
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultra-low volume oxygen tolerant photoinduced Cu-RDRP†
Evelina
Liarou
 ,
Athina
Anastasaki
,
Richard
Whitfield
,
Carmelo E.
Iacono
,
Georgios
Patias
,
Nikolaos G.
Engelis
,
Arkadios
Marathianos
,
Glen R.
Jones
and
David M.
Haddleton
*
,
Athina
Anastasaki
,
Richard
Whitfield
,
Carmelo E.
Iacono
,
Georgios
Patias
,
Nikolaos G.
Engelis
,
Arkadios
Marathianos
,
Glen R.
Jones
and
David M.
Haddleton
*
University of Warwick, Department of Chemistry, Library Road, Coventry, CV4 7AL, UK. E-mail: d.m.haddleton@warwick.ac.uk
First published on 14th January 2019
Abstract
We introduce the first oxygen tolerant ultra-low volume (as low as 5 μL total reaction volume) photoinduced copper-RDRP of a wide range of hydrophobic, hydrophilic and semi-fluorinated monomers including lauryl and hexyl acrylate, poly(ethylene glycol methyl ether acrylate) and trifluoroethyl (meth)acrylates. In the absence of any external deoxygenation, well-defined homopolymers can be obtained with low dispersity values, high end-group fidelity and near-quantitative conversions. Block copolymers can be efficiently synthesized in a facile manner and the compatibility of the system to larger scale polymerizations (up to 0.5 L) is also demonstrated by judiciously optimizing the reaction conditions. Importantly, the online monitoring of oxygen consumption was also conducted through an oxygen probe and the role of each component is identified and discussed.
Introduction
The development of reversible-deactivation radical polymerization (RDRP) has amplified the field of polymer synthesis providing access to the synthesis of well-defined polymeric materials, in high yields, with different architectures and unlimited functionality.1–12 Typically, all RDRP techniques (e.g. atom-transfer radical polymerization (ATRP),13,14 single electron transfer-living radical polymerization (SET-LRP),8,15 reversible addition–fragmentation chain-transfer polymerization (RAFT),16–18 nitroxide-mediated polymerization (NMP),19,20etc.) rely on intensive deoxygenation techniques including the use of glove box equipment, freeze–pump–thaw cycles or inert gas sparging to reduce (and ideally eliminate) the presence of oxygen. Oxygen is reported to be detrimental for radical polymerizations since it acts as an efficacious radical scavenger, rapidly reacting with carbon-centred radicals (both initiating and propagating radicals), and eventually leading to peroxy radicals and hydroperoxides which are inefficient at reinitiating the polymerization.21–24Although traditional deoxygenation techniques are efficacious for the complete removal of oxygen, they can also be disadvantageous in some cases. For instance, the use of glove box equipment is a sophisticated, albeit expensive and time-consuming approach which also requires extensive training prior to use. Freeze–pump–thaw is another costly and time-consuming deoxygenation method which can also be problematic when proteins or enzymes are involved, leading to denaturation and loss of their secondary structure.25
Finally, inert gas sparging with either argon or nitrogen (so-called “bubbling”) is perhaps the most popular and convenient method to remove oxygen from polymerization solutions. However, sparging can lack reproducibility as it may alter the concentration of volatiles and precludes the use of low sample volumes. In addition, all the existing deoxygenation methods may not be available in all laboratories and/or add complexity to the set-up, thus restricting the accessibility to non-experts.
