
Poly(ethylene oxide) (PEO), often referred to as poly(ethylene glycol), is a FDA-approved polymer for clinical use because of its unique properties such as its chemical stability, its hydrophilicity, its biocompatibility and above all its non-recognition by the immune system (stealth effect).1 The presence of functional groups at chain ends allows the conjugation of biologically active molecules with PEO (PEGylation). Thus, so-called PEGylated cargos could be transported to the target site without being recognized by the immune system. To get longer circulation time and better steric shielding effect, the hydrodynamic size of conjugates after PEGylation should be above 6–8 nm, the threshold of glomerular filtration, to avoid renal clearance. However, due to its non-degradability, the molar mass of PEO used does generally not exceed 40 kg mol−1 owing to its potential bioaccumulation in vivo.2To overcome this issue, much effort has been devoted to impart degradability to the chains of PEO by incorporating degradable linkages within their backbone.3 The most common strategy was through polycondensation of PEO telechelics, involving the incorporation of esters,4 disulfide,5 acetal,6 oxime, imine,7 carbonate8 linkages into PEO polycondensates. However, the latter PEO derivatives exhibited very broad distribution and ill-defined structures.
A classical strategy is to anionically copolymerize ethylene oxide with other monomers, and then introduce within PEO backbone degradable linkages. For instance, Lynd and coworkers copolymerized EO and epichlorohydrin followed by an efficient elimination reaction to generate degradable methylene ethylene oxide (MEO) repeat units within a PEO backbone.9 Similarly, Worm et al. first copolymerized EO with 3,4-epoxy-1-butene (EPB) via anionic ring-opening polymerization (AROP), and then isomerized the allyl moieties of EPB into pH-cleavable vinyl ethers.10 An alternate strategy involves post-oxidation of the prepared or commercially available PEO to generate hydrolysable linkages along the backbone. For instance hemiacetals were randomly introduced into the backbone of PEOs by Elisseeff et al. through Fenton reaction by hydrogen peroxide and ferric chloride at a neutral pH.11 Likewise, Liu and Bielawski reported a ruthenium-catalyzed post-polymerization oxyfunctionalization of PEGs generating acid-degradable poly[(ethylene glycol)-co-(glycolic acid)] copolymers.12 Although these last two approaches do give rise to degradable PEG with well-defined structures and low dispersities, they suffer from the following drawbacks: many steps are needed for the synthesis and compatibility issues with functional groups should be overcome during the post-polymerization step.
Polylactide (PLLA) is another important polymer, being widely utilized in the biomedical area due to its biocompatibility and degradability, as well as its availability from bioresources.13 However, because of its high crystallinity, hydrophobic nature and degradability PLLA has found biomedical applications different from those of PEO. Copolymerization of LLA with other monomers represents a general strategy to tune its physical properties for various biomedical applications. For instance di- or triblock copolymers have been obtained by sequential polymerization of various monomers and LLA.14 With respect to epoxide monomers and namely ethylene oxide, only a limited number of investigations are reported in the literature describing their copolymerizations with LLA. Besides the attempt to grow PLLA blocks from a PEO macroinitiator, Gross et al. mentioned the use of various Al and Sn–Al bimetallic catalysts to prepare LLA-EO multiblock copolymers exhibiting broad distributions.15 Lynd et al. resorting to the classical Vandenberg catalysts obtained random copolymers of LLA and EO of high molar mass.16 In the two above studies the main aim was to investigate the “copolymerizability” of LLA and epoxides using various coordinating catalysts and to characterize the type of copolymers eventually obtained: multiblock in the first case and random in the second case.
The objective of our work is different: it is to incorporate a low to very low percentage of LLA units within PEO chains by anionic copolymerization of EO with LLA, in order to impart degradability to these PEO chains without modifying their intrinsic properties of hydrophilicity, crystallinity, etc. In particular we studied the role of triethylborane (TEB) in the copolymerization of EO with LLA. Such boron-activated copolymerization of EO and LLA allows the synthesis of well-defined P(EO-co-LLA) samples exhibiting low dispersity value and a tunable content of EO and LLA units (Scheme 1).
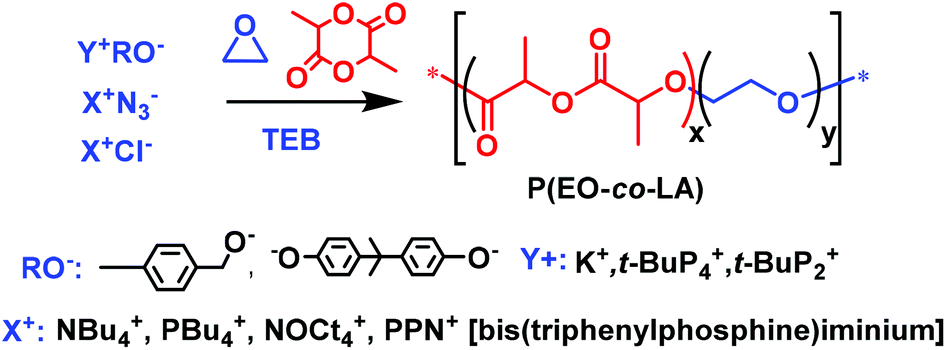 | ||
| Scheme 1 Anionic ring opening copolymerization of EO/LLA using TEB as activator. | ||
Experimental section
General methods
