
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect comparison of solution and solid phase synthesis of sequence-defined macromolecules†
Joshua O.
Holloway‡
 a,
Katharina S.
Wetzel‡
ab,
Steven
Martens
a,
Filip E.
Du Prez
*a and
Michael A. R.
Meier
*b
a,
Katharina S.
Wetzel‡
ab,
Steven
Martens
a,
Filip E.
Du Prez
*a and
Michael A. R.
Meier
*b
aPolymer Chemistry Research Group, Centre of Macromolecular Chemistry (CMaC), Department of Organic and Macromolecular Chemistry, Faculty of Sciences, Ghent University, Krijgslaan 281 S4bis, Campus Sterre, Ghent, B-9000, Belgium. E-mail: Filip.DuPrez@UGent.be
bKarlsruhe Institute of Technology (KIT, Institute of Organic Chemistry (IOC)), Materialwissenschaftliches Zentrum für Energiesyteme (MZE), Straße am Forum 7, 76131 Karlsruhe, Germany. E-mail: m.a.r.meier@kit.edu
First published on 5th June 2019
Abstract
The synthesis of perfectly defined, monodisperse macromolecules is one of the challenges faced by polymer chemists today. Such precision synthesis requires a fundamentally different approach to conventional polymer synthesis, but in turn can unlock the door to many new applications. Therefore, we introduce here the combination of ultra-fast “click” reactions using 1,2,4-triazoline-3,5-diones (TAD) with the highly efficient and versatile Passerini three-component reaction. This new approach not only resulted in the synthesis of monodisperse, sequence-defined macromolecules of high purity and molecular weight (>7000 Da), but also offered new insights into the iterative synthesis of sequence-defined macromolecules in general, as we present a detailed comparative study of the same chemistry protocols carried out on solid phase as well as in solution.
Introduction
Over the last decade, the field of sequence-control in polymer chemistry has evolved to become a hot research topic, as researchers worldwide strive to take inspiration from nature to synthesise perfectly defined, monodisperse macromolecules.1–6 As such, this field is now rapidly growing and resulting in various new routes towards precision macromolecular synthesis and potential applications thereof.7–21 In this context, it is important to distinguish between the terms “sequence-controlled” and “sequence-defined”, whereby only the latter offers unique, monodisperse macromolecules, while sequence-controlled systems still show dispersity in many aspects.22,23 Most sequence-defined macromolecules tend to be oligomers as opposed to polymers and thus extending reachable degrees of polymerisation as well as scalability are issues of high relevance. Conventional polymerisation techniques on the other hand are scalable, but lead to irregularity and dispersity and thus, at best, can be considered to be sequence-controlled, depending on the polymerisation mechanism.6,24A common route towards sequence-defined oligomers, offering full control over each monomer unit, is the iterative synthesis approach.7,25–32 This step-by-step growth of the macromolecule is necessary to ensure a perfectly defined sequence as well as monodispersity. Whilst many different approaches exist, the use of multi-component reactions seems to be a logical choice within this area and such reactions have indeed been shown to be highly effective tools for the synthesis of sequence-defined macromolecules.17,18,33–40 In particular, the Passerini three-component reaction (P-3CR, Scheme 1, top left) between an acid, an aldehyde and an isocyanide, first discovered in 1921,41 has been reported to be a highly effective technique in sequence-defined oligomer synthesis.33–36
 | ||
| Scheme 1 Two-step iterative reaction cycle consisting of the P-3CR and the TAD Diels–Alder reaction, which can be applied to synthesise sequence-defined macromolecules on solid phase and in solution. Box top left: General reaction scheme of the P-3CR. Box top right: Irreversible reactions of TAD. | ||
On the other hand, “click” chemistry, a term first introduced by Sharpless and coworkers in 2001,42 is also an ideal concept for sequence-defined synthesis.1,2 Its main principles of high yields, being wide in substrate scope, production of inoffensive by-products and easy purification are all desirable to an iterative approach towards synthesising highly defined macromolecules. One notable example of click chemistry is 1,2,4-triazoline-3,5-dione (TAD) chemistry.43 The fast and efficient irreversible reactions of TAD (Scheme 1, top right), signified by a visual feedback on account of the vivid red colour of the TAD molecule,43–45 make it an ideal component of click chemistry for a wide range of applications46,47 and also a suitable reaction partner for sequence-defined chemistry as demonstrated here for the first time. Depending on the substrate, the irreversible reactions of TAD can vary from less than one second to several hours.43,44 Thus, by synthesising a carboxylic acid functionalised TAD compound, this click chemistry – in this case via the Diels–Alder reaction (Scheme 1, top right) – could be combined with the P-3CR. This has enabled us to present here a highly effective, novel strategy for the synthesis of sequence-defined macromolecules. The use of the Diels–Alder reaction in sequence-defined chemistry remains very limited to date, with only a few examples existing.16,40,48,49 Not only will we report on the powerful combination of multicomponent reactions with click chemistry, but simultaneously we will describe, for the first time in this research area, a comprehensive comparative study of solution and solid phase chemistry approaches (see Scheme 1). These two techniques are undoubtedly both valuable tools for chemists in the pursuit of precise macromolecular control sequence-definition and their direct comparison is long overdue. We discuss here the advantages and disadvantages of both techniques, using these two chemistries as a specific example and demonstrate that the combination of TAD Diels–Alder chemistry with the P-3CR provides a powerful tool for achieving high molecular weight, high purity, multi-functional, monodisperse macromolecules. Moreover, we show here that the introduction of TAD chemistry to the P-3CR has had a profound effect on both its efficiency and speed of the synthesis.