To mitigate this arduous task of conventional deoxygenation, many groups have exploited the use of enzymes to deoxygenate controlled radical polymerization.26–30 For example, Chapman et al. were the first to use glucose oxidase (GOx) to remove oxygen in a RAFT polymerization where narrow molecular weight distributions were achieved even when the experiments were performed in open vessels.31,32 In the case of ATRP, Matyjaszewski and co-workers subsequently reported the first controlled aqueous ATRP in an open vessel which was coined as “breathing ATRP”.29 In their system, GOx was employed to continuously remove oxygen from the polymerization solution. Alternative strategies to deoxygenate controlled radical polymerizations include the use of various reducing agents such as triethylamine,33,34 hydrazine35,36 and ascorbic acid.37,38
Among the various oxygen tolerant polymerization strategies, photoinduced electron-transfer (PET)-RAFT is arguably one of the most popular.39 The use of light, in particular, has attracted considerable interest due to the polymerizations proceeding under conditions milder than conventional thermal approaches, it is non-invasive and environmentally benign, and gives the potential for spatiotemporal control.19,40–42 Boyer and co-workers first reported the oxygen tolerance of PET-RAFT in which the oxygen can be consumed by either a photocatalyst or a reducing agent. Examples of photocatalysts include (fac-[Ir(ppy)3]),43 zinc tetraphenylporphine (ZnTPP)44 and Eosin Y,38 while ascorbic acid37,38 and triethylamine45 are the most commonly employed reducing agents. The strong reducing ability of the photocatalysts facilitates the oxygen removal prior to polymerization.24,46,47 This is particularly important for low-scale polymerizations and combinatorial synthesis. The ability to conduct polymerizations in extremely low volumes (typically from 20 μL to 500 μL) is an area of growing academic interest as it allows for the inexpensive, fast and high throughput screening of a wide range polymeric materials.24 To date, PET-RAFT is the main controlled radical polymerization method that has been utilized for the high throughput synthesis of a range of polymeric materials.39 However, examples of oxygen tolerant photoinduced ATRP are very limited.33,48–51 To the best of our knowledge, there is no report on the use of any copper-mediated controlled radical polymerization method to afford the controlled synthesis of polymeric materials at very low volumes. This is a significant oversight given the high efficiency of Cu-RDRP to synthesize a wide range of complex polymeric materials with controlled functionality, dispersity and architecture (e.g. stars, multiblocks etc.).1,4,6 In addition, the key role of each component in oxygen consumption during photoinduced copper mediated radical polymerization has not been identified and clarified.
In this contribution, photoinduced Cu-RDRP is conducted in the absence of any external deoxygenation methods. Subsequently, the polymerization of a range of monomers with different polarity is carried out at very low volumes (Scheme 1), and the capability of the system to afford chain extensions is explored. The scalability of this approach to higher volume polymerizations (up to 0.5 L) is also reported. Finally, an oxygen probe for the in situ online monitoring of oxygen consumption during photoinduced controlled radical polymerization is employed, aiming to identify the role of the reaction components in oxygen consumption. Finally, the scalability of our approach to higher volume polymerizations (up to 0.5 L) is explored.