All reactions were carried out under a dry and oxygen-free argon atmosphere in a Braun Labmaster glovebox. Ethylene oxide (EO, ≥99.9%), L-lactide (LLA, 99%), diethylene glycol monomethyl ether, p-methyl benzyl alcohol (PMBA), bisphenol A (BPA), t-BuP4 (1.0 M in hexane), t-BuP2 (2.0 M in THF), triethylborane (1.0 M in THF), tetra butyl ammonium chloride (TBACl), bis(triphenylphosphine)iminium chloride (PPNCl), tetraoctyl ammonium chloride (TOACl), tetrabutyl phosphonium chloride (TBPCl), tetrabutyl ammonium azide (TBAA), allyl alcohol (Allyl A) were purchased from Aldrich. Tetrahydrofuran (THF) and toluene (Tol) were distilled over sodium/benzophenone mixture before used. 1,4-Dioxane was distilled over CaH2 after stirring two days. Ethylene oxide was purified by stirring over CaH2 for one day and distilled into flask containing n-BuLi, stirred for couple of hours followed by further distillation. LLA was purified by two times recrystallization from ethyl acetate followed by lyophilization from dry dioxane. Diethylene glycol monomethyl ether and allyl alcohol were purified by azeotropic distillation from benzene. PMBA and BPA were lyophilized from dioxane. Onium salt initiators were all dried over P2O5 under vacuum. All 1H and 13C NMR spectra were recorded on a Bruker AVANCE III-400 Hz instrument in CDCl3. GPC sample (3 mg mL−1 in THF) analysis was recorded by VISCOTEK VE2001 equipped with 2 Styragel HR2 THF (1 mL min−1) as eluent and RI as detector. Narrow Mw polystyrene standards were used to calibrate the instrument. DSC measurements were performed with a Mettler Toledo DSC1/TC100 under air. The samples were first heated from RT to 200 °C in order to erase the thermal history, then cooled to −100 °C, and finally heated again to 200 °C at a heating/cooling rate of 10 °C min−1. This cycle was repeated until constant melting and cooling temperatures (Tm and Tc) were recorded.Representative procedure for the synthesis of P(EO-co-LLA) using tetrabutylammonium chloride (TBACl) as initiator
A pre-dried 30 mL glass Schlenk tube (80 mm × 28 mm) composed of rotaflo stopcock and equipped with magnetic stirring bar was used to carry out this reaction. Under argon atmosphere, 86 μL of triethylborane (0.086 mmol) was first added to a solution of TBACl (4.8 mg, 0.017 mmol) in toluene (0.5 mL) in the glass Schlenk tube. In a side neck fitted with a septum LLA (100 mg, 0.69 mmol), ethylene oxide (150 mg, 3.47 mmol) and 1 mL of toluene were introduced, the formed solution was then added through stopcock into initiator-borane system. The polymerization was carried out at room temperature (25 °C) for 4 hours under stirring. Then the reaction was quenched with a few drops of 5% HCl in methanol and the polymer was precipitated in cold diethyl ether. The obtained polymer after filtration was dried in vacuum oven and characterized by GPC and NMR.Representative procedure for the synthesis of P(EO-co-LLA) using t-BuP4 initiator
A pre-dried 30 mL glass Schlenk tube (80 mm × 28 mm) composed of rotaflo stopcock and equipped with magnetic stirring bar was used to carry out this reaction. Under argon atmosphere, to a solution of PMBA (4.3 mg, 0.035 mmol) in toluene (0.5 mL), t-BuP4 solution (35 μL, 0.035 mmol) was charged into reaction flask. After stirring the medium for a few minutes under RT, triethylborane (176 μL, 0.176 mmol) was added. In a side neck fitted with a septum a solution of LLA (75 mg, 0.520 mmol) and ethylene oxide (152 mg, 3.47 mmol) in toluene (1 mL) was introduced similarly to the case of TBACl through stopcock into the initiator-borane system and the polymerization was carried out at room temperature for 1 hour under stirring. The reaction was quenched with a few drops of 5% HCl in methanol and precipitated in cold diethyl ether. The polymer obtained after filtration was dried in vacuum oven and characterized by GPC and NMR.Results and discussion
Since EO and LLA exhibit very different reactivities and since the monomer unit corresponding to LLA is prone to transesterification reactions under anionic conditions, they could never be copolymerized using even a mild base as initiator. It takes for instance 3 days to complete the polymerization of EO in the presence of an alcohol and a mild base such as t-BuP2 whereas only 1 minute is necessary to achieve the full conversion of LLA under the same conditions.17 This has been the main reason for using coordinative chemistry to copolymerize these two monomers. Catalytic processes which imply a necessary coordination step of the monomer have their advantages such as the production of long chains but they also have drawbacks such as chains that are not necessarily well-defined and are of broad molar mass distribution.15 In this study we propose a novel approach for the copolymerization of EO with LLA which is based not on purely anionic species but on an ate complex involving a Lewis acid, namely triethylborane, and a base typically an alkoxide.18 We actually resorted to similar ate complexes for the successful copolymerization of epoxides and CO2 without the formation of cyclic carbonates that are generally obtained by purely ionic species.19 Likewise boron-based ate complexes were found very efficient for initiating and bringing about a controlled polymerization of glycidyl azide, an epoxide monomer that could never be polymerized before.20 In each of the two above examples in addition to the boron-based ate complex, free trialkylboron had to be added to activate the monomer for the polymerization to occur as the growing ate complex was generally not nucleophilic enough.We first attempted to homopolymerize EO and LLA using PMBA/t-BuP4 as initiator system and TEB as Lewis acid to form the ate complex responsible for the polymerization. Both homopolymerizations were carried out in the presence of an excess of 5 eq. TEB to activate the monomer. In the case of EO the homopolymerizations occurred as expected affording samples with the expected molecular weights either in THF or toluene: clearly the presence of free TEB was essential to trigger the polymerization (entries 1 and 2, Table 1). In contrast, hardly any homopolymerization could be observed in the case of LLA (Scheme 2), in spite of the presence of 5 eq. of excess of TEB (conversion below 1%) and addition of further excess of TEB didn't help to increase the conversion of LLA (entries 3 and 4, Table 1).
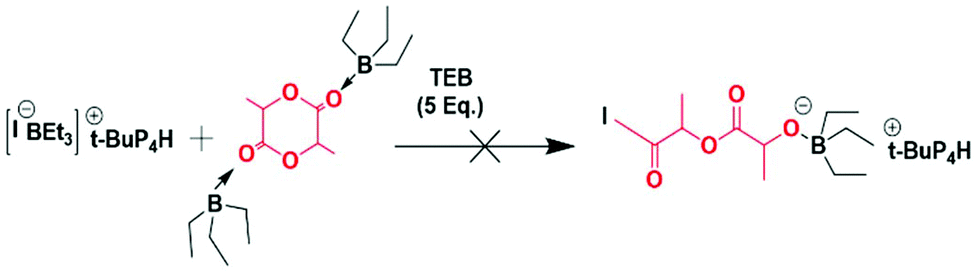 | ||
| Scheme 2 Unsuccessful attempts at homopolymerizing LLA in the presence of excess triethylborane, I representing the initiator. | ||
| Entry no. | EO/LLA (feeding) | [EO + LLA]/[initiator] | Time (min) | Solvent | Yield (%) | F LLA (%) | M n(theo) | M n(NMR) | M n(GPC) | PDI |
|---|---|---|---|---|---|---|---|---|---|---|
| a All the copolymerizations were performed at room temperature with molar ratio of 5 of TEB to initiator, PMBA: p-methyl benzyl alcohol, P4: t-BuP4, P2: t-BuP2. MDEG: diethylene glycol monomethyl ether, BPA: bisphenol A, Allyl A: allyl alcohol, TBACl: tetrabutyl ammonium chloride, TOACl: tetraoctyl ammonium chloride, TBPCl: tetrabutyl phosphonium chloride, TBAA: tetrabutyl ammonium azide. b M n(theo) = (mp/NI), mp = total weight of polymer recovered, NI = mole of initiator. c Ester content and Mn(NMR) calculated based on 1H NMR. d GPC determined with THF as eluent and calibrated by polystyrene standards. | ||||||||||
| 1 | 600/0 | PMBA/P4 (600/1) | 10 | THF | 54 | 0 | 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 800 |
15100 |
13100 |
1.28 |
| 2 | 500/0 | PMBA/P4 (500/1) | 30 | Tol | 95 | 0 | 19400 |
24000 |
21000 |
1.20 |
| 3 | 0/100 | PMBA/P4 (100/1) | 960 | THF | 0 | 0 | — | — | — | — |
| 4 | 0/100 | PMBA/P4 (100/1) | 960 | Tol | 0 | 0 | — | — | — | — |
| 5 | 87/13 | MDEG/P4 (300/1) | 180 | THF | 40 | 7.1 | 9200 | 15100 |
5900 | 1.17 |
| 6 | 88/12 | PMBA/P4 (57/1) | 15 | Tol | 53 | 1.1 | 1700 | 2700 | 5200 | 1.19 |
| 7 | 87/13 | PMBA/P4 (115/1) | 45 | Tol | 65 | 1.2 | 4100 | 4700 | 7000 | 1.11 |
| 8 | 86/14 | PMBA/P4 (175/1) | 75 | Tol | 63 | 2.0 | 6400 | 6600 | 9500 | 1.18 |
| 9 | 87/13 | PMBA/P4 (230/1) | 120 | Tol | 80 | 3.6 | 10000 |
10600 |
11100 |
1.14 |
| 10 | 87/13 | PMBA/P4 (345/1) | 150 | Tol | 70 | 2.9 | 12000 |
12200 |
14700 |
1.13 |
| 11 | 83/17 | PMBA/P4 (360/1) | 360 | Tol | 66 | 7.2 | 13500 |
11600 |
10900 |
1.11 |
| 12 | 88/12 | PMBA/P4 (570/1) | 180 | Tol | 80 | 3.0 | 21000 |
23800 |
25600 |
1.10 |
| 13 | 88/12 | BPA/P4 (570/1) | 180 | Tol | 68 | 3.1 | 21200 |
20100 |
22000 |
1.26 |
| 14 | 93/7 | BPA/P4 (1290/1) | 960 | Tol | 80 | 4.3 | 52200 |
48500 |
50700 |
1.05 |
| 15 | 87/13 | PMBA/P2 (115/1) | 120 | Tol | 64 | 5.2 | 4200 | 5700 | 7000 | 1.17 |
| 16 | 83/17 | TBACl (240/1) | 840 | Tol | 91 | 14 | 13000 |
— | 13100 |
1.17 |
| 17 | 83/17 | TBACl (240/1) | 120 | Tol | 25 | 7.2 | 3500 | — | 6900 | 1.20 |
| 18 | 83/17 | PPNCl (360/1) | 180 | Tol | 38 | 5.1 | 8100 | — | 9700 | 1.19 |
| 19 | 83/17 | TOACl (360/1) | 420 | Tol | 37 | 8.8 | 7600 | — | 11800 |
1.11 |
| 20 | 83/17 | TBPCl (360/1) | 210 | Tol | 20 | 8.7 | 3800 | — | 6800 | 1.17 |
| 21 | 83/17 | TBAA (360/1) | 300 | Tol | 39 | 11 | 8800 | — | 9500 | 1.12 |
| 22 | 83/17 | Allyl A/P4 (240/1) | 150 | Tol | 67 | 3.0 | 9000 | 7800 | 14800 |
1.13 |
Interestingly a monomer like LLA which homopolymerizes very fast when subjected to purely ionic species stayed “put” in the presence of ate complexes and failed to undergo ring-opening when added to boron-based ate complexes. We then investigated the copolymerization of LLA with EO in the presence of 5 eq. of TEB. 15–20 mol% of LLA were fed in the reaction medium in order to see whether a low content of ester could be incorporated into the PEO backbone. In all the following experiments, toluene, an apolar solvent, was used in the copolymerizations of EO with LLA. After polymerization the reaction mixture was poured in cold ether to collect all the produced polymer and characterized by GPC and NMR. A representative 1H NMR spectrum is shown in Fig. 1. The characteristic peaks of LLA and of EO units could be clearly detected at 5.2, 1.5 ppm (peaks a, b) and at 3.5 ppm (peak c) indicating the incorporation of the ester units. The peak g at 4.3 ppm indeed corresponds to the methylene protons of EO and to the methine protons of LLA connected to LLA units and to EO units CH2CH2O(O)CCH(CH3)O(O)CCH(CH3)O–, respectively. The peak h at 4.10 ppm corresponds to the methine protons of LLA units connected to both LLA and EO units –(O)CCH(CH3)O(O)CCH(CH3)OCH2CH2O–, as shown in Fig. 1, and unambiguously interpreted and assigned by Gross et al.15 The presence of connection peaks characteristic of PLLA units flanked by two EO units indicates the formation of a random copolymer. Further characterization by 13C NMR (Fig. S1†) reveals the absence of any signal in the range of 174–175 ppm [terminal carbonyl atom HOCH(CH3)C(O)OCH(CH3)CO–],21 indicating the absence of homopolymerization of LLA and its homopolymer PLLA; the correlation spectra of heteronuclear single quantum coherence (HSQC) and heteronuclear multiple bond correlation (HMBC) also confirm the formation of copolymer PEO-co-PLLA (Fig. S2 and 3†). In addition DOSY characterization clearly indicates the absence of homopolymers and the copolymer nature of the polymer chains formed (Fig. S4†). The integral ratio of peak g to h in Fig. 1 is close to 3, indicating negligible transesterifications of LLA. Based on the NMR data, the content of LLA units could then be calculated and the molar mass of the obtained copolymer estimated using the peaks of p-methylbenzene alcohol (d, e, f at 4.5, 7.1, 2.3 ppm) initiator as reference (please refer to related data listed in Table 1). The average segment length of PLLA was determined to be equal to 1.98 by the equation LLLA = (SI5.21 ppm + 2SI4.10 ppm)/2SI4.10 ppm,15 where SI is the integral intensity of the respective peaks. This means that on average less than 2 units of LLA could be found adjacent along the polymer backbone, indicative of a very low value of the reactivity ratio of LLA, rLLA (Fig. 1). Following the same procedure, the polymerization was initiated by the system PMBA/t-BuP4 in the presence of TEB and different molar masses were targeted (entries 6–14, Table 1). The values of molar mass obtained from NMR for the various samples are close to the theoretical ones; a molar mass as high as 50 kg mol−1 (entry 14, Table 1) could be reached and ester contents in all cases were kept around 5%. Upon changing the feeding ratio of LLA to EO, the content in ester in the obtained copolymer could be varied (entry 11, Table 1). Analysis by GPC of the copolymer samples obtained shows unimodal traces with a narrow distribution of molar masses (Fig. 2); the latter being close the theoretical values and to ones generated from NMR calculations. We could thus demonstrate that under these conditions EO and LLA could be copolymerized in a “controlled” manner and transesterification could be totally suppressed.
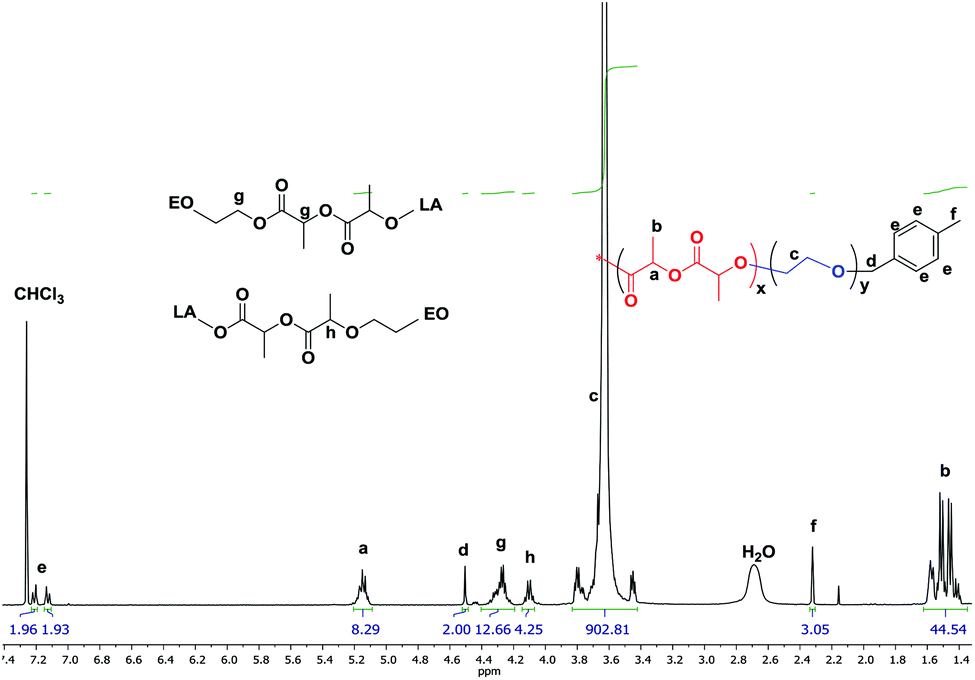 | ||
| Fig. 1 Representative 1H NMR spectrum of P(EO-co-LLA) random copolymer (entry 9 of Table 1). | ||
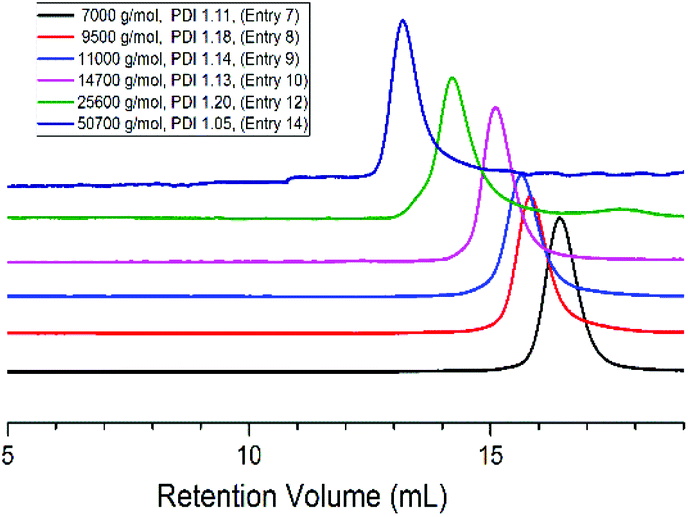 | ||
| Fig. 2 GPC traces of various copolymer samples targeted ethylene oxide from 100 DP to 1200 DP (Table 1). | ||
To our knowledge this is not only the first successful attempt at copolymerizing monomers under “controlled” conditions as different as EO and LLA but this investigation also proves that very small amounts of ester linkages can be incorporated in polyether chains, a feat never achieved so far. When carried out in THF (entry 5, Table 1), a slightly polar solvent, we observed a loss of control of the molar mass of copolymer sample, indicating the occurrence of transesterification reactions and thus very likely of back-biting reactions.
