DOI:
10.1039/C9PY00438F
(Paper)
Polym. Chem., 2019,
10, 2716-2722
Monodisperse, sequence-defined macromolecules as a tool to evaluate the limits of ring-closing metathesis†
Received
22nd March 2019
, Accepted 22nd April 2019
First published on 30th April 2019
Abstract
Sequence-defined macromolecules of uniform size unlock the door to many new applications in polymer chemistry, such as structure/property or structure/activity relationship investigations, which cannot be conducted accurately, if the investigated macromolecules exhibit dispersity. We herein demonstrate a first example by reporting the efficient and template-free synthesis of monodisperse, sequence-defined cyclic oligomers that are significantly larger than conventional large macrocycles (here >150 backbone atoms). Linear monodisperse precursors were utilized to evaluate the limits of ring-closing metathesis (RCM), manifesting clear trends depending on the ring size and introduced side chains. Furthermore, this work is the first example of a sequence-defined synthesis of a polymer architecture other than linear macromolecules.
Introduction
The field of sequence-control and definition in polymer chemistry has evolved to become one of the hottest topics in polymer chemistry today, as chemists worldwide seek more control over the molecular structure of macromolecules, and it has allowed to develop new applications for well-defined polymers and to correlate structure–property relationships.1 Over the last decade, numerous new approaches toward defined macromolecules have been developed,1a,2 which have paved the road for applications like data storage,3 anti-counterfeit tags4 or enzyme mimicking catalysts.5 As Lutz, Ouchi and Sawamoto defined in 2013, it is crucial to distinguish between the two terms “sequence-controlled” and “sequence-defined” in this context, since only the latter are unique macromolecules (i.e. not showing dispersity in the sequence or molecular weight).6 Sequence-controlled polymers, on the other hand, still exhibit distributions in both the building block sequence and macromolecule size (Đ > 1).7
Most synthetic approaches toward sequence-defined macromolecules are based on iterative strategies, which form oligomers in a stepwise fashion, either by strategic combination of orthogonal reactions, or by using protecting groups.2e,8 Multicomponent reactions, like the Passerini three-component reaction (P-3CR) used in this work, have been shown to be a highly efficient tool to introduce various functionalities to the oligomeric backbone due to their modular character.2c,d,3a,9 P-3CR, which was first discovered in 1921, is a robust and efficient one-pot reaction between an acid, an aldehyde and an isocyanide, providing nearly quantitative yields (see Scheme 1B).10
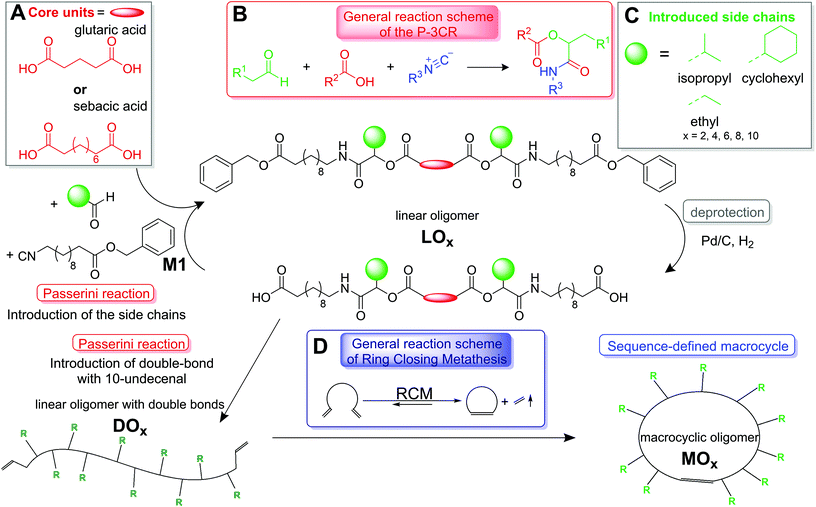 |
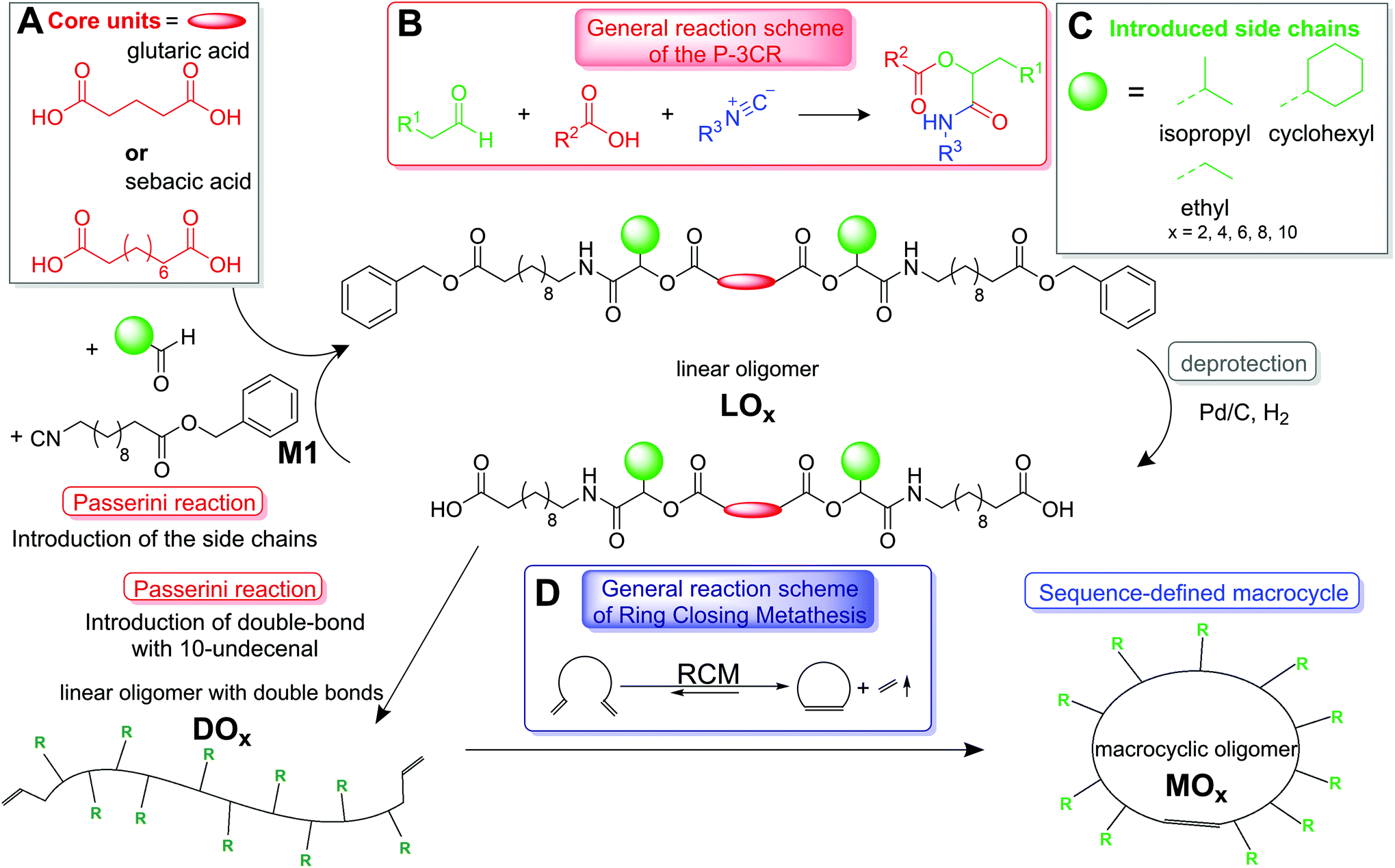
| | Scheme 1 General reaction scheme for the synthesis of large, monodisperse and sequence-defined macrocycles using bifunctional acids (panel A) as core units. The three different side chains (displayed in panel C) are introduced to the linear macromolecules. The general reaction scheme of the P-3CR, which is used to form linear oligomers, is shown in panel B and the general reaction scheme of the RCM, which is used for macrocyclization, is shown in panel D. | |
Synthetic investigations on polymer architectures, such as linear polymers, polymer brushes, star polymers, ladder polymers, dendrimers, hyperbranched polymers, and network polymers, have led to an understanding on how the material properties are fundamentally affected by the respective molecular architecture.11 Oligomeric, monodisperse macrocycles have gained growing interest due to their characteristic behavior compared to their linear analogs, because the end groups of non-cyclic polymers have demonstrated a significant influence on their properties.12 The synthesis of cyclic polymers is classified into two main methods: end-to-end