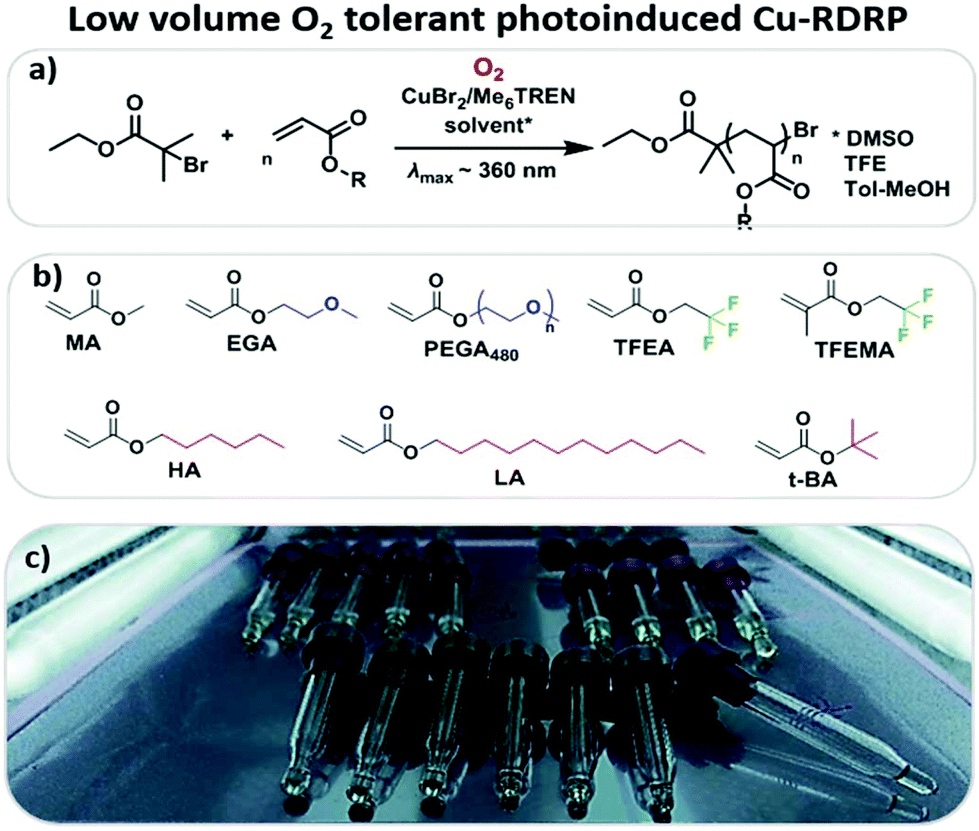 | ||
| Scheme 1 a) Typical reaction scheme for the low volume oxygen tolerant photoinduced-RDRP, (b) different hydrophobic, hydrophilic and semi-fluorinated monomers employed and (c) low volume reaction setup utilizing commercially available glass inserts and a UV nail lamp with broad band λmax ∼ 360 nm. | ||
Results and discussion
Initially, we explored the possibility of photoinduced Cu-RDRP to operate in a controlled manner, in the absence of external deoxygenation. For this study, methyl acrylate (MA) was employed as the monomer, dimethyl sulfoxide (DMSO) as the solvent, tris(2-(dimethylamino)ethyl)-amine (Me6Tren) as the ligand, ethyl α-bromoisobutyrate (EBiB) as the initiator with Cu(II)Br2 as the copper source, following the ratio [MA]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[I]:[CuBr2]:[Me6Tren] = [50]:[1]:[0.02]:[0.12] under a UV “nail lamp” with a broad band λmax ∼ 360 nm. Preliminary experiments were conducted on an 8 mL reaction scale with 50% v/v DMSO. Inspired by our previous investigation using Cu(0) wire-mediated RDRP where the elimination of headspace was the crucial step to achieve an oxygen tolerant system,52 we left the polymerization to commence in a fully filled vial, capped with a lid. In the absence of any type of commonly applied deoxygenation method and without any externally added reducing agents, 98% monomer conversion was achieved, leading to PMA50 with narrow molecular weight distribution (Đ ∼ 1.08) and good agreement between the theoretical (Mn,th = 4400) and the experimental molecular weight (Mn,SEC = 4900) (Fig. 1a). Importantly, the resulting polymer exhibited negligible differences when compared to an identical experiment in which N2 sparging was applied prior to polymerization (Fig. 1b). The slight deviation in the final molecular weights (<10%) can be attributed to the small amount of dissolved oxygen within the polymerization solution which is consistent with previous literature.52 A detailed description of the oxygen consumption is provided in the final section of this manuscript.
:[I]:[CuBr2]:[Me6Tren] = [50]:[1]:[0.02]:[0.12] under a UV “nail lamp” with a broad band λmax ∼ 360 nm. Preliminary experiments were conducted on an 8 mL reaction scale with 50% v/v DMSO. Inspired by our previous investigation using Cu(0) wire-mediated RDRP where the elimination of headspace was the crucial step to achieve an oxygen tolerant system,52 we left the polymerization to commence in a fully filled vial, capped with a lid. In the absence of any type of commonly applied deoxygenation method and without any externally added reducing agents, 98% monomer conversion was achieved, leading to PMA50 with narrow molecular weight distribution (Đ ∼ 1.08) and good agreement between the theoretical (Mn,th = 4400) and the experimental molecular weight (Mn,SEC = 4900) (Fig. 1a). Importantly, the resulting polymer exhibited negligible differences when compared to an identical experiment in which N2 sparging was applied prior to polymerization (Fig. 1b). The slight deviation in the final molecular weights (<10%) can be attributed to the small amount of dissolved oxygen within the polymerization solution which is consistent with previous literature.52 A detailed description of the oxygen consumption is provided in the final section of this manuscript.