Apart from the polymerizations initiated by the system PMBA/tBuP4, other organic initiators ammonium and phosphonium halides like TBACl, TBPCl were also utilized in the presence of TEB for the copolymerizations. Similar results were obtained, but the ester content within the isolated copolymer tends to be slightly higher than for the copolymers generated from the alkoxide/tBuP4 system (entries 16 to 20, Table 1). This may be due to the difference of reactivity between EO and LLA in the presence of the various cations associated with alkoxides (vide infra). Using various initiators, we also prepared difunctional and hetero-difunctional copolymers. For instance, upon choosing bisphenol A as initiator, two hydroxyl-ended PEO could be obtained including 3–4% ester content with a molar mass up to 50.7 kg mol−1 (entries 13 and 14 of Table 1); upon starting from allyl alcohol and tetrabutyl azide as initiators (entries 21 and 22, Table 1), we could generate copolymer samples carrying vinyl and azide end groups (Fig. S5†), which are interesting and powerful functional groups for biological conjugation and applications.
To identify the nature of the copolymer formed, kinetic data were collected and monomer conversions were measured under the same initial feeding ratio, using PMBA/tBuP4, TBACl and PPNCl as initiators. The related polymerization data are listed in Table 2. With increasing polymerization time, the ester content gradually increased indicating the formation of gradient-type copolymers as EO was consumed much faster than LLA. The reactivity ratio rEO and rLLA could be calculated and the tendency of self-propagation or incorporation of the other monomer could be determined by terminating the polymerization at different intervals and analyzing the composition of the corresponding copolymer. Various methods of determination of reactivity ratios are available including Mayo–Lewis,22 Fineman–Ross23 and Kelen–Tüdos24etc., but these methods are only applicable at low conversion. Given the low reactivity of LLA and thus the very modest incorporation of the latter monomer in the copolymer at low conversions, we decided not to use these so-called “instantaneous” non-integrated models due to the inaccuracy of the results obtained. We thus resorted to integrated methods of data analysis such as the non-terminal model of Beckingham et al. (BSL)25 and the terminal model of Meyer–Lowry (ML)26 under ideal (gradient category) condition for the determination of the reactivity ratios rEO and rLLA of the two monomers. The measurement of reactivity ratios with these two models was well summarized very recently by Frey et al.27 The non-terminal model assumes that the reactivity of the propagating species only depends on the reactivity of the incoming monomer, and ignores the nature of the last monomer featuring the active species; in contrast the terminal model assumes that the reactivity is influenced by the terminal monomer unit. Both models are applicable up to full conversion. As recommended in a recent contribution of Lynd et al.25b we simultaneously determined the reactivity ratios of EO and LLA using both BSL and ML equations. The reactivity ratios calculated according to BSL equation of the non-terminal model yield the following values as shown in Table 3: rLLA = 0.17 ± 0.04, rEO = 5.37± 0.4 for P4+ as counter cation, rLLA = 0.49 ± 0.08, rEO = 2.07± 0.25 for TBA+ and rLLA = 0.14 ± 0.01, rEO = 6.61± 0.67 for PPN+ (see ESI† and Table 3). In the three cases investigated with three different cations (P4+, TBA+, PPN+) the product of reactivity ratios rEO × rLLA is very close to one, confirming the character by 1H NMR which indicated the formation of gradient copolymers. We also resorted to the terminal model of ML for the determination of the reactivity ratios, rEO and rLLA. Assuming that the copolymers formed are of gradient nature with no tendency to blockiness or alternation. We derived a simple relation of reactivity ratios as function of the conversion upon starting from generic, conversion dependent version of the copolymer equation of Meyer and Lowry (see ESI† for the derivation). The terminal model of ML thus affords for the copolymerization of EO with LLA in the presence of TEB the following values of reactivity ratios: rLLA = 0.19 ± 0.02, rEO = 5.15 ± 0.56 for P4+, rLLA = 0.50 ± 0.07, rEO = 2.03 ± 0.27 for TBA+ and rLLA = 0.15 ± 0.01, rEO = 6.49 ± 0.46 for PPN+ (Table 3). The graphs of conversion vs. fEO were plotted using the Meyer–Lowry equation which could be mathematically simplified after assuming the formation of gradient copolymers (see Fig. S6–S8†).26 As shown in Table 3, the reactivity ratios obtained from these two models are quite close and consistent. We can then conclude, following the recommendation of Lynd et al. in their recent publication,25b that the non-terminal model (BSL) describes adequately the copolymerization of EO with LLA in the presence of TEB. This is not surprising as the non-terminal model is known to be appropriate for ionic or pseudo-ionic mechanism.
| Entry no. | EO/LLA (feeding) | [EO + LLA]/initiator | Time (min) | Yield (%) | Esterc (%) | M n(theo) | M n(NMR) | M n(GPC) | PDI |
|---|---|---|---|---|---|---|---|---|---|
| a All the copolymerizations were performed in toluene at room temperature with molar ratio of 5 of TEB to initiator, PMBA: p-methyl benzyl alcohol, P4: t-BuP4, TBACl: tetrabutyl ammonium chloride, PPNCl: bis(triphenylphosphine)iminium chloride. b M n(theo) = (mp/NI), mp = total weight of polymer recovered, NI = mole of initiator. c Ester content and Mn(NMR) calculated based on 1H NMR. d GPC determined with THF as eluent and calibrated by polystyrene standards. | |||||||||
| 1 | 87/13 | PMBA/P4 (575/1) | 60 | 20 | 2.5 | 11900 |
10900 |
14500 |
1.04 |
| 2 | 87/13 | PMBA/P4 (575/1) | 120 | 33 | 3.1 | 13600 |
12600 |
16800 |
1.13 |
| 3 | 87/13 | PMBA/P4 (575/1) | 180 | 50 | 4.1 | 15600 |
14000 |
17300 |
1.10 |
| 4 | 87/13 | PMBA/P4 (575/1) | 300 | 61 | 5.0 | 20500 |
20900 |
22300 |
1.12 |
| 5 | 87/13 | PMBA/P4 (575/1) | 540 | 75 | 6.1 | 22400 |
22800 |
24700 |
1.14 |
| 6 | 83/17 | TBACl (240/1) | 120 | 35 | 9.2 | 3500 | — | 6900 | 1.15 |
| 7 | 83/17 | TBACl (240/1) | 240 | 48 | 10.1 | 7000 | — | 9800 | 1.19 |
| 8 | 83/17 | TBACl (240/1) | 360 | 61 | 11.5 | 8800 | — | 11100 |
1.09 |
| 9 | 83/17 | TBACl (240/1) | 480 | 71 | 13.1 | 10300 |
— | 11500 |
1.18 |
| 10 | 83/17 | TBACl (240/1) | 840 | 84 | 14.5 | 13000 |
— | 13100 |
1.17 |
| 11 | 83/17 | PPNCl (240/1) | 90 | 38 | 3.5 | 5500 | — | 7000 | 1.07 |
| 12 | 83/17 | PPNCl (240/1) | 180 | 51 | 4.5 | 7500 | — | 9100 | 1.19 |
| 13 | 83/17 | PPNCl (240/1) | 300 | 62 | 6.0 | 9000 | — | 11000 |
1.17 |
| 14 | 83/17 | PPNCl (240/1) | 360 | 70 | 7.1 | 10200 |
— | 11500 |
1.11 |
| 15 | 83/17 | PPNCl (240/1) | 450 | 74 | 8.1 | 10500 |
— | 12200 |
1.16 |
| No. | Initiator | BSL model | ML model | ||
|---|---|---|---|---|---|
| r EO | r LLA | r EO | r LLA | ||
| 1 | PMBA/P4 | 5.37 ± 0.40 | 0.17 ± 0.04 | 5.15 ± 0.56 | 0.19 ± 0.02 |
| 2 | TBACl | 2.07 ± 0.25 | 0.49 ± 0.08 | 2.03 ± 0.27 | 0.50 ± 0.07 |
| 3 | PPNCl | 6.61 ± 0.67 | 0.14 ± 0.01 | 6.49 ± 0.46 | 0.15 ± 0.01 |
As proposed in Scheme 3, in the presence of an excess of TEB, the propagating ate complex is preferably attacking the activated EO; on the other hand, LLA is not activated enough by TEB, and therefore fails to extensively react with the growing ate complex. Copolymers P(EO-co-LLA) containing low to moderate ester LLA linkages are thus formed.
 | ||
| Scheme 3 Copolymerization mechanism of ethylene oxide and L-lactide in the presence of an excess of triethylborane (I representing the initiator). | ||
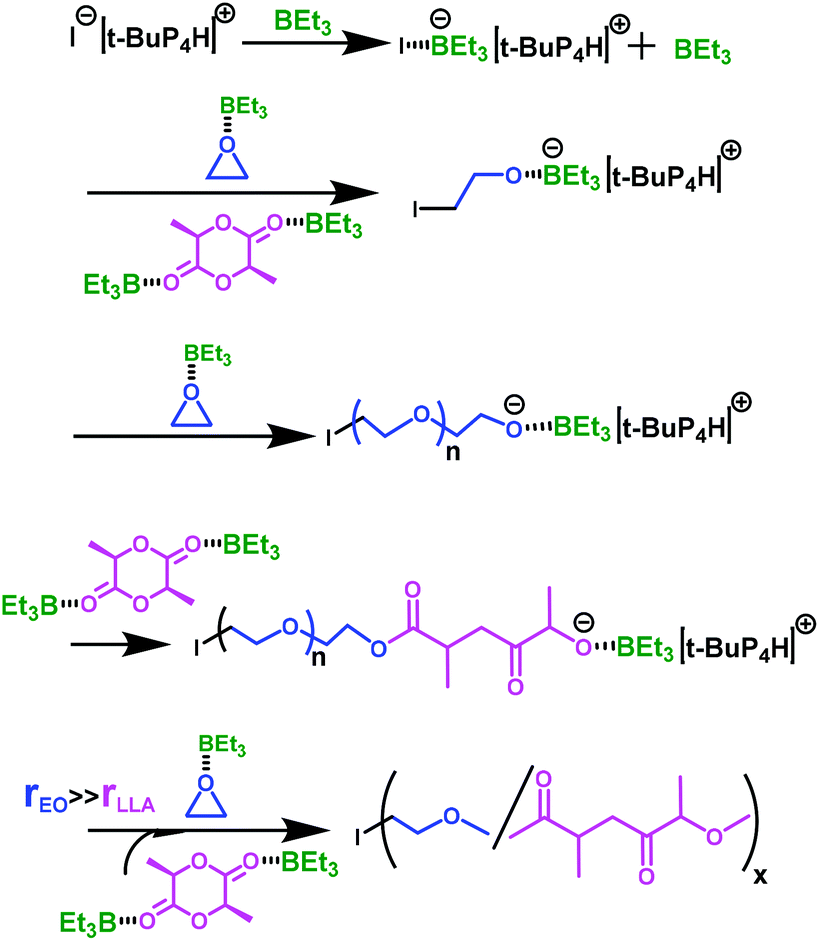
Since one of the aim of this investigation was to control as precisely as possible the incorporation of ester units in the PEO chains and if possible to a limited percentage (∼5%) to impart PEO chains with degradability, we decided to check the thermal properties of the copolymer samples obtained by DSC. The melting transitions of PEO were all detected, and compared to those of pure PEO; the melting temperature (Tm) of the copolymers obtained gradually decreased with a higher incorporation of ester units into the PEO backbone as expected. With the incorporation of 3% of ester Tm reduced to 52.3 °C from 58.9 °C. Upon further increasing the content of ester linkages to 7% Tm decreased to 38.5 °C and then to 28.6 °C for 14% incorporation of ester units (Fig. 3). Especially, in the latter case due to the presence of more ester units, a pronounced cold crystallization transition28 at −17.6 °C was detected. However, no melting transition of PLLA was detected, even for the sample containing 14% of ester, indicating that very short PLLA segments were incorporated along the PEO backbone. As a comparison, a melting temperature (Tm) due to PLLA was clearly detected in the case of Gross’ multiblock P(EO-co-LLA) copolymers which contained 17% of ester units.15
 | ||
| Fig. 3 DSC traces of copolymers P(EO-co-LLA) with different ester compositions. | ||
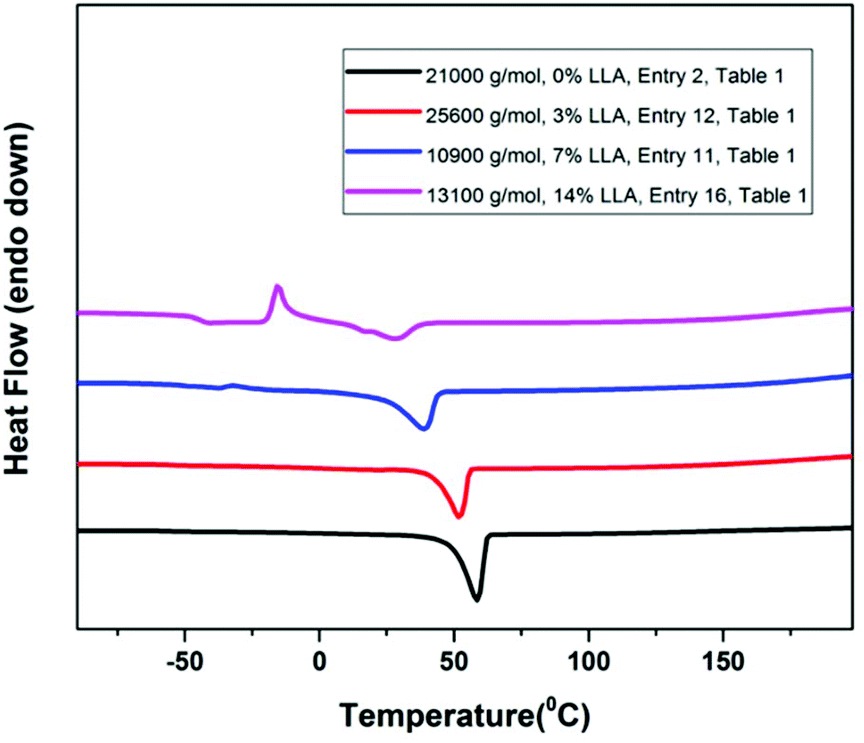
Lastly the degradation of P(EO-co-LLA) was performed to check the average length of PEO segments after hydrolysis. The copolymer was dissolved in 40:60 methanol:water solution containing 0.5 M NaOH, and stirred for two days to break the ester linkages. The polymer recovered through extraction by dichloromethane after such treatment was characterized by 1H NMR which clearly showed the complete degradation and disappearance of ester linkages (Fig. S9†). The molar mass of PEO after degradation was directly concentrated from the reaction mixture and analyzed by GPC. As exemplified in Fig. 4, the copolymer samples exhibiting an initial molar mass of 50.7 kg mol−1, 25.6 kg mol−1 and 14.7 kg mol−1 (Fig. 2) were respectively reduced to 3 kg mol−1, 3 kg mol−1 and 2.6 kg mol−1 after degradation.
 | ||
| Fig. 4 GPC traces overlay of copolymer P(EO-co-LLA) before and after degradation (entries 10, 12 and 14, Table 1). | ||
Conclusions
Through metal-free ring-opening copolymerization of EO and LLA, degradable poly(ethylene oxide)s could be directly prepared in a controlled way with a low dispersity value and a well-defined structure. The presence of TEB selectively increased the reactivity of EO, and suppressed transesterification reactions. This general method can be applied not only to synthesize functionalized linear PEOs, but also branched PEOs with high molar mass without concern of the degradability issue. In addition, a metal-free synthesis gives more credit to this approach for biomedical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work is supported by KAUST under baseline funding (BAS/1/1374-01-01).Notes and references
- (a) K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed; (b) J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed; (c) A. A. D'souza and R. Shegokar, Expert Opin. Drug Delivery, 2016, 13, 1257–1275 CrossRef PubMed.
- R. Webster, E. Didier, P. Harris, N. Siegel, J. Stadler, L. Tilbury and D. Smith, Drug Metab. Dispos., 2007, 35, 9–16 CrossRef CAS PubMed.
- C. Dingels and H. Frey, in Hierarchical Macromolecular Structures: 60 Years after the Staudinger Nobel Prize II, ed. V. Percec, Springer International Publishing, Cham, 2013, pp. 167–190, DOI:10.1007/12_2013_235.
- A. Mero, O. Schiavon, G. Pasut, F. M. Veronese, E. Emilitri and P. Ferruti, J. Bioact. Compat. Polym., 2009, 24, 220–234 CrossRef CAS.
- (a) J. Lee, M. K. Joo, J. Kim, J. S. Park, M.-Y. Yoon and B. Jeong, J. Biomater. Sci., Polym. Ed., 2009, 20, 957–965 CrossRef CAS PubMed; (b) W. Na, D. Anjie, R. Maciej and S. Youqing, J. Biomed. Mater. Res., Part A, 2008, 84, 148–157 Search PubMed; (c) B. Alena, P. Michal, L. Richard and U. Karel, Macromol. Chem. Phys., 2007, 208, 2642–2653 CrossRef.
- (a) C. Dingels, S. S. Müller, T. Steinbach, C. Tonhauser and H. Frey, Biomacromolecules, 2013, 14, 448–459 CrossRef CAS PubMed; (b) X. Feng, E. L. Chaikof, C. Absalon, C. Drummond, D. Taton and Y. Gnanou, Macromol. Rapid Commun., 2011, 32, 1722–1728 CrossRef CAS PubMed; (c) R. Tomlinson, M. Klee, S. Garrett, J. Heller, R. Duncan and S. Brocchini, Macromolecules, 2002, 35, 473–480 CrossRef CAS.
- J. Collins, Z. Xiao and L. A. Connal, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3826–3831 CrossRef CAS.
- L. Meabe, H. Sardon and D. Mecerreyes, Eur. Polym. J., 2017, 95, 737–745 CrossRef CAS.
- P. Lundberg, B. F. Lee, S. A. van den Berg, E. D. Pressly, A. Lee, C. J. Hawker and N. A. Lynd, ACS Macro Lett., 2012, 1, 1240–1243 CrossRef CAS PubMed.
- M. Worm, D. Leibig, C. Dingels and H. Frey, ACS Macro Lett., 2016, 5, 1357–1363 CrossRef CAS.
- B. Reid, S. Tzeng, A. Warren, K. Kozielski and J. Elisseeff, Macromolecules, 2010, 43, 9588–9590 CrossRef CAS PubMed.
- D. Liu and C. W. Bielawski, Macromol. Rapid Commun., 2016, 37, 1587–1592 CrossRef CAS PubMed.
- (a) J. Pretula, S. Slomkowski and S. Penczek, Adv. Drug Delivery Rev., 2016, 107, 3–16 CrossRef CAS PubMed; (b) A. Michalski, M. Brzezinski, G. Lapienis and T. Biela, Prog. Polym. Sci., 2019, 89, 159–212 CrossRef CAS.
- (a) K. K. Bawa and J. K. Oh, Mol. Pharm., 2017, 14, 2460–2474 CrossRef CAS PubMed; (b) J. K. Oh, Soft Matter, 2011, 7, 5096–5108 RSC.
- X. Chen, S. P. McCarthy and R. A. Gross, Macromolecules, 1997, 30, 4295–4301 CrossRef CAS.
- M. Chwatko and N. A. Lynd, Macromolecules, 2017, 50, 2714–2723 CrossRef CAS.
- J. Zhao, D. Pahovnik, Y. Gnanou and N. Hadjichristidis, Polym. Chem., 2014, 5, 3750–3753 RSC.
- (a) C. Billouard, S. Carlotti, P. Desbois and A. Deffieux, Macromolecules, 2004, 37, 4038–4043 CrossRef CAS; (b) C. J. Zhang, H. Y. Duan, L. F. Hu, C. H. Zhang and X. H. Zhang, ChemSusChem, 2018, 11, 4209–4213 CrossRef CAS PubMed; (c) Y. Chen, J. Shen, S. Liu, J. Zhao, Y. Wang and G. Zhang, Macromolecules, 2018, 51, 8286–8297 CrossRef CAS.
- D. Zhang, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, J. Am. Chem. Soc., 2016, 138, 11117–11120 CrossRef CAS PubMed.
- S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, Nat. Commun., 2019, 10, 293 CrossRef PubMed.
- (a) I. Rashkov, N. Manolova, S. M. Li, J. L. Espartero and M. Vert, Macromolecules, 1996, 29, 50–56 CrossRef CAS; (b) J. L. Espartero, I. Rashkov, S. M. Li, N. Manolova and M. Vert, Macromolecules, 1996, 29, 3535–3539 CrossRef CAS.
- F. R. Mayo and F. M. Lewis, J. Am. Chem. Soc., 1944, 66, 1594–1601 CrossRef CAS.
- M. Fineman and S. D. Ross, J. Polym. Sci., 1950, 5, 259–262 CrossRef CAS.
- T. Kelen and F. Tüdos, J. Macromol. Sci., Part A, 1975, 9, 1–27 CrossRef.
- (a) B. S. Beckingham, G. E. Sanoja and N. A. Lynd, Macromolecules, 2015, 48, 6922–6930 CrossRef CAS; (b) N. A. Lynd, R. C. Ferrier and B. S. Beckingham, Macromolecules, 2019, 52, 2277–2285 CrossRef CAS.
- (a) V. E. Meyer and G. G. Lowry, J. Polym. Sci., Part A: Gen. Pap., 1965, 3, 2843–2851 CrossRef CAS; (b) F. T. Wall, J. Am. Chem. Soc., 1944, 66, 2050–2057 CrossRef CAS.
- J. Blankenburg, E. Kersten, K. Maciol, M. Wagner, S. Zarbakhsh and H. Frey, Polym. Chem., 2019 10.1039/C9PY00500E.
- S. Kripotou, C. Psylla, K. Kyriakos, K. N. Raftopoulos, J. Zhao, G. Zhang, S. Pispas, C. M. Papadakis and A. Kyritsis, Macromolecules, 2016, 49, 5963–5977 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data, IR, reactivity ratio calculation, Fig. S1–S9. See DOI: 10.1039/c9py00605b |
| This journal is © The Royal Society of Chemistry 2019 |