
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Identification of β scission products from free radical polymerizations of butyl acrylate at high temperature†
Marco
Drache
 a,
Maria
Stehle
a,
Jonas
Mätzig
a,
Katrin
Brandl
a,
Marcel
Jungbluth
a,
Jan C.
Namyslo
b,
Andreas
Schmidt
b and
Sabine
Beuermann
*a
a,
Maria
Stehle
a,
Jonas
Mätzig
a,
Katrin
Brandl
a,
Marcel
Jungbluth
a,
Jan C.
Namyslo
b,
Andreas
Schmidt
b and
Sabine
Beuermann
*a
aClausthal University of Technology, Institute of Technical Chemistry, Arnold-Sommerfeld-Straße 4, 38678 Clausthal-Zellerfeld, Germany. E-mail: sabine.beuermann@tu-clausthal.de
bClausthal University of Technology, Institute of Organic Chemistry, Leibnizstraße 6, 38678 Clausthal-Zellerfeld, Germany
First published on 13th March 2019
Abstract
Acrylate polymerizations are associated with intra- and intermolecular H transfer to polymer and the formation of tertiary midchain radicals. At high temperatures β scission reactions of the midchain radicals may occur. To obtain more insights into the reaction mechanism in free radical butyl acrylate polymerizations at high temperature batch and semi-batch reactions were carried out in two solvents. The polymerizations were thermally initiated either by the monomer itself or a peroxide initiator. Molar mass distributions determined by size-exclusion chromatography (SEC) indicate the presence of low molar mass species in all products. To facilitate further analyses each sample was fractionated via preparative SEC. The separated low molar mass species were characterized via electro spray ionization – mass spectrometry and various nuclear magnetic resonance spectroscopy methods. Macromonomer species of chain lengths two to twelve were identified. The data are indicative of significant radical migration along the backbone prior to β scission.
Introduction
The knowledge of detailed polymerization kinetics and mechanisms is an essential prerequisite for reliable modeling of polymerization reactions with respect to optimizing the polymerization process and to synthesize material with tailored properties. In free radical polymerizations this is particularly important since the polymer architecture and properties are directly influenced by the polymerization kinetics. A recent review article by Ballard and Asua gives an overview on the relevant literature regarding radical polymerizations of acrylate monomers.1 In recent years the application of pulsed laser polymerization methods led to a good understanding of the propagation kinetics and the termination kinetics.2–10 The latter includes a detailed understanding of the chain length dependence of the termination rate coefficients, kt, and the conversion dependence of kt.11–13 Early investigations into the acrylate propagation kinetics revealed that significant contributions from chain transfer reactions have to be considered.14,15 Particularly important is the so-called backbiting reaction, an intramolecular chain transfer reaction at the chain end. The reaction is considered to constitute a 1,5 H-shift via a six-membered ring structure. The experimental data is supported by DFT calculations.16 Further, the chain transfer to a polymer chain has to be considered. Both reactions lead to the formation of tertiary radicals, the so-called midchain radicals (MCR). As shown by electron paramagnetic resonance (EPR) spectroscopy already at temperatures as low as 50 °C more than 90% of all radicals are MCRs.17 Due to the lower reactivity of these tertiary radicals the associated propagation step is by three orders of magnitude lower than that of the chain end radical (CER).11,18,19 Of course, MCRs may undergo other reactions, e.g., termination or transfer, as well. The complete reaction scheme is given for example in ref. 1. MCRs do not only influence the polymerization kinetics, they also affect the polymer architecture, because MCRs generate branching points once subsequent propagation steps occur.In addition to the above-described reactions, the midchain radical may also undergo a β scission reaction, which is particularly important to account for at high temperatures.20–32 As illustrated in Scheme 1, β scission yields a molecule with a terminal double bond and a chain end radical. If the midchain radical is formed by a backbiting reaction – as shown in Scheme 1 – the chain lengths of both species will be very different. The unsaturated species is either a macromolecular molecule (PBA![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ) or a trimer given by MM3CH2 and the radical either short or long. In this contribution the notations MMnCH2 and MMnH are used for unsaturated and saturated species, respectively, that may be identified in the ESI-MS spectra up to m/z of 1800 Da. A second aspect that needs to be considered is that the tertiary radicals generated via backbiting were reported to migrate along the polymer backbone. To identify macromonomer formation as a consequence of migration and subsequent β scission, firstly, model compounds were considered. Vandenbergh and Junkers reported on the preparation of macromonomers using poly(butyl acrylate) (PBA) obtained via atom transfer radical polymerization or nitroxide mediated polymerization. The polymer was activated at 140 °C and the products were analyzed via electron spray ionization – mass spectrometry (ESI-MS).33,34 The spectra clearly showed a distribution of macromonomers of different chain length rather than a single m/z obtained via β scission at one distinct monomer unit of the macromonomer. The finding is in line with EPR results reported by Kajiwara for acrylate model compounds.35,36 By now migration was reported for acrylate reversible-deactivation radical polymerization37,38 and just recently for a BA free radical polymerization.39 Mass spectrometry analyses and in particular ESI-MS proved to be important for getting more insight into the β scission and subsequent migration mechanism.28,33,34,39–41
) or a trimer given by MM3CH2 and the radical either short or long. In this contribution the notations MMnCH2 and MMnH are used for unsaturated and saturated species, respectively, that may be identified in the ESI-MS spectra up to m/z of 1800 Da. A second aspect that needs to be considered is that the tertiary radicals generated via backbiting were reported to migrate along the polymer backbone. To identify macromonomer formation as a consequence of migration and subsequent β scission, firstly, model compounds were considered. Vandenbergh and Junkers reported on the preparation of macromonomers using poly(butyl acrylate) (PBA) obtained via atom transfer radical polymerization or nitroxide mediated polymerization. The polymer was activated at 140 °C and the products were analyzed via electron spray ionization – mass spectrometry (ESI-MS).33,34 The spectra clearly showed a distribution of macromonomers of different chain length rather than a single m/z obtained via β scission at one distinct monomer unit of the macromonomer. The finding is in line with EPR results reported by Kajiwara for acrylate model compounds.35,36 By now migration was reported for acrylate reversible-deactivation radical polymerization37,38 and just recently for a BA free radical polymerization.39 Mass spectrometry analyses and in particular ESI-MS proved to be important for getting more insight into the β scission and subsequent migration mechanism.28,33,34,39–41
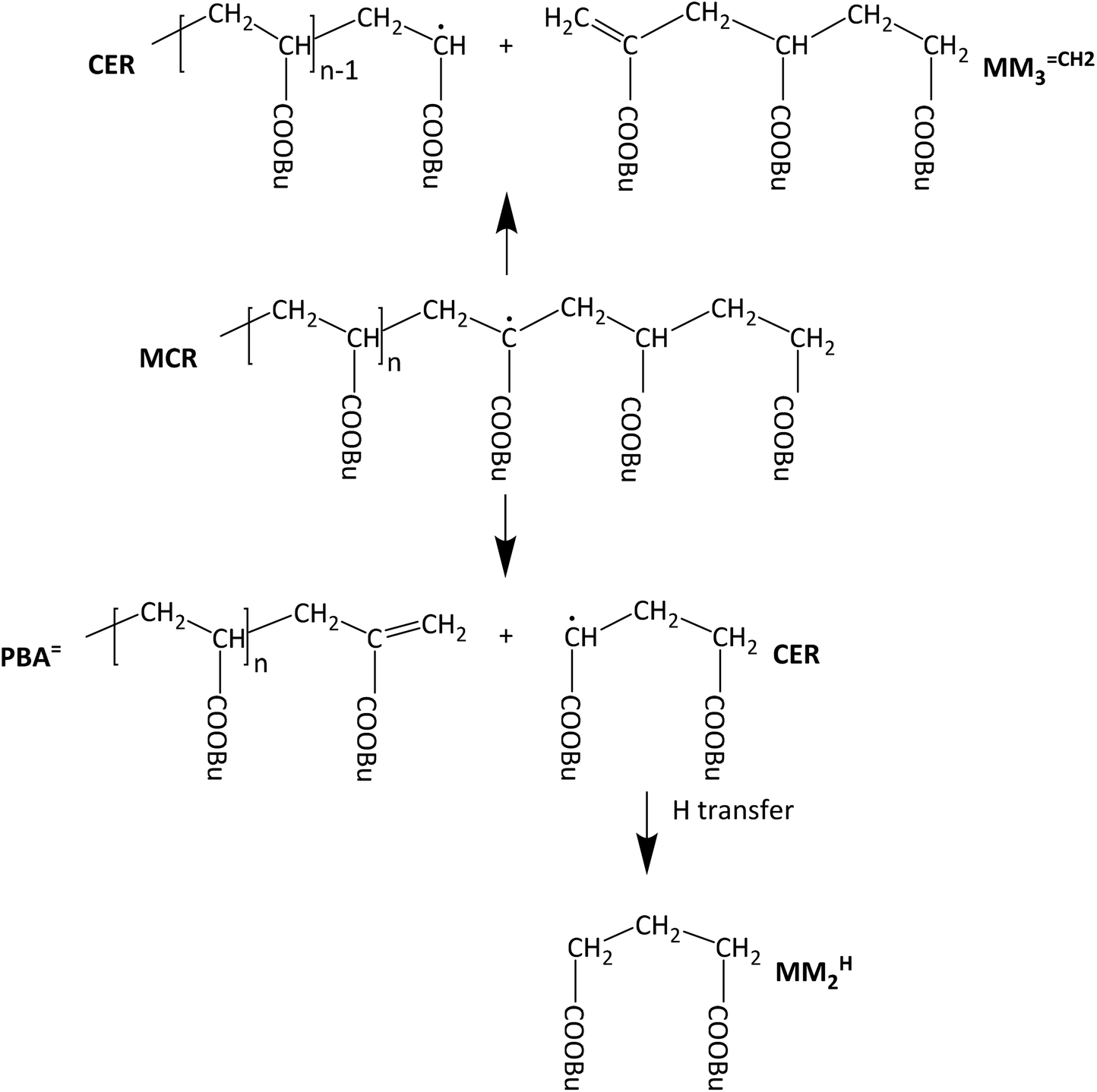 | ||
| Scheme 1 β scission reactions of a midchain radical obtained from backbiting via 1,5 H shift. | ||
The goal of this work was to carefully analyze the products obtained from free radical acrylate polymerizations at temperatures of at least 120 °C and to identify the low molar mass scission products. Studying free radical polymerizations appeared particularly important due to its high technical relevance. There are several challenges associated with high temperature acrylate polymerizations: firstly, reaction conditions have to be established that allow for isothermal polymerization at temperatures of at least 120 °C. Secondly, the low molar mass species have to be identified in a sample that contains large amounts of macromolecular species, making the identification very difficult. In order to overcome these challenges the following experimental approach was pursued: polymerizations were carried out with thermal initiation by the monomer butyl acrylate itself to achieve low radical concentrations. Further, semi-batch polymerizations with BA feed, associated with a low BA concentration, were performed using di-tert butyl peroxide as initiator. To allow for identification of the low molar mass species the samples were fractionated with a preparative size-exclusion chromatography set-up. Then, the low molar mass species were analyzed by ESI-MS and various NMR experiments in the absence of high molar mass polymeric species.
Experimental
Materials
The monomer n-butyl acrylate (BA, ≥99%, stabilized with 10–60 ppm monomethyl ether hydroquinone, Sigma Aldrich) was distilled at 5 mbar to remove the inhibitor. The initiator di-tert butyl peroxide (DTBP, Luperox®, 98%, Sigma-Aldrich) was used as received. The solvents 1,4 dioxane (99.8%, water free, Sigma Aldrich) and xylene (technical mixture of isomers, 98%, VWR) were used as received. Tetrahydrofuran (THF, 99%, Grüssing) was used as eluent for size-exclusion chromatography (SEC) analyses. Hydroquinone (99%, Riedel) dissolved in methanol (98.5%, CG Chemikalien Hannover) is used to stop the polymerization. Acetone-d6 (99.96%d, Deutero) and acetonitrile (ROTISOLV® HPLC Gradient, 99.9%, Roth) were used as solvents for NMR and ESI-MS analyses, respectively.Batch polymerization
Batch polymerizations of BA were carried out at temperatures ranging from 120 to 140 °C at slightly elevated pressure of at most 5 bar in an optical high-pressure cell equipped with sapphire windows allowing for quantitative in-line FT-NIR spectroscopy. The reaction cell was heated to the reaction temperature and vacuum was applied. The reaction mixture consisting of monomer and dioxane was purged with nitrogen to remove oxygen. Then, the mixture was introduced into the reaction cell, which was placed inside the sample compartment of the FT-NIR spectrometer and recording of spectra was started directly after adding the reaction mixture. Monomer conversions were calculated from an absorption band at 6145 cm−1 assigned to the first overtone of the olefinic C–H stretching vibration. Details of the reaction cell and quantitative in-line FT-NIR spectroscopy were given elsewhere.42,43Table 1 lists details of the individual experiments. The reactions were stopped after 1 h and the reaction mixture removed from the reaction cell. A solution of 4 mg hydroquinone in 1 mL of methanol was added to stop the polymerization. At room temperature BA and dioxane were evaporated. The remaining sample was dissolved in THF for SEC analysis.| Sample | T/°C | c BA,0/(mol L−1) | M n/(g mol−1) | M w/(g mol−1) | D | x |
|---|---|---|---|---|---|---|
| #1 | 120 | 2.92 | 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 683 683 |
47650 |
2.6 | 0.75 |
| #2 | 120 | 3.69 | 14201 |
82472 |
5.8 | 0.80 |
| #3 | 130 | 2.06 | 4708 | 18702 |
4.0 | 0.88 |
| #4 | 130 | 2.92 | 7383 | 32091 |
4.3 | 0.85 |
| #5 | 130 | 3.69 | 7782 | 50118 |
6.4 | 0.89 |
| #6 | 140 | 2.92 | 4227 | 15130 |
3.6 | 0.90 |
| #7 | 140 | 3.69 | 6427 | 30795 |
4.8 | 0.92 |
Semi-batch polymerization
The polymerizations were carried out in a 250 mL double walled stainless steel reactor (Büchi) equipped with temperature control (Beckmann + Egle) and a stirrer (büchi-drive 075 black line). All chemicals and the reactor were purged with nitrogen prior to the reaction. The reactor was charged with 87 g of xylene and heated to 130 °C while stirring with 240 rpm. Then, 114 mg DTBP dissolved in 5.12 g BA was added to the reactor with a syringe. At the same time continuous feed of BA to a reactor using a syringe pump (Landgraf LA 110) with a feed of 0.17 mL min−1 was started. Every 10 min a sample of 4 mL was taken from the reactor to determine monomer conversion gravimetrically. After 1 h the reaction was stopped by adding a solution of hydroquinone in methanol. The monomer and solvent were evaporated at ambient pressure.Characterization
Analytical SEC analyses were carried out using a Waters 515 HPLC pump, an autosampler from Knauer Marathon, a Knauer Smartline refractive index detector, and SEC columns from PSS (100 Å, 1000 Å, and 10000 Å). The samples (8 mg dissolved in 4 mL THF overnight) were passed through a syringe filter (45 μm) and analyzed at room temperature at a flow rate of 1 mL min−1. The SEC set-up was calibrated with polystyrene standards ranging from 162 to 3053000 g mol−1 from PSS.
Preparative SEC separation was performed with a set-up consisting of Knauer ceramics pump, an Isco fraction collector Foxy 200, a Knauer refractive index detector K-2401, two PLgel 10 μm MIXED-D columns from Agilent, and a Ultrastyragel 500 Å column from Waters. 100 mg of the dry polymer were dissolved in 10 mL THF overnight, passed through a syringe filter (45 μm), and injected into the sample loop. The fractionation was carried out at room temperature at a flow rate of 10 mL min−1 yields 10 fractions, each of them consisting of 10 mL solution. Fractions 6 and 7 were combined and used for further analyses via ESI-MS and NMR.
The high resolution mass spectra were obtained with a Bruker Impact II TOF mass spectrometer equipped with a direct inlet and an electrospray ionization source. Samples were dissolved in anhydrous acetonitrile and sprayed from the same solvent using a flow of 3.0 L min−1 of nitrogen as drying gas at a temperature of 200 °C. The capillary, end plate offset, and charging voltages were set to 3100 V, −400 V, and 2000 V, respectively. Mass spectra were taken in the positive ion detection mode applying an ion energy of 5.0 eV, a collision energy of 7.0 eV and transfer and prepulse storage times of 100.0 μs and 5.0 μs, respectively.
NMR spectra were recorded on a Bruker Avance III 600 (Bruker, Ettlingen, Germany) with 600 MHz proton frequency equipped with a room temperature BBO probe head with z–gradient. The spectrometer was additionally provided with the NMR thermometer hardware (Bruker) that ensured highly stable temperature conditions. The calibration of gradient strength was conducted by means of a deuterium oxide sample containing H2O traces44 and was found to be 0.548 T m−1. 1H-NMR spectra were referenced to the residual solvent signal of deuteroacetone at 2.04 ppm. 13C-NMR spectra refer to the solvent signal center of the methyl groups at 29.3 ppm. 2D-NMR spectra (i.e. DQF–1H,1H–COSY, 1H,13C–HSQC, and 1H,13C–HMBC were measured applying the non–uniform sampling mode (NUS) in Bruker's TopSpin 3.5 NMR software (10% NUS data).
The diffusion ordered spectra (DOSY) were conducted in high–quality 5 mm NMR tubes (Wilmad 535) in an adequate sample spinner provided with a mass-ring under stabilized temperature conditions, i.e. (298 ± 0.1) K. The probe head nitrogen gas flow was adjusted to 500 L h−1. Acceptable measurement time was made possible using a non–optimized pulse repetition delay of 3 seconds instead of about 22 seconds that resulted from T1 inversion-recovery measurements. Sample spinning (20 Hz) was used in order to avoid convection.45 The applied pulse sequence was the DOSY Oneshot experiment46 performed as a pseudo–2D measurement containing 12 one–dimensional proton spectra with squared increase of the gradient field strength stepped up from 10 to 80%. The diffusion time (parameter d20, Bruker) was set to 100 ms and the corresponding gradient lengths (p30, δ/2) was set to 1250 μs.47 The DOSY data were analyzed applying the DOSY Toolbox of Nilsson for exponential fitting and calculation of diffusion coefficients.48 Thereby, all single FIDs were treated in the exponential multiplication step with an appropriate line broadening factor of 1.0 Hz. The DOSY Toolbox calculations from the mono–exponential decay resulted in diffusion coefficients (D) with errors of about 2%.
DFT calculations
All density-functional theory (DFT)-calculations were carried out with the current Spartan Software49 running on an Intel® Core™ i7-6950X decacore system equipped with 64 GB RAM main memory and sufficient solid-state disc space. MMFF optimized structures were used as starting geometries. Then, the B3LYP density functional and the 6–31G* basis set was used. The final geometry was proven to be a true minimum by the absence of imaginary frequencies. Additional NMR shift calculations were performed applying the ωB97X–D/6–31G* Density Functional Model.FT-NIR spectra were recorded on a Bruker Vertex 70 spectrometer using a halogen lamp, a Si-coated CaF2 beamsplitter, and a liquid N2 cooled InSb detector. The spectra were recorded with a resolution of 2 cm−1. 20 scans were co-added.
Results and discussion
Investigations into the β scission reaction and its reaction products requires high polymerization temperatures of at least 120 °C. Since radical polymerizations are exothermic it is a challenge to establish isothermal reaction conditions.27 To control the temperature rather low radical concentrations are advantageous. Therefore, rather than using a thermally decomposing initiator the monomer itself provided radicals in a thermally initiated polymerization. In addition, polymerizations were performed at reduced BA concentrations between 2.1 and 3.7 mol L−1 in dioxane. Due to the rather low volatility of the solvent and monomer in the closed reactor the pressure will exceed ambient pressure, thus, an optical high-pressure cell was used. The main advantage of using this reaction cell is that the stainless steel cell body has a high heat conduction coefficient and the cell mass is 5.34 kg, while the mass of the reaction mixture is only around 2 g. Thus, carrying out the polymerizations in this special reaction cell promotes a constant temperature. A thermocouple placed directly at the reaction volume showed that the temperature varied by at most 0.1 °C. In addition, the optical cell allowed for in-line FT-NIR spectroscopy and, thus, monitoring of monomer conversion during the reaction.Thermally initiated polymerizations were carried out in the temperature range from 120 to 140 °C for 1 h. The products were analyzed with the analytical SEC set-up. The results are given in Table 1. In addition, monomer conversions are listed. Due to the significant fraction of oligomeric species seen in every molar mass distribution, in the following Mw values are considered for comparison. As expected the molar masses are lowered with decreasing initial BA concentration, cBA,0, at a given temperature. Further, it is clearly seen that Mw, are lowered upon increasing temperature at a given cBA,0. This finding may be explained by a higher radical concentration at higher temperatures and consequently a higher termination probability. In addition, at higher temperatures more β scission reactions occur, that lead to a further lowering of Mw. Moreover, at a given cBA,0 the dispersities increase in going from 120 to 130 °C polymerization temperature. A further increase in temperature to 140 °C leads to a lowering of dispersity. The molar mass distributions (MMDs) for polymers obtained at 130 °C are shown in Fig. 1. The distributions are shifted to higher molar masses the higher cBA,0. All MMDs feature a rather broad high molar mass peak and a small distinct peak at logM ∼ 2.4 with the intensity being highest for the lowest BA concentration. The MMDs for polymers obtained at 120 and 140 °C given as Fig. S1 and S2 in the ESI† show similar features upon changing the BA concentration.
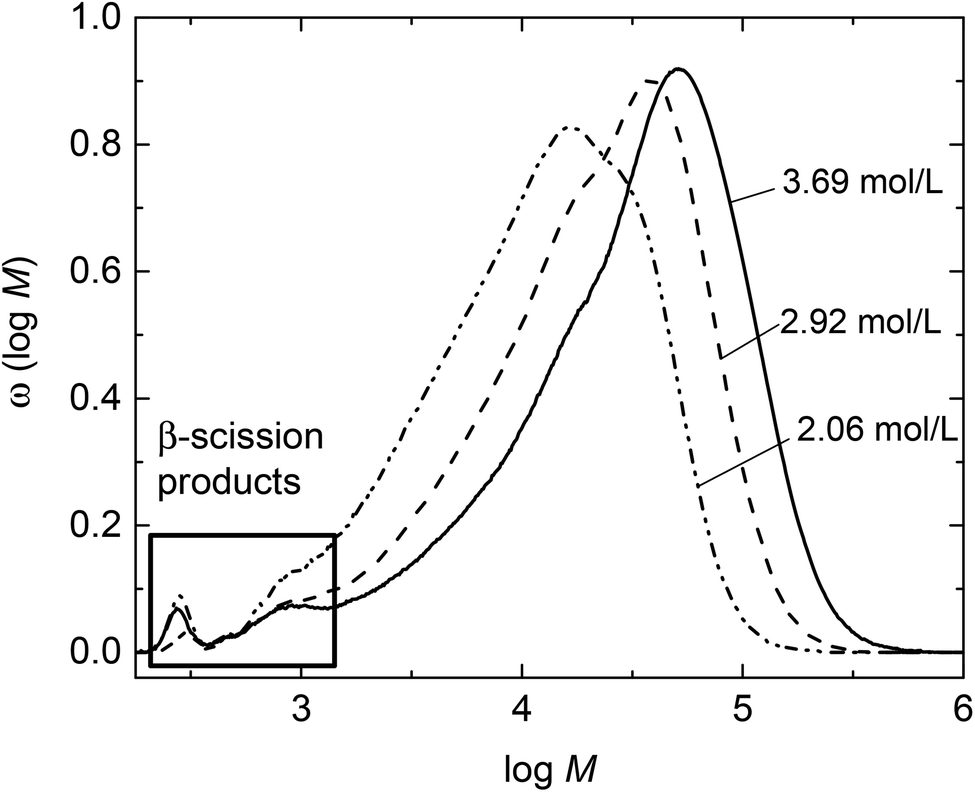 | ||
| Fig. 1 Molar mass distributions for polymer samples obtained from thermally initiated batch BA polymerizations at 130 °C. Initial BA concentrations are assigned to the MMDs. | ||
Fig. 1 indicates that the amount of low molar mass material is very small compared to the high molar mass polymers and, thus, hampering further analyses of these low molar mass species by ESI-MS or NMR. If these species may be separated from the high molar mass polymer further analyses will be facilitated. For this purpose the polymerization products require fractionation. Since the batch polymerizations of BA described so far yielded at most 250 mg of material prior to work-up, a different type of polymerization giving access to higher amounts of product allowing for advanced NMR analyses was required. For this reason, semi-batch polymerizations with BA feed were carried out. Due to the at all times low monomer concentrations between 0.4 and 0.5 mol L−1 inside the reactor the contributions from thermal initiation by the monomer itself are expected to be minor compared to initiation via initiator decomposition. In addition, the low monomer concentration should favor midchain radical migration occurring in competition to monomer addition to the midchain radical.
The semi-batch polymerizations were initiated by thermal decomposition of DTBP, which decomposes rather slowly between 120 and 140 °C. Further, DTBP was selected since the decomposition mechanism is known from literature.50 Upon decomposition DTBP generates two tert-butoxy radicals that start chain propagation. In addition, if elimination of acetone occurs, methyl radicals may be formed. This simple decomposition mechanism was expected to be advantageous for the assignment of the various species identified via ESI-MS. The semi-batch polymerizations were carried out in xylene. It was anticipated that transfer to two different solvents may be helpful with respect to assignment of the species to be identified by ESI-MS. Firstly, the material obtained from a semi-batch polymerization at 130 °C with a reaction time of 1 h was analyzed by analytical SEC. Due to the low monomer concentration the product has a low Mn of 2148 g mol−1. The molar mass distribution given by the green curve in Fig. 2 is rather broad and shows a distinct peak at low molar masses.
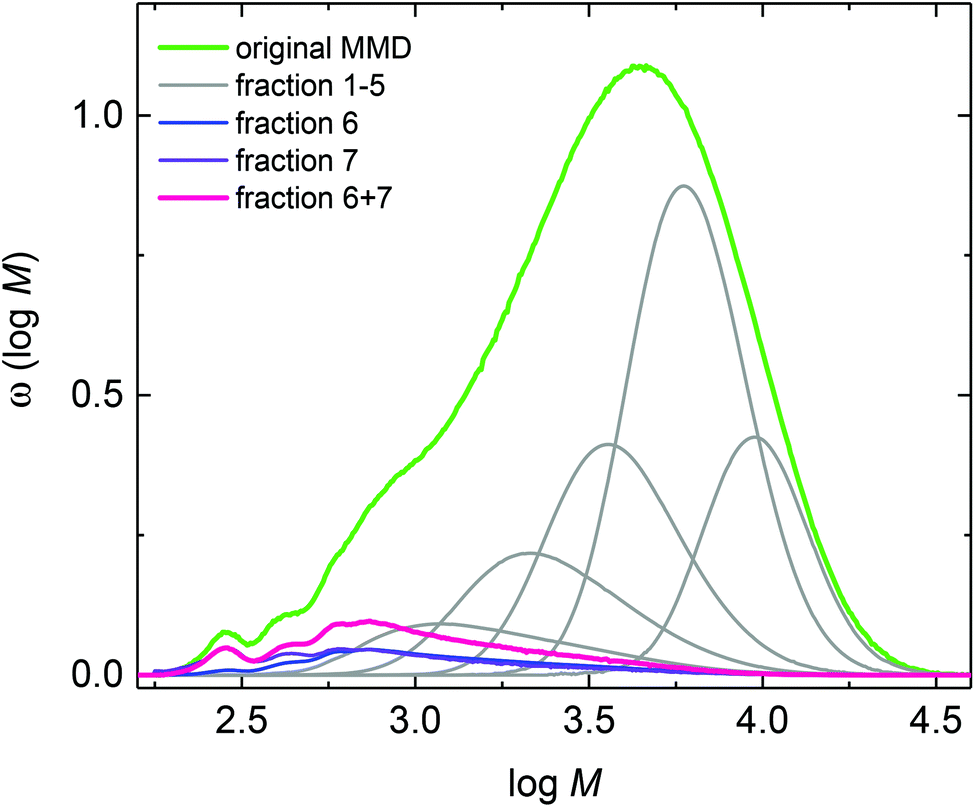 | ||
| Fig. 2 Molar mass distributions measured for fractions 1 to 7 obtained via preparative SEC and the original MMD (green) measured prior to fractionation of the material obtained from a semi-batch polymerization at 130 °C after 1 h in xylene. | ||
The product from the semi-batch polymerization was fractionated in the preparative SEC. As an example, Fig. 3 gives the corresponding elution curves for nine fractions of the above-mentioned experiment. Curves 8 and 9 indicate that the residual monomer is well separated from all other fractions. To obtain the individual molar mass distributions of fractions 1 to 7 they were analyzed via analytical SEC. The resulting MMDs are contained in Fig. 2 and the average molar masses are given in Table S1 of the ESI.†Fig. 2 indicates that the high molar mass material was successfully separated in fractions 1 to 5. For further analyses with respect to the low molar mass species fractions 6 and 7 were combined prior to ESI-MS and NMR analyses. Integration of the individual peaks for fractions 1 to 7 in Fig. 3 yields the relative amount of each fraction in the material. For this purpose the fractions from the preparative SEC were directly injected into the analytical SEC to allow for quantification of the species. This data listed in Table S1† was used to scale the distributions in Fig. 2. Fractions 6 and 7 constitute about 7% of the entire material. Firstly, the ESI-MS data of the combined fractions 6 and 7 is presented in Fig. 4.
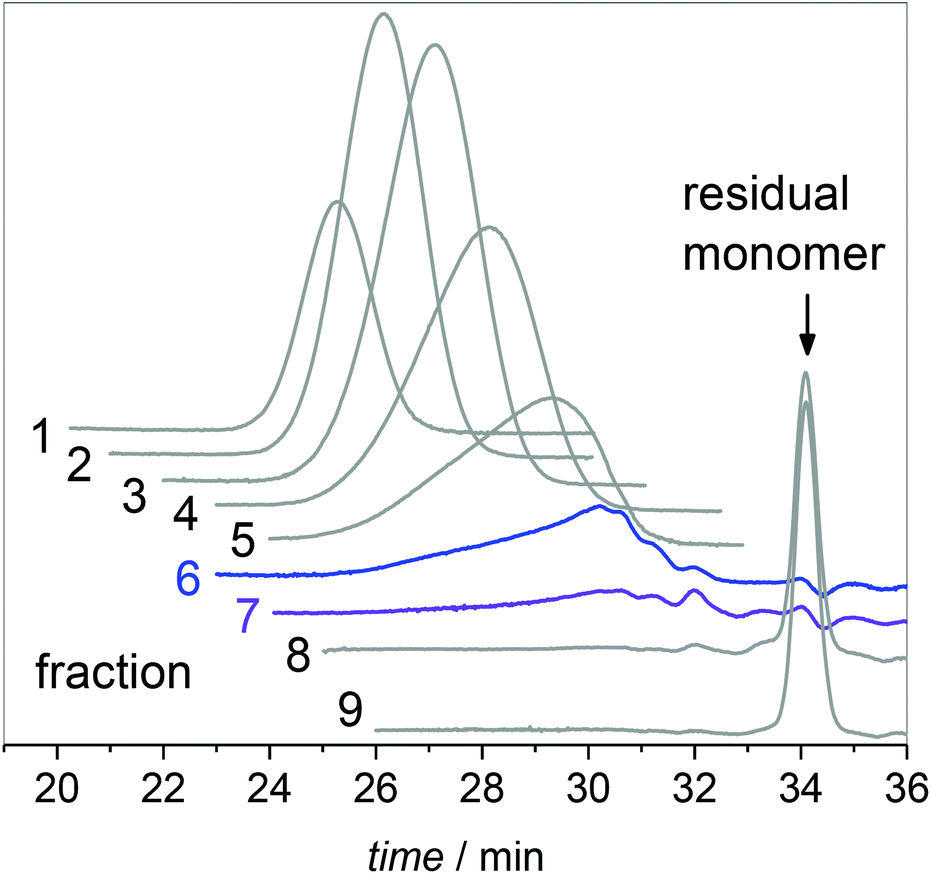 | ||
| Fig. 3 Elution curves obtained via preparative SEC for material from semi-batch polymerization at 130 °C. | ||
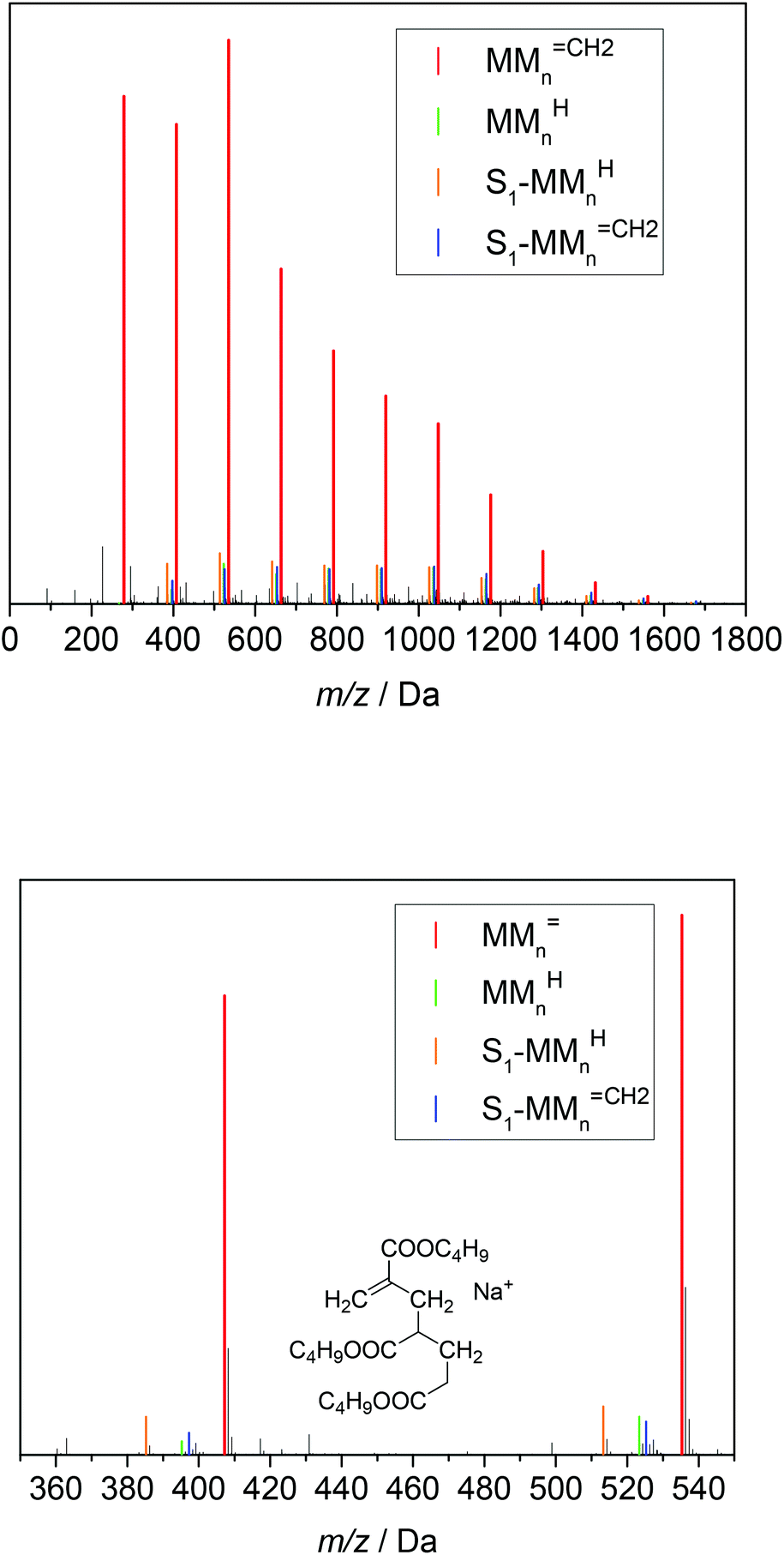 | ||
| Fig. 4 ESI-MS data of the combined fractions 6 and 7. The series of the following β scission products are identified: MMnCH2, MMnH, S1-MMnCH2 and S1-MMnH. S1 represents a xylene end group. The chemical structure of the unsaturated trimer MM3CH2 is displayed. For peak assignments refer to the text and to Table 2. | ||
The upper part of Fig. 4 shows that the material consists of a major series at (279.1567 + n·128.0837) Da. The increments of 128.0837 Da correspond to the BA monomer. The highest intensities are observed up to 536 Da for m/z. For higher m/z the peak intensities decrease steadily. These major peaks may be assigned to BA macromonomer species carrying a double bond, which were obtained by β scission of a midchain radical and are denoted as MMnCH2 (see Scheme 1). For example, the peaks at m/z of 279.1567 Da, 407.2404 Da, and 535.3240 Da refer to dimeric, trimeric, and 4-meric species, respectively.
Three additional series of rather low intensity were identified, which are visible in the enlargement given in the lower part of Fig. 4. Again, the increment between the elements of a given series is 128.0837 Da. The series at (267.1568 + n·128.0837) Da may be assigned to MMnH, which may be obtained, if the chain end radical obtained via β scission undergoes H transfer. The other two low intensity series seen in Fig. 4 may be assigned to species that carry a xylene end group, which originates from H transfer reactions of the propagating radical to the solvent. The solvent radicals start propagation of a new chain. The notation S1-MMnCH2 and S1-MMnH refers to unsaturated and saturated species, respectively. The structures and corresponding m/z values are contained in Table 2.
| Semi-batch polymerization in xylene | Batch polymerization in dioxane |
|---|---|
| MM2CH2: 279.1567 Da |
|
| MM2H: 267.1567 Da | |
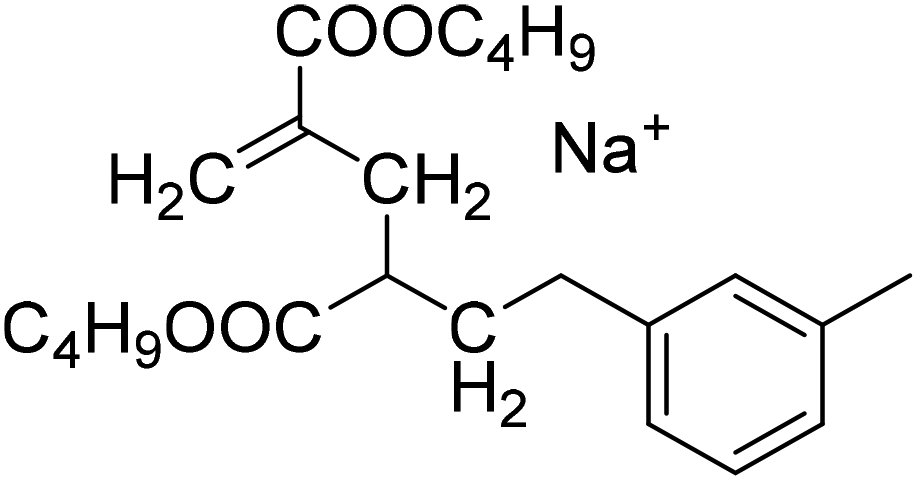
|
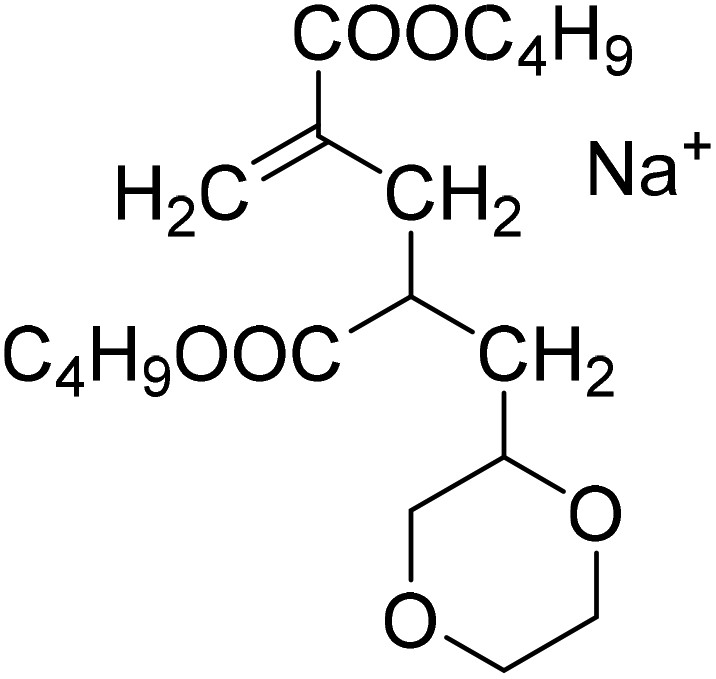
|
| S1-MM2CH2 |
S2-MM2CH2 |
| 397.2349 Da | 379.2091 Da |
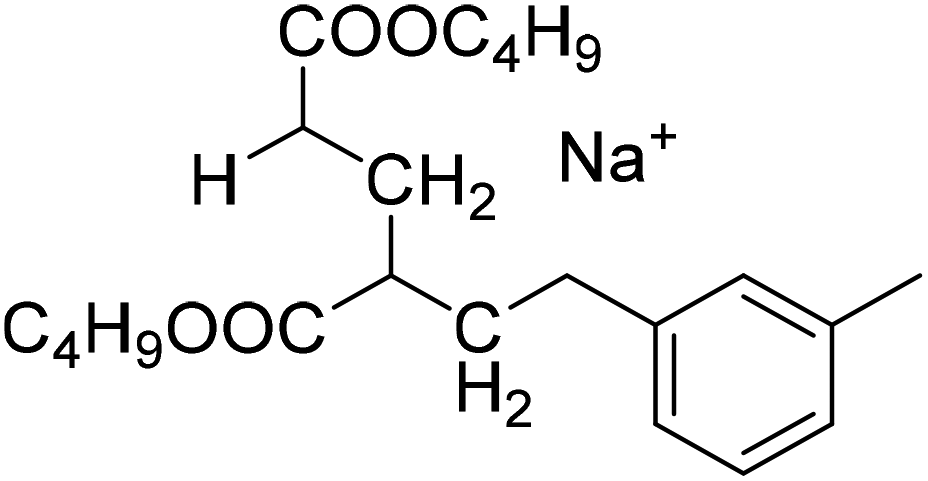
|
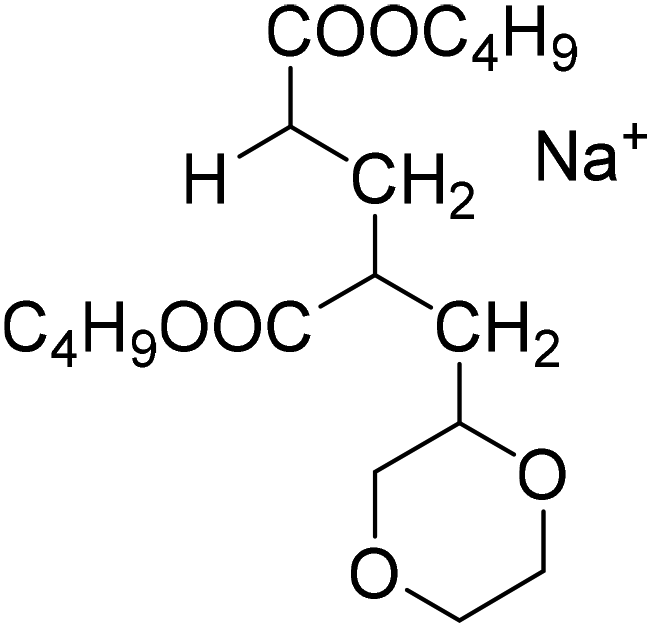
|
| S1-MM2H | S2-MM2H |
| 385.2349 Da | 367.2091 Da |
Backbiting via 1,5 H shift at the propagating chain end and subsequent β scission yields trimeric unsaturated species. Thus, the occurrence of higher order unsaturated species in the ESI-MS spectra given in Fig. 4 strongly suggests that the radical functionality may migrate along the backbone. The migration process is illustrated in Scheme 2. The red arrows indicate migration of the radical functionality from an MCR obtained via backbiting (red monomer unit) over two or four monomer moieties. Upon β scission 5-meric and 7-meric macromonomers are obtained.
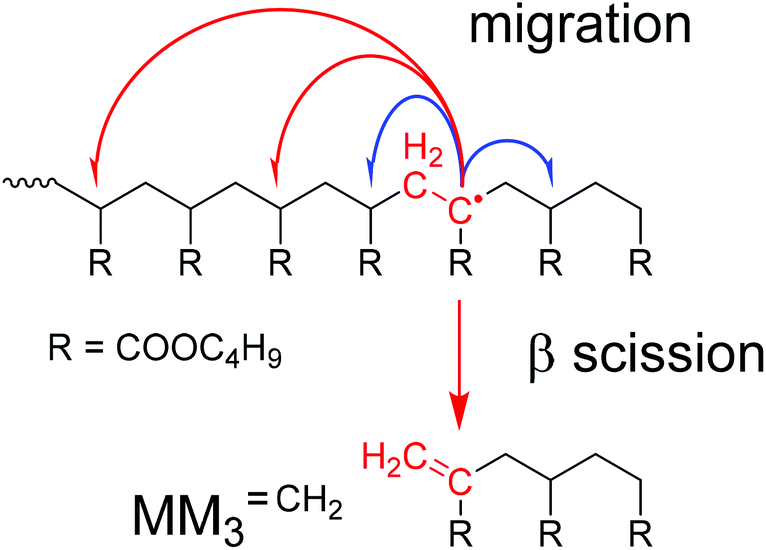 | ||
| Scheme 2 Illustration of migration and β scission observed for midchain radicals. | ||
The occurrence of dimeric unsaturated species at m/z of 279.1567 Da and higher order species with an even number of monomer units in all samples analyzed is surprising and cannot be explained to be due to backbiting via 1,5 H shift, migration of the radical functionality over two monomer units, and subsequent β scission. This type of species could result from migration of the radical functionality of an MCR to the neighboring monomer unit (blue arrow in Scheme 2) followed by β scission. Unsaturated Species carrying either a tert butoxy group or a methyl group originating from DTBP decomposition and subsequent addition of two monomer molecules are associated with a m/z of 365.2298 Da or 307.1880 Da, respectively. Trimeric species would occur at 493.3135 Da or 435.2718 Da. However, these species cannot be detected in Fig. 4.
In addition, all products obtained from batch polymerizations listed in Table 1 were fractionated by preparative SEC and analyzed via ESI-MS. As an example, Fig. 5 gives the ESI-MS data referring to a batch polymerization at 130 °C with cBA,0 = 2.92 mol L−1. As reported in Fig. 4 there is one major series at (279.1567 + n·128.0837) Da referring to MMnCH2 and three series of low intensity assigned to MMnH and the species being initiated by the solvent radical, in this case dioxane. Of course, MMnCH2 and MMnH occur at identical m/z values for both systems, whereas differences are seen for the species containing a solvent molecule. The structures and associated masses of S2-MMnCH2 and S2-MMnH are contained in Table 2. As mentioned above the dimeric unsaturated species cannot be due 1,5 H shift and subsequent β scission. Since these dimeric species are found in both types of reactions with different initiation it is highly unlikely that they are due to the initiation process.
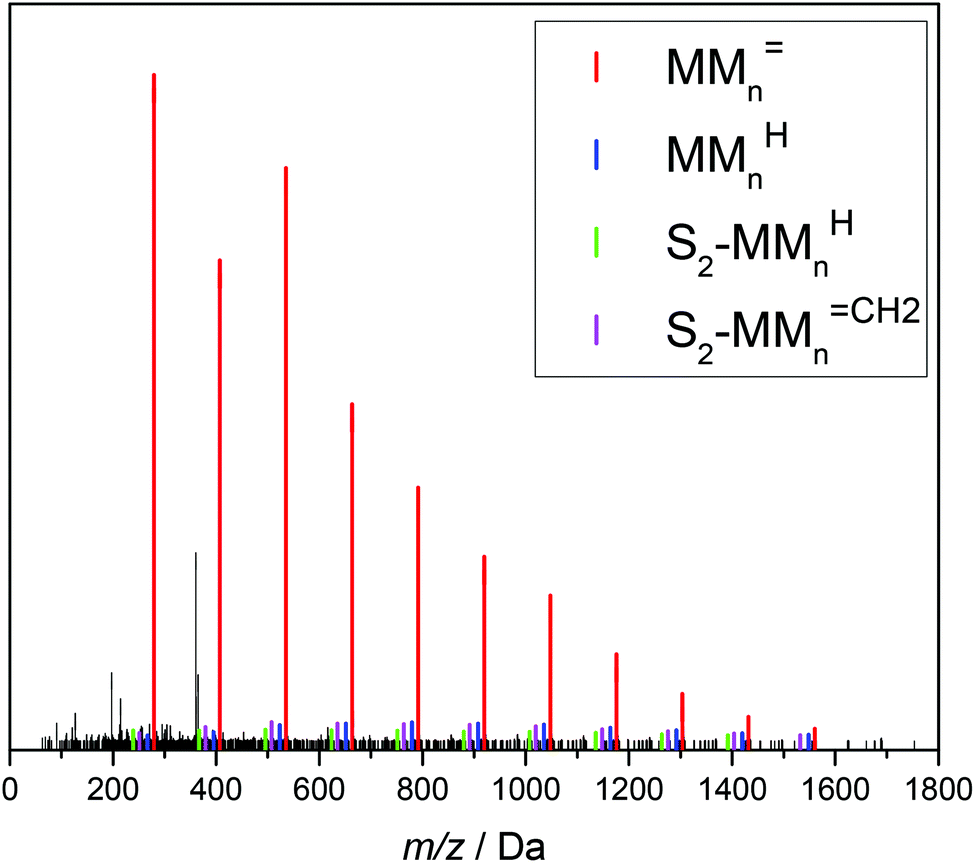 | ||
| Fig. 5 ESI-MS data for material obtained from batch polymerization at 130 °C with cBA,0 = 2.92 mol L−1. The series of the following β scission products are identified: MMnCH2, MMnH, S2-MMnCH2 and S2-MMnH. S2 represents a dioxane end group. For peak assignments refer to the text and to Table 2. | ||
All results of the ESI-MS analyses are summarized in the ESI in Fig. S3 to S10† and the associated tables. For the unsaturated species MMnCH2 a total of 88 signals was identified by ESI-MS. With respect to the theoretical values of m/z a standard deviation of 0.0020 Da was calculated. A slightly higher standard deviation of 0.0027 Da was determined from 251 signals of the species MMnH, S1-MMnCH2, S1-MMnH, S2-MMnCH2 and S2-MMnH.
Close inspection of the data in Fig. 5 shows alternating intensities of the first 3 peaks. Similar features were seen in the ESI-MS spectra obtained for both samples from batch polymerizations at 120 °C (samples #1 and #2 in Table 1) and for two samples from batch polymerization at 130 °C (samples #4 and #5 in Table 1), which are given as Fig. S5 to S8 in the ESI.† ESI-MS spectra obtained for material obtained at 140 °C in batch polymerizations show a steady decrease in peak intensities (ESI, Fig. S9 and S10†). A publication by the group of Junkers for material obtained from reversible addition fragmentation transfer (RAFT) BA polymerization at 150 °C and higher monomer concentration shows an alternating intensity of the ESI-MS peaks assigned to the macromonomers.37 The variations were explained by the fact that migration along the backbone favors positioning of the radical at every second monomer unit. The higher peak intensities were suggested to occur for species with an odd number of monomer units. The literature data37 and the data presented here follow this trend. The discussion of the ESI-MS data indicates that uncertainties concerning the mechanism of backbiting and migration still exist. Thus, it is of high importance to obtain detailed information on the exact structure of the species. For this purpose, in the next section structural elucidation via comprehensive NMR measurements and DFT calculations is presented.
Prior to comprehensive NMR measurements, a DFT calculation of the above-mentioned unsaturated trimeric molecule MM3CH2 was performed in order to obtain an appropriate chemical shift prediction. These calculations applying current Spartan software49 were started with a geometry optimization using the B3LYP functional and the 6–31G* basis set. The absence of imaginary IR frequencies confirms that a proper result was obtained. Based on this optimized structure, proton and carbon NMR shift predictions were carried out with the ωB97X–D functional and the 6–31G* basis set. All calculated shifts are given in Fig. 6.
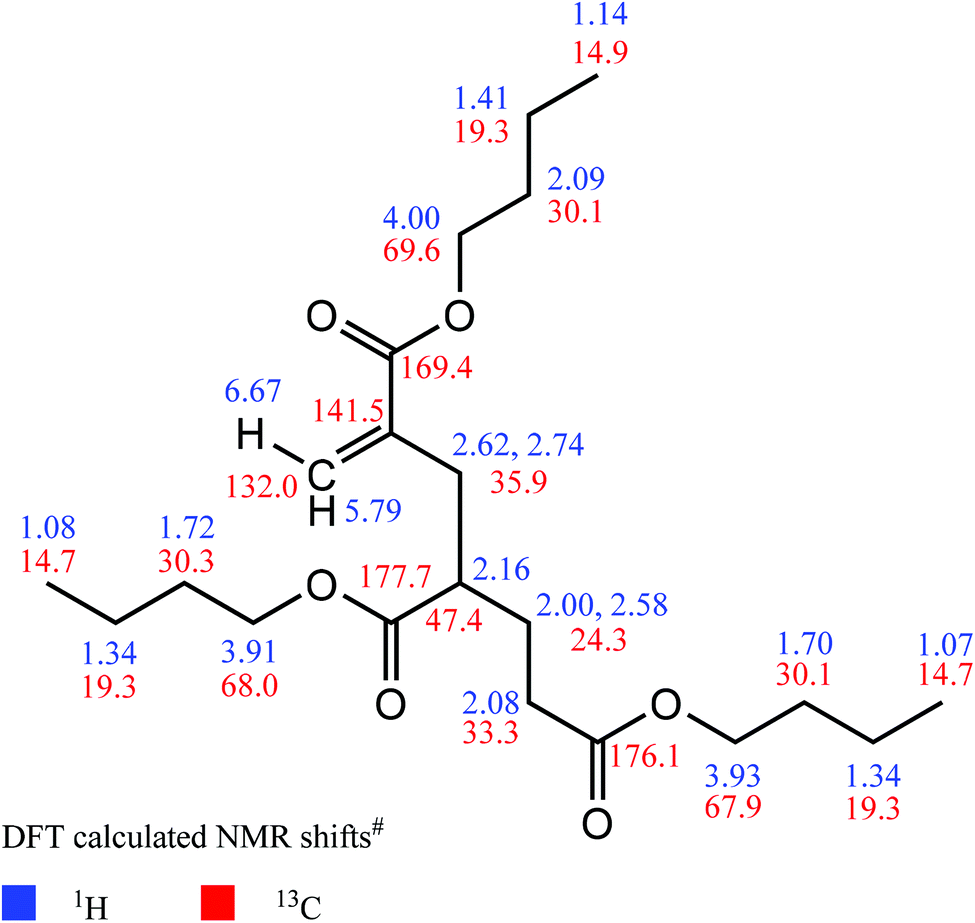 | ||
| Fig. 6 DFT calculations of NMR chemical shift values (#obtained from ωB97X-D functional and 6-31G* basis set, molecule MM3CH2). | ||
Expectedly, small shift differences were found in the corresponding measured NMR spectra due to usage of an NMR solvent (deuteroacetone) in contrast to vacuum-based DFT calculations. With the help of an entire 1D and 2D NMR data set, the latter comprising the 1H,13C–HSQC experiment for detection of protons which are directly attached to carbon atoms, the 1H,1H–COSY for two- and three–bond proton–proton connectivities, and the 1H,13C–HMBC for proton–carbon multiple bond connectivities, significant NMR signals of the proposed olefinic substructure were found and were unambiguously assigned as described below. The corresponding NMR spectra are given in Fig. S11 to S16 in the ESI.†
In detail, the cis–olefinic proton in β–position to the carbonyl carbon atom of the α–alkylated acrylic–type moiety (see Fig. 7) was slightly shifted downfield to about 6.13 ppm compared to the trans–oriented β–proton that showed its resonance at about 5.60 ppm. The olefinic β–carbon (exo-methylene) itself is detected at 125.0 ppm. The HMBC revealed a cross–signal that arises from vicinal coupling of this cis–olefinic proton and the acrylic carbonyl carbon atom which is characteristically shielded to about 166.3 ppm due to its α,β–unsaturation. In contrast to this, the ester carbonyl atoms of the saturated acrylate substructures are shifted downfield to about 172.5 and 174.5 ppm, respectively. These shift differences between ester carbonyl atoms of α,β-unsaturated carbonyls in comparison to their saturated counterparts are in accordance with the DFT calculation and with literature values.51
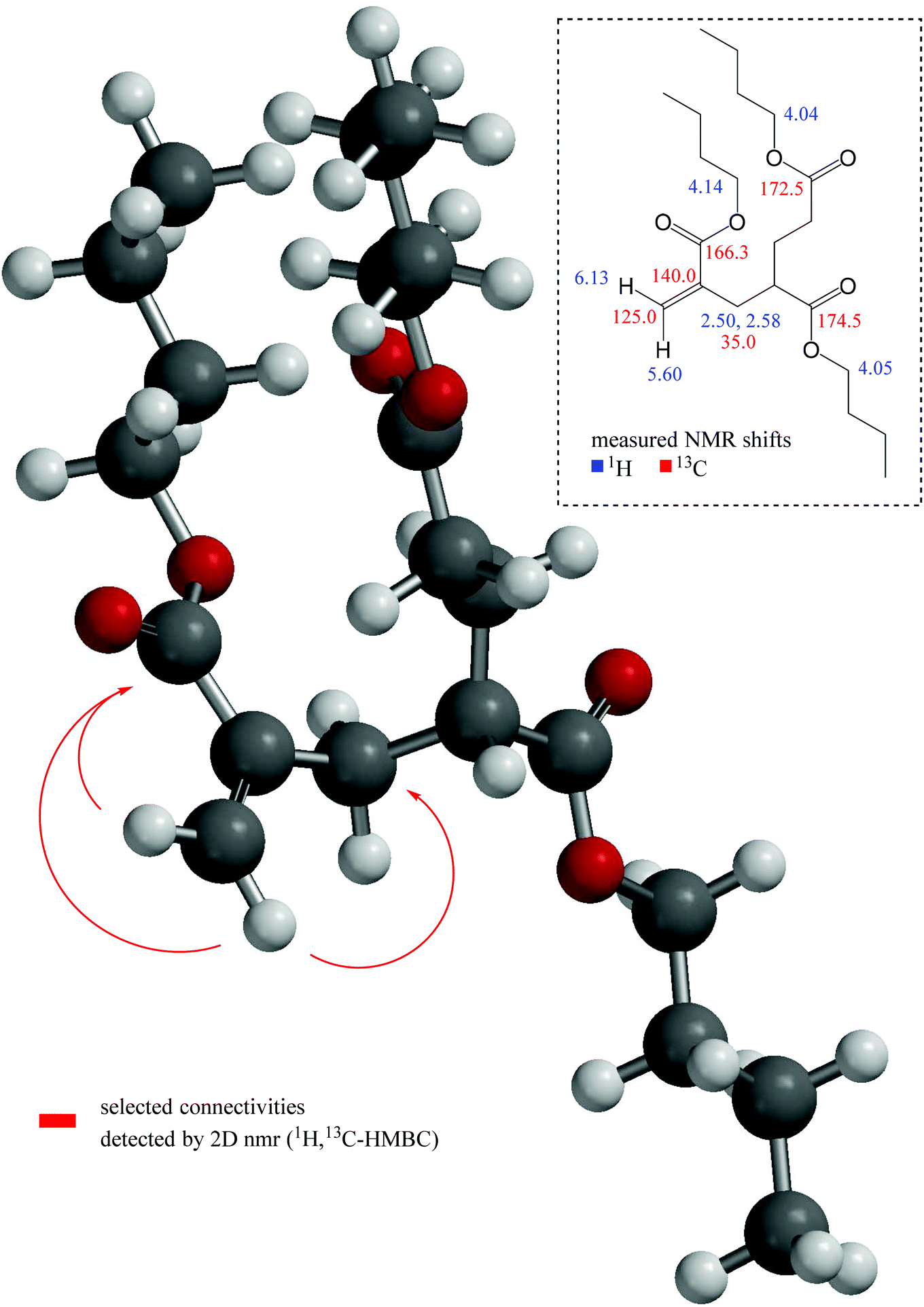 | ||
| Fig. 7 DFT-calculated geometry of molecule MM3CH2 with pivotal connectivities detected via HMBC NMR (red arrows) and significant measured shift values. | ||
A further, highly diagnostic NMR cross–peak within the HMBC is the signal that proves the connectivity over three chemical bonds between the above-mentioned trans–oriented β–proton and the methylene carbon adjacent to the olefinic α–position at 35.0 ppm (refer to Fig. 7). Thereby, this sp3–CH2 group represents the first position of the α–alkylation and thus ties up the acrylic substructure to the saturated backbone (consisting of two further monomeric units in case of molecule MM3CH2). The chemical shift values of the diastereotopic methylene protons in this position are 2.50 and 2.58 ppm, respectively. For clarity, the latter mentioned particular atom sequence Hβ,trans(olefin)–Cα(olefin)–CCH2 as well as further selected connectivities that consistently proves our structural assumption are depicted in Fig. 7 with the help of red arrows.
Due to the explicit separation of the NMR resonance of the OCH2 protons in the acrylic ester substructure at about 4.14 ppm proton shift from their O-methylene counterparts of the saturated acid esters, which are found at about 4.04 ppm, an integration is feasible in order to obtain the ratio of unsaturated to saturated substructures. Corresponding integral values yielding a 1.0:6.8 ratio can be found in Fig. 8.
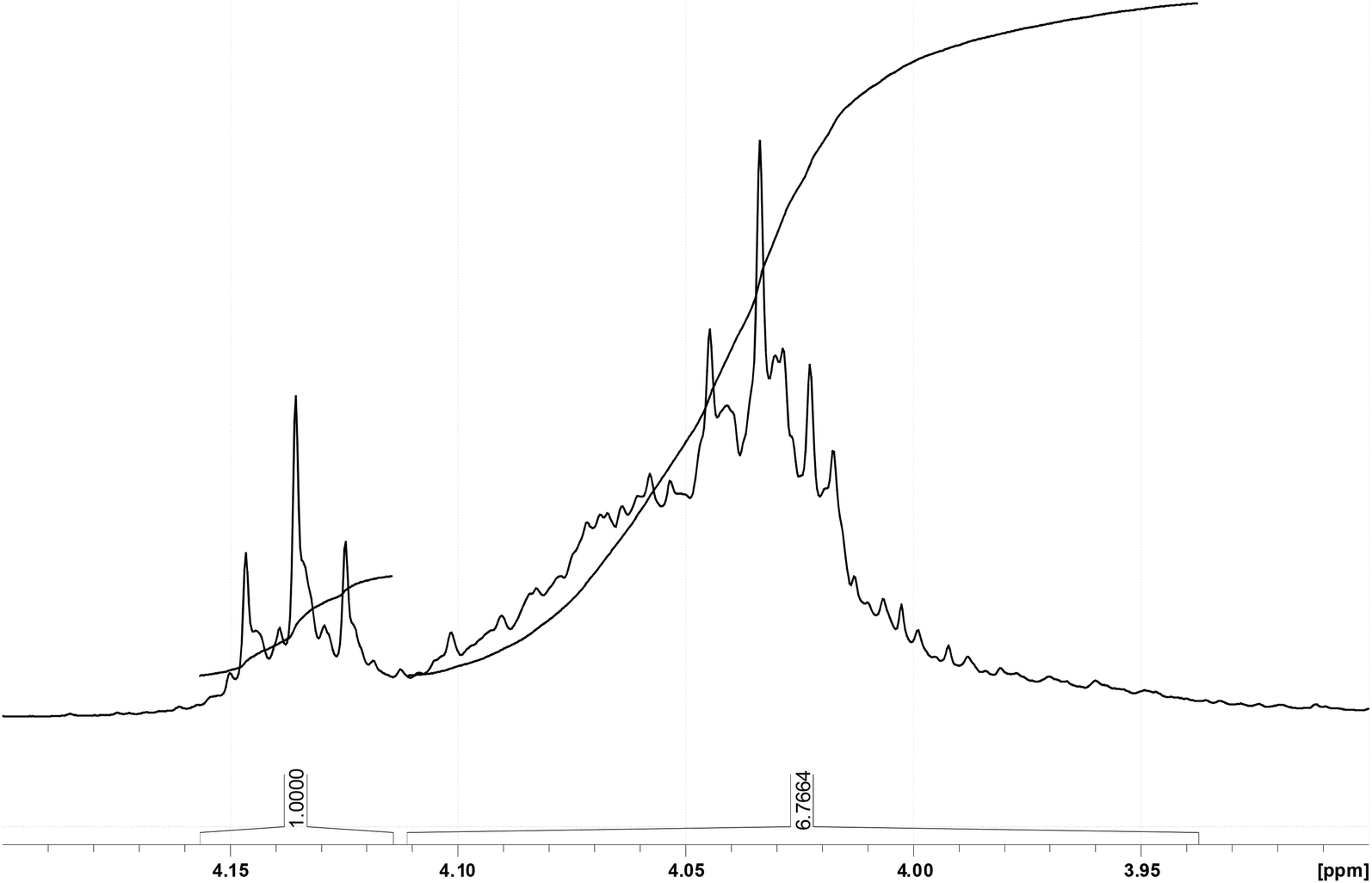 | ||
| Fig. 8 Ratio of acrylic ester OCH2 at 4.14 ppm vs. saturated acid ester OCH2 at about 4.04 ppm. | ||
As found in Fig. 4 and 5 higher mass species are present in the sample. Thus, it appeared interesting to perform also diffusion ordered NMR spectroscopy (DOSY)52 for the above discussed compound MM3CH2 and three further main components bearing the unsaturated acrylic-type substructure. The DOSY data were obtained from a series of attenuated 1D proton spectra, whereby from one spectrum to another a varied pair of gradient pulses was provided in order to at first encode and subsequently decode diffusing molecules.53 The inter–gradient delay Δ in the Stejskal–Tanner equation given below corresponds to the diffusion time and the gradient length is the value for δ in this equation. From the observed mono-exponential decay of the individual signal intensities I within the series of attenuated spectra the diffusion coefficient D of each single component is calculated upon an appropriate curve fitting by means of this Stejskal–Tanner equation:
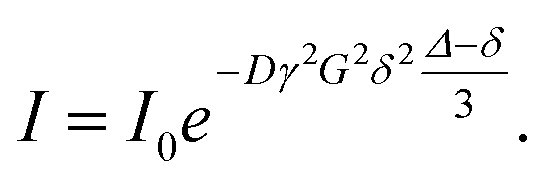
Further parameter for the DOSY measurement are the z–gradient strength G and I0, which reflects the signal intensity without diffusion. γ is the gyromagnetic ratio. The applied pulse program was the so–called Oneshot sequence published by Morris and co-workers.46
As a main result of the DOSY measurement, the above-mentioned four unsaturated compounds each appeared as a comparatively sharp, well separated signal of its cis–olefinic proton in β–position to the carbonyl carbon atom of the α-alkylated acrylic-type structural unit as discussed above. The different molar masses of these compounds were reflected by their individual diffusion coefficients D. The calculation of these D values was performed by means of the so–called DOSY Toolbox published by Nilsson.48 The pseudo–2D DOSY plot of the β–olefinic proton region depicts these four signals as well as the corresponding diffusion coefficients (see Fig. 9).
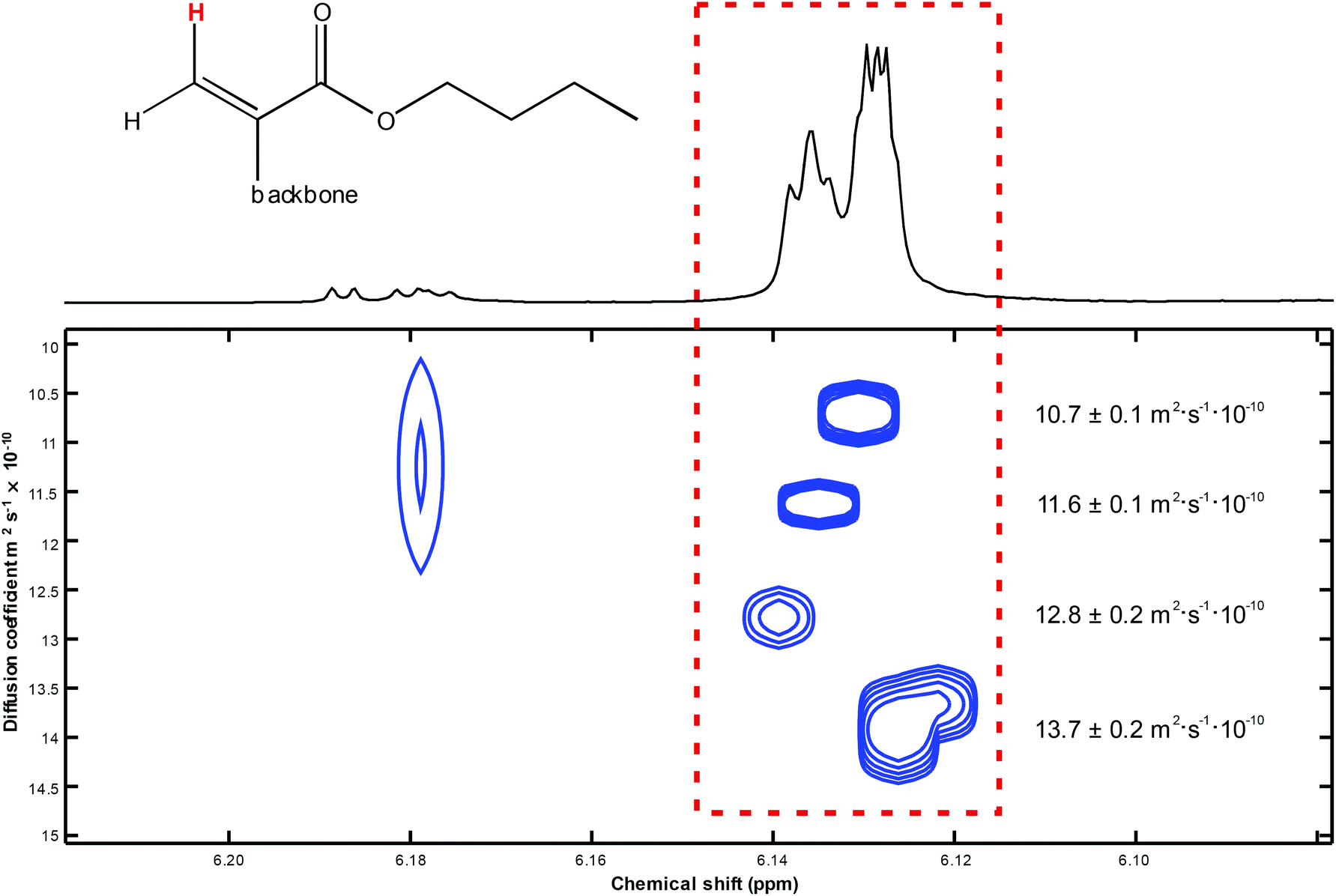 | ||
| Fig. 9 DOSY plot of the cis-oriented β-proton within the acrylic substructure of four suchlike species with different backbone/molar mass ratios. | ||
With respect to the obtained diffusion coefficients it has to be mentioned that the real shapes of the polymeric compounds that we have investigated are undetermined, even though the hydrodynamic radius r in the common Stokes–Einstein equation (D = kT/(6πεr)) presumes a spherical structure. Albeit intensities of calculated DOSY signals as well as relative peak intensities in mass spectra – even if the measured structures are very similar – are to treat with caution, in the current case they basically correspond to each other (see Table 3).
| DOSY signal no. (top to bottom) | Diffusion coefficient D/m2 s−1 × 10−10 | Rel. signal intensity from DOSY NMR | Rel. signal intensity from mass spectrometry |
|---|---|---|---|
| 1 | 10.7 ± 0.1 | 1.00 | 1.00 |
| 2 | 11.6 ± 0.1 | 1.39 | 1.68 |
| 3 | 12.8 ± 0.2 | 0.98 | 1.43 |
| 4 | 13.7 ± 0.2 | 2.04 | 1.52 |
The excellent agreement of the results from different analytical methods is particularly pleasing to note. NMR spectroscopy reveals that the ratio of saturated BA moieties and unsaturated moieties is 6.8 to 1. If predominantly unsaturated low molar mass species are contained in the sample – as indicated by ESI-MS-data, a degree of polymerization, DPn, of 7.8 corresponding to Mn(NMR) of 999 g mol−1 is derived from NMR. For the same sample SEC analyses yields an Mn(SEC)of 757 g mol−1, which is associated with a DPn of 5.9. The agreement with the NMR derived number is very good, considering that single detector SEC yields molar masses relative to the polymer standards used for calibration. Formally, the ESI-MS data may be used to calculate a DPN of 4.6 for the unsaturated molar mass macromonomer. The deviation is minor keeping in mind that the ionization is less effective for higher molar masses, thus, underestimating the fraction of these species.
In the past it was generally considered that β scission yields a short radical and long unsaturated species, despite the fact that to the best of our knowledge clear experimental evidence for the formation of long chain macromonomers is lacking. One reason for this assumption may be seen in the fact that only in the last two or three years radical migration in acrylate radical polymerizations is considered to be relevant. Thus, in earlier studies it was expected that backbiting is followed by β scission and consequently trimeric macromonomers should be formed in the scission reaction. Since these species were not identified, e.g., in a supernatant after precipitation or filtration of polymer, it was assumed that polymeric unsaturated species were formed. Clear evidence for radical migration prior to β scission37–39 and the ESI-MS data presented in this contribution strongly suggest that β scission may lead to the formation of low molar mass macromonomeric species, too. As pointed out in the discussion of the batch data preparative SEC of the polymer obtained via BA batch polymerization at 130 °C the fraction of low molar mass species (fractions 6 and 7 in Fig. 3) amounts to 7% of the entire material. The ESI-MS data indicate that the vast majority of these species is unsaturated. The NMR spectra show that these species contain a terminal CH2 moiety. On the contrary, NMR spectra of the high molar mass material show no significant peaks for olefinic protons. Integration of the low molar mass peak of the MMDs obtained for PBA obtained via batch polymerization yields low molar mass fractions of up to 6% for polymerizations at 140 °C with 20 wt% BA.
The data presented here indicate that independent of the reaction conditions (e.g. batch vs. semi-batch, solvent, type of initiator, radical concentration, monomer concentration, temperature) the same low molar mass unsaturated species are identified. Careful ESI-MS and NMR analyses indicate the formation of a large fraction of macromonomers (up to 7% of the mass of the entire sample) that are suggested to originate from migration of midchain radicals and subsequent β scission. To further support the generation of significant amounts of low molar mass macromonomers upon β scission in the future further experiments will be carried out at higher temperatures and low BA concentrations. Ideally, the molar masses will be limited to allow for a detailed analysis of all species formed. Further, modeling of the reactions will be performed to identify whether the occurrence of the large fraction of macromonomers may be achieved with physically meaningful rate coefficients.
Conclusions
BA batch and semi-batch polymerizations were carried out at high temperatures to get more insights into the β scission reaction. SEC analyses of the products clearly indicate that a significant fraction of low molar mass species is formed. Further, the data suggests that the size of these species does not depend on the polymerization conditions, such as type of initiation and solvent choice. Moreover, the polymerization process, batch vs. semi-batch, and the associated differences in monomer concentration do not lead to significant changes in the ESI-MS spectra. In order to safely analyze the β scission products the samples were fractionated via preparative SEC and the fractions of the low molar mass products were analyzed via ESI-MS and advanced NMR spectroscopy. Both analytical tools show that unsaturated species were formed. Further, ESI-MS data indicate that the unsaturated species have chain lengths ranging from 2 to 12. In case of midchain radical formation via intramolecular H transfer from the chain end to the penultimate unit (backbiting) and subsequent β scission trimeric unsaturated species were expected to be generated. Thus, the identification of longer unsaturated species is in line with recent literature reports on radical migration via H transfer along the backbone of the macromonomer. The structure of the unsaturated macromonomer species was confirmed by NMR spectroscopy. ESI-MS literature data also shows that trimeric and higher order macromonomers were obtained.Despite recent progress in understanding high temperature acrylate radical polymerizations, the underlying mechanisms are not yet fully understood. For example, radical migration was reported to occur on every second monomer unit rather on every monomer unit, which should favor the formation of species with odd monomer units. However, in our own work and in literature reports all macromonomers with odd and even chain lengths were identified. The data indicates that the understanding of high temperature acrylate polymerizations is improving largely, however, there are still some questions to be answered.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledge Deutsche Forschungsgemeinschaft (DFG) for co-financing the ESI-MS instrument.References
- N. Ballard and J. M. Asua, Prog. Polym. Sci., 2018, 79, 40 CrossRef CAS.
- C. Barner-Kowollik, M. Buback, M. Egorov, T. Fukuda, A. Goto, G. T. Russell, P. Vana, B. Yamada and P. B. Zetterlund, Prog. Polym. Sci., 2005, 30, 605 CrossRef CAS.
- M. Buback and S. Beuermann, Prog. Polym. Sci., 2002, 27, 191 CrossRef.
- M. Buback, H. Schroeder and H. Kattner, Macromolecules, 2016, 49, 3193 CrossRef CAS.
- B. Wenn and T. Junkers, Macromol. Rapid Commun., 2016, 37, 781 CrossRef CAS PubMed.
- C. Plessis, G. Arzamendi, J. R. Leiza, H. A. S. Schoonbrood, D. Charmot and J. M. Asua, Macromolecules, 2000, 33, 4 CrossRef CAS.
- A. M. van Herk, Macromol. Rapid Commun., 2001, 22, 687 CrossRef CAS.
- J. Barth, M. Buback, C. Barner-Kowollik, T. Junkers and G. T. Russell, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4740 CrossRef CAS.
- J. M. Asua, S. Beuermann, M. Buback, P. Castignolles, B. Charleux, R. G. Gilbert, R. A. Hutchinson, J. R. Leiza, A. N. Nikitin, J.-P. Vairon and A. M. van Herk, Macromol. Chem. Phys., 2004, 205, 2151 CrossRef CAS.
- C. Barner-Kowollik, S. Beuermann, M. Buback, P. Castignolles, B. Charleux, M. L. Coote, R. A. Hutchinson, T. Junkers, I. Lacík, G. T. Russell, M. Stach and A. M. van Herk, Polym. Chem., 2015, 5, 204 RSC.
- T. Junkers, A. Theis, M. Buback, T. P. Davis, M. H. Stenzel, P. Vana and C. Barner-Kowollik, Macromolecules, 2005, 38, 9497 CrossRef CAS.
- J. Barth, M. Buback, P. Hesse and T. Sergeeva, Macromolecules, 2010, 43, 4023 CrossRef CAS.
- M. Buback, P. Hesse, T. Junkers, T. Theis and P. Vana, Aust. J. Chem., 2007, 60, 779 CrossRef CAS.
- R. A. Lyons, J. Hutovic, M. C. Piton, D. I. Christie, P. A. Clay, B. G. Manders, S. H. Kable and R. G. Gilbert, Macromolecules, 1996, 29, 1918 CrossRef CAS.
- S. Beuermann, D. A. Paquet, J. H. McMinn and R. A. Hutchinson, Macromolecules, 1996, 29, 4206 CrossRef CAS.
- D. Cuccato, E. Mavroudakis, M. Dossi and D. Moscatelli, Macromol. Theory Simul., 2013, 22, 127 CrossRef CAS.
- J. Barth, M. Buback, P. Hesse and T. Sergeeva, Macromol. Rapid Commun., 2009, 30, 1969 CrossRef CAS PubMed.
- A. N. Nikitin, R. A. Hutchinson, M. Buback and P. Hesse, Macromolecules, 2007, 40, 8631 CrossRef CAS.
- M. Buback, T. Junkers and M. Müller, Polymer, 2009, 50, 3111 CrossRef CAS.
- N. F. Peck and R. A. Hutchinson, Macromolecules, 2004, 37, 5944 CrossRef.
- W. Wang, A. N. Nikitin and R. A. Hutchinson, Macromol. Rapid Commun., 2009, 23, 2022 Search PubMed.
- J. Chiefari, J. Jeffrey, R. T. A. Mayadunne, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1999, 32, 7700 CrossRef CAS.
- W. Wang and R. A. Hutchinson, Chem. Eng. Technol., 2010, 33, 1745 CrossRef CAS.
- C. Quan, M. Soroush, M. C. Grady, J. E. Hansen and W. J. Simonsick, Macromolecules, 2005, 38, 7622 CrossRef.
- F. S. Rantow, M. Soroush, M. C. Grady and G. A. Kalfas, Polymer, 2006, 47, 1423 CrossRef CAS.
- A. N. Nikitin, R. A. Hutchinson, W. Wang, G. A. Kalfas, J. R. Richards and C. Bruni, Macromol. React. Eng., 2010, 4, 691 CrossRef CAS.
- S. Hamzehlou, N. Ballard, Y. Reyes, A. Aguirre and J. M. Asua, Polym. Chem., 2016, 7, 2069 RSC.
- S. P. S. Koo, T. Junkers and C. Barner-Kowollik, Macromolecules, 2009, 42, 62 CrossRef CAS.
- T. Junkers and C. Barner-Kowollik, Macromol. Theory Simul., 2009, 18, 421 CrossRef CAS.
- J. E. S. Schier, M. Zhang, M. C. Grady and R. A. Hutchinson, Macromol. React. Eng., 2018, 12, 1800008 CrossRef.
- A. B. Vir, Y. W. Marien, P. H. M. Van Steenberge, C. Barner-Kowollik, M. F. Reyniers, G. B. Marin and D. R. D'hooge, React. Chem. Eng., 2018, 3, 807 RSC.
- M. Busch and M. Müller, Macromol. Symp., 2004, 206, 399 CrossRef CAS.
- J. Vandenbergh and T. Junkers, Macromolecules, 2012, 45, 6850 CrossRef CAS.
- J. Vandenbergh and T. Junkers, Macromolecules, 2013, 46, 3324 CrossRef CAS.
- A. Kajiwara, Macromol. Symp., 2007, 248, 50 CrossRef CAS.
- A. Kajiwara, Controlled/Living Radical Polymerization: Progress in ATRP; ACS Symposium Series 1023, ed. K. Matyjaszewski, American Chemical Society: Washington, DC, 2009, ch. 4, pp. 49–59 Search PubMed.
- J. J. Haven, N. Zaquen, M. Rubens and T. Junkers, Macromol. React. Eng., 2017, 11, 1700016 CrossRef.
- A. Kajiwara, Polymer, 2015, 72, 253 CrossRef CAS.
- N. Ballard, A. Veloso and J. M. Asua, Polymers, 2018, 10, 765 CrossRef.
- P. M. V. Steenberge, J. Vandenbergh, T. Junkers, D. R. D'hooge and G. B. Marin, Macromolecules, 2017, 50, 2625 CrossRef.
- J.-B. Lena, M. Deschamps, N. F. Sciortino, S. L. Masters, M. A. Squire and G. T. Russell, Macromol. Chem. Phys., 2018, 219, 17000579 CrossRef.
- M. Buback, Angew. Chem., Int. Ed. Engl., 1991, 30, 641 CrossRef.
- S. Beuermann, M. Buback, C. Schmaltz and F.-D. Kuchta, Macromol. Chem. Phys., 1998, 199, 1209 CrossRef CAS.
- M. Holz and H. Weingärtner, J. Magn. Reson., 1991, 92, 115 CAS.
- J. Lounila, K. Oikarinen, P. Ingman and J. Jokisaari, J. Magn. Reson., Ser. A, 1996, 118, 50 CrossRef CAS.
- M. D. Pelta, G. A. Morris, M. J. Stchedroff and S. J. Hammond, Magn. Reson. Chem., 2002, 40, 147 CrossRef.
- P. Stilbs, Anal. Chem., 1981, 53, 2135–2137 CrossRef CAS.
- M. Nilsson, J. Magn. Reson., 2009, 200, 296–302 CrossRef CAS PubMed . DOSYToolbox, Version 2.5 (12 December 2014), Matlab version for Matlab2014b downloaded on 08 December 2015 from http://nmr.chemistry.manchester.ac.uk..
- Spartan'18, Wavefunction, Inc., Irvine, CA. Available from: http://www.wavefun.com.
- M. Buback, B. Fischer, S. Hinrichs, S. Jauer, J. Meijer and J. Sandmann, Macromol. Chem. Phys., 2007, 208, 772 CrossRef CAS.
- See for example: H. O. Kalinowski, S. Berger and S. Braun, 13C-NMR-Spektroskopie, G. Thieme, Stuttgart, 1984 Search PubMed.
- K. F. Morris and C. S. Johnson Jr., J. Am. Chem. Soc., 1992, 114, 3139 CrossRef CAS.
- E. O. Stejskal and J. E. Tanner, J. Chem. Phys., 1965, 42, 288 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py00103d |
| This journal is © The Royal Society of Chemistry 2019 |