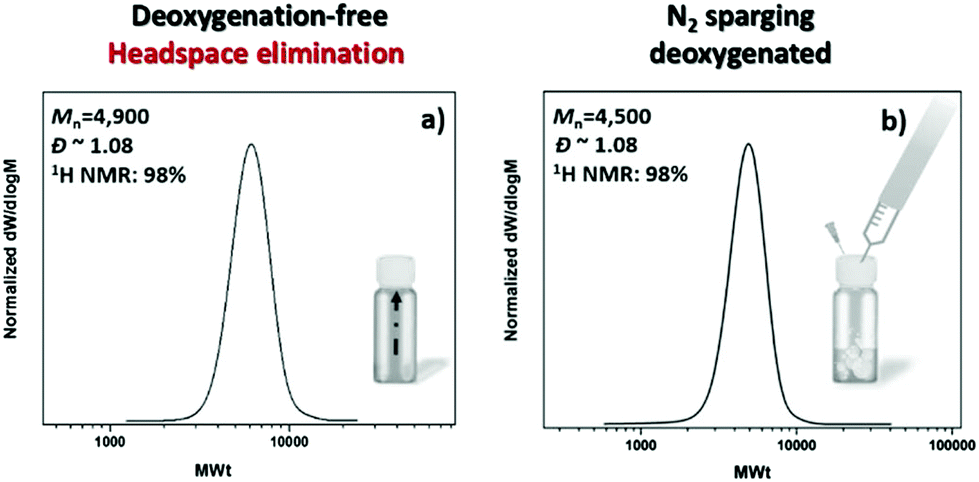 | ||
| Fig. 1 SEC traces of (a) the non-deoxygenated PMA50 and (b) the N2 sparging-deoxygenated PMA50 both synthesized via photoinduced Cu-RDRP with [MA]:[EBiB]:[Cu(II)Br2]:[Me6Tren] = [50]:[1]:[0.02]:[0.12] under a UV nail lamp with broad band λmax ∼ 360 nm. | ||
The possibility of conducting controlled polymerizations in the presence of oxygen can potentially enable the high throughput synthesis of a wide range of polymers at low reaction volumes. In order to test this hypothesis, commercially available vial inserts with a full capacity of 200 μL were utilized and all the reactions were sealed with lids (Fig. S1†). To eliminate the headspace, the vial insert was initially fully filled with the reaction solution (200 μL/200 μL) (Fig. 2a). Under these conditions, PMA with degree of polymerization (DP) 50 was targeted, yielding well-defined PMA, with near-quantitative monomer conversion (96%, Fig. S2†), low dispersity and symmetrical size exclusion chromatography (SEC) traces (Đ ∼ 1.07, Mn,exp ∼ 5200).
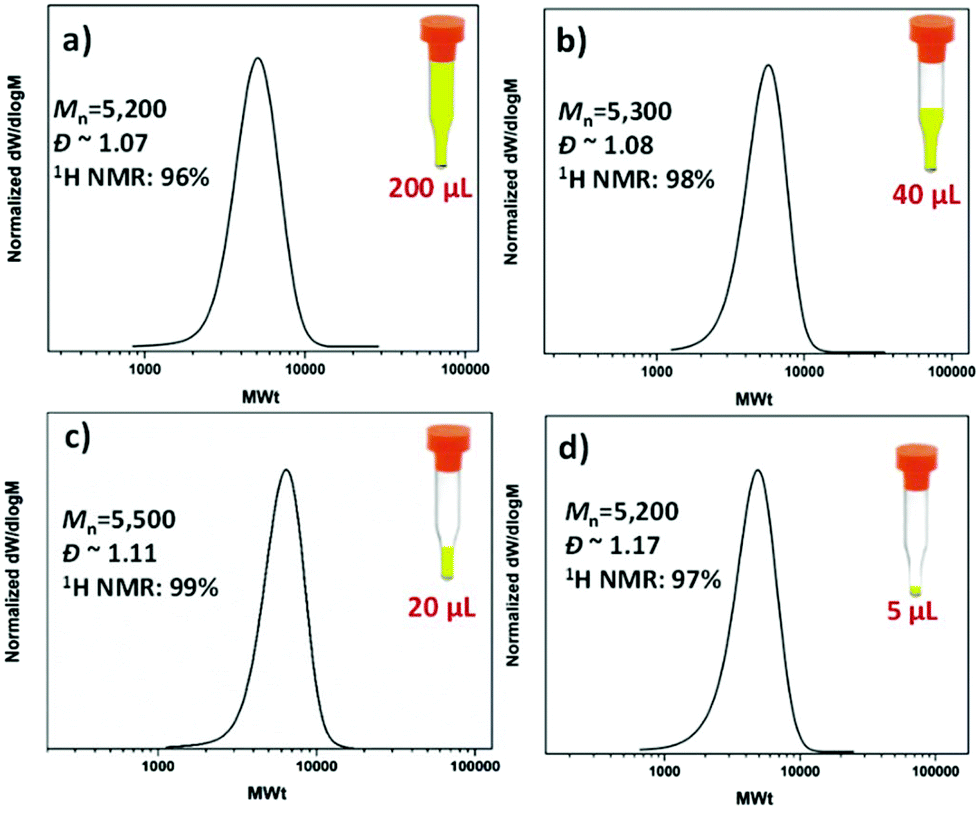 | ||
| Fig. 2 SEC traces for PMA50 with (a) 200 μL (b) 40 μL, (c) 20 μL and (d) 5 μL reaction volume synthesized via oxygen tolerant photoinduced Cu-RDRP with [MA]:[EBiB]:[Cu(II)Br2]:[Me6Tren] = [50]:[1]:[0.02]:[0.12] using a UV lamp with broad band λmax ∼ 360 nm. | ||
Encouraged by these initial findings, we then lowered the reaction volume from 200 μL to 60, 40, 20, 10 and 5 μL (Fig. 2(a–d) and Fig. S3†). For these experiments, we maintained the 200 μL commercially available vial inserts, in which the headspace was increased from zero to 140, 160, 180, 190 and 195 μL, respectively. Despite the increase of the headspace, all polymerizations reached near-quantitative conversions (>96%) without compromising the control over the molecular weight and the molecular weight distributions (Đ ∼ 1.1). In all cases, comparable initiator efficiencies were observed (Mn,SEC ∼ 5000–5500) indicating that in DMSO, the headspace has only negligible effects, if any, on the targeted molecular weight (Fig. 2 and Table 1). This is in contrast with other copper-mediated approaches where the headspace has been reported to play an important role.52 This may be attributed to the absence of stirring in this system which limits the diffusion of oxygen into the polymerization solution. It should also be highlighted that even at ultra-low volumes (i.e. 5 μL), the polymerization proceeds efficiently, although with slightly higher dispersity values (Đ ∼ 1.17). To the best of our knowledge, this is the lowest scale reported for controlled radical polymerization to date. The low volume experiments were also conducted with ∼300 ppm of copper. Importantly, we could further reduce the concentration to 37 ppm without significantly compromising the control over the molecular weight distributions (Fig. S4†).
| Scale (μL) | Headspace (μL) | Conversion 1H NMR (%) | M n, theory (g mol−1) | M n,SEC | Đ |
|---|---|---|---|---|---|
|
a In all polymerizations, the volume ratio of monomer to solvent was maintained 1:1 and conversion was calculated via1H NMR.
b Determined by THF SEC analysis and expressed as molecular weight equivalents to PMMA narrow molecular weight standards.
|
|||||
| 200 | 0 | 96 | 4300 | 5200 | 1.07 |
| 60 | 140 | 98 | 4400 | 5400 | 1.08 |
| 40 | 160 | 98 | 4400 | 5300 | 1.08 |
| 20 | 180 | 99 | 4500 | 5500 | 1.11 |
| 10 | 190 | 99 | 4500 | 5000 | 1.11 |
| 5 | 195 | 97 | 4400 | 5200 | 1.17 |
To explore the extent of control over higher molar masses, a range of different degrees of polymerization (100–400) were investigated. All polymerizations were performed on a 60 μL scale. Well-defined PMAs up to DP = 400 were obtained with final Mn,SEC = 38500 and a dispersity of 1.19 at high monomer conversions (>82%) (Fig. 3 and Table 2). Unfortunately, targeting higher DPs (i.e. 600 and 800) resulted in no conversion, even when the reaction was left to proceed overnight. This was attributed to the low initiator and catalyst concentrations and will be further discussed in the mechanistic section of this paper. We were then interested in expanding the scope of the low volume photoinduced Cu-RDRP to a wide range of hydrophobic, hydrophilic and semi-fluorinated monomers. Given the tolerance of the methodology in the presence of large headspace when DMSO was used as a solvent, ethylene glycol methyl ether acrylate (EGA) was polymerized efficiently at 10 μL scale with Mn,SEC = 7300 and Đ ∼ 1.17 at 99% conversion (Fig. 4a, Fig. S5† and Table 3). Polymerization of the hydrophilic poly(ethylene glycol) methyl ethyl acrylate (PEGA480)20 also afforded a well-defined polymer at high conversion (>99%) with narrow molecular weight distributions (Đ ∼ 1.18) (Fig. 4b, Fig. S6† and Table 3). These results further highlight the versatility of DMSO to enable the synthesis of controlled polymers at very high conversions and ultra-low reaction volumes,53 even in the presence of headspace.
 | ||
| Fig. 3 SEC traces for the low-scale oxygen-tolerant photoinduced Cu-RDRP of MA with targeted DPs 50–400 and [MA]:[EBiB]:[Cu(II)Br2]:[Me6Tren] = [DPn]:[1]:[0.02]:[0.12] under a UV lamp with broad band λmax ∼ 360 nm. | ||
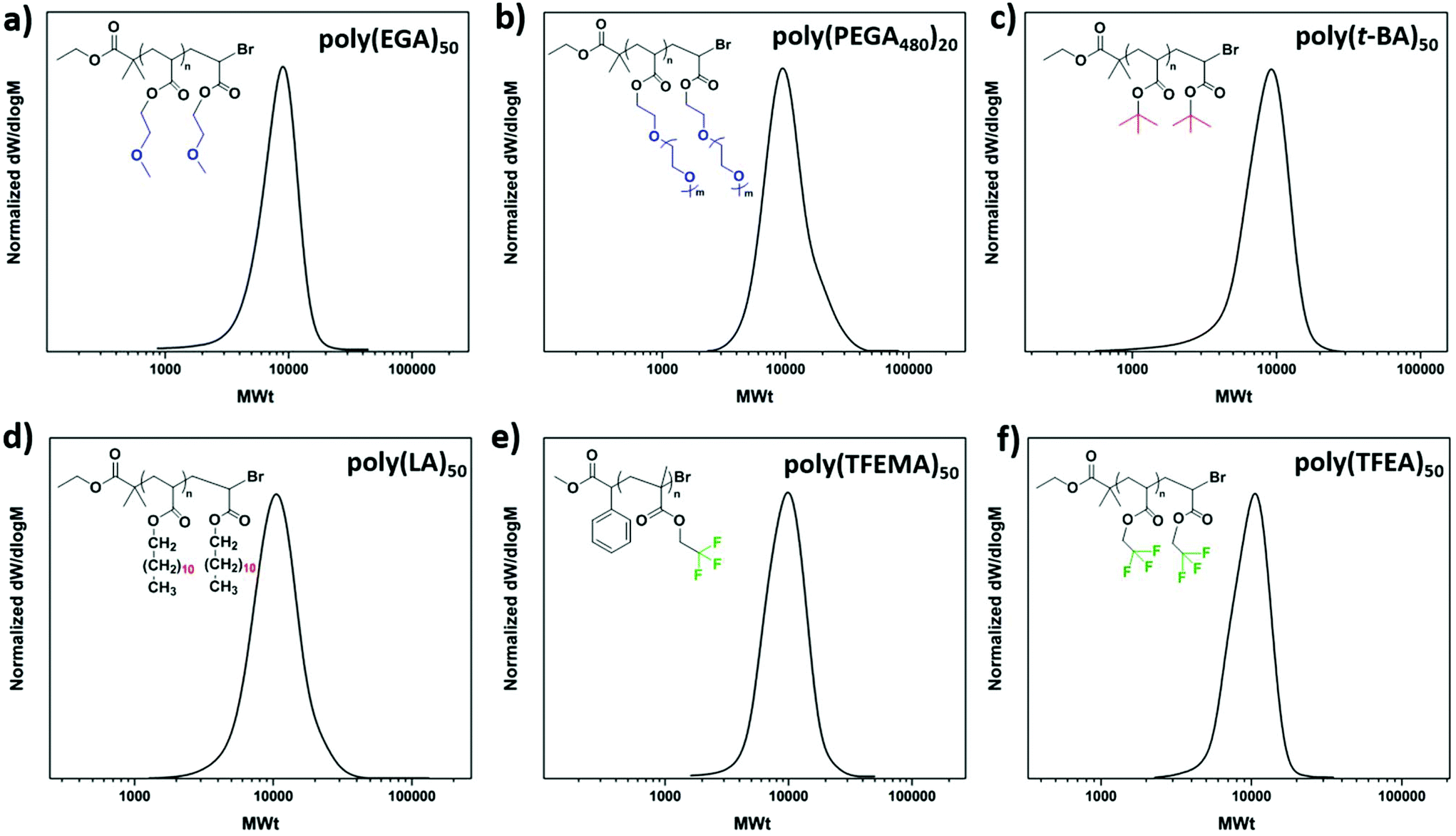 | ||
| Fig. 4 SEC traces for (a) poly(EGA)50, (b) poly(PEGA480)20, (c) poly(t-BA)50, (d) poly(LA)50, (e) poly(TFEMA)50 and (f) poly(TFEA)50 obtained through low-volume deoxygenation-free photoinduced Cu-RDRP. | ||
| DP | Conversion 1H NMR (%) | M n, th. (g mol−1) | M n,SEC | Đ |
|---|---|---|---|---|
|
a In all polymerizations the volume ratio of monomer to solvent was maintained 1:1 and conversion was calculated via1H NMR.
b Determined by THF SEC analysis and expressed as molecular weight equivalents to PMMA narrow molecular weight standards.
|
||||
| 50 | 98 | 4400 | 5400 | 1.08 |
| 100 | 99 | 8700 | 11000 |
1.18 |
| 200 | 92 | 16000 |
23000 |
1.19 |
| 400 | 82 | 24800 |
38500 |
1.19 |
| Polymer | Scale (μL) | Solvent | DP | Conversion 1H NMR, % | M n, theory (g mol−1) | M n,SEC | Đ |
|---|---|---|---|---|---|---|---|
|
a In all polymerizations the volume ratio of monomer to solvent was maintained 1:1 and conversion was calculated via1H NMR.
b Determined by THF SEC analysis and expressed as molecular weight equivalents to PMMA narