
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBidirectional ROMP of paracylophane-1,9-dienes to tri- and penta-block p-phenylenevinylene copolymers†
Venukrishnan
Komanduri
 ,
Dharam R.
Kumar
,
Daniel J.
Tate
,
Raymundo
Marcial-Hernandez
,
Benjamin J.
Lidster
and
Michael L.
Turner
*
,
Dharam R.
Kumar
,
Daniel J.
Tate
,
Raymundo
Marcial-Hernandez
,
Benjamin J.
Lidster
and
Michael L.
Turner
*
Organic Materials Innovation Centre (OMIC), The School of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: michael.turner@manchester.ac.uk
First published on 24th May 2019
Abstract
Dialkoxy and dialkyl substituted paracyclophane-1,9-dienes undergo bidirectional ring opening metathesis polymerisation (ROMP) on addition of bifunctional Hoveyda–Grubbs initiators. The living nature of the polymerisation was demonstrated for different [monomer]/[initiator] ratios and block copolymers were prepared by sequential ROMP and ROMP-ATRP to give fully conjugated rod–rod–rod and partially conjugated coil–rod–coil ABA and BAB triblock copolymers, respectively. These strategies were successfully extended to the synthesis of the corresponding pentablock copolymers. Optical and electrochemical properties of cis/trans and all trans homo and block copolymers are discussed.
Introduction
π-Conjugated polymers have found extensive application as active layers in the fabrication of optoelectronic and electronic devices such as OLEDs,1 OFETs,2,3 OPVs4,5 and sensors.6,7 Key advantages for the use of polymeric materials include improved flexibility, mechanical robustness, lighter weight and ease of processing.8 The performance of these devices is greatly influenced by the morphology of the active layer, and this is strongly influenced by the kinetics of thin film formation during device fabrication. An attractive approach to obtaining reproducible thin film morphologies is to utilise the thermodynamically driven phase separation of block copolymers, as these materials form well defined nanostructures that can be self-assembled in biphasic media.9,10 Block copolymers with conjugated segments can either be fully conjugated rod–rod systems, in a diblock or rod–coil systems where the conjugated block is coupled to a saturated polymer backbone.11 There are only a few polymerisation methods that give well-defined fully conjugated block copolymers. These include Kumada catalyst-transfer polymerisation (KCTP also known as GRIM),12,13 a nickel catalysed Negishi type polymerisation14 and a palladium catalyst transfer Suzuki–Miyaura polycondensation.15 The synthesis of rod–coil systems has been more extensively reported, including the ROMP of cyclooctadiene followed by ATRP of the resulting macroinitiator.16 Phenylenevinylene (PPV) block copolymers can be prepared by the Siegrist polycondensation or by complex multistep syntheses.17,18,18 However these approaches lead to poor control of the molecular weight distribution and result in polymers with broad polydispersities (Đm) and significant backbone defects.19Ring opening metathesis polymerisation (ROMP) of strained paracyclophanedienes20–24,25 and cyclophanetrienes26,27 gives well-defined PPV homopolymers and fully conjugated diblock copolymers.28,29 Rod-coil PPV diblock copolymers can be prepared by the sequential one-pot ROMP of tetraalkoxy paracyclophanediene and oxanorbornene monomers.30 This sequential ROMP strategy was adopted by Weck et al. using di, tetraalkoxy cyclophanedienes and norbornene ester monomers to access PPV tri and tetrablock copolymers.31,32 A step-wise strategy using monotelechelic PPV blocks as macroinitiators for ATRP of methylmethacrylate gives rod–coil PPV-PMMA diblock copolymers.33 Rod–coil PPV block copolymers have also been prepared through the preparation of PPV macroinitiators using anionic polymerisation followed by the introduction of the coil unit using a radical polymerisation.34 However, to date there have been very few reports on the preparation of fully and partially conjugated PPV triblock copolymers. These include an ADMET polymerisation/Wittig olefination strategy to prepare fully conjuagted ABA and ABCBA type PPV block copolymers35 and the synthesis of donor–acceptor–donor triblock copolymers by a Knoevenagel condensation followed by a Nickel catalysed Yamamoto coupling by Tu et al.36 Coil–rod–coil ABA triblock copolymers with pendent PEG chains have been prepared by a Heck coupling strategy37 and ferrocene containing ABC triblock copolymers have been made by an anionic polymerisation-Wittig olefination approach.38 These strategies all involve complex multistep syntheses and lead to poor control over the molecular weight distribution.
In this study we report a precisely controlled approach to the synthesis of conjugated triblock copolymers,23 by the ROMP of cyclophanediene monomers M1 and M2 with bifunctional ruthenium complex 1 to form the telechelic polymer intermediate [I] (Fig. 1). Trapping of this intermediate with a second cyclophanediene gives the fully conjugated rod–rod–rod ABA triblock copolymer. Quenching of the intermediate [I] with an α-bromoester functionalised vinylether E followed by ATRP of the resulting macroinitiator generates coil–rod–coil ABA triblock copolymers. This approach can be extended to the synthesis of the corresponding pentablock copolymers. To our knowledge this is the first bidirectional ROMP39 of paracyclophanedienes using bifunctional ruthenium carbene initiators towards the synthesis of conjugated multiblock copolymers (Fig. 1).
 | ||
| Fig. 1 A bidirectional ROMP of strained paracyclophanediene monomers to both partially and fully conjugated tri and pentablock copolymers. | ||
Results and discussion
Initial optimisation studies focussed on the ROMP of monomers M1 and M2 using the bifunctional catalyst 1. The required cyclophane-1,9-diene monomers M1 (R = O-2-ethylhexyl)33 and M2 (R = n-octyl)40 were prepared using standard synthetic procedures. The bifunctional catalyst 1 was synthesised by cross metathesis of Grubbs second generation catalyst G2 with 2,5-dimethoxy-1,4-divinylbenzene as reported.41The first ROMP experiment was performed using dialkoxy monomer M1 ([M1]/[1] = 10) in DCM-d2 at room temperature. The polymerisation was followed by in situ1H NMR spectroscopy and complete monomer consumption was observed after 63 h. However, reaction times were reduced to 10 h when the ROMP was performed at 60 °C in TCE-d2 (Fig. S3†). After consumption of the monomer the polymerisation was quenched with excess ethylvinylether and stirred at rt for 4 h. Precipitation of the crude product (MeOH/Celite), followed by elution with chloroform gave telechelic PPV 2a (Mn(calc.) = 4.7 kDa, Mn(obs) = 5.7 kDa, Đm = 1.40) in 95% yield (entries 1 and 2, Table S1†).
Bidirectional ROMP of the dialkyl monomer M2 ([M2]/[1] = 10) in DCM-d2 at room temperature was complete in 1.5 h but gave poor control over the molecular weight distribution (Mn(calc.) = 4.4 kDa, Mn(obs) = 79 kDa, Đm = 1.8) due to incomplete initiation and fast propagation. The higher propagation rates are due to the absence of Lewis basic sites in monomer M2 that can ligate to the intermediate 14 electron ruthenium complex formed after initiation. Therefore, the ROMP was performed in the presence of an external ligand 3-BrPy (40 mol%) at 40 °C in TCE-d2 for 20 h (Fig. S4†) to slow down propagation. This gave PPV 3a with significant control over molecular weight (Mn(obs.) ∼ 7.1 kDa vs. 79 kDa in the absence of 3-BrPy, entries 3 and 4, Table S1†). Further control over the molecular weight and an improved yield was obtained (Mn(obs.) ∼ 5.4 kDa) when the ROMP was performed at 40 °C using anhydrous degassed DCM as solvent (entries 4 and 5, Table S2†). This could be due to the better solubility of 1 in DCM. The living nature of the polymerisation was demonstrated by varying the [monomer]/[initiator] ratio to obtain polymers 2a–c (96–93% yield) and 3a–c (95–81% yield) as shown in Fig. 2 (also, see Table S2†). The [M]/[I] ratios given for the monomers M1 and M2 are different because ROMP of M1 under the optimal conditions gives poor control of the molecular weight and dispersity beyond ratios of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
 | ||
| Fig. 2 Bidirectional ROMP of monomers M1 and M2 with initiator 1. (a) and (c) dependence of Mn of the polymers 2a–c and 3a–c on [M]/[1] ratio, (b) and (d) molecular weight distribution of polymers 2a–c and 3a–c respectively. | ||
A powerful application of this bidirectional ROMP methodology was demonstrated by the synthesis of both rod–rod–rod and coil–rod–coil triblock copolymers. Triblock conjugated copolymers have found interesting application as the active layer in OPV and OLED devices42 but only limited synthetic methods have been reported. AB and ABA block copolymers with PPV units are known to self-assemble into nanostructures, the morphology of which depends on the mole fraction of the individual blocks.9,43
The α-bromoester end capped PPV macroinitaitors 4a and 5a were prepared in excellent yields (95–98%) by bidirectional ROMP of monomers M1 and M2 followed by quenching with α-bromoester phenyl methylvinyl ether E (f = 89% and 75%, see Table S2†). Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) of polymer 4a and 5a showed major peaks corresponding to the desired polymers with α-bromoester end groups (●), the major series of peaks separated by the mass of the polymer repeat unit 461.4 Da and 428.9 Da corresponding to the molecular weights of monomers M1 and M2 respectively (Fig. 3, S19†).44
 | ||
| Fig. 3 MALDI-TOF-MS of macroinitiator 4a (●) peaks correspond to α-bromoester end groups (✲) phenol with elimination of HBr and (✦) PPV with elimination of HBr. | ||
These macroinitiators (4a and 5a) were subjected to CuBr mediated atom transfer radical polymerisation (ATRP)33 with methyl methacrylate (MMA) in hot toluene for 3 h. Aliquots of the reactions were terminated at different time intervals (30, 90, 180 min) by exposing to air followed by precipitation into excess methanol. The solids were redissolved in chloroform and precipitated by addition of diethyl ether. Upon filtration, PMMA-b-PPV-b-PMMA triblock polymers 6a–c and 7a–c were obtained as orange/yellow solids with dispersities ranging from 1.23 to 1.33 and a unimodal molar mass distribution by GPC analysis (Scheme 1, Fig. S21 and Table S3†).
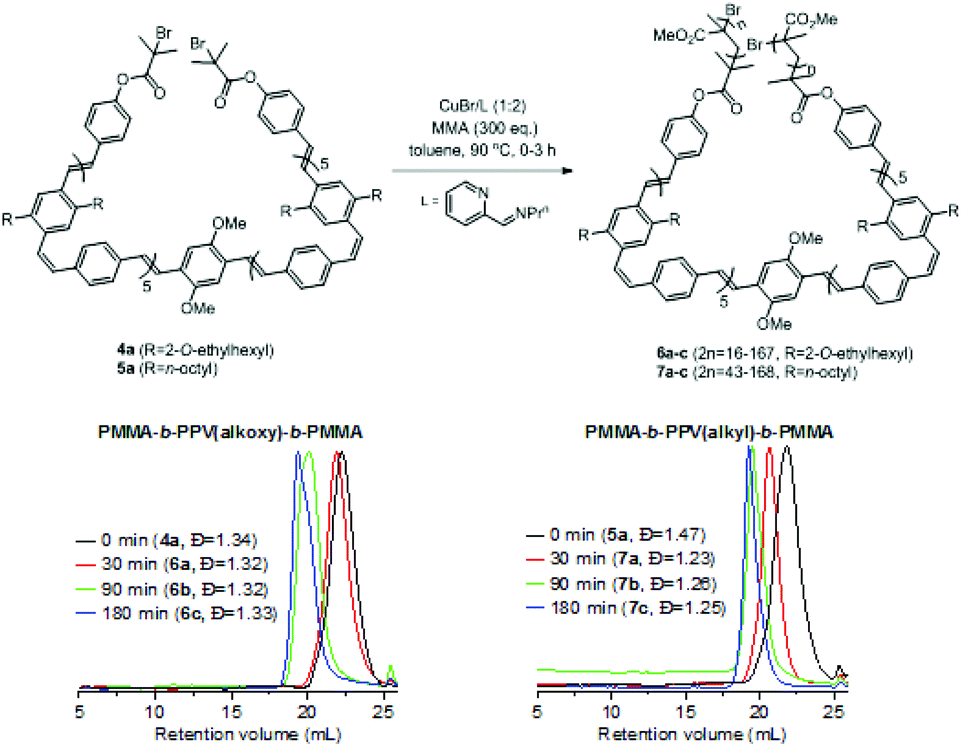 | ||
| Scheme 1 ROMP-ATRP strategy to prepare coil–rod–coil ABA phenylenevinylene triblock copolymers 6a–c and 7a–c (GPC in THF). | ||
The MMA content in these triblock copolymers was determined from the 1H NMR spectra by a comparison of the integration of the OMe group of MMA (δ = 3.98–3.17 ppm) against the combined phenyl and vinylene protons for 6a–c (δ = 7.93–6.34 ppm) and methylene protons for 7a–c (δ = 2.86–2.16 ppm) respectively (Fig. S22–27, Table S3†).
Bidirectional living polymerisation of cyclophanedienes in a sequential manner gave fully conjugated triblock copolymers. ROMP of monomer M1 with initiator 1 ([M1]/[1] = 10) in TCE at 60 °C was monitored by GPC and TLC analyses (Fig. S47†). After complete consumption (t = 8 h) of monomer M1 an external ligand, 3-BrPy (40 mol%), and a solution of the second monomer M2 in TCE were added. The reaction mixture was stirred at 40 °C until the second monomer M2 was consumed (t = 12, 16 and 18 h) and the polymerisation was quenched with excess ethylvinylether. Purification by precipitation (Celite/MeOH) and elution with chloroform afforded fully conjugated ABA triblock polymers 8a–c in 80–88% yields with dispersities ranging from 1.87–1.97. The higher polydispersities and the shoulder peak observed in the GPC traces of block copolymer 8c can be assigned to partial isomerisation of the cis-alkenes (Scheme 2, left and Table S4†). The sequential ROMP strategy was more efficient (yields 93–97% and Đm = 1.35–1.87) when the monomer addition sequence was reversed to give BAB triblock copolymers 9a–c containing alkyl PPV segments at the center and alkoxy PPV units on either side (Fig. S48,†Scheme 2, right and Table S4†).
 | ||
| Scheme 2 Sequential ROMP strategy to fully conjugated rod–rod–rod ABA (8a–c) and BAB (9a–c) phenylenevinylene triblock copolymers (GPC in THF). | ||
The triblock copolymers 8a–c and 9a–c were characterised by 1H NMR spectroscopy and the ratio of monomer units M1 and M2 incorporated was calculated by combined integration of the alkoxy methylene protons corresponding to M1 and the methoxy protons of 1 (δ 4.12–3.77 ppm) against the benzyl protons corresponding to M2 (δ 2.72–2.20 ppm) (Fig. S31–41†). The individual block content ratios determined from the 1H NMR spectra were in good correlation with the calculated values based on the monomer to initiator ratio (Table S4†).
1H NMR spectral analysis of the BAB triblock copolymers 9a–c (Fig. S37†) indicated high vinyl end-capping efficiency (f > 80%) suggesting that further functionalisation/elaboration could be possible. Therefore the sequential ROMP was quenched with the α-bromoester substituted phenylmethyl ether E instead of ethyl vinylether to give the bifunctional triblock PPV macroinitiator 10 in excellent yield with excellent end capping efficiency (95% yield, f > 87%). Under similar ATRP reaction conditions to those in Scheme 1 the macroinitiator 10 and MMA gave coil–rod–rod–rod–coil CBABC pentablock copolymers 11a–c with molecular weights ranging from 20.6–36.5 kDa and dispersities in the range 1.49–1.25. The slightly broader dispersity of 11a is due to partial isomerisation of the cis-alkenes (Scheme 3, Table S3 and Fig. S28–30†).
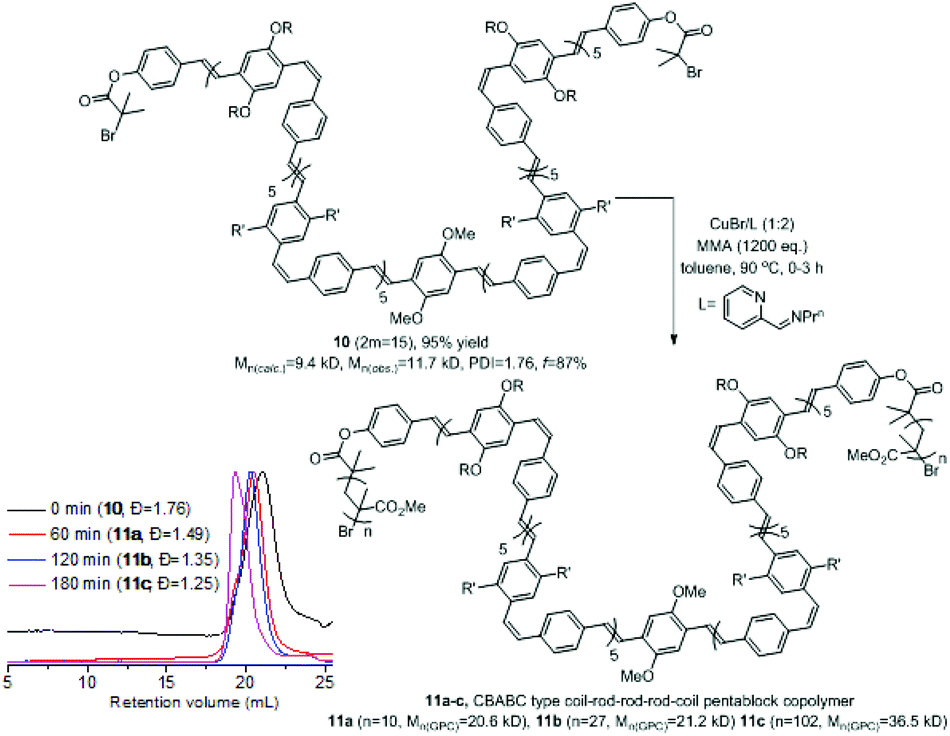 | ||
| Scheme 3 Sequential ROMP-ATRP strategy to coil–rod–rod–rod–coil CBABC pentablock copolymers 11a–c (GPC in THF). | ||
Furthermore the sequential ROMP approach can be used to synthesise fully conjugated pentablock copolymers as shown in Scheme 4. Dialkyl monomer M2 was subjected to ROMP ([M2]/[1] = 10) in the presence of 3-BrPy ligand and upon complete consumption the dialkoxy monomer M1 was added and stirred for 15 h, complete consumption of this monomer (M1) was followed by addition of either M2 or a mixture of the isomeric tetraalkyl monomers M3, M4 (M4 being unreactive). The polymerisation reaction was eventually quenched by adding excess ethylvinyl ether and the fully conjugated ABABA and CBABC type pentablock copolymers 12 and 13 were isolated by precipitation (Scheme 4, S43 & S45†).
 | ||
| Scheme 4 Sequential ROMP strategy to fully conjugated rod ABABA 12 and CBABC 13 pentablock copolymers (GPC in THF). | ||
Dilute solutions of conjugated polymers 2a–c, 3a–c, 8a–c, 9a–c, 12 and 13 in degassed anhydrous dichloromethane were subjected to photoisomerisation under UV irradiation at 365 nm for 48–72 h. 1H NMR analysis of the polymers obtained indicated complete isomerisation and an all trans geometry of the backbone vinylenes (Fig. 4a, also see ESI†). In the GPC a lower retention time was observed for the all trans isomers as expected because of the higher hydrodynamic volume of this polymer (Fig. 4b).
 | ||
| Fig. 4 Photoisomerisation of triblock copolymer 8a, (a) 1H NMR spectra of 8a and trans-8a in DCM-d2, (b) GPC traces of 8a and trans-8a in CHCl3. | ||
The optical properties of the parent homopolymers were measured in solution (Fig. S49 and Table S5†) and the block copolymers were recorded in both solution and as thin films (Fig. 5, S50–54, and Table S5†). The absorption spectra of the fully conjugated tri and pentablock copolymers 8a–c, 9a–c, 12 and 13 showed λmax values around 450 nm and 380 nm correlating to absorptions of the individual blocks 2a–c and 3a–c. Increasing the content of monomer M2 in polymers 8a to 8c increased the intensity at 370 nm in direct correlation with the ratio of the individual blocks (Fig. 5a). For polymers 9a–c the absorption maxima were red shifted going from 9a to 9c with increasing alkoxy monomer content (Fig. 5b). Excitation of the block copolymer 8a–c, 9a–c, 12 and 13 at 370 nm and the corresponding all trans isomers at 440 nm resulted in emission maxima approximately at 520 nm (Table S5†).
 | ||
| Fig. 5 Absorption and emission profiles of rod (a) ABA triblock (b) BAB triblock and (c) CBABC pentablock copolymers. | ||
The absorption and emission spectra of rod–coil block copolymers 6a–c, 7a–c and 11a–c showed absorption maxima at 460 nm (6a–c), 400 nm (7a–c), 370 nm (11a–c) and emission maxima at 527 nm (6a–c), 491 nm (7a–c) and 520 nm (11a–c). These values agree well with those of parent macroinitiators 4a, 5a and 10, indicating that the PMMA block has no influence on the optical properties of these materials. The ΦPL of the macroinitiators 4a, 5a and 10 and the block copolymers 7a–c, 8a–c and 11a–c are identical and this suggests that the radical polymerisation did not perturb the π-conjugated backbone (Table S5†). A small red shift in emission maxima values (∼20 nm) was observed in the solid state spectra of the block copolymers consistent with previous studies on PPV polymers (Fig. S53–54 and Table S6†).28
The electrochemical properties of the homopolymers (2a–c and 3a–c) and fully conjugated block copolymers (8a–c, 9a–c, 12 and 13) were studied by cyclic voltammetry in dichloromethane using [(C4H9)4N]PF6 (0.1 M solution in DCM) as the electrolyte in conjunction with a platinum working electrode. The calculated HOMO and LUMO energy levels for 2a–c are consistent with the reported values (HOMO = −5.14 and LUMO = −2.56 eV) for these polymers.28 The homopolymers 3a–c have deeper HOMO levels (∼−5.36 eV) (Table 1, entries 1 & 3 and Table S7†).
| Entry | Polymer | HOMOa (eV) | LUMOb (eV) |
E
g(elc)c,d (eV) |
|---|---|---|---|---|
| a HOMO = (Eonsetox − Fcox) + 4.8. b LUMO = (Eonsetred − Fcox) + 4.8. c E g (elc.) = HOMO–LUMO. d Optical band gap values in parenthesis (Eg (opt.) = 1240/λonset). | ||||
| 1 | 2a | −5.14 | −2.66 | 2.48 (2.30) |
| 2 | 2a (E) | −5.01 | −2.84 | 2.17 (2.28) |
| 3 | 3a | −5.36 | −2.58 | 2.78 (2.50) |
| 4 | 3a (E) | −5.10 | −2.81 | 2.29 (2.46) |
| 5 | 8a | −5.23 | −2.77 | 2.46 (2.31) |
| 6 | 8b | −5.12 | −2.69 | 2.43 (2.27) |
| 7 | 8c | −5.05 | −2.78 | 2.27 (2.27) |
| 8 | 8b (E) | −5.03 | −2.70 | 2.33 (2.29) |
| 9 | 9a | −5.10 | −2.67 | 2.43 (2.33) |
| 10 | 9b | −5.14 | −2.66 | 2.48 (2.32) |
| 11 | 9c | −5.14 | −2.60 | 2.54 (2.30) |
| 12 | 9a (E) | −5.02 | −2.75 | 2.27 (2.30) |
| 13 | 12 | −5.14 | −2.64 | 2.50 (2.34) |
| 14 | 12 (E) | −5.04 | −2.69 | 2.35 (2.29) |
| 15 | 13 | −5.19 | −2.79 | 2.40 (2.31) |
| 16 | 13 (E) | −5.05 | −2.67 | 2.38 (2.28) |
The HOMO and LUMO energy levels of the tri- and penta-block copolymers are consistent with those of the parent homopolymers. The cis/trans ABA triblock BCPs 8a–c exhibited reversible oxidation E1/2 values in the range of 0.6–0.4 V, with slight increase in HOMO energy levels (−5.23 to −5.05 eV) but the BCPs 9a–c showed the same HOMO value (∼−5.14 eV) indicating localisation of the HOMO on the dialkoxyphenylene vinylene block in 9a–c. The LUMO energy levels for BCPs 8a–c and 9a–c are in a similar range, −2.69 to −2.78 eV for 8a–c and −2.60 to −2.67 eV for 9a–c (Table S7† and Table 1, entries 5–7 & 9–11). The cis/trans pentablock copolymers 12 and 13 showed reversible oxidation and reduction potentials E1/2 at 0.5–0.6 V and −1.99, –1.80 V with the HOMO energy level approximately −5.14 eV but slightly deeper LUMO level (−2.79 eV) for BCP 13 (Table S7† and Table 1, entries 13 & 15). In general, the all trans homo and block copolymers have a higher lying HOMO when compared to their cis/trans forms due to extended conjugation in the trans form. Also, a consistent deepening of LUMO levels was observed for all the trans polymers except for trans-13 (Table 1, entries 15 & 16). Finally, a clear trend in bandgap reduction was observed for the all trans polymers due to extended delocalisation and both the electrochemical and optical bandgap values for all the polymers are comparable (Table 1).
Conclusions
In summary, the first bidirectional ROMP of strained paracyclophane-1,9-dienes using a bifunctional Hoveyda–Grubbs initiator is reported. This methodology was used in the controlled synthesis of rod–rod and rod–coil tri- and pentablock copolymers through sequential ROMP and ROMP-ATRP reactions. The initially formed cis,trans polymers can be photoisomerised to the corresponding all trans forms. The optical properties of the block copolymers directly correlate with those of the individual polymer blocks and complete energy transfer between blocks is observed in the fluorescence emission spectra of thin films. Fluorescence quantum efficiency measurements of the rod–coil block copolymers showed no damage to π-conjugated backbone under radical ATRP reaction conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the European Commission for the award of a Marie-Curie International Fellowship (CyclAr) to VK, the Indian High Commission Education Wing, London for PhD studentship to DRK (11015/16/2012-SCD-V) and the EPSRC for a studentship award to BJL. The authors would also like to thank the EPSRC for funding of the NMR spectrometers under grant EP/K039547/1 and Dr Ralph Adams, Mr Carlo Bawn for technical advice and Mr Adam Woodward for his technical support in measuring fluorescence quantum yields.References
- A. C. Grimsdale, K. Leok Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed
.
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633–12665 CrossRef CAS PubMed
- Y. He, W. Hong and Y. Li, J. Mater. Chem. C, 2014, 2, 8651–8661 RSC
- L. Ying, F. Huang and G. C. Bazan, Nat. Commun., 2017, 8, 14047 CrossRef CAS PubMed
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS PubMed
- D. Khim, G.-S. Ryu, W.-T. Park, H. Kim, M. Lee and Y.-Y. Noh, Adv. Mater., 2016, 28, 2844–2844 CrossRef
- C. Zhang, P. Chen and W. Hu, Chem. Soc. Rev., 2015, 44, 2087–2107 RSC
- J. R. Sheats and P. F. Barbara, Acc. Chem. Res., 1999, 32, 191–192 CrossRef CAS
- M. Lee, B.-K. Cho and W.-C. Zin, Chem. Rev., 2001, 101, 3869–3892 CrossRef CAS PubMed
- F. Mathias, A. Fokina, K. Landfester, W. Tremel, F. Schmid, K. Char and R. Zentel, Macromol. Rapid Commun., 2015, 36, 959–983 CrossRef CAS PubMed
- M. He, F. Qiu and Z. Lin, J. Mater. Chem., 2011, 21, 17039–17048 RSC
- S. Miyane, H. Mori and T. Higashihara, Microsyst. Technol., 2016, 22, 3–10 CrossRef CAS
- J. Wang, M. Ueda and T. Higashihara, ACS Macro Lett., 2013, 2, 506–510 CrossRef CAS
- M. Verswyvel, J. Steverlynck, S. Hadj Mohamed, M. Trabelsi, B. Champagne and G. Koeckelberghs, Macromolecules, 2014, 47, 4668–4675 CrossRef CAS
- K. B. Woody, B. J. Leever, M. F. Durstock and D. M. Collard, Macromolecules, 2011, 44, 4690–4698 CrossRef CAS
- C. W. Bielawski, T. Morita and R. H. Grubbs, Macromolecules, 2000, 33, 678–680 CrossRef CAS
- G. Qu, F. Jiang, S. Zhang and S. Usuda, Mater. Lett., 2007, 61, 3421–3424 CrossRef CAS
- H. Wang, W. You, P. Jiang, L. Yu and H. H. Wang, Chem. – Eur. J., 2004, 10, 986–993 CrossRef CAS PubMed
- L. zur Borg, A. L. Domanski, R. Berger and R. Zentel, Macromol. Chem. Phys., 2013, 214, 975–984 CrossRef CAS
- C.-Y. Yu and M. L. Turner, Angew. Chem., 2006, 118, 7961–7964 CrossRef
- A. M. Spring, C.-Y. Yu, M. Horie and M. L. Turner, Chem. Commun., 2009, 2676–2678 RSC
- C.-Y. Yu, M. Helliwell, J. Raftery and M. L. Turner, Chem. – Eur. J., 2011, 17, 6991–6997 CrossRef CAS PubMed
- B. J. Lidster, D. R. Kumar, A. M. Spring, C.-Y. Yu and M. L. Turner, Polym. Chem., 2016, 7, 5544–5551 RSC
- D. R. Kumar, B. J. Lidster, R. W. Adams and M. L. Turner, Polym. Chem., 2017, 8, 3186–3194 RSC
- S.-W. Chang and M. Horie, Chem. Commun., 2015, 5, 9113–9116 RSC
- D. Mäker, C. Maier, K. Brödner and U. H. F. Bunz, ACS Macro Lett., 2014, 415–418 CrossRef
- F. Menk, M. Mondeshki, D. Dudenko, S. Shin, D. Schollmeyer, O. Ceyhun, T.-L. Choi and R. Zentel, Macromolecules, 2015, 48, 7435–7445 CrossRef CAS
- C.-Y. Yu, M. Horie, A. M. Spring, K. Tremel and M. L. Turner, Macromolecules, 2009, 43, 222–232 CrossRef
- C. Y. Yu, J. W. Kingsley, D. G. Lidzey and M. L. Turner, Macromol. Rapid Commun., 2009, 30, 1889–1892 CrossRef CAS PubMed
- F. Menk, S. Shin, K.-O. Kim, M. Scherer, D. Gehrig, F. Laquai, T.-L. Choi and R. Zentel, Macromolecules, 2016, 49, 2085–2095 CrossRef CAS
- E. Elacqua, G. T. Geberth, D. A. Vanden Bout and M. Weck, Chem. Sci., 2019, 10, 2144–2152 RSC
- S. K. Pomarico, D. S. Lye, E. Elacqua and M. Weck, Polym. Chem., 2018, 9, 5655–5659 RSC
- B. J. Lidster, J. M. Behrendt and M. L. Turner, Chem. Commun., 2014, 50, 11867–11870 RSC
- I. Cosemans, J. Vandenbergh, L. Lutsen, D. Vanderzande and T. Junkers, Polym. Chem., 2013, 4, 3471–3479 RSC
- K. Nomura, T. Haque, T. Onuma, F. Hajjaj, M. S. Asano and A. Inagaki, Macromolecules, 2013, 46, 9563–9574 CrossRef CAS
- G. Tu, H. Li, M. Forster, R. Heiderhoff, L. J. Balk and U. Scherf, Macromolecules, 2006, 39, 4327–4331 CrossRef CAS
- H. Wang, H. H. Wang, V. S. Urban, K. C. Littrell, P. Thiyagarajan and L. Yu, J. Am. Chem. Soc., 2000, 122, 6855–6861 CrossRef CAS
- F. He, T. Gädt, M. Jones, G. D. Scholes, I. Manners and M. A. Winnik, Macromolecules, 2009, 42, 7953–7960 CrossRef CAS
- P. Lu, N. M. Alrashdi and A. J. Boydston, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2977–2982 CrossRef CAS
- B. J. Lidster, D. R. Kumar, A. M. Spring, C.-Y. Yu, M. Helliwell, J. Raftery and M. L. Turner, Org. Biomol. Chem., 2016, 14, 6079–6087 RSC
- K. Grudzień, M. Malinska and M. Barbasiewicz, Organometallics, 2012, 31, 3636–3646 CrossRef
- R. A. Segalman, B. McCulloch, S. Kirmayer and J. J. Urban, Macromolecules, 2009, 42, 9205–9216 CrossRef CAS
- N. Sary, L. Rubatat, C. Brochon, G. Hadziioannou, J. Ruokolainen and R. Mezzenga, Macromolecules, 2007, 40, 6990–6997 CrossRef CAS
- It is common that the Mn values obatined from GPC do not correlate with MALDI-TOF-MS data especially for polydisperse polymers, see: H. Rashidzadeh and B. Guo, Anal. Chem., 1998, 70, 131–135 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py00147f |
| This journal is © The Royal Society of Chemistry 2019 |