As mentioned above, sequence-defined iterative synthesis protocols can be carried out in two ways. One option is via a Merrifield inspired50 solid phase approach, whereby polymer resins are functionalised with a typically acid sensitive cleavable linker, thus allowing the sequence to be grown on the resin before being recovered at the end.7 Although usually limited in scale, this approach is particularly advantageous because of simple purification by washing and the potential to automate such a method.7,31,51 The other option is solution phase chemistry. Whilst this more scalable approach facilitates the characterisation and thus optimisation and understanding of each reaction step, purification can be more time-consuming. Alternatively, synthesis can be carried out using soluble polymer supports,52–54 such as polystyrene and purified each time via precipitation. Also, the sequence can be synthesised in solution without any support and purification can be done, for example, by column chromatography.17,33–35,55 Whilst a few reports of the use of similar chemistries on both solid and solution phase approaches exist, to the best of our knowledge, none make a full and direct comparison of the two, showcasing both the advantages and disadvantages.52,56–58 We believe that this is a much needed study as the choice of whether to opt for solid or solution phase synthesis when referring to sequence-defined protocols is always difficult, because it is highly dependent on the end goal of the work and the desired scale, purity and length of the macromolecule.
Results and discussion
Functional AB-type linker molecules
To combine the P-3CR with TAD Diels–Alder reactions, two AB-type linker molecules (L1 and L2) were synthesised (see ESI† for experimental details) and applied in a two-step iterative cycle (see Scheme 1), resulting in sequence-defined macromolecules. L1, equipped with an isocyanide and a conjugated diene, was synthesised via a four-step protocol (see Scheme S1†). The synthesis was carried out on 15-gram scale with an overall yield of 65%. L2, containing both the TAD and carboxylic acid moieties, was synthesised on 2-gram scale, also via a four-step synthesis (see Scheme S2†) with an overall yield of 27% of the corresponding urazole compound (the pre-cursor to TAD). TAD is formed from the oxidation of its respective bench-stable urazole compound. Thus, L2 was obtained by oxidising the urazole in small batches, as required, to avoid any unwanted degradation, as the stability of the TAD-functionality can vary from hours to months, depending on its nature and purity.43 The linker molecules both contained one moiety that reacts in the Diels–Alder reaction (TAD and a diene) and a second one that is active in the P-3CR (carboxylic acid and isocyanide), allowing a protecting group-free, iterative approach, benefiting from the two orthogonal reactions. As third component in the P-3CR, three aldehydes were used from commercially available sources to provide different functionalities and thus resulting in the desired sequence-definition. As it was already demonstrated previously by our group that a large variety of side chains can be introduced to the oligomeric backbone by varying the aldehyde component in the P-3CR, this study was limited to three different aldehydes to generate “[ABC]x”-sequences.29,30,34,35Using L1 and L2, a two-step, iterative cycle was developed, consisting of a TAD-based Diels–Alder reaction, followed by the P-3CR (Scheme 1). Both reactions reached quantitative conversions and yields and were carried out both on the solid phase and in solution. As a result, a sequence-defined dodecamer and nonamer were obtained, respectively, via the two different methods. Apart from the starting block, almost identical sequences were synthesised in both cases. Stearic acid was used as the starting molecule for the solution phase synthesis to make subsequent purifications easier. For the solid phase, the resin was first loaded with hexadiene-1-ol to provide a suitable reaction site for the first Diels–Alder reaction. The obtained products were carefully compared regarding yield, purity, reaction time, degree of polymerisation, purification method and scale. An overview of the synthesis strategy is provided in Scheme 1.