cyclization of linear precursors, which can be further divided into unimolecular reactions of α,ω-heterofunctional polymers13 and bimolecular reactions of α,α′-homodifunctional polymers,14 as well as ring-expansion polymerizations.15 However, regardless of the mentioned methodology, the macrocycles obtained were either disperse in size,12b or monodisperse and small in size (ca. 3–13 bonds along the backbone).11 In organic chemistry, ring-closing metathesis (RCM) is mostly applied for the preparation of small rings bearing five to seven ring atoms.16 However, also larger rings of eight to eleven ring atoms, which are referred to as medium sized rings, are formed by RCM17 and furthermore so-called large macrocycles of 12 to 15 and more ring atoms17 or more complex polycyclic systems are obtained.18 These monodisperse cycles obtained by RCM exhibit versatile structures17,19 and the yields vary from very low to nearly quantitative, depending on the substrates.17,19 Several examples of macrocyclic biomacromolecules are also known, for instance macrocyclic peptides or DNA are reported.20 In cyclic peptides, typically smaller cycles consisting of four to six amino acids are formed,21 while examples of ultra large DNA cycles are reported.22 Interestingly, such cycles are formed using rigid precursors with an appropriate geometry, which lead to a preorganization and thus easier cyclization. Preorganization of linear precursors by ligands, hydrogen bonds or rigid architectures is a very useful and widely applied concept to achieve an efficient macrocylization.23 In contrast, macrocycles with a molecular weight in the range of polymers (for example cyclic polythiolactones with a molecular weight of up to 13 kDa24) are obtained if disperse polymeric or oligomeric precursors, obtained by controlled radical polymerization (i.e. ATRP polystyrene oligomers with a relatively low PDI < 1.225), are cyclized. The synthesis of macrocycles, that are both large and monodisperse, in contrast, was especially developed for catenanes,26 where the use of a metal ion template, that serves as a ligand coordination site for the linear components, was inevitable.27 One interesting exception reported by Alabi and coworkers is an iterative approach towards defined macrocycles consisting of up to 56 ring atoms.28
Results and discussion
Synthesis of symmetric linear oligomers (LOX)
We herein present a new, efficient and fast template-free approach toward large sequence-defined macrocycles (up to 152 ring atoms) of uniform size, bearing flexible backbones and tailored side chains. To form sequence-defined macrocycles, linear and symmetric oligomers bearing tailored side chains were first synthesized as precursors. For this purpose, a synthesis protocol, which was previously developed by our group in order to synthesize a sequence-defined icosamer,9d was adapted. The oligomers were formed in a two-step iterative cycle consisting of P-3CR and a deprotection step. Apart from the monomer (M1), which bears an isocyanide and a benzyl ester protected acid, and which is synthesized in three steps from the corresponding amino acid (see the ESI† for details),9d all used substances are commercially available. We have previously shown that this strategy leads to the straightforward and high-yielding synthesis of a sequence-defined icosamer.9d Here, a bifunctional carboxylic acid as the starting material (see Scheme 1A, i.e. bidirectional growth) reduced the overall reaction time. As such, linear oligomers of different lengths (LO2,4,6,8,10, see Fig. 1) carrying three different side chains (namely ethyl, cyclohexyl or isopropyl, see Scheme 1C) were synthesized. Just as a note for clarity, the formed stereocenters remain undefined and thus a mixture of stereoisomers is formed, as is typical in this young field of research. In the future, it would be of course of high relevance and importance to increase the molecular definition even further by stereoselective synthesis.
 |
| | Fig. 1 SEC analysis of the linear oligomers (LO2–8) carrying isopropyl side chains. The SEC traces clearly verify the uniform size distribution of the obtained products. | |
Initially, glutaric acid was used as the starting material, but the yield in the first P-3CR was relatively low (69–86%, depending on the side chain). Thus, glutaric acid was later replaced with the longer sebacic acid leading to significantly increased yields of 97–99%. Four different oligomers were synthesized: two with glutaric acid as the starting material and ethyl or cyclohexyl side chains and two with sebacic acid as the core unit carrying cyclohexyl or isopropyl side chains (see Scheme 1). The P-3CRs were conducted in dichloromethane (DCM) at a concentration of 0.5 M. Monomer M1 and the aldehyde components (propanal, cyclohexane carboxaldehyde or isobutyraldehyde) were added in a small excess (1.5 eq.) relative to the acid groups (3.0 eq. of these components relative to the diacid). After a reaction time of 24 hours, the crude products were purified by column chromatography to obtain the pure dimers in nearly quantitative yields (i.e. 100% by NMR spectroscopy, 98–99% purity by SEC, see Fig. 1). The oligomer synthesis was performed on a gram scale (i.e. product LO8 was obtained with a yield of 2.5 g). The products were characterized after each reaction step by 1H, 13C NMR, and IR spectroscopy, mass spectrometry and size exclusion chromatography (SEC) to verify the structure of the obtained products and their dispersity (compare Fig. 1 and see the ESI† for the respective set of analytical data of all the obtained products). In the second reaction of the iterative cycle, the benzyl ester protecting groups were removed by hydrogenolysis in a quantitative manner. Upon complete deprotection, a second P-3CR can be performed on account of the newly formed dicarboxylic acid. By applying this iterative strategy, symmetric oligomers of a length up to ten units were synthesized. In the third P-3CR, the reaction time was extended to 48 hours to ensure full conversion, while for the deprotection step, the concentration was decreased with increasing length of the oligomers to address the increased viscosity and ensure full conversion (see the ESI† for experimental details).
Introduction of terminal double bonds to form DOX
At different stages of the synthesis, small amounts (300–900 mg) of the deprotected oligomers (LOx) were separated from the product and were used in one last P-3CR with 10-undecenal, in order to introduce two terminal double bonds to the final linear oligomer (DOx). The introduction of the double bonds always marked the final reaction before macrocyclization for the respective oligomer DOx. The remaining LOx was used to continue the synthesis towards higher molecular weight oligomers, with the introduction of the double bonds taking place at a later stage. Thus, symmetric monodisperse dimers, tetramers, hexamers, octamers, and decamers, containing tailored side chains and terminal double bonds (DOx), were obtained.
Ring-closing metathesis to form MOX
The introduced terminal double bonds were exploited for the formation of sequence-defined macrocycles (MOx) via RCM (see Scheme 1D). Grubbs 1st generation catalyst was chosen, since it catalyzes the RCM reaction, while minimizing the ring-opening polymerization of the formed cycles.29 Furthermore, it does not show olefin isomerization side reactions, which are very pronounced for the 2nd generation catalyst and can only be suppressed to some extent (and would thus lead to dispersity in our system).30 Initially, the RCM reaction of a DO8 carrying cyclohexyl side chains was investigated and the crude product (MO8) was analyzed by SEC-ESI-MS and a relatively low conversion towards the macrocycle of around 32% was determined. The reaction was thus optimized by adding the same amount (10 mol%) of catalyst several times during the reaction, thus increasing the total amount of catalyst to 40 mol%. This catalyst concentration is relatively high, if compared with typical catalyst concentrations of 1–5 mol% for the synthesis of smaller cycles; however, if larger or highly functionalized molecules were cyclized, also higher catalyst concentrations of 30 or even 60 mol% are reported.17a After this optimization, all RCM reactions were performed under identical conditions in order to evaluate the influence of the side chains or the oligomer length on the conversion towards the macrocycle. The reactions were carried out in high dilution (ca. 5 × 10−4 mol L−1 in chloroform) in order to prevent acyclic diene metathesis (ADMET), using Grubbs 1st generation catalyst, which was added in four aliquots in the course of the reaction (4 × 10 mol%). The desired macrocycles were obtained after five hours at 45 °C. Remarkably, the reactivity of the catalyst was sufficient to achieve high conversion even at such low concentrations.17a
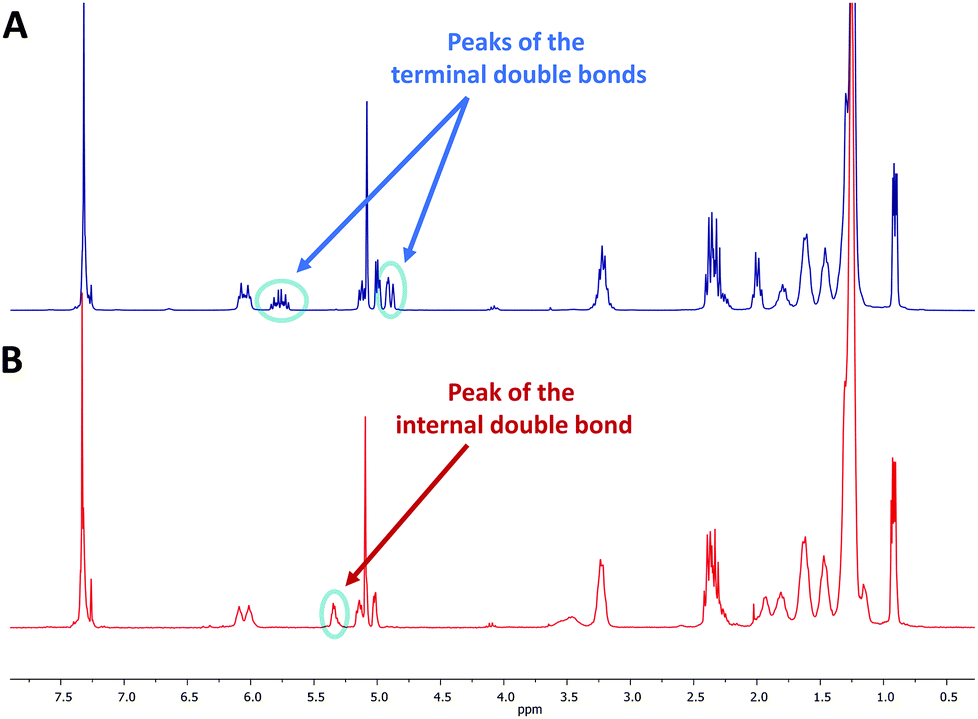
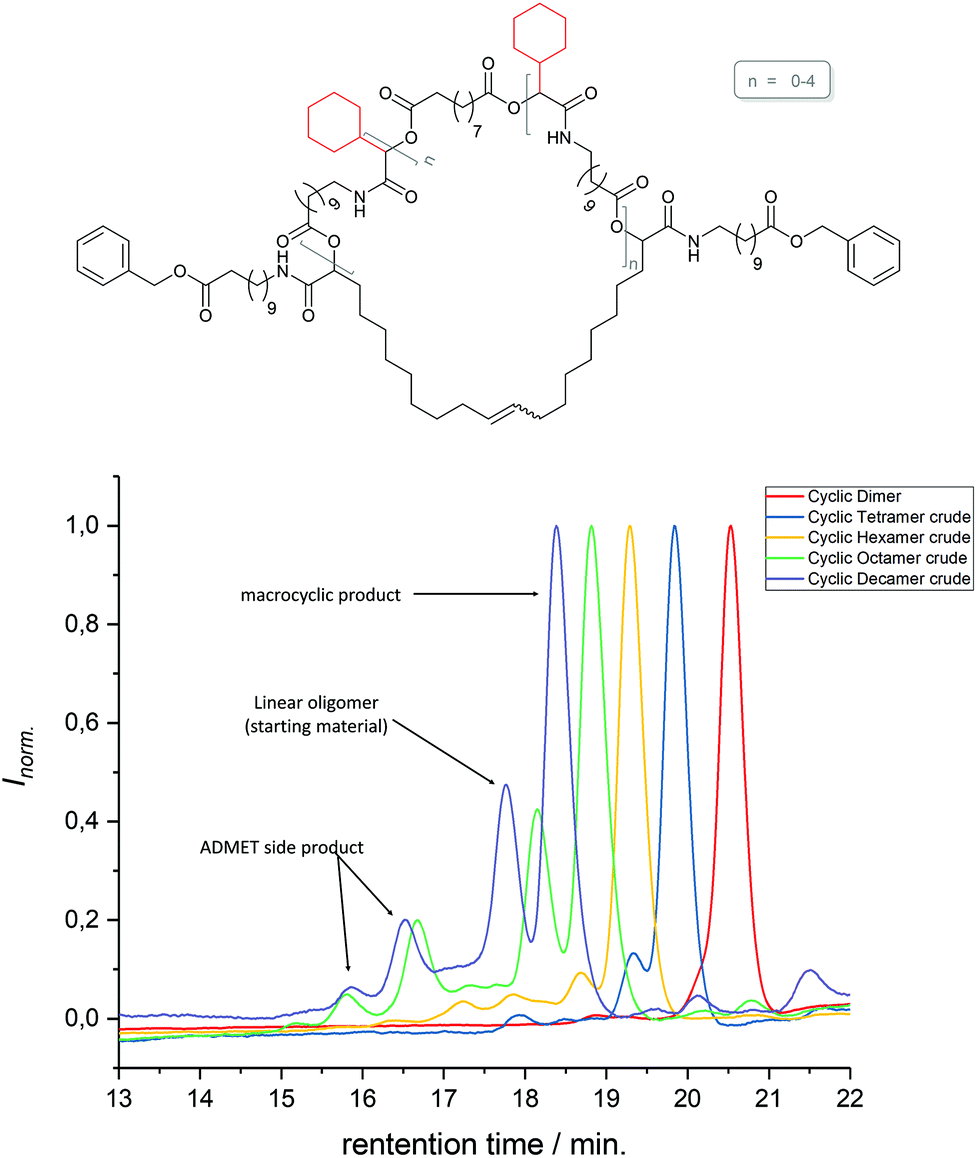
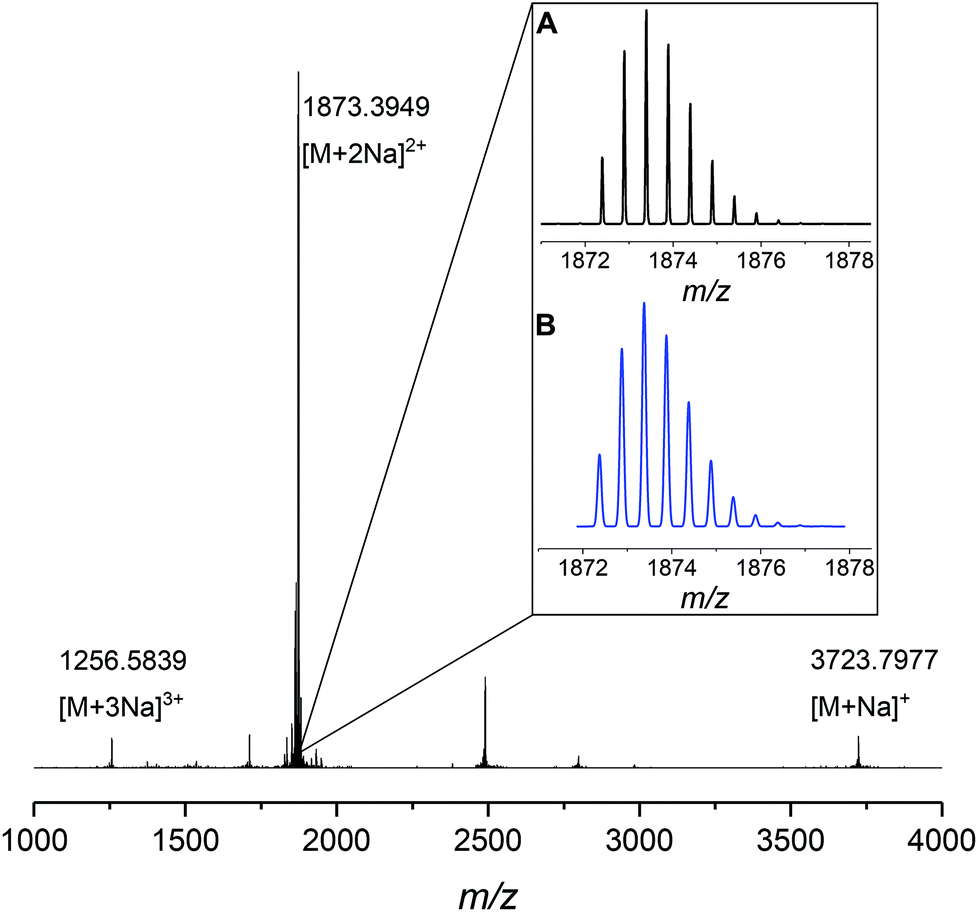
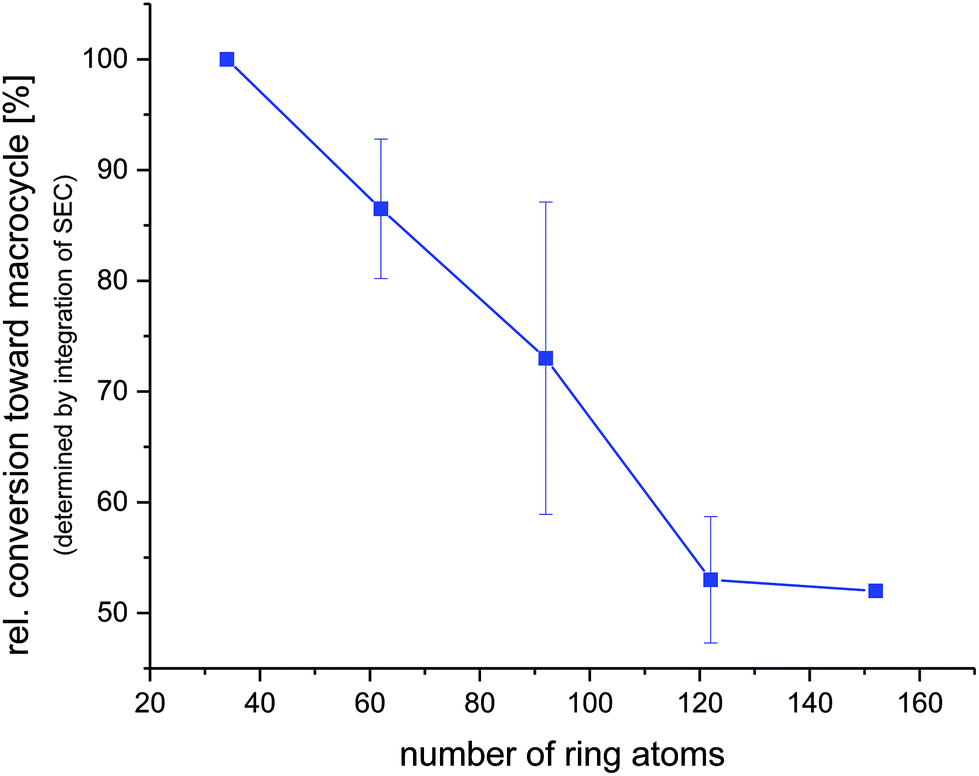
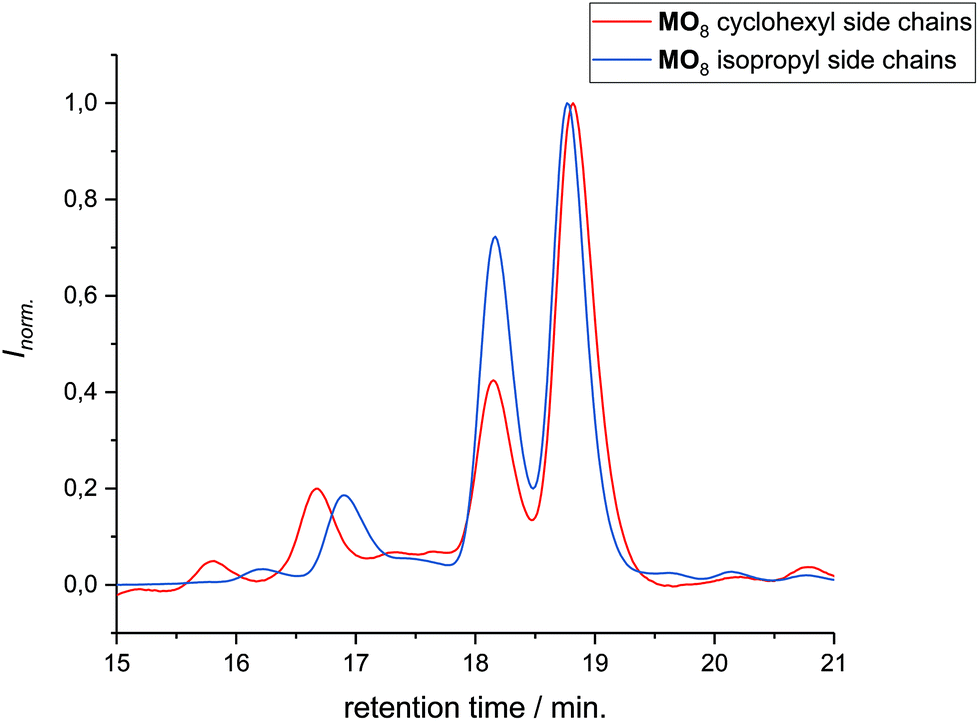
The 1H NMR spectra of MO4, carrying isopropyl side chains, as well as its precursor, DO4, are exemplarily depicted in Fig. 2. Spectrum A, showing MO4 before the cyclization reaction, shows the characteristic signals of the terminal double bonds, which disappear completely in spectrum B after cyclization, indicating full conversion toward the desired product. SEC analysis, on the other hand, revealed that the conversion of DO4 toward MO4 was only 91%. Besides the product, traces of the ADMET side product are present as well as unreacted DO4, which remained in the crude product. The comparison of the NMR and SEC results thus clearly demonstrated the importance of SEC characterization for macromolecules exhibiting uniform size distribution, because only the latter provides sufficient resolution to prove the absence of tiny amounts of impurities, which is essential and can otherwise not be provided by any other characterization technique. The crude macrocyclic products MOX were thus characterized and compared by SEC-ESI-MS analysis. Surprisingly, the side chains did not influence the conversion significantly. The length of the oligomers, on the other hand, resulted in a considerable difference in conversion as observed by SEC (see Fig. 3): for the dimer (DO2), nearly quantitative conversion towards the desired macrocycle (MO2) was obtained, whereas lower conversions were observed when longer oligomers underwent the RCM reaction. For the decamer (MO10), 51% conversion towards the macrocycle was achieved, which is the lowest conversion compared to the smaller macrocycles (MO2–8). Furthermore, ADMET oligomerization could be fully prevented in the case of (DO2), whereas significant amounts of the ADMET side product were obtained in the RCM reaction of (DO10) (ca. 17%, according to SEC analysis). Isolation of the macrocyclic product (from the linear oligomer and the ADMET side product) by column chromatography was not successful due to the similar polarity of the compounds. We also considered purification by preparative SEC, however the resolution was low compared to that of the oligomer-specific SEC columns used for characterization. Nonetheless, characterization of the crude macrocyclic products by SEC-ESI-MS analysis was found to be adequate not only to determine the ratio between the linear starting material, macrocyclic product, and ADMET side product, but also to support the successful formation of the desired sequence-defined macrocycles arising from the advantageous combination of the ESI-MS and SEC data. For the decamer, for example, three peaks were observed, the one at a retention time of 14.36 belonging to the ADMET side product and the one at 15.33 minutes belonging to DO10. The ESI-MS spectrum of the product fraction at 15.86 minutes showed a single isotopic distribution with a maximum at 1873.3949 m/z, which corresponds to the Na2+ adduct of the cyclized MO10 ([C220H372O34N10Na2]2+, see Fig. 4) It is noteworthy that large macrocycles, composed of up to 152 ring backbone atoms, were obtained (see Fig. 5). In Fig. 6, the RCM towards two MO8 with different side chains are compared regarding conversion toward the desired macrocycle and the ADMET side product, revealing low dependence on the side-chains. Furthermore, Fig. 5 reveals that the conversion toward the macrocycle is relatively independent of the applied side-groups for different ring sizes. On the other hand, the conversion strongly depends on the size of the macrocycle. These trends confirm theoretical calculations.31 Interestingly, such conclusions could not be drawn if disperse oligomers were studied for their cyclization tendency,32 as (i) resolution in SEC would certainly be too poor to allow for proper integration (compare Fig. 3 and Fig. 6) and (ii) molecular weight dispersity would introduce a significant error in the abscissa of Fig. 5. Considering, for example, a typical dispersity of controlled radical polymerization of Đ ∼ 1.2, a mass of 3724 m/z with 152 ring atoms (MO10) would translate to a dispersity in ring size of 35 ring atoms. Macromolecules of uniform size, on the other hand, allow for easy analysis by integration of the SEC traces if oligomer-specific columns with sufficient resolution are utilized.
 |
| | Fig. 2
1H NMR comparison of a DO4 (A) and a MO4 (B) carrying isopropyl side chains. In the first 1H NMR spectrum, the signals of the terminal double bonds are clearly observed, whereas in the spectrum on the bottom, the signal of the newly formed internal double bond appears, and the two signals of the terminal double bond disappear completely, indicating full conversion towards MO4. | |
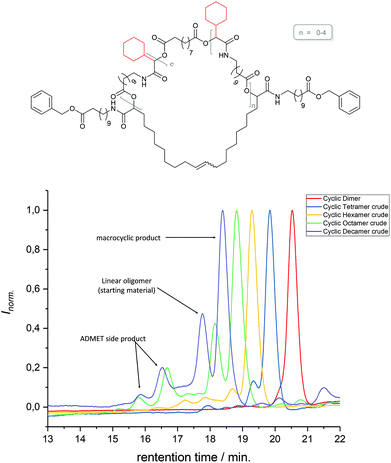 |
| | Fig. 3 Schematic representation of the structure of macrocycles with cyclohexyl side chains and sebacic acid as the core unit in different sizes (dimer – decamer), and the cooresponding SEC analysis results, revealing the influence of the size of the oligomer on the conversion towards the macrocycle. For the decamer, a detailed description of the obtained products is depicted exemplary. The different species were identified by SEC-ESI-MS analysis. | |
 |
| | Fig. 4 SEC-ESI-MS analysis of the macrocyclic decamer bearing cyclohexyl side chains. The single, double and triple charged sodium cations are clearly observed. The expanded region (panel A) shows the apparent isotopic pattern (black) which was found to be in good agreement with the calculated one (blue, panel B), obtained by the program mmas. | |
 |
| | Fig. 5 Dependence of the oligomer size on the conversion toward the macrocycle. The mean values and standard deviation of the conversions for MO2,4,6,8,10 are displayed; the line is only drawn to guide the eye. MO2 and MO10 do not have a standard deviation since they were not synthesized with different side chains. | |
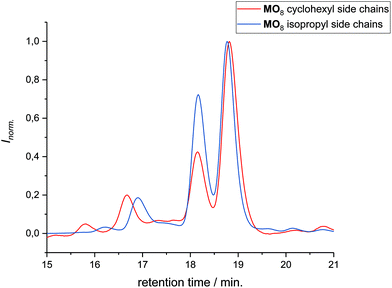 |
| | Fig. 6 Comparison of the conversion toward the macrocycles MO8 carrying different side chains, namely cyclohexyl and isopropyl by SEC analysis. The conversion is 57 and 49% for MO8 carrying cyclohexyl and isopropyl side chains, respectively. The peaks were identified by SEC-ESI-MS to be MO8, DO8, and the two ADMET side products (DO8)2 and (DO8)3, from higher to lower retention time. | |
Conclusions
In summary, we have shown the first example of a template-free approach towards sequence-defined, large macrocycles. This is furthermore the first example of sequence-defined macromolecules that are employed for the synthesis of a polymer architecture other than linear macromolecules. The synthetic procedure allowed the introduction of different tailored side chains and long aliphatic backbones to the macrocyclic backbone. During RCM, clear trends were observed, offering the possibility to identify the limits of RCM regarding conversion and to determine clear structure-activity relationships, here the dependence of RCM on the length of the linear oligomer and on the side chains. Indeed, the side chains did not influence the conversion significantly, whereas the length of the oligomer greatly influenced the reaction selectivity. This study serves as a model and example of application possibilities of sequence-defined macromolecules of uniform size to determine quantitative structure property/activity relationships, which cannot be analyzed accurately if disperse systems are utilized. More precisely, we have demonstrated herein the limits and possibilities of RCM, which will be of interest to polymer and organic chemists, not only for metathesis reactions, but also for other ring-closing approaches.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Funding from the Deutsche Forschungsgemeinschaft (German Research Council, DFG) within the SFB 1176 “Structuring of Soft Matter” (project A3) is gratefully acknowledged. We thank Maximiliane Frölich, Rieke Schulte and the analytical team from KIT for analytical support, Lea Bayer for synthetic support, and Prof. Barner-Kowollik (KIT) and his group for access to SEC-ESI-MS equipment.
Notes and references
-
(a) S. C. Solleder, R. V. Schneider, K. S. Wetzel, A. C. Boukis and M. A. R. Meier, Macromol. Rapid Commun., 2017, 38, 1 Search PubMed;
(b)
S. Martens, J. O. Holloway and F. E. Du Prez, in Sequence-Controlled Polymers, ed. J.-F. Lutz, Wiley VCH, Weinheim, 2018, p. 379 Search PubMed;
(c) N. Badi and J.-F. Lutz, Chem. Soc. Rev., 2009, 38, 3383 RSC;
(d) J.-F. Lutz, J.-M. M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 1 Search PubMed.
-
(a) L. Charles, C. Laure, J.-F. Lutz and R. K. Roy, Macromolecules, 2015, 48, 4319 CrossRef CAS;
(b) A. Al Ouahabi, M. Kotera, L. Charles and J.-F. Lutz, ACS Macro Lett., 2015, 4, 1077 CrossRef CAS;
(c) S. C. Solleder and M. A. R. Meier, Angew. Chem., Int. Ed., 2014, 53, 711 CrossRef CAS PubMed;
(d) S. C. Solleder, K. S. Wetzel and M. A. R. Meier, Polym. Chem., 2015, 6, 3201 RSC;
(e) S. Martens, J. Van Den Begin, A. Madder, F. E. Du Prez and P. Espeel, J. Am. Chem. Soc., 2016, 138, 14182 CrossRef CAS PubMed;
(f) S. Martens, A. Landuyt, P. Espeel, B. Devreese, P. Dawyndt and F. Du Prez, Nat. Commun., 2018, 9, 4451 CrossRef PubMed.
-
(a) A. C. Boukis, K. Reiter, M. Frölich, D. Hofheinz and M. A. R. Meier, Nat. Commun., 2018, 9, 1439 CrossRef PubMed;
(b) A. C. Boukis and M. A. R. Meier, Eur. Polym. J., 2018, 104, 32 CrossRef CAS;
(c) J.-F. Lutz, Acc. Chem. Res., 2013, 46, 2696 CrossRef CAS PubMed;
(d) H. M. Colquhoun and J.-F. Lutz, Nat. Chem., 2014, 6, 455 CrossRef CAS PubMed;
(e) J.-F. Lutz, Macromolecules, 2015, 48, 4759 CrossRef CAS.
-
(a) R. K. Roy, A. Meszynska, C. Laure, L. Charles, C. Verchin and J.-F. Lutz, Nat. Commun., 2015, 6, 7237 CrossRef PubMed;
(b) D. Karamessini, S. Poyer, L. Charles and J.-F. Lutz, Macromol. Rapid Commun., 2017, 38, 1700426 CrossRef PubMed.
- C. Gerke, M. F. Ebbesen, D. Jansen, S. Boden, T. Freichel and L. Hartmann, Biomacromolecules, 2017, 18, 787 CrossRef CAS PubMed.
-
(a) J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 1238149 CrossRef PubMed;
(b) J.-F. Lutz, ACS Macro Lett., 2014, 3, 1020 CrossRef CAS PubMed.
-
(a)
J.-F. Lutz, in Sequence-Controlled Polymers, ed. J.-F. Lutz, Wiley VCH, Weinheim, 1st edn, 2018, p. 1 Search PubMed;
(b) J.-F. Lutz, Macromol. Rapid Commun., 2017, 1700582 CrossRef PubMed.
-
(a) P. Espeel, L. L. G. Carrette, K. Bury, S. Capenberghs, J. C. Martins, F. E. Du Prez and A. Madder, Angew. Chem., Int. Ed., 2013, 52, 13261 CrossRef CAS PubMed;
(b)
C. Alabi, in Sequence-Controlled Polymers, ed. J.-F. Lutz, Wiley VCH, Weinheim, 1st edn., 2018, p. 159 Search PubMed;
(c) T. T. Trinh, C. Laure and J.-F. Lutz, Macromol. Chem. Phys., 2015, 216, 1498 CrossRef CAS;
(d) J. C. Barnes, D. J. C. Ehrlich, A. X. Gao, F. A. Leibfarth, Y. Jiang, E. Zhou, T. F. Jamison and J. A. Johnson, Nat. Chem., 2015, 7, 810 CrossRef CAS;
(e) J. O. Holloway, C. Mertens, F. E. Du Prez and N. Badi, Macromol. Rapid Commun., 2018, 1800685 Search PubMed.
-
(a) S. C. Solleder and M. A. R. Meier, Angew. Chem., Int. Ed., 2014, 53, 711 CrossRef CAS;
(b) R. Kakuchi, Angew. Chemie – Int. Ed., 2014, 53, 46 CrossRef CAS;
(c) L. Yang, Z. Zhang, B. Cheng, Y. You, D. Wu and C. Hong, Sci. China: Chem., 2015, 58, 1734 CrossRef CAS;
(d) S. C. Solleder, D. Zengel, K. S. Wetzel and M. A. R. Meier, Angew. Chemie – Int. Ed., 2016, 55, 1204 CrossRef CAS PubMed;
(e) S. C. Solleder, S. Martens, P. Espeel, F. E. Du Prez and M. A. R. Meier, Chem. – Eur. J., 2017, 23, 13906 CrossRef CAS;
(f) Y.-H. Wu, J. Zhang, F.-S. Du and Z.-C. Li, ACS Macro Lett., 2017, 6, 1398 CrossRef CAS;
(g) Z. Zhang, Y. You and C. Hong, Macromol. Rapid Commun., 2018, 1800362 CrossRef PubMed;
(h) W. Konrad, F. R. Blößer, K. S. Wetzel, A. C. Boukis, M. A. R. Meier and C. Barner-Kowollik, Chem. – Eur. J., 2018, 24, 3413 CrossRef CAS PubMed.
- M. Passerini, Gazz. Chim. Ital., 1921, 51, 126 CAS.
- B. A. Laurent and S. M. Grayson, Chem. Soc. Rev., 2009, 38, 2202 RSC.
-
(a) Y. Tezuka and H. Oike, J. Am. Chem. Soc., 2001, 123, 11570 CrossRef CAS PubMed;
(b)
K. Endo, in New Frontiers in Polmer Science, ed. S. Kobayashi, Springer-Verlag, Berlin Heidelberg, 2008, p. 121 Search PubMed.
-
(a) M. Schappacher and A. Deffieux, Makromol. Chem., Rapid Commun., 1991, 12, 447 CrossRef CAS;
(b) L. Rique-Lubet, M. Schappacher and A. Deffieux, Macromolecules, 1994, 27, 6318 CrossRef;
(c) Y. Tezuka and F. Ohashi, Macromol. Rapid Commun., 2005, 26, 608 CrossRef CAS;
(d) L. Wang, M. O. Vysotsky, A. Bogdan, M. Bolte and V. Böhmer, Science, 2004, 304, 1312 CrossRef CAS PubMed.
-
(a) D. Geiser and H. Höcker, Macromolecules, 1980, 13, 653 CrossRef CAS;
(b) G. Hild, A. Kohler and P. Rempp, Eur. Polym. J., 1980, 16, 525 CrossRef CAS;
(c) J. Roovers and P. M. Toporowski, Macromolecules, 1983, 16, 843 CrossRef CAS.
-
(a) Y. Li, X. Qiu and Z.-X. Jiang, Org. Process Res. Dev., 2015, 19, 800 CrossRef CAS;
(b) X. Lv, X. Zheng, Z. Yang and Z.-X. Jiang, Org. Biomol. Chem., 2018, 16, 8537 RSC;
(c) H. Jacobson and W. H. J. Stockmayer, Chem. Phys., 1950, 18, 1600 CAS;
(d) C. W. Bielawski, D. Benitez and R. H. Grubbs, Science, 2002, 297, 2041 CrossRef CAS PubMed;
(e) C. W. Bielawski, D. Benitez and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 8424 CrossRef CAS PubMed.
- R. H. Grubbs, S. J. Miller and G. C. Fu, Acc. Chem. Res., 1995, 28, 446 CrossRef CAS.
-
(a) A. Deiters and S. F. Martin, Chem. Rev., 2004, 104, 2199 CrossRef CAS PubMed;
(b) C. Heinis, Nat. Chem. Biol., 2014, 10, 696 CrossRef CAS PubMed.
- S. K. Armstrong, J. Chem. Soc., Perkin Trans. 1, 1998, 371 RSC.
- I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127 CrossRef CAS PubMed.
-
(a) J. N. Lambert, J. P. Mitchell and K. D. Roberts, J. Chem. Soc., Perkin Trans. 1, 2001, 471 RSC;
(b) S. A. Kates, N. A. Solé, C. R. Johnson, D. Hudson, G. Barany and F. Albericio, Tetrahedron Lett., 1993, 34, 1549 CrossRef CAS;
(c) J. J. Storhoff and C. A. Mirkin, Chem. Rev., 1999, 99(7), 1849 CrossRef CAS PubMed;
(d) M. Göritz and R. Krämer, J. Am. Chem. Soc., 2005, 127, 18016 CrossRef PubMed.
- E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608 CrossRef CAS PubMed.
- J. Shi and D. E. Bergstrom, Angew. Chem., Int. Ed. Engl., 1997, 36, 111 CrossRef CAS.
- L. Zhang, A. J. Stephens, A. L. Nussbaumer, J. F. Lemonnier, P. Jurček, I. J. Vitorica-Yrezabal and D. A. Leigh, Nat. Chem., 2018, 10, 1083 CrossRef CAS PubMed.
- R. D. Adams, M. Huang, W. Huang and J. A. Queisser, J. Am. Chem. Soc., 1996, 118, 9442 CrossRef CAS.
- B. A. Laurent and S. M. Grayson, J. Am. Chem. Soc., 2006, 128, 4238 CrossRef CAS PubMed.
-
(a) M. Mayor and C. Didschies, Angew. Chem., Int. Ed., 2003, 42, 3176 CrossRef CAS PubMed;
(b) D. V. Kondratuk, L. A. Perdigão, A. M. Esmail, J. N. O'shea, P. H. Beton and H. L. Anderson, Nat. Chem., 2015, 7, 317 CrossRef CAS PubMed.
-
(a) E. Wasserman, J. Am. Chem. Soc., 1960, 82, 4433 CrossRef CAS;
(b) G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, Angew. Chem., 2015, 127, 6208 CrossRef;
(c) M. Raza Shah, S. Duda, B. Müller, A. Godt and A. Malik, J. Am. Chem. Soc., 2003, 125, 5408 CrossRef PubMed;
(d) S. Duda and A. Godt, Eur. J. Org. Chem., 2003, 2003, 3412 CrossRef;
(e) A. Godt, Eur. J. Org. Chem., 2004, 2004, 1639 CrossRef.
- M. Porel, D. N. Thornlow, N. N. Phan and C. A. Alabi, Nat. Chem., 2016, 8, 590 CrossRef CAS PubMed.
-
(a) J. A. Berrocal, S. Albano, L. Mandolini and S. Di Stefano, Eur. J. Org. Chem., 2015, 34, 7504 CrossRef;
(b) G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746 CrossRef CAS PubMed.
-
(a) P. A. Fokou and M. A. R. Meier, J. Am. Chem. Soc., 2009, 131, 1664 CrossRef CAS PubMed;
(b) P. A. Fokou and M. A. R. Meier, Macromol. Rapid Commun., 2010, 31, 368 CrossRef CAS PubMed.
-
(a) S. Di Stefano and G. Ercolani, Adv. Phys. Org. Chem., 2016, 50, 1 CrossRef;
(b) S. Di Stefano and L. Mandolini, Chem. Phys. Phys. Chem., 2019, 21, 955 RSC.
- M. A. R. Meier and C. Barner-Kowollik, Adv. Mater., 2018, 1806027 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data. See DOI: 10.1039/c9py00438f |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*