
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymer grafted graphitic carbon nitrides as precursors for reinforced lubricant hydrogels†
Baris
Kumru
a,
Valerio
Molinari
a,
Markus
Hilgart
b,
Florian
Rummel
 b,
Michael
Schäffler
b and
Bernhard V. K. J.
Schmidt
*ac
b,
Michael
Schäffler
b and
Bernhard V. K. J.
Schmidt
*ac
aMax-Planck Institute of Colloids and Interfaces, Department of Colloid Chemistry, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: bernhard.schmidt@mpikg.mpg.de
bAnton Paar Germany GmbH, Hellmuth-Hirth-Strasse 6, 73760 Ostfildern, Germany
cSchool of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK
First published on 22nd May 2019
Abstract
Hydrogels constitute an important class of polymeric materials, mainly due to their promising applications in the biomedical field. Particularly, mechanical properties are a limiting factor for applications, and reinforcement and improved wear resistance are major directions in current research. In here, graphitic carbon nitride (g-CN)-based hydrogels are presented. The hydrogels are formed in a two-step procedure. In the first step, a prepolymer is synthesized that leads to a significantly improved g-CN dispersibility and stability of g-CN dispersions. Hence, the g-CN content in the final hydrogels can be increased to obtain tough hydrogels. More importantly, hydrogels from charged monomers can be formed as well, which leads to lubricant properties. As such, the investigated route opens up new opportunities to fabricate functional hydrogels with significant toughness, compressibility and lubricity.
Introduction
Water is an essential component of biological tissues that encompass soft properties or tough properties, e.g. in the case of skin or cartilage, respectively. Articular cartilage, for example, consists of 70% water and collagen type II fibers which can mainly be found in joints.1 Despite the fact that the composition sounds simple, articular cartilage has amazing mechanical properties, e.g. it can withstand enormous amounts of cyclic compressions arising from the mobility in daily life.2 The stress load on joints depends on the type of mobility, such as walking or running that result in different amounts of load.3 A smart solution provided by nature for extraordinary mechanical performance is not just the stiffness of articular cartilage, but also its lubricity. The composition of joint materials comprises charged surfaces due to proteoglycans that provide ultra-low friction between the layers in synovial fluid. Hence, joints have significant wear resistance. Unfortunately, the ultra-low friction properties do not last forever as aging and diseases like osteoarthritis decrease the lubricity in joints which causes significant damage and affects the daily life routine negatively.4 Therefore, joint replacement became an important issue, which requires materials with similar mechanical performance to articular cartilage.Current technology makes use of ultra-high molecular weight polyethylene in combination with metal alloys. However the life-time of current solutions is limited as the material wears off and revision treatment is needed.5 At this point, hydrogels are proposed to be one of the main candidates for artificial cartilage replacement due to their water content and adjustable mechanical properties.6 In order to fabricate hydrogels with adequate properties, reinforcement of the usually soft materials is needed, e.g. via addition of a reinforcer,7–9 use of tetra-PEG systems,10,11 fiber addition,12,13 and use of slide-ring gels14,15 or double network systems.16,17 The double network approach provides rigid and tough hydrogels with compressive strength in the range of a few MPas by using two independent networks.18 The employment of combination of monomers (neutral or charged) as well as the addition of a reinforcer provides a library of materials with different mechanical properties and lubricity.19,20 However, there is a long way to go before a material that mimics articular cartilage perfectly is obtained.
Graphitic carbon nitride (g-CN), a metal-free photocatalyst, emerged as a promising material owing to its absorption in the UV and visible light range.21 g-CN is composed of repeating triazine or tri-s-triazine units which can be formed from low cost and abundant precursors while its properties are tunable.22–24 Moreover, the g-CN surface can be functionalized via several chemical approaches, e.g. photochemical, nucleophilic substitution or cross-coupling.25–28 The aromatic semiconductor structure forms excitons and holes upon visible light irradiation, making it possible to apply photo-based applications such as water splitting,29,30 dye degradation,31,32 sensors33 and CO2 reduction.34–36 Recently, g-CN was introduced in polymer chemistry. As a heterogeneous catalyst, g-CN can be utilized as a photoinitiator in free radical or reversible deactivation radical polymerization systems such as atom transfer radical polymerization and reversible addition fragmentation chain transfer polymerization.37–39 Continuous aerosol photopolymerization utilizing g-CN creates spherical poly(butylacrylate) composites via a hard template approach.40
Contrarily, utilizing g-CN in aqueous systems requires good dispersions and results in interesting materials such as photoactive hydrogels and smart sensors.41–43 Hence, g-CN (0.6 wt%) was reported to be a photoinitiator and a reinforcer for hydrogel systems showing a significant increase in mechanical properties.44 Radicals created on the surface via photoexcitation result in covalent bonding between g-CN and monomers, such as in the case of hydrogel reinforcement. Due to the 2-D sheet-like structure, g-CN can be utilized for hydrogel formation without commercial crosslinkers yielding self-standing soft hydrogels. Strong π–π interactions between g-CN sheets lead to the self-assembly and formation of weak hydrogels in water even without a monomer and crosslinker.45 The increase in the g-CN amount up to 4 wt% in hydrogels via the formation of organohydrogel intermediates showed promising results for tough aquamaterials.46 Moreover, the utilization of sulfonic acid functionalized g-CN enabled the formation of soft hydrogels with extraordinary compressibility, showing that the surface properties of g-CN are a key to access hydrogels with a variety of properties.47 Our group recently introduced a g-CN based prepolymer via photografting 2-hydroxyethyl methacrylate in water–ethylene glycol (EG) media, which can further be thermally crosslinked by citric acid for photoactive robust film formation.48 In such a way, the prepolymer approach facilitates improved colloidal stability of g-CN dispersions and control over g-CN weight content. In addition, a combination of g-CN and biomacromolecules for the formation of hydrogels was presented, e.g. with peptides and polysaccharides.49,50
Another significant property of hydrogels is lubricity. As mentioned before, lubricity is of crucial importance for various applications, especially in the biomedical field. Inspired by natural lubricant materials that are based on charged surfaces, researchers have focused on charged brushes on surfaces to obtain ultra-low friction behavior.51,52 Hydrogels with low friction properties have been investigated as well, e.g. a double network hydrogel by Gong and coworkers.53 Another efficient solution might be the combination of g-CN-based hydrogels with charged polymers. Finding a balance between toughness and lubricity in hydrogels utilizing g-CN might provide a novel class of materials for joint replacement. So far CN-based hydrogel fabrication could not give access to charged monomers and lubricity due to the immediate sedimentation of charged g-CN particles in dispersion after the addition of charged monomers.
Herein, a solution to the problem of colloidal stability of g-CN dispersions in a high ionic strength environment is presented. An injectable g-CN based prepolymer with advanced colloidal stability is prepared and utilized for hydrogel formation. In the first step, synthesis and analysis of the g-CN based prepolymer is performed. The prepolymer formulation can be mixed with a monomer, water and crosslinker of desired amounts, without an external photoinitiator for hydrogel formation under visible light, providing a diverse class of novel tough aquamaterials (Scheme 1). The employment of charged monomers, for the first time for g-CN dispersions, results in tough and lubricant hydrogels that are carefully evaluated regarding friction coefficient (γ) and mechanical properties.
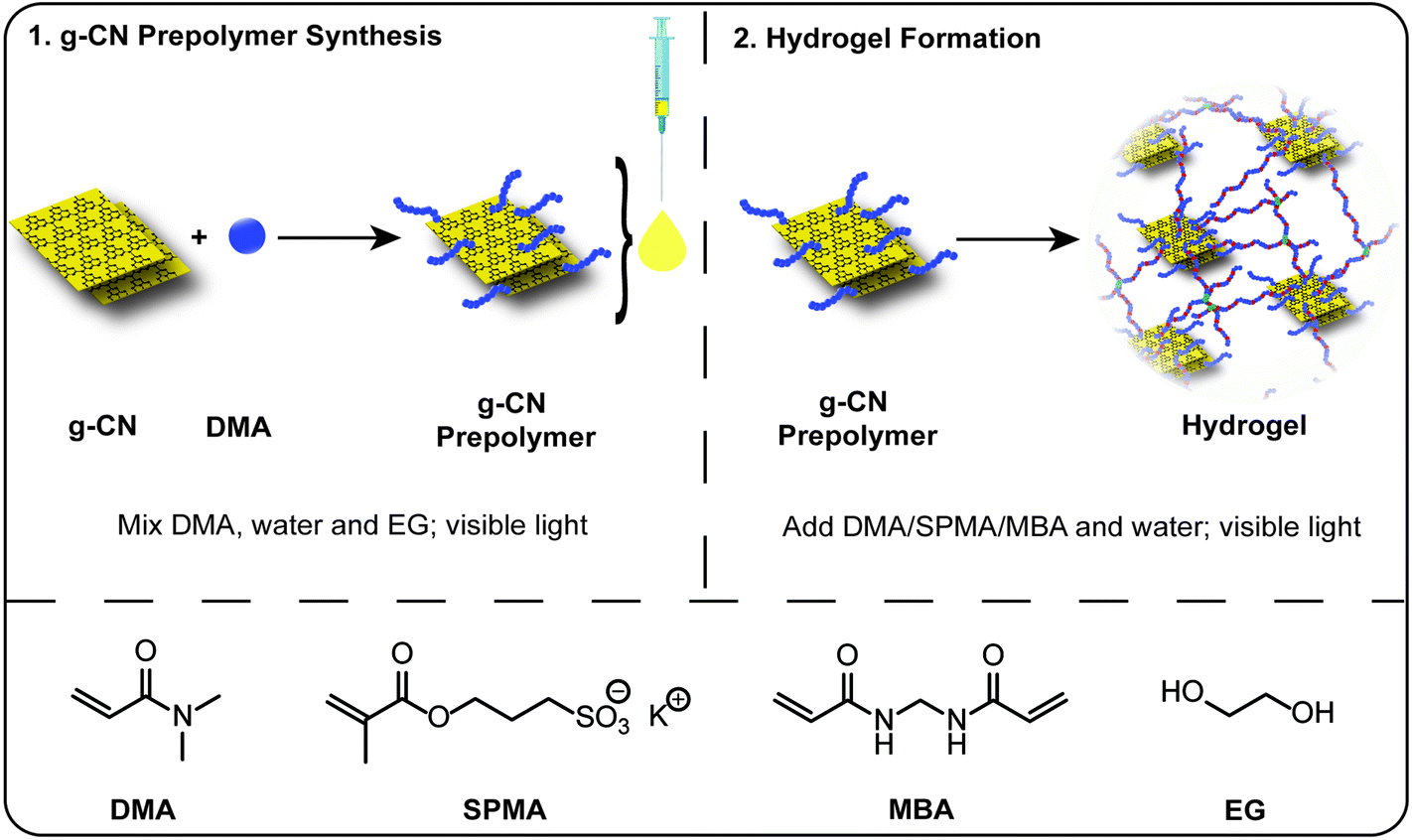 | ||
| Scheme 1 Schematic overview for the synthesis of a g-CN based prepolymer and its utilization as a precursor in hydrogel fabrication (abbreviations: DMA: N,N-dimethylacrylamide, SPMA: 3-sulfopropyl methacrylate potassium salt, MBA: N,N′-methylenebis(acrylamide) and EG: ethylene glycol). | ||
Experimental
Materials
Al2O3 basic (Sigma Aldrich), ascorbic acid (AscA, 99%, Sigma Aldrich), cyanuric acid (98%, Sigma Aldrich), ethylene glycol (EG, 99%, Fluka), hydrogen peroxide (30% in water, Sigma Aldrich), melamine (99%, Sigma Aldrich), N,N′-methylenebis(acrylamide) (MBA, 99%, Sigma Aldrich) and 3-sulfopropyl methacrylate potassium salt (SPMA, 98%, Sigma Aldrich) were used as purchased. N,N-Dimethylacrylamide (DMA, 98%) was purchased from Acros and passed through a basic alumina column prior to use to remove the inhibitor. 50 W LED chips (Foxpic High Power 50 W LED Chip Bulb Light DIY White 3800LM 6500 K) were connected to a self-made circuit and cooling system. g-CN was synthesized via a cyanuric acid–melamine complex as a precursor according to the literature.54Synthesis of g-CN based prepolymers
200 mg g-CN was dispersed in a mixture of 4.5 g water and 4.5 g EG and ultrasonicated at 50% amplitude for 20 minutes (10 × 2 minute portions) to yield a g-CN dispersion. Afterwards, 0.8 g DMA was added to the dispersion and the mixture was placed between two 50 W LED daylight sources (20 cm apart from each other) and reacted for certain times (1, 3, 6 and 12 hours, coded later as g-CN pre1h, g-CN pre3h, g-CN pre6h, g-CN pre12h) with mild stirring. For the synthesis of g-CN 2pre3h, 1.6 g DMA were employed in the reaction instead.Exemplary synthesis of tough organohydrogel (touA)
1 g g-CN pre3h (obtained after 3 h of irradiation) was mixed with 1 g water, 2 g DMA, and 100 mg MBA and stirred for 10 minutes. Subsequently, the mixture was flushed with nitrogen for 3 minutes and the mixture was placed between two 50 W LED daylight sources (20 cm apart from each other) for 1 hour to obtain opaque crosslinked organohydrogel materials.Synthesis of lubricant organohydrogel (lubA)
1 g g-CN pre3h was mixed with 2 g water, 1 g DMA, 0.8 g SPMA, and 75 mg MBA and stirred until the dissolution of the solid monomer. Subsequently, the mixture was flushed with nitrogen for 3 minutes and the mixture was placed between two 50 W LED daylight sources (20 cm apart from each other) for 1 hour to obtain opaque crosslinked organohydrogel materials.Removal of EG from organohydrogels
All tough organohydrogel samples with different compositions were first immersed in 30 mL acetone overnight and then in 50 mL deionized water for 2 days by changing water 2 times per day. Lubricant organohydrogels can be treated in the same way to yield highly swollen hydrogels, or they can be freeze dried after the reaction, swollen in water overnight and freeze dried again. Certain amounts of water can be added to the dry hydrogel for a desired amount of swelling.Freeze drying of hydrogel samples
Hydrogels were cut into smaller pieces, transferred into a flask and frozen via liquid nitrogen. In this frozen form, they were immediately put into a Lyotech GT 2E freeze dryer overnight. The resulting products are stiff hydrogels which retain their pores on the microscopic level.Characterization
Zeta potential measurements of colloidal dispersion of g-CN were performed with a Zetasizer Nano ZS90 from Malvern. X-ray diffraction (XRD) patterns were obtained using a Bruker D8 Advance X-ray diffractometer via Cu-Kα radiation. Scanning electron microscopy (SEM) was performed using a JSM-7500F (JEOL) equipped with an Oxford Instruments X-MAX 80 mm2 detector for the determination of the morphology of g-CN. Fourier transform-infrared (FT-IR) spectra were obtained on a Nicolet iS 5 FT-IR spectrometer. Solid state ultraviolet-visible (UV-Vis) spectroscopy was performed via a Cary 500 Scan spectrophotometer equipped with an integrating sphere. Thermogravimetric analysis (TGA) was performed via a TG 209 Libra from Netzsch under a nitrogen atmosphere with a heating rate of 10 K min−1 using an aluminum crucible for samples. Ultrasonication was performed at 50% amplitude (Branson D450). Elemental analysis of g-CN was performed via a Vario Micro device. To obtain the swelling ratios, 100 mg (Wd) freeze dried hydrogel samples were put into a flask which contains 5 mL distilled water and left to stand for 24 hours. Swollen gels or hydrogels are weighed (Ws) and the swelling ratio was calculated with the formula: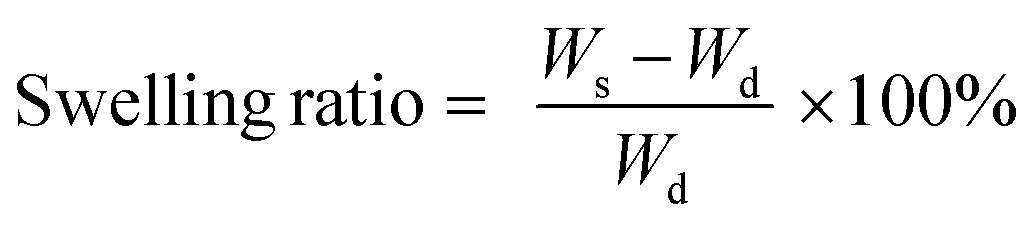
For rheological investigations of g-CN prepolymers, an Anton Paar MCR 301 rheometer equipped with a cone plate 12 (CP-12) was used. Measurements were performed at a constant angular frequency (10 rad s−1) with a strain range from 0.1–100% with 31 measuring points and 0.02 mm gap. Frequency dependent measurements were performed at a constant strain (0.1%) with changing frequency in the range of 1–100 rad s−1. Viscosity measurements were performed at ambient temperature with changing shear rates between 1–20 s−1. Compression was measured on a Zwick mechanical tester zwickiLine Z2.5 equipped with a loadcell of 1 kN. Measurements were recorded after a preload of 0.1 N (speed pre-load 40 mm s−1) and the test was performed at 0.05 mm s−1. The cycling tests were performed until 0.5 MPa maximum force and recovered until 1 mm of strain (on specimens of 10 mm in height) before the stress was removed to restart the cycle (50 times). Elastic modulus was manually calculated at 10% of strain before break and at break for every specimen. All the compression measurements were recorded using the software TestXpert II V3.71.
Measurement of friction coefficient (γ)
First of all, lubRef, lubA and lubB samples were chosen to investigate the tribological properties. Each sample was prepared in a syringe and cut as cylinders with a diameter of 1.5 cm. In addition, each sample was prepared in a Petri dish (60 mm diameter and 3 mm height) as well. Organohydrogel samples were transformed into pure hydrogels via washing with water as described previously. Finally, the friction of hydrogel cylinders against a hydrogel disc was measured after the addition of water to obtain complete coverage (Fig. S1†). Stribeck curves were measured with a T-PID/44 Pin-on-disc sample holder on an MCR 302 Tribometer from Anton Paar. The measurement was performed at a normal force of 1 N according to 1/3 N per contact in the sliding velocity range of 10−7 to 0.1 m s−1 in the rotation mode at ambient temperature. A total of 91 data points were collected with a logarithmic distribution of measurement time per data point from 10 s (at 10−7 to 0.1 m s−1) to 3 s (at 0.1 m s−1).Results and discussion
Synthesis and characterization of g-CN based prepolymers
At first, g-CN was synthesized according to the literature54 and characterized via UV-Vis, powder XRD, FT-IR, surface zeta potential as well as elemental analysis (Fig. S2 and Table S1†). Previous work showed that g-CN forms radicals on the surface during visible light illumination,25 and the 2-D sheet like structure provides crosslinking points for polymeric networks. Combining the radical formation with significant dispersibility of g-CN in a water–EG mixture, g-CN grafted with PDMA (g-CN prepolymer) was synthesized via the addition of DMA to g-CN water–EG dispersion and irradiation with visible light (Fig. 1a). After 3 hours a viscoelastic and injectable g-CN-based material was obtained that exhibited increased viscosity (g-CN pre3h) but could be transferred into a sol state after dilution. Furthermore, TEM was utilized to image the formed g-CN-PDMA-hybrids, showing the dispersed g-CN particles in clusters (Fig. S3†). TGA investigations with g-CN pre3h indicate a mass loss profile due to the evaporation of water–EG solvents (up to 150 °C) and loss of organic networks (around 420 °C) (Fig. S4†), which is expected due to the composition of the g-CN-precursor polymer. For understanding the prepolymer formation, irradiation was applied for different time intervals and the conversion of DMA was elucidated via1H NMR. A DMA conversion of 12% and 32% was observed after 3 and 6 h respectively, which shows the gradual increase of DMA conversion with time. To gain insight into the properties of PDMA grafted g-CN, 1 mL g-CN pre3h sample was diluted with 10 mL water and centrifuged at 6000 rpm for 30 minutes. The sedimented PDMA-g-CN hybrid was collected and dried overnight in vacuo at 60 °C. Elemental analysis results show a significant increase in the C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N ratio, which can be attributed to the grafting of carbon-rich DMA to g-CN sheets (Table S2†). Surface zeta potential of DMA grafted g-CN particles remained similar to the initial g-CN, as no charges were introduced through grafting (Table S2†). Light absorption of PDMA grafted g-CN particles showed a similar pattern to parental g-CN, indicating the likely photoactivity of the precursor (Fig. S5a†). In addition, FT-IR spectra present the appearance of new peaks arising from PDMA (Fig. S5b†) for example the carbonyl stretching peak around 1700 cm−1.
:N ratio, which can be attributed to the grafting of carbon-rich DMA to g-CN sheets (Table S2†). Surface zeta potential of DMA grafted g-CN particles remained similar to the initial g-CN, as no charges were introduced through grafting (Table S2†). Light absorption of PDMA grafted g-CN particles showed a similar pattern to parental g-CN, indicating the likely photoactivity of the precursor (Fig. S5a†). In addition, FT-IR spectra present the appearance of new peaks arising from PDMA (Fig. S5b†) for example the carbonyl stretching peak around 1700 cm−1.
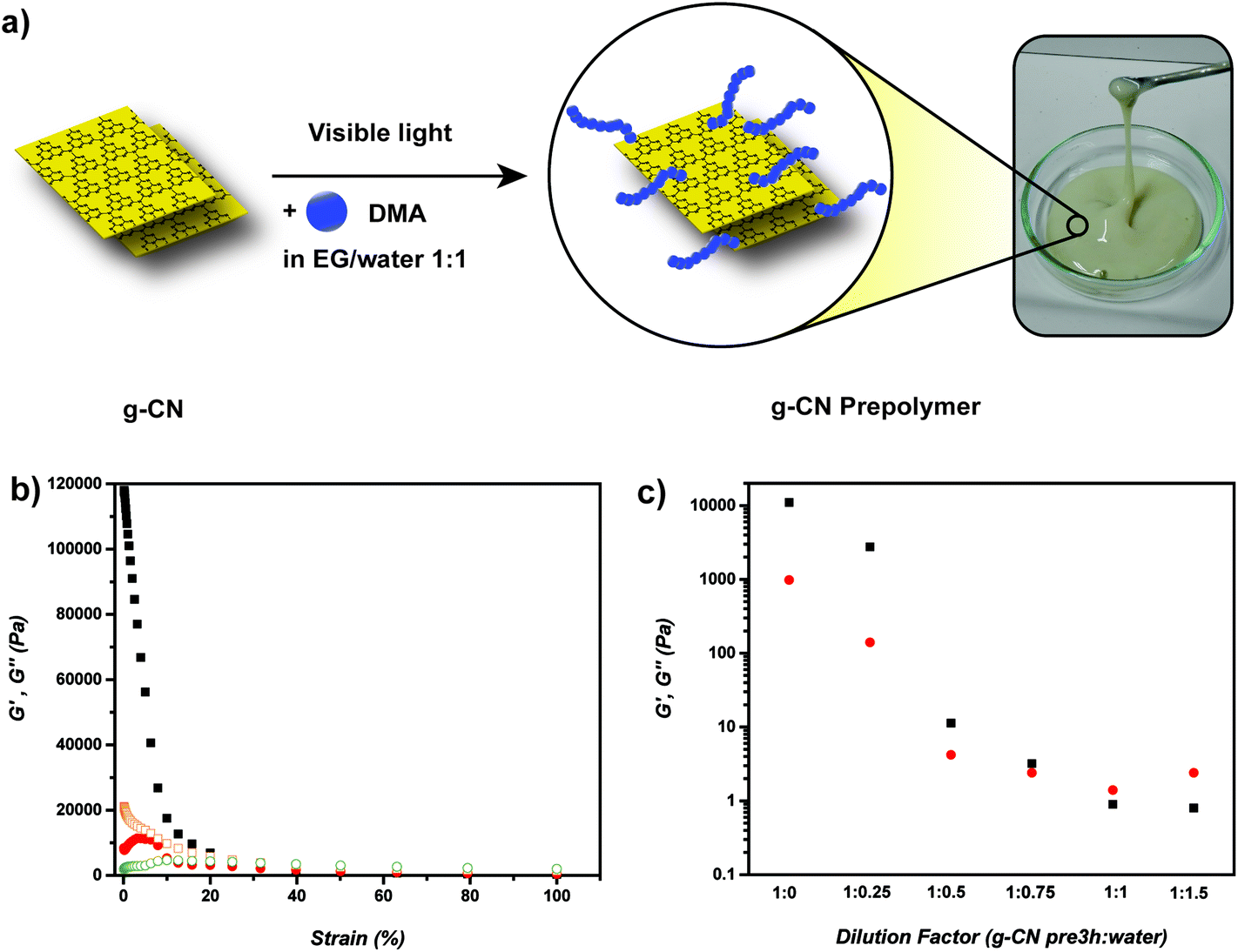 | ||
| Fig. 1 (a) Schematic representation of g-CN precursor formation, (b) storage and loss moduli values of g-CN pre3h against strain and (c) storage and loss moduli values of g-CN pre3h under dilution with water at 0.1% strain. | ||
Furthermore, the rheology of samples from different time points of polymerization was investigated (1, 3, 6 and 12 h). Storage moduli values (G′) increased dramatically during the reaction, i.e. more than 100 times between 1 h and 12 h reaction time, yet similar strain dependency was observed in all cases (Table 1, Fig. 1b, Fig. S6–8†). The increase can be attributed to the grafting and growth of PDMA chains on the surface of g-CN particles that leads to increased entanglement of chains and probably also increased crosslinking. In order to investigate the aforementioned effect, the prepolymer with a double amount of DMA was prepared, irradiated for 3 hours and its rheology was investigated (g-CN 2pre3h; Fig. S9†). In comparison with the previous recipe, increasing the organic content in prepolymer formulation resulted in a much stronger material where G′ exhibits a value of 118 kPa at 0.1% strain (10 times increased compared to g-CN pre3h). The increased value might be due to increased particle–particle crosslinking and indeed an organohydrogel formation. Increased DMA amounts (g-CN 2pre3h) result in stronger network formation, which diminishes the opportunity to utilize g-CN 2pre3h as a precursor for further reactions. As the grafting time increases, the active sites and accessibility of g-CN sheets decrease, which hinders the capacity for the gelation in the next step and therefore g-CN pre3h is chosen as an ideal precursor to be utilized in the next step.
| Prepolymer | Irradiation time (h) | G′c (Pa) | G′′c (Pa) | Viscosityd (Pa s) |
|---|---|---|---|---|
| a Composed of 2 wt% g-CN, 45 wt% water and EG, 8 wt% DMA. b Composed of 2 wt% g-CN, 41 wt% water, 41 wt% EG, 16 wt% DMA. c At 0.1% strain. d At 1 s−1 shear rate. | ||||
| g-CN pre1ha | 1 | 267 | 41 | 0.88 |
| g-CN pre3ha | 3 | 11100 |
870 | 5.32 |
| g-CN pre6ha | 6 | 28200 |
2370 | 23.9 |
| g-CN pre12ha | 12 | 36050 |
2340 | 163 |
| g-CN 2pre3hb | 3 | 118000 |
9500 | 770 |
Furthermore, frequency dependent rheology measurements were investigated for g-CN pre3h. Frequency sweeps showed a slight increase in G′ values with increasing frequency, confirming the stable covalent incorporation of the polymers (Fig. S10†). Due to the intention of identifying a suitable – and at best injectable – precursor for further processing, the viscosity of the prepolymers was investigated. As grafting takes place, viscosity of the prepolymer increases as well. In accordance with the G′ values, higher viscosity values of the prepolymer are obtained for longer irradiation times as indicated from viscosity measurements (Table 1, Fig. S11–15†). Particularly, doubling the concentration of the DMA monomer resulted in more than 100 fold increase in viscosity. The increase is attributed to polymer formation, entanglement and grafting to the g-CN surface. From a mechanistic point of view, the formation of photoinitiated g-CN prepolymers suggests a covalent connection between g-CN and polymer chains. The covalent incorporation of g-CN into hydrogels after light irradiation has been stressed before, e.g. in hydrogel formation without additional crosslinkers, hydrogelation in the illuminated area and no radical transfer to area in the dark. g-CN acts as a photoinitiator triggering radical polymerization which is initiated from the g-CN surface, therefore forming a covalent bond with vinyl molecules.
One of the main aspects is the colloidal stability of the prepolymer. Although hydrogen bonding and π–π interactions are present, no sedimentation was observed in the g-CN based prepolymer after 2 months of standing (Fig. S16†), which can be attributed to prepolymer formation that is based on the grafting of hydrophilic polymers onto well dispersed g-CN corresponding to a steric stabilization mechanism. As g-CN is negatively charged, overall an electrosteric stabilization mechanism is present. Such a significant colloidal stability of g-CN-based compounds could be highly practical for industrial applications. The addition of water to the prepolymer is expected to result in changes in the rheological profile and a transition from the gel to sol state. Therefore, the prepolymer mixture was diluted with water in different ratios to determine the concentration of transition from gel to sol. As expected, the addition of water caused a significant decrease in G′ values in g-CN pre3h where a 1:1 dilution led to a transition to the sol state (G′′ > G′) (Fig. 1c, Fig. S17–21†), which corresponds to a composition of 72.5 wt%, 22.5 wt% and 5 wt% for water, EG and g-CN-PDMA, respectively. Overall, a homogeneous, highly dispersible (Fig. 1a), injectable (Fig. S22†) and highly stable g-CN based viscoelastic prepolymer was formulated and employed for the exemplary formation of tough hydrogels in the next step.
Synthesis of tough hydrogels employing g-CN based prepolymer
Hydrogel synthesis using a g-CN prepolymer was chosen as an exemplary application using g-CN pre3h. Previously, our group reported the integration of g-CN particles as a reinforcer and a visible light photoinitiator in hydrogel synthesis.44 The utilization of a prepolymer as a primary network and further mixing it with a monomer and a crosslinker is a similar approach to double network hydrogels, where the first network is a non-solid viscous prepolymer. To show the versatility of the approach three concentrations were chosen (touA, touB, touC, touRef, Table S3†). The main advantage of the prepolymer approach is the presence of dispersed g-CN particles that can initiate a second photopolymerization without the addition of an extra photoinitiator, which also confirms the retained photoactivity of the synthesized g-CN prepolymers. The prepolymer was mixed with water, DMA and MBA in different concentrations and organohydrogel formation took place via short term visible light irradiation (approx. 20 minutes). The remaining EG can be easily removed from the organohydrogel network via washing with water to afford pure hydrogels.46After freeze drying of hydrogels, solid state analyses of touA were performed. As g-CN shows a characteristic absorption pattern in UV spectra (Fig. S2a†), similar absorption was observed, while the reference hydrogel shows no significant absorption (Fig. S23a†). Moreover, g-CN is composed of a crystalline structure due to interplanar tectonics and intraplanar domains. Thus, powder XRD was investigated for hydrogels (Fig. S23b†). The reference hydrogel shows a broad peak between 12–21° which might be attributed to the undefined porous network, where the g-CN based hydrogel possesses a pronounced broad peak between 13–25° which is due to sheet–sheet interactions. The FT-IR spectra of both reference and g-CN prepolymer based hydrogel present a strong carbonyl stretching band around 1700 cm−1 and C–N stretching around 1200 cm−1, and it is possible to observe triazine motifs around 800 cm−1 for the g-CN prepolymer based hydrogel (Fig. S23c†). The SEM image of the freeze dried g-CN prepolymer based hydrogel shows small particles on the smooth polymeric surface which might be related to g-CN particles within the polymer structure (Fig. S23d†). In order to gain a deeper insight into the network structure, swelling was probed. The swelling ratio of touA was slightly lower than the swelling of the reference sample (touRef) as g-CN particles in touA provide extra crosslinking resulting in a decreased amount of water uptake (Fig. S24†).
In the next step, both organohydrogels and hydrogels were characterized via mechanical compression tests (Fig. 2, Fig. S25 and Table S4†). The sample touA showed the highest strength against compression up to 2.67 MPa and a compression of 34%, while the compressibility decreased with increasing water content. A reference organohydrogel without g-CN incorporation showed the weakest performance (up to 0.14 MPa). In addition, elastic moduli, Emod, were determined at 10% strain before break. For the touA organohydrogel (with the highest organic content) the highest Emod of 11.9 MPa was obtained as expected, while doubling of the water content decreased Emod down to 6 MPa. Similar compression tests were also performed on hydrogels after EG removal. Commonly in the case of all samples hydrogels after EG removal deform at lower elongations compared to organohydrogels. In the case of swollen hydrogels, the touA hydrogel showed the highest Emod values at 9.9 MPa (10% before break) and a maximum stress of 2.36 MPa, whereas hydrogels with other compositions were much weaker. The incorporation of EG influences the mechanical properties of hydrogels, mainly in elasticity as it was observed in previous studies.46 EG enables deformation at higher elongation rates with higher Emod values compared to hydrogels. In the literature several types of reinforced tough hydrogels are described, for example double network hydrogels or gels containing hydrophobic domains.16,55,56 Albeit, a fair comparison of hydrogels in the literature originating from different methods is difficult to undertake, e.g. due to differences in the solid content, our hydrogels perform in a similar region to the common compressible hydrogels (fracture stresses and Emod in the MPa range). Overall, the first step of employing a g-CN based prepolymer strategy opens up a new platform for the photopolymerization based synthesis of tough hydrogels. A broad range of mechanical properties can be adjusted with this approach of hydrogel formation utilizing tailored compositions, which would allow targeting a variety of properties for various biomechanical systems in the future.
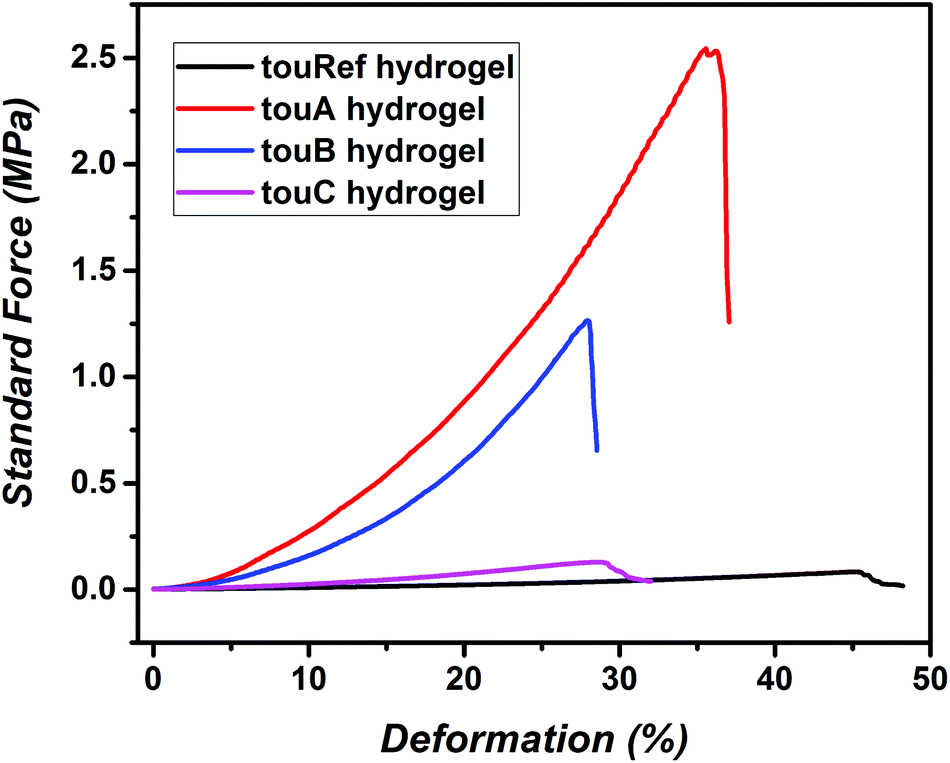 | ||
| Fig. 2 Compression test results of swollen reference hydrogels and g-CN pre3h based hydrogels with different compositions. | ||
Synthesis of lubricant hydrogels employing a g-CN based prepolymer
Extending the functionality of hydrogels beyond toughness has been of significant interest.57 Inspired from the composition of articular cartilage, lubricity should be accompanied by the toughness for the fabrication of functional hydrogels. Lubricity could be achieved via utilization of charged monomers in hydrogels. Extensive research has been conducted and charged monomers were investigated to provide lubricity in hydrogel systems. Significant knowledge about the friction mechanism between the charged monomer and water interface was obtained.53 Previous attempts to utilize charged monomers in g-CN dispersions failed as colloidal stability of g-CN in water was diminished upon the addition of charged compounds. As shown above, g-CN based prepolymers provide excellent colloidal stability, which enables the study of lubricity in g-CN based hydrogels. For the synthesis of g-CN pre3h, DMA and SPMA were applied (Fig. 3a, Table S5;† lubA, lubB, lubC, lubRef). g-CN pre3h based mixtures were illuminated via visible light and gelation was achieved in 1 hour. Moreover, EG can easily be removed via washing with water or two washing–freeze drying cycles.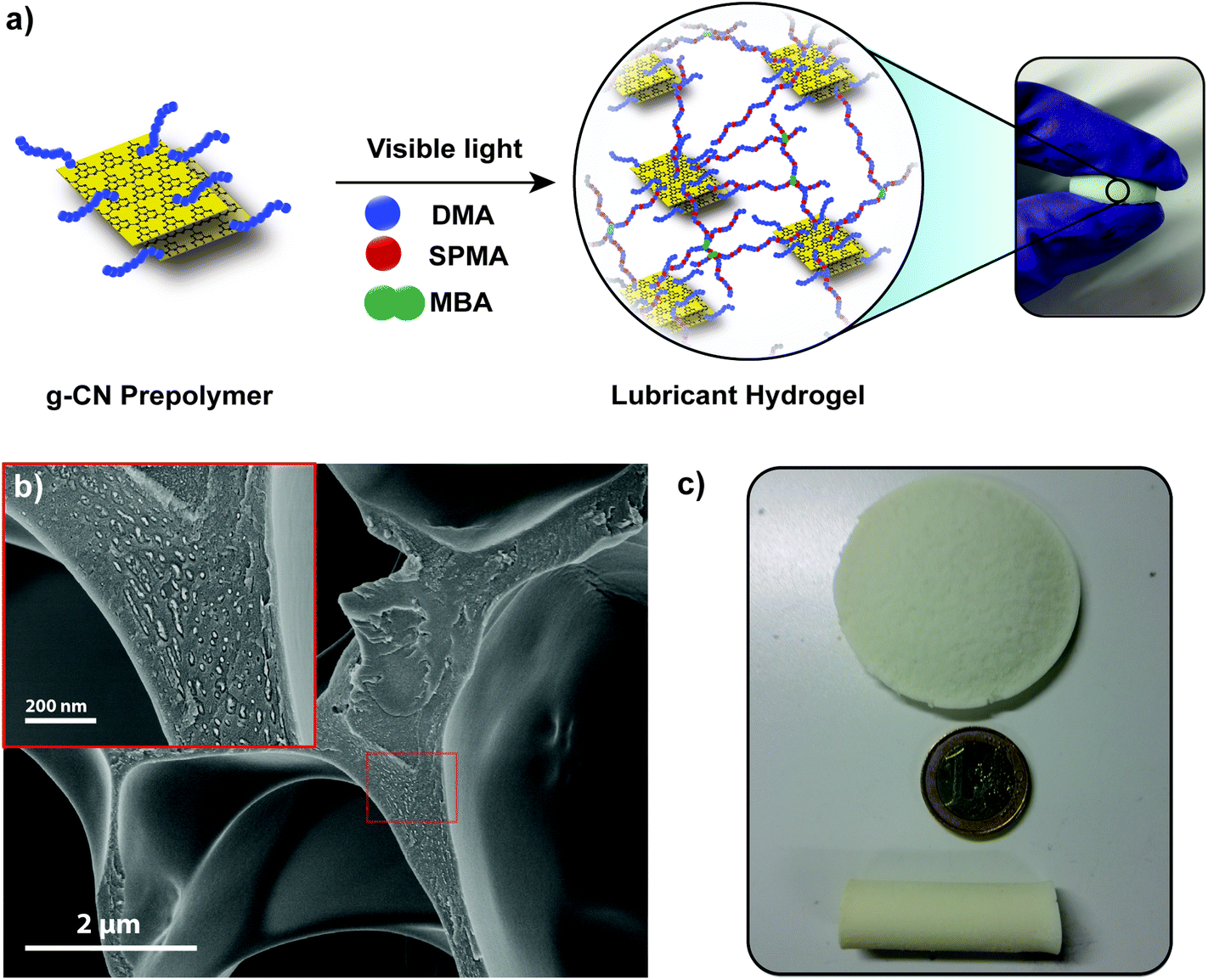 | ||
| Fig. 3 (a) Schematic overview of lubricant hydrogel formation, (b) SEM image of freeze dried lubA hydrogels and (c) images of lubA hydrogels fabricated in different shapes and sizes. | ||
After freeze drying of hydrogels, analyses of the dried samples were performed (exemplary with the sample lubA). The absorption pattern of parent g-CN can be observed after lubricant hydrogel synthesis, which indicates g-CN incorporation and photophysical properties (Fig. S26a†). XRD shows insignificant crystallinity of the freeze dried reference gel, i.e. lubA hydrogels show a peak at 27° which might be arising from g-CN sheets in the structure (Fig. S26b†). The FT-IR spectra of both reference and g-CN pre3h-based hydrogel present a strong carbonyl stretching band around 1700 cm−1 and the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band around 1200–1300 cm−1 corresponding to the sulfonate, where triazine peaks of g-CN can be observed around 800 cm−1 for the g-CN pre3h-based hydrogel (Fig. S26c†). The SEM image of the freeze dried lubA hydrogel shows a highly porous network with a smooth surface and at higher magnification, particles can be observed within the polymeric network that might correspond to incorporated g-CN (Fig. 3b). The swelling ratio of the reference hydrogel (lubRef) was slightly higher than that of lubA, as g-CN particles in lubA provide additional crosslinking points which result in a lower amount of water uptake (Fig. S24†), yet the lubricant hydrogel shows superhydrophilic character due to the charged SPMA units in the structure. Visible light initiation and synthesis from abundant and accessible chemicals allow scale-up synthesis conditions where lubricant hydrogels can be synthesized in any shape and size such as disc-like or tube-like (Fig. 3c).
O stretching band around 1200–1300 cm−1 corresponding to the sulfonate, where triazine peaks of g-CN can be observed around 800 cm−1 for the g-CN pre3h-based hydrogel (Fig. S26c†). The SEM image of the freeze dried lubA hydrogel shows a highly porous network with a smooth surface and at higher magnification, particles can be observed within the polymeric network that might correspond to incorporated g-CN (Fig. 3b). The swelling ratio of the reference hydrogel (lubRef) was slightly higher than that of lubA, as g-CN particles in lubA provide additional crosslinking points which result in a lower amount of water uptake (Fig. S24†), yet the lubricant hydrogel shows superhydrophilic character due to the charged SPMA units in the structure. Visible light initiation and synthesis from abundant and accessible chemicals allow scale-up synthesis conditions where lubricant hydrogels can be synthesized in any shape and size such as disc-like or tube-like (Fig. 3c).
Subsequently, organohydrogels and hydrogels were characterized via compression tests to obtain mechanical properties (Fig. 4a and b, Fig. S27 and 28†). Investigating the compression of organohydrogels, lubB which consists of only DMA as a secondary network shows the highest Emod values (around 3.1 MPa) and lubC (consisting of only SPMA as a secondary network) shows the lowest (around 1.27 MPa). The performance of lubA, by means of Emod value, falls exactly between them (around 2.52 MPa), which shows the adjustable properties of lubricant g-CN-based organohydrogels (Table S6†). After EG is removed from the organohydrogel networks, further investigations were performed to understand the mechanical properties of pure hydrogels. The pure lubA hydrogel consists of two different monomers, namely DMA and SPMA. DMA was chosen to adjust toughness, while SPMA was chosen to enable and adjust lubricity. The compression test results of hydrogels showed similar profiles to the tough hydrogels. The reference hydrogel formed without g-CN incorporation (lubRef) shows inferior compression behavior compared to hydrogels based on g-CN incorporation (maximum stress of 0.29 MPa), which clearly shows the reinforcer role played by g-CN in the system. Thus, as an example the pure hydrogel sample of lubA shows a significant compressibility with a maximum stress of 0.47 MPa (Fig. 4a, Fig. S28†). It is important to note that the lubC hydrogel which consists of SPMA as a secondary network was slimy and almost non-self-standing. Thus, no reliable compression measurements could be performed with lubC. Except that, the lubB hydrogel can withstand the highest amount of load, as expected, and the lubA hydrogel has a performance that lies between lubB and lubC. Accordingly, elastic moduli values of the hydrogels point in the same direction (Table S6†). First of all, compared to the lubRef hydrogel, the lubA hydrogel loses its flexibility and breaks at lower strain. lubB shows the highest Emod values at any elongation rate, and lubA presents much higher Emod values at break compared to the reference. Overall, the compression tests support the necessity to include g-CN as a reinforcer and DMA as a mechanical adjuster in the composite hydrogel system. As durability against continuous compression is important in cartilage like systems, cyclic compression was performed with the lubA hydrogel (Fig. 4b). Therefore, cyclic compression was performed up to a strain 5% below break, which corresponds to a load of 0.5 MPa. No signs of fracture were observed after 100 cycles and the compression curve shows only a minor shift, indicating the durability of the synthesized hydrogel.
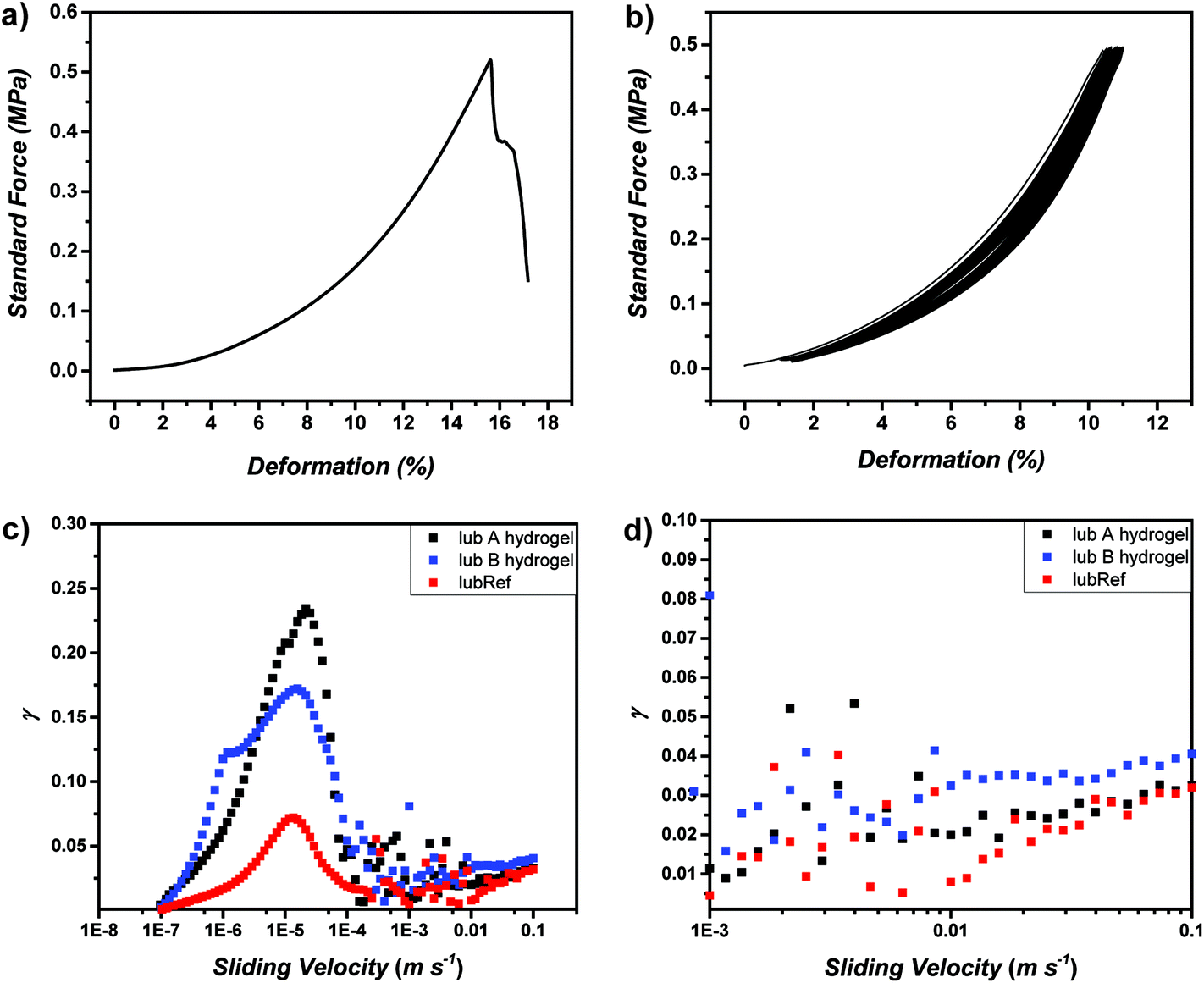 | ||
| Fig. 4 (a) Compression profiles of lubA hydrogels, (b) cyclic compression (100 times) profiles of lubA hydrogels, (c) and (d) Stribeck curves for lubricant hydrogels. | ||
The incorporation of SPMA endows the hydrogel with low friction properties albeit rather strong mechanical properties are retained. The lubricity of the lubA hydrogel can be observed directly by its sliding properties on an inclined plane compared to a reference hydrogel without SPMA incorporation (lubB) (refer to Videos S1 and S2†). In addition, tribological measurements were performed to quantify the lubrication properties, i.e. expressed in the friction coefficient (γ). Lubricity was investigated via extended Stribeck curves in the sliding velocity range of 10−7 to 0.1 m s−1. At 0.1 m s−1γ of 0.03 was observed for lubA and lubRef reference hydrogels, while γ of 0.04 was observed for the lubB hydrogel. The obtained values are in good agreement with the literature materials.58,59 Notably, the low friction properties are combined with high compressibility in the lubA material. Also the shape of the extended Stribeck curve is in agreement with the literature60,61 and can be explained with the adsorption–repulsion model.62 In the present case, the friction of hydrogel on hydrogel was investigated (Fig. S27†). The increase of γ at low sliding velocities can be explained with the elastic friction arising from elastic stretching due to attraction of the dangling polymer chains between the two hydrogel surfaces. At higher sliding velocities, the hydrogel surface polymer chains that were adsorbed first, desorb and the movement is too fast to form new adsorption sites. Therefore, hydrodynamic lubrication takes over, which increases with velocity in a monotone way. The local maximum at a sliding velocity of 10−6 m s−1 in the extended Stribeck curve of sample lubB is another literature known effect, which has been attributed to the onset of macroscopic motion.61 Overall, for the first time, charged monomers can be employed together with g-CN for visible light induced gelation which allows a variety of functions to be investigated in future.
Conclusions
Herein, g-CN was introduced into colloidally stable dispersed systems via the synthesis of g-CN based prepolymers. An injectable prepolymer was synthesized via visible light irradiation and possesses high stability over long periods as well as facile processing. As an exemplary application, the g-CN based prepolymer was employed as the first network in hydrogel synthesis yielding tough hydrogels where g-CN acts as a reinforcer and photoinitiator. Colloidal stability of the g-CN based prepolymer allows the introduction of charged monomers as well, yielding tough and lubricant hydrogels via a photoinitiated reaction without an external initiator. Initial attempts to synthesize g-CN based cartilage-like tough and lubricant hydrogels were successful and further attempts to investigate lubricity, accelerate the gelation rate and bio-related studies are underway. These might reveal visible light photo-polymerization based injectable cartilage-replacement formation, which would entirely change the treatment for such problems from periodical and painful surgeries to affordable and facile injection methods.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors thank Max Planck Society for financial support and Dr Kerstin Blank and Dr Alberto Sanz de Leon for rheometer access.Notes and references
- T. Murakami, S. Yarimitsu, K. Nakashima, N. Sakai, T. Yamaguchi, Y. Sawae and A. Suzuki, Proc. – Inst. Mech. Eng., 2015, 229, 864–878 CrossRef PubMed.
- T. Murakami, JSME Int. J., Ser. III, 1990, 33, 465–474 Search PubMed.
- B. Winkeljann, A. B. Bussmann, M. G. Bauer and O. Lieleg, Biotribology, 2018, 14, 11–18 CrossRef.
- M. Brittberg, A. H. Gomoll, J. A. Canseco, J. Far, M. Lind and J. Hui, Acta Orthop., 2016, 87, 26–38 CrossRef PubMed.
- S. Yarimitsu, S. Sasaki, T. Murakami and A. Suzuki, Biosurf. Biotribol., 2016, 2, 40–47 CrossRef.
- T. Li, B. Kumru, N. Al Nakeeb, J. Willersinn and B. V. K. J. Schmidt, Polymers, 2018, 10, 576 CrossRef PubMed.
- M. Liu, Y. Ishida, Y. Ebina, T. Sasaki, T. Hikima, M. Takata and T. Aida, Nature, 2015, 517, 68–72 CrossRef CAS PubMed.
- G. Gao, G. Du, Y. Cheng and J. Fu, J. Mater. Chem. B, 2014, 2, 1539 RSC.
- J. Djonlagic, A. Lancuski, M. S. Nikolic, J. Rogan, S. Ostojic and Z. Petrovic, J. Appl. Polym. Sci., 2017, 134, 44535 CrossRef.
- K. Fujii, H. Asai, T. Ueki, T. Sakai, S. Imaizumi, U.-i. Chung, M. Watanabe and M. Shibayama, Soft Matter, 2012, 8, 1756–1759 RSC.
- M. Fukasawa, T. Sakai, U.-i. Chung and K. Haraguchi, Macromolecules, 2010, 43, 4370–4378 CrossRef CAS.
- A. Agrawal, N. Rahbar and P. D. Calvert, Acta Biomater., 2013, 9, 5313–5318 CrossRef CAS PubMed.
- S. E. Bakarich, R. Gorkin 3rd, M. in het Panhuis and G. M. Spinks, ACS Appl. Mater. Interfaces, 2014, 6, 15998–16006 CrossRef CAS PubMed.
- T. Murakami, B. V. K. J. Schmidt, H. R. Brown and C. J. Hawker, Macromolecules, 2015, 48, 7774–7781 CrossRef CAS.
- K. Ito, Polym. J., 2007, 39, 489–499 CrossRef CAS.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- J. P. Gong, Soft Matter, 2010, 6, 2583 RSC.
- Q. Chen, H. Chen, L. Zhu and J. Zheng, J. Mater. Chem. B, 2015, 3, 3654–3676 RSC.
- B. G. Cooper, R. C. Stewart, D. Burstein, B. D. Snyder and M. W. Grinstaff, Angew. Chem., Int. Ed., 2016, 55, 4226–4230 CrossRef CAS PubMed.
- X. Zhang, J. Wang, H. Jin, S. Wang and W. Song, J. Am. Chem. Soc., 2018, 140, 3186–3189 CrossRef CAS PubMed.
- J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Fu, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 441–444 CrossRef CAS PubMed.
- H. Li, L. Wang, Y. Liu, J. Lei and J. Zhang, Res. Chem. Intermed., 2015, 42, 3979–3998 CrossRef.
- J. Liu, H. Wang and M. Antonietti, Chem. Soc. Rev., 2016, 45, 2308–2326 RSC.
- J. Barrio and M. Shalom, ChemCatChem, 2018, 10, 5573–5586 CrossRef CAS.
- B. Kumru, M. Antonietti and B. V. K. J. Schmidt, Langmuir, 2017, 33, 9897–9906 CrossRef CAS.
- B. Kumru, D. Cruz, T. Heil, B. V. K. J. Schmidt and M. Antonietti, J. Am. Chem. Soc., 2018, 140, 17532–17537 CrossRef CAS.
- J. Sun, R. Phatake, A. Azoulay, G. Peng, C. Han, J. Barrio, J. Xu, X. Wang and M. Shalom, Chem. – Eur. J., 2018, 24, 14921–14927 CrossRef CAS PubMed.
- J. K. Kim, S. Park, R. J. Yoo, H. J. Jeong, J. Oh, Y. J. Lee, S. Park and D. W. Kim, Chem. – Eur. J., 2018, 24, 3506–3511 CrossRef CAS PubMed.
- C. Butchosa, P. Guiglion and M. A. Zwijnenburg, J. Phys. Chem. C, 2014, 118, 24833–24842 CrossRef CAS.
- K. Striegler, Modified graphitic carbon nitrides for photocatalytic hydrogen evolution from water: copolymers, Springer, 2015 Search PubMed.
- Q. Zheng, D. P. Durkin, J. E. Elenewski, Y. Sun, N. A. Banek, L. Hua, H. Chen, M. J. Wagner, W. Zhang and D. Shuai, Environ. Sci. Technol., 2016, 50, 12938–12948 CrossRef CAS PubMed.
- J. Barrio and M. Shalom, ACS Appl. Mater. Interfaces, 2018, 10, 39688–39694 CrossRef CAS PubMed.
- J. Ji, J. Wen, Y. Shen, Y. Lv, Y. Chen, S. Liu, H. Ma and Y. Zhang, J. Am. Chem. Soc., 2017, 139, 11698–11701 CrossRef CAS PubMed.
- R. Kuriki, K. Sekizawa, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2015, 54, 2406–2409 CrossRef CAS PubMed.
- J. Lin, Z. Pan and X. Wang, ACS Sustainable Chem. Eng., 2014, 2, 353–358 CrossRef CAS.
- J. Mao, T. Peng, X. Zhang, K. Li, L. Ye and L. Zan, Catal. Sci. Technol., 2013, 3, 1253 RSC.
- S. Dadashi-Silab, M. A. Tasdelen, B. Kiskan, X. Wang, M. Antonietti and Y. Yagci, Macromol. Chem. Phys., 2014, 215, 675–681 CrossRef CAS.
- Q. Fu, Q. Ruan, T. G. McKenzie, A. Reyhani, J. Tang and G. G. Qiao, Macromolecules, 2017, 50, 7509–7516 CrossRef CAS.
- M. Al-Naji, B. Puértola, B. Kumru, D. Cruz, M. Bäumel, B. V. K. J. Schmidt, N. Tarakina and J. Pérez-Ramírez, ChemSusChem, 2019 DOI:10.1002/cssc.201900418.
- J. Poostforooshan, A. Badiei, M. Kolahdouz and A. P. Weber, ACS Appl. Mater. Interfaces, 2016, 8, 21731–21741 CrossRef CAS PubMed.
- Z. Huang, F.-W. Yan and G. Yuan, RSC Adv., 2017, 7, 1318–1325 RSC.
- B. Ye, C. Yao, M. Yan, H. Zhang, F. Xi, J. Liu, B. Li and X. Dong, Macromol. Mater. Eng., 2019, 304, 1800500 CrossRef.
- C. Hu, Y. R. Lin and H. C. Yang, ChemSusChem, 2019, 12, 1794–1806 CrossRef CAS PubMed.
- B. Kumru, M. Shalom, M. Antonietti and B. V. K. J. Schmidt, Macromolecules, 2017, 50, 1862–1869 CrossRef CAS.
- Y. Zhang, Z. Zhou, Y. Shen, Q. Zhou, J. Wang, A. Liu, S. Liu and Y. Zhang, ACS Nano, 2016, 10, 9036–9043 CrossRef CAS PubMed.
- B. Kumru, V. Molinari, M. Shalom, M. Antonietti and B. V. K. J. Schmidt, Soft Matter, 2018, 14, 2655–2664 RSC.
- B. Kumru, V. Molinari, R. Dunnebacke, K. G. Blank and B. V. K. J. Schmidt, Macromol. Rapid Commun., 2019, 40, 1800712 Search PubMed.
- B. Kumru, J. Barrio, J. Zhang, M. Antonietti, M. Shalom and B. V. K. J. Schmidt, ACS Appl. Mater. Interfaces, 2019, 11, 9462–9469 CrossRef CAS PubMed.
- J. W. Ko, W. S. Choi, J. Kim, S. K. Kuk, S. H. Lee and C. B. Park, Biomacromolecules, 2017, 18, 3551–3556 CrossRef CAS PubMed.
- P. He, X. Tang, L. Chen, P. Xie, L. He, H. Zhou, D. Zhang and T. Fan, Adv. Funct. Mater., 2018, 28, 1801121 CrossRef.
- U. Raviv, S. Giasson, N. Kampf, J.-F. Gohy, R. Jerome and J. Klein, Nature, 2003, 425, 163–165 CrossRef CAS PubMed.
- M. Kobayashi, Y. Terayama, N. Hosaka, M. Kaido, A. Suzuki, N. Yamada, N. Torikai, K. Ishihara and A. Takahara, Soft Matter, 2007, 3, 740–746 RSC.
- D. Kaneko, T. Tada, T. Kurokawa, J. P. Gong and Y. Osada, Adv. Mater., 2005, 17, 535–538 CrossRef CAS.
- M. Shalom, S. Inal, C. Fettkenhauer, D. Neher and M. Antonietti, J. Am. Chem. Soc., 2013, 135, 7118–7121 CrossRef CAS PubMed.
- D. Gu, S. Tan, C. Xu, A. J. O'Connor and G. G. Qiao, Chem. Commun., 2017, 53, 6756–6759 RSC.
- Q. Chen, H. Chen, L. Zhu and J. Zheng, J. Mater. Chem. B, 2015, 3, 3654–3676 RSC.
- M. M. Blum and T. C. Ovaert, Mater. Sci. Eng., C, 2013, 33, 4377–4383 CrossRef CAS PubMed.
- P. E. Milner, M. Parkes, J. L. Puetzer, R. Chapman, M. M. Stevens, P. Cann and J. R. T. Jeffers, Acta Biomater., 2018, 65, 102–111 CrossRef CAS PubMed.
- Y. Ohsedo, R. Takashina, J. P. Gong and Y. Osada, Langmuir, 2004, 20, 6549–6555 CrossRef CAS PubMed.
- F. Rummel, K. S. Pondicherry, M. Ruge and V. Fronk, Tribol. Schmierungstech., 2018, 65, 36–43 Search PubMed.
- K. S. Pondicherry, F. Rummel and J. Laeuger, Biosurf. Biotribol., 2018, 4, 34–37 CrossRef.
- T. Kurokawa, T. Tominaga, Y. Katsuyama, R. Kuwabara, H. Furukawa, Y. Osada and J. P. Gong, Langmuir, 2005, 21, 8643–8648 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental procedures, measurement data and video materials. See DOI: 10.1039/c9py00505f |
| This journal is © The Royal Society of Chemistry 2019 |