Solid phase approach
For the solid phase reactions, a 2-chlorotrityl chloride functionalised resin was used. This resin has very mild cleavage conditions (1% TFA in DCM), thus preventing unwanted side reactions such as degradation of ester bonds formed by the P-3CR, which would be problematic. Functionalisation of this resin with an alcohol was adapted from an earlier reported method.7 The conjugated diene structure is a suitable reaction partner for the irreversible TAD Diels–Alder reaction as it is known to be extremely fast and efficient.43,44 For subsequent cycles, the conjugated diene, necessary for this step, was provided by the diene-isocyanide linker molecule L1. Starting from this functionalised resin and applying the protocol outlined in Scheme 1, a monomer and a sequence-defined dimer and trimer were first synthesised separately to confirm the success of the protocol, as confirmed by Liquid Chromatography Mass Spectroscopy (LCMS) analysis (Fig. 1a). Furthermore, the synthesis of these three oligomers enabled complete characterisation of an ABC-type sequence by NMR and HRMS (see ESI, section 5 and 6†). The protocol was then repeated in order to synthesise longer sequences, thus resulting in an [ABC]3-nonamer and an [ABC]4-dodecamer. Propanal, isobutyraldehyde and cyclohexanecarboxaldehyde were used alternatively to introduce the side groups.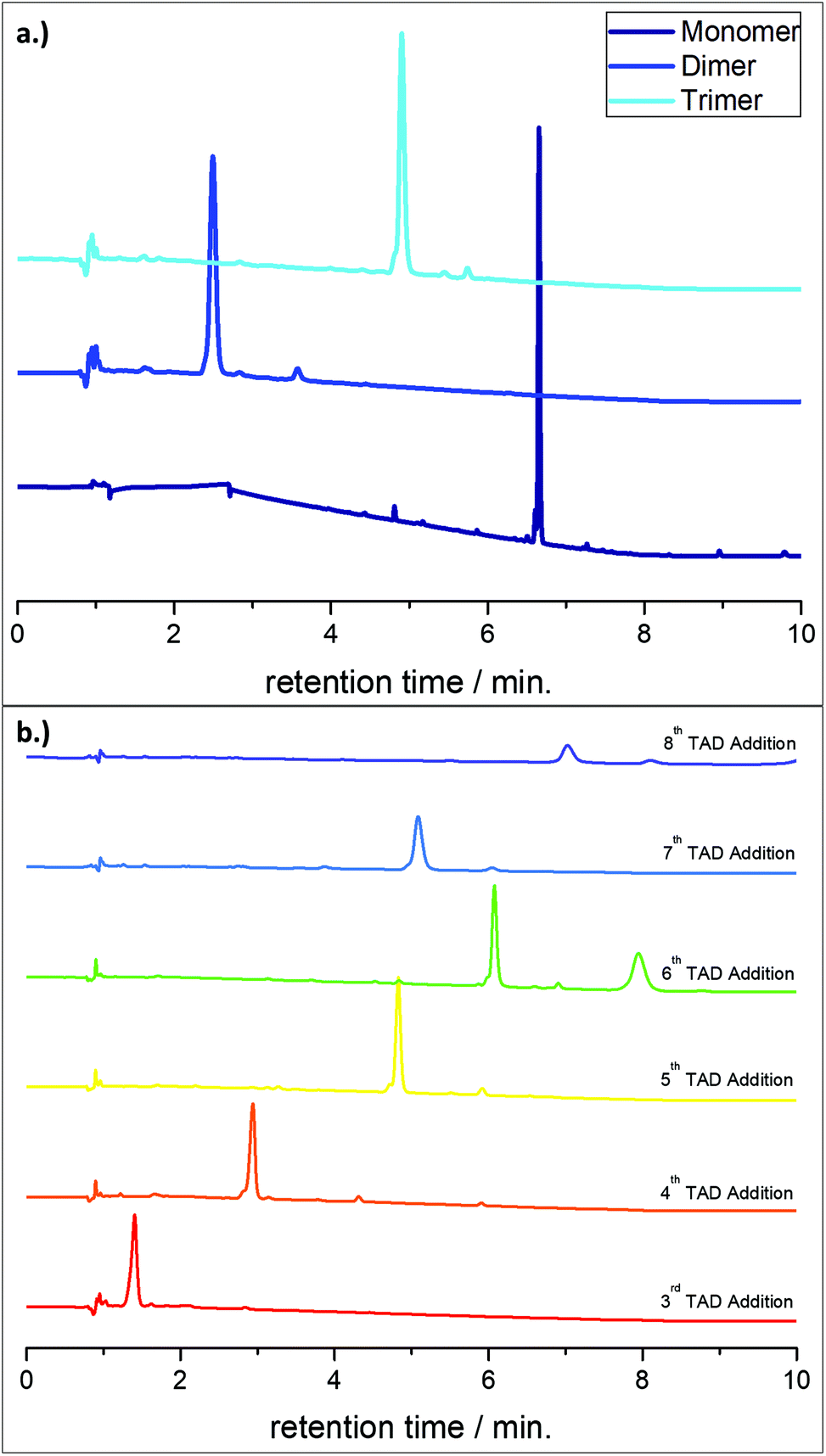 | ||
| Fig. 1 (a) LCMS chromatograms at λ = 214 nm of PhTAD capped monomer, dimer and trimer, demonstrating excellent conversion and purity. The monomer was processed at a solvent gradient of 0 → 100% acetonitrile → water, the dimer and trimer were processed at a 75 → 100% gradient on account of their decreased polarity. The reduced gradient results in a lower retention time for the molecule. (b) LCMS chromatograms at λ = 214 nm of the TAD molecule (L2) addition step. The increased polarity resulting from the carboxylic acid end-group made this step much easier to analyse by LCMS than the P-3CR step. By following the same step in the cycle, one can see a shift in retention time as the molecular weight increases. It should be noted that this does not continue in a liner fashion because the solvent gradient was changed from 75–100% to 90–100% after the 6th cycle (see Fig. S3† for LCMS chromatograms of each step in the cycle). | ||
The main advantage of solid phase chemistry is the ability to work with large excesses of reagents to ensure 100% conversion, as unwanted starting material can be simply washed away, thus massively simplifying purification. This is particularly advantageous for sequence-definition, as long sequences require a high number of iterative synthesis steps. It can also be a time or labour-saving approach as the purification can be much faster than, for example, column chromatography and one can, in principle, automate the reactions.7,51 For the TAD addition step, the high reactivity of TAD molecules required only 2 equivalents (notably low for solid phase synthesis, compared to other reported methods7,25,31) with shaking for 5 minutes at room temperature to ensure 100% conversion, as quantified by LCMS (Fig. S1†).
Despite previous reports of the P-3CR on the solid-phase,59–61 the speed of the synthesis was of great importance here, thus the P-3CR step in the cycle was followed kinetically by LCMS. These results showed that the reaction was complete within 30 minutes (Fig. S2†) and was compared with the reaction kinetics in solution (vide infra). The reaction was much faster than anticipated and up to twelve times faster than in solution. The equivalents of reagents were increased 10-fold with respect to the solution phase experiments and in line with previous solid phase reactions of our group.7,25,31 The speed of the solid phase reactions is a direct result of the high excess of reagents. The reaction time was increased every 3–4 cycles by 30 minutes in response to observations from LCMS analysis after each cycle, which showed a slowing down of the reaction as the oligomer grew larger. This was to be expected as it was known from the solution phase kinetics by online-IR (vide infra) that the reaction slows down because of increasing oligomer length.
One disadvantage of solid phase chemistry is scale, although large-scale (∼10 kg) solid phase synthesis has been reported.62 Reactions here were typically carried out on 50 mg of resin, yielding 14.4 mg (in the case of the dodecamer) of oligomer, once cleaved from the resin. Automation of this approach could circumvent problems with scale, as the use of a peptide-synthesiser allows up to 72 reactions to be carried out in parallel.7,19,51 However, the small scale used in this work resulted in a limited range of characterisation techniques available to use, which could be difficult when optimising reaction conditions compared to the solution phase approach. However, as previously reported by Du Prez and coworkers,7,31 LCMS was used to unambiguously follow the progress of the reaction after each step (see Fig. 1b). This technique only requires 2 mg of resin to be taken from the reaction vessel for quantitative analysis. The LCMS sample itself can be diluted further and used for HRMS analysis, too. Nonetheless, because of the high molecular weight oligomers achieved here (7200 Da), compared to our previous reported works (ca. 4000 Da),7,25,31,33 the limitations of the LCMS equipment used here became clear, as displayed in Fig. 1b, by the 7th TAD addition step (ca. 3800 Da). In general, LCMS was much more useful after the TAD-COOH addition reaction in the iterative cycle, as the more polar end-group resulted in a lower retention time in the LC. By comparison, after the P-3CR step of the 4th cycle, analysis by LCMS became too difficult as the long, apolar carbon chain from the linker molecule L1 resulted in a too high retention time for accurate and quantitative analysis, even with a reduced solvent gradient from acetonitrile to water of 75–100% and eventually 90–100% instead of the usual 0–100%. Thus, the sequence was eventually continued without intermediate analysis and the final dodecamer obtained was fully analysed by IR, NMR and SEC. Half of the reaction sample (25 mg resin) at the nonamer stage was taken for intermediate analysis and the reaction was continued to the dodecamer with the remaining 25 mg of resin.
Periodic analysis of the solid phase synthesised oligomers was also conducted by SEC for reasons of comparison with the solution approach (see Fig. 2). This was the preferred method of analysis for the oligomers synthesised in solution, as there was more material after each cycle for such analysis and the above-mentioned problems of the LCMS analysis could be overcome. The SEC measurements were performed using refractive index detectors and the columns used were specifically designed for low molecular weight molecules (100–60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da). Thus, one could easily compare both the purity and monodispersity of the obtained oligomers. In order not to lose material during the synthesis of the dodecamer, a separate trimer was synthesised for SEC analysis, as this could be quickly done within just three hours via the solid phase approach. With each of the oligomers, the conjugated diene chain end was “end-capped” with phenyl-TAD (Ph-TAD) before analysis, to prevent any further side reaction or cleavage of the ester bond induced by acid hydrolysis during cleavage from the solid phase resin. A small amount of impurity could be seen in the SEC trace of the nonamer and dodecamer as a result of a small percentage of dead chains (14%) and a polymerisation side-product (12%). Thus, the final product was obtained with a purity of 74%. An additional monomer, dimer and trimer were also separately synthesised, and the structures were fully resolved by 1H and 13C NMR techniques with the aid of 2D analysis (see ESI, section 6†). This, together with SEC and (LC)MS techniques ensured complete proof of the sequence-defined oligomers obtained and aided the final analysis of the dodecamer by NMR.
000 Da). Thus, one could easily compare both the purity and monodispersity of the obtained oligomers. In order not to lose material during the synthesis of the dodecamer, a separate trimer was synthesised for SEC analysis, as this could be quickly done within just three hours via the solid phase approach. With each of the oligomers, the conjugated diene chain end was “end-capped” with phenyl-TAD (Ph-TAD) before analysis, to prevent any further side reaction or cleavage of the ester bond induced by acid hydrolysis during cleavage from the solid phase resin. A small amount of impurity could be seen in the SEC trace of the nonamer and dodecamer as a result of a small percentage of dead chains (14%) and a polymerisation side-product (12%). Thus, the final product was obtained with a purity of 74%. An additional monomer, dimer and trimer were also separately synthesised, and the structures were fully resolved by 1H and 13C NMR techniques with the aid of 2D analysis (see ESI, section 6†). This, together with SEC and (LC)MS techniques ensured complete proof of the sequence-defined oligomers obtained and aided the final analysis of the dodecamer by NMR.
 | ||
| Fig. 2 Evolution of the solid phase synthesised oligomer from trimer through to nonamer and dodecamer. In the nonamer, and even more in the dodecamer, a minor high molecular weight side-product is observed (left of the main peak), while a dead chain from the trimer level of the synthesis (right of the main peak) is also present. SEC measurement was performed at Ghent University. See ESI† for detailed device information. | ||
The side-reaction was later observed to a much greater extent in the solution phase (vide infra) and was identified to be an unwanted polymerisation between excess TAD species and the aldehyde and isocyanide of the multicomponent reaction. Therefore, the focus was shifted there to optimise the synthesis to obtain 100% purity. Performing the reactions in solution was crucial to further understanding the observed side-reactions (vide infra) and to optimise the procedure, because the larger scale of the solution phase facilitated full characterisation after each reaction step. After having optimised the reaction in solution (vide infra), the optimised conditions were then applied to the solid phase approach. A new nonamer was synthesised to verify that the optimisation (quenching any excess un-reacted TAD with 2,3-dimethylbutene) worked for both approaches. A significant increase in the purity was observed by SEC (see Fig. 3). Additionally, the product was analysed by SEC-ESI-MS analysis to further confirm the structure. The purity of the sequence-defined nonamer increased to 84% (cf. 74% from earlier) as a result of applying the optimisation conditions. The previously observed polymerisation side reaction was successfully prevented. However, minor impurities were still observed and could be attributed to the fact that whilst theoretically solid-phase synthesis facilitates 100% conversion, in practice it is often slightly lower, depending on the shaking and handling of the resin. That, combined with the multi-step, iterative nature of this protocol, a negligible amount of lower molecular weight products (dead chains) were still present, as can be seen in Fig. 3. One could potentially attribute this to human error, as it has been reported that automation introduces more consistency into iterative approaches, thus improving purity and reproducibility.51
 | ||
| Fig. 3 SEC analysis of the solid phase synthesised oligomers, depicting the 3.5 mer, 6.5 mer and nonamer. The P-3CR polymerisation side product was significantly reduced following the successful application of the optimisation conditions (compare Fig. 2). SEC measurement was performed at KIT in Karlsruhe. See ESI† for detailed device information. | ||
Solution phase approach
For the solution phase synthesis, stearic acid was used as starting substrate. This acid compound was first reacted with an aldehyde and with the isocyanide of linker molecule L1 in a P-3CR. The same three aldehydes as used in the solid-phase approach were used alternatively in the sequences to build monodisperse [ABC]x structures.First, the kinetics of the P-3CR were investigated. Earlier works on the P-3CR never investigated this and typically reported reactions for 24 hours to ensure full conversion.33–35 To study this, the three different aldehyde compounds were reacted with stearic acid and linker molecule L1 and the reactions were followed by online IR (see Fig. 4 and Fig. S8–11†). The peak of the isocyanide at around 2145 cm−1 was the most significant one in the spectrum and thus used for monitoring. Since the isocyanide was used in excess, full conversion was indicated when the intensity of the isocyanide peak reached a plateau. After four to six hours of reaction time, the plateau was reached for all aldehydes. The Passerini product was purified by column chromatography, whereby a yield of 98% was achieved. In further P-3CRs, the reaction was thus typically stirred for eight to ten hours to ensure full conversion, thus offering a significant time advantage over previous approaches.
 | ||
| Fig. 4 Top: Example of an online IR measurement during a P-3CR, highlighting the isocyanide absorption band at 2145 cm−1. Bottom: By zooming in, the decrease of the absorbance of the isocyanide peak is clearly observed. | ||
Subsequently, the first TAD Diels–Alder reaction was performed. Since TAD compounds have an intense pink/red colour, which disappears as they are consumed, a visual feedback was observed during the reaction. Hence, linker molecule L2 was added in small stoichiometric excess to the reaction mixture and the reaction was conducted as a titration. As soon as the colour slightly remained, the conversion was considered to be complete and the crude product was directly used for the subsequent P-3CR. This also resulted in a significant advance in time and ease of procedure. Most importantly, a purification step is saved in the new combination of TAD with P-3CR. By iterating this cycle several times, sequence-defined oligomers were synthesised.
During the fifth reaction cycle, the online IR measurement was repeated (Fig. S11†), confirming an expected slowing-down of the reaction because of possible increasing chain entanglement and thus less accessible end groups. The reaction was complete after 16 hours, so for further P-3CRs, the reaction time was extended to 24 and eventually to 48 hours for higher molecular weight oligomers. By applying these reaction conditions, a sequence-defined nonamer was obtained. However, SEC analysis (Scheme 2) revealed that a side reaction had occurred. After each P-3CR, the obtained product was carefully characterised by 1H and 13C NMR, IR spectroscopy, mass spectrometry, SEC, SEC-MS and LCMS analysis. SEC was the most crucial technique to confirm the (mono)dispersity and thus purity of the products, as the side reaction was not observable by NMR nor LCMS analysis, whereas even traces of impurities were clearly visible in the SEC chromatogram. Starting from the trimer stage, side products of lower retention time, thus higher molecular weight, were obtained, which could not be separated from the product. Over the course of the following reactions, the amount of polymeric side product increased drastically and at the final stage the reaction product consisted of 46% side product and only 54% sequence-defined nonamer. This side reaction was also observed, albeit to a much lesser extent, on the solid phase synthesised oligomer (as already discussed above), but initial focus was on optimising the solution phase reactions as the impurity was much more pronounced there, before later applying this to the solid-phase approach. Furthermore, the larger scale in solution allowed for full characterisation after each reaction step, which helped to identify and understand the reaction as well as the recurring side reactions. The side product was analysed by SEC-ESI-MS and was identified to be the product of a P-3CR polymerisation, occurring because of the small excess of TAD compound present after each reaction cycle (Scheme 2). Despite the absence of the pink colour, a trace of TAD-COOH was still present after evaporation of the solvent from the reaction mixture. The two linker molecules L1 and L2 underwent a click reaction with each other, forming a new monomer carrying a carboxylic acid and isocyanide moiety, which reacted in a P-3CR polymerisation, together with the aldehyde compounds and the sequence-defined oligomer.
 | ||
| Scheme 2 Side reaction that occurred between the excess TAD-COOH molecule, L2 and the diene isocyanide linker molecule, L1 to afford an uncontrolled Passerini-3CR polymerisation with present aldehydes; and the SEC traces of the obtained P-3CR polymerisation products. The solution phase synthesis enabled successful identification of the side reaction, which could later be avoided in both solid and liquid phase approaches. Please see Fig. 5 for the colour key of the SEC traces. | ||
To prevent this side reaction, the reaction conditions were adjusted by adding a non-functional alkene (20 μL of dimethylbut-2-ene) after the TAD addition reaction to quench the excess of linker L2via an Alder-ene type reaction.43,44 This resulted in an immediate disappearance of the remaining pink colour and the reaction was then continued with the subsequent P-3CR. Because of the better scalability of the reactions in solution, the optimisation was first performed for the solution phase approach, as it offered the possibility to check the dispersity after every iterative cycle, and afterwards this optimisation was transferred to the solid phase synthesis. By applying these optimised reaction conditions, the side reaction was prevented and a sequence-defined nonamer of very high purity was successfully synthesised in solution. This oligomer, with a molecular weight of 5340.02 g mol−1, was synthesised in 17 reaction steps with an overall yield of 18% (180 mg). SEC analysis verified the high purity of the final product (Fig. 5). In SEC-ESI-MS analysis, the doubly (m/z 2670.97), triply (m/z 1780.99) and quadruply (m/z 1335.86) protonated masses as well as the sodium ions were observed and the isotopic pattern was compared with the calculated one, confirming the structure of the product. The product was further analysed by NMR- and IR spectroscopy, as well as by high resolution mass spectrometry, all confirming the high purity of the macromolecule.
 | ||
| Fig. 5 Top: Structure of the [ABC]3 sequence-defined nonamer with three different side chains. Bottom left: SEC analysis of the obtained products from the optimised iterative synthesis cycle. The SEC results show the successful prevention of the side reaction and verify the high purity of the products. Bottom right: SEC-ESI-MS analysis of the nonamer showing the chromatogram and the corresponding mass spectrum at a retention time of 14 min 30 s. The assigned peaks correspond to the mass plus two, three and four protons. | ||
Conclusions
In summary, a new approach towards sequence-defined macromolecules is introduced, combining the advantages of the P-3CR with the very efficient and ultra-fast TAD chemistry. A careful comparison of this approach carried out in solution as well as on solid phase is given in Table 1. A dodecamer, was synthesised in 25 steps with an overall yield of 5% (14.4 mg) on the solid phase. The synthesis was performed using 50 mg of loaded resin and the final product was first obtained with a purity of 74%, because of a P-3CR polymerisation as a side reaction. This side reaction was originally observed to a much greater extent with the solution phase approach. However, simple reaction optimisation showed how this could be successfully prevented, leading to a very practical and quick build-up of oligomers. The optimisation was later successfully applied to the solid phase approach to synthesise a sequence-defined nonamer, where the purity was significantly increased to 84%. The most important advantage of the solid phase approach is the required time, not only for the reactions themselves, but also for the purification. P-3CRs were performed within 30 to 120 minutes, while TAD Diels–Alder reactions reached full conversions in less than five minutes. The products were purified by simple washing procedures. Thus, working on a solid support simplifies and accelerates the synthesis and workup procedure significantly. Whilst the synthesis of a nonamer in solution requires approximately three weeks, whereas the same molecule synthesised on a solid support can be obtained within two days. One can also, in principle, automate this using a peptide synthesiser and thus can not only make vast libraries of different sequences in tandem but also obtain the product in gram-scale.| Solid phase | Solution phase | |
|---|---|---|
| a Prior to optimisation, a degree of polymerisation of 12 was achieved. b Reaction time for TAD Diels–Alder reaction. c Reaction time for P-3CR. | ||
| Yield (%) | 5 | 18 |
| Purity (%) | 84 | >99% |
| Scale (mg) | 50 | 200 |
| Degree of polymerisation | 9a | 9 |
| Purification method | Washing | Column chromatography |
| Reaction time | <5 min,b 30–120 minc | 5 min,b 8–48 hc |
| Overall required time | 2 days | 3 weeks |
In solution, on the other hand, following the successful application of the optimisation conditions, 180 mg of a monodisperse nonamer with a purity of >99% was obtained in 17 reaction steps, with an overall yield of 18%. P-3CRs were carried out over a reaction time of 8 to 48 hours, while TAD Diels–Alder reactions were complete in less than 5 minutes. The products were purified by column chromatography, which is effective, but time consuming. Here, the reactions were typically carried out in 200 mg scale but, theoretically, the synthesis could easily be scaled up to multigram scale.
The comprehensive study presented here clearly demonstrates the power of combining click chemistry with multicomponent reactions. This combination leads to an ideal situation for iterative growth and multifunctionalisation of macromolecules, significantly improving already reported procedures in terms of purity, time and transferability between approaches for scalability. Through this comparative study, we have also demonstrated that many different and versatile chemistries can be carried out on both the solid phase and in solution. The user choice for the appropriate procedure should be guided by decisions of synthesis speed, potential for library synthesis and necessary scale.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. S. W. and M. M. would like to acknowledge funding by the German Research Council (DFG) in the context of the SFB 1176 (project A3). J. O. H would like to acknowledge funding of his PhD Scholarship by the European Union's Horizon 2020 Research and Innovation Program under the Marie Skłodowska-Curie Actions Grant Agreement No. 642083 and also funding by the Ghent University Special Research Fund-ITN (BOF-ITN). S. M. would like to acknowledge funding of his PhD scholarship by the Research Foundation Flanders (FWO). F. D. P. gratefully acknowledges project support from the FWO under EOS-project 30650939.The authors gratefully acknowledge Bernhard De Meyer, Jan Goeman, Timothee Courtin from UGent, Maximiliane Frölich, Rieke Schulte and the analytical team from KIT for analytical support, Laetitia Vlaminck, Bastiaan Dhanis and Hannes Houck from UGent for the help in the development of TAD-compounds, Prof. Barner-Kowollik (KIT) and his group for access to SEC-ESI-MS equipment and Leonie Golka (KIT) for her help with the evaluation of the online IR measurements.
Notes and references
- S. Martens, J. O. Holloway and F. E. Du Prez, in Sequence-Controlled Polymers, ed. J.-F. Lutz, Wiley VCH, Weinheim, 2018, pp. 379–416 Search PubMed.
- S. Martens, J. O. Holloway and F. E. Du Prez, Macromol. Rapid Commun., 2017, 38, 1700469 CrossRef PubMed.
- S. C. Solleder, R. V. Schneider, K. S. Wetzel, A. C. Boukis and M. A. R. Meier, Macromol. Rapid Commun., 2017, 38, 1600711 CrossRef PubMed.
- N. Badi and J.-F. Lutz, Chem. Soc. Rev., 2009, 38, 3383–3390 RSC.
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 1–14 Search PubMed.
- J. A. De Neve, J. J. Haven, L. Maes and T. Junkers, Polym. Chem., 2018, 9, 4692–4705 RSC.
- S. Martens, J. Van Den Begin, A. Madder, F. E. Du Prez and P. Espeel, J. Am. Chem. Soc., 2016, 138, 14182–14185 CrossRef CAS PubMed.
- C. Gerke, F. Jacobi, L. E. Goodwin, F. Pieper, S. Schmidt and L. Hartmann, Macromolecules, 2018, 51, 5608–5619 CrossRef CAS.
- C. Fu, Z. Huang, C. J. Hawker, G. Moad, J. Xu and C. Boyer, Polym. Chem., 2017, 8, 4637–4643 RSC.
- B. N. Norris, S. Zhang, C. M. Campbell, J. T. Auletta, P. Calvo-Marzal, G. R. Hutchison and T. Y. Meyer, Macromolecules, 2013, 46, 1384–1392 CrossRef CAS.
- H. Zhang, X. Li, Q. Shi, Y. Li, G. Xia, L. Chen, Z. Yang and Z.-X. Jiang, Angew. Chem., Int. Ed., 2015, 54, 3763–3767 CrossRef CAS PubMed.
- Y. Hibi, M. Ouchi and M. Sawamoto, Nat. Commun., 2016, 7, 11064 CrossRef PubMed.
- D. de Rochambeau, M. Barłóg, T. G. W. Edwardson, J. J. Fakhoury, R. S. Stein, H. S. Bazzi and H. F. Sleiman, Polym. Chem., 2016, 7, 4998–5003 RSC.
- A. Al Ouahabi, M. Kotera, L. Charles and J.-F. Lutz, ACS Macro Lett., 2015, 4, 1077–1080 CrossRef CAS.
- M. Porel, D. N. Thornlow, N. N. Phan and C. A. Alabi, Nat. Chem., 2016, 8, 590–596 CrossRef CAS PubMed.
- N. Zydziak, F. Feist, B. Huber, J. O. Mueller and C. Barner-Kowollik, Chem. Commun., 2015, 51, 1799–1802 RSC.
- S. C. Solleder, K. S. Wetzel and M. A. R. Meier, Polym. Chem., 2015, 6, 3201–3204 RSC.
- A. C. Boukis, K. Reiter, M. Frölich, D. Hofheinz and M. A. R. Meier, Nat. Commun., 2018, 9, 1439 CrossRef PubMed.
- S. Martens, A. Landuyt, P. Espeel, B. Devreese, P. Dawyndt and F. E. Du Prez, Nat. Commun., 2018, 9, 4451 CrossRef PubMed.
- R. L. Kanasty, A. J. Vegas, L. M. Ceo, M. Maier, K. Charisse, J. K. Nair, R. Langer and D. G. Anderson, Angew. Chem., Int. Ed., 2016, 55, 9529–9533 CrossRef CAS PubMed.
- J. W. Grate, K. F. Mo and M. D. Daily, Angew. Chem., Int. Ed., 2016, 55, 3925–3930 CrossRef CAS PubMed.
- J.-F. Lutz, in Sequence-Controlled Polymers, ed. J.-F. Lutz, Wiley VCH, Weinheim, 1st edn, 2018, pp. 1–26 Search PubMed.
- J.-F. Lutz, Macromol. Rapid Commun., 2017, 1700582, 1–12 Search PubMed.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505–2514 CrossRef PubMed.
- P. Espeel, L. L. G. Carrette, K. Bury, S. Capenberghs, J. C. Martins, F. E. Du Prez and A. Madder, Angew. Chem., Int. Ed., 2013, 52, 13261–13264 CrossRef CAS PubMed.
- C. Alabi, in Sequence-Controlled Polymers, ed. J.-F. Lutz, Wiley VCH, Weinheim, 1st edn, 2018, pp. 159–181 Search PubMed.
- T. T. Trinh, C. Laure and J.-F. Lutz, Macromol. Chem. Phys., 2015, 216, 1498–1506 CrossRef CAS.
- J. C. Barnes, D. J. C. Ehrlich, A. X. Gao, F. A. Leibfarth, Y. Jiang, E. Zhou, T. F. Jamison and J. A. Johnson, Nat. Chem., 2015, 7, 810–815 CrossRef CAS PubMed.
- D. Van Lysebetten, S. Felissati, E. Antonatou, L. Carrette, P. Espeel, E. Foquet, F. E. Du Prez and A. Madder, ChemBioChem, 2018, 19, 641–646 CrossRef CAS PubMed.
- D. Chan-Seng, J. Louwsma, J.-F. Lutz and S. Joly, Macromol. Rapid Commun., 2018, 38, 1700764 CrossRef PubMed.
- J. O. Holloway, S. Aksakal, F. E. Du Prez and C. Remzi Becer, Macromol. Rapid Commun., 2017, 38, 1700500 CrossRef PubMed.
- M. R. Golder, Y. Jiang, P. E. Teichen, H. V.-T. Nguyen, W. Wang, N. Milos, S. A. Freedman, A. P. Willard and J. A. Johnson, J. Am. Chem. Soc., 2018, 140, 1596–1599 CrossRef CAS PubMed.
- S. C. Solleder, S. Martens, P. Espeel, F. E. Du Prez and M. A. R. Meier, Chem. – Eur. J., 2017, 23, 13906–13909 CrossRef CAS PubMed.
- S. C. Solleder and M. A. R. Meier, Angew. Chem., Int. Ed., 2014, 53, 711–714 CrossRef CAS PubMed.
- S. C. Solleder, D. Zengel, K. S. Wetzel and M. A. R. Meier, Angew. Chem., Int. Ed., 2016, 55, 1204–1207 CrossRef CAS PubMed.
- Y.-H. Wu, J. Zhang, F.-S. Du and Z.-C. Li, ACS Macro Lett., 2017, 6, 1398–1403 CrossRef CAS.
- R. Kakuchi, Angew. Chem., Int. Ed., 2014, 53, 46–48 CrossRef CAS.
- L. Yang, Z. Zhang, B. Cheng, Y. You, D. Wu and C. Hong, Sci. China: Chem., 2015, 58, 1734–1740 CrossRef CAS.
- Z. Zhang, Y. You and C. Hong, Macromol. Rapid Commun., 2018, 1800362, 1800362 CrossRef PubMed.
- W. Konrad, F. R. Blößer, K. S. Wetzel, A. C. Boukis, M. A. R. Meier and C. Barner-Kowollik, Chem. – Eur. J., 2018, 24, 3413–3419 CrossRef CAS PubMed.
- M. Passerini, Gazz. Chim. Ital., 1921, 51, 126–129 CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- K. De Bruycker, S. Billiet, H. A. Houck, S. Chattopadhyay, J. M. Winne and F. E. Du Prez, Chem. Rev., 2016, 116, 3919–3974 CrossRef CAS PubMed.
- S. Billiet, K. De Bruycker, F. Driessen, H. Goossens, V. Van Speybroeck, J. M. Winne and F. E. Du Prez, Nat. Chem., 2014, 6, 815–821 CrossRef CAS.
- P. Espeel and F. E. Du Prez, Macromolecules, 2015, 48, 2–14 CrossRef CAS.
- O. Roling, K. De Bruycker, B. Vonhören, L. Stricker, M. Körsgen, H. F. Arlinghaus, B. J. Ravoo and F. E. Du Prez, Angew. Chem., Int. Ed., 2015, 54, 13126–13129 CrossRef CAS PubMed.
- H. A. Houck, F. E. Du Prez and C. Barner-Kowollik, Nat. Commun., 2017, 8, 1869 CrossRef PubMed.
- X. Elduque, A. Sánchez, K. Sharma, E. Pedroso and A. Grandas, Bioconjugate Chem., 2013, 24, 832–839 CrossRef CAS PubMed.
- N. Zydziak, W. Konrad, F. Feist, S. Afonin, S. Weidner and C. Barner-Kowollik, Nat. Commun., 2016, 7, 1–10 Search PubMed.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- J. O. Holloway, C. Mertens, F. E. Du Prez and N. Badi, Macromol. Rapid Commun., 2018, 40, 1800685 CrossRef PubMed.
- M. I. Amrane, D. Chouikhi, N. Badi and J.-F. Lutz, Macromol. Chem. Phys., 2014, 215, 1984–1990 CrossRef CAS.
- A. Meszynska, N. Badi, H. G. Börner and J.-F. Lutz, Chem. Commun., 2012, 48, 3887–3889 RSC.
- S. Pfeifer, Z. Zarafshani, N. Badi and J.-F. Lutz, J. Am. Chem. Soc., 2009, 131, 9195–9197 CrossRef CAS.
- R. Dong, R. Liu, P. R. J. Gaffney, M. Schaepertoens, P. Marchetti, C. M. Williams, R. Chen and A. G. Livingston, Nat. Chem., 2019, 11, 136–145 CrossRef CAS.
- G. Li, X. Wang, J. Li, X. Zhao and F. Wang, Tetrahedron, 2006, 62, 2576–2582 CrossRef CAS.
- J.-J. Hwang and J. M. Tour, Tetrahedron, 2002, 58, 10387–10405 CrossRef CAS.
- M. Landa, M. Kotera, J. S. Remy and N. Badi, Eur. Polym. J., 2016, 84, 338–344 CrossRef CAS.
- R. W. Armstrong, A. P. Combs, P. A. Tempest, S. D. Brown and T. A. Keating, Acc. Chem. Res., 1996, 29, 123–131 CrossRef CAS.
- L. Banfi, A. Basso, G. Guanti and R. Riva, Mol. Divers., 2003, 6, 227–235 CrossRef CAS PubMed.
- A. Basso, L. Banfi, R. Riva, P. Piaggio and G. Guanti, Tetrahedron Lett., 2003, 44, 2367–2370 CrossRef CAS.
- B. L. Bray, Nat. Rev. Drug Discovery, 2003, 2, 587–593 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures, experimental details, synthesis and analysis of compounds and materials and analytical methods used. See DOI: 10.1039/c9py00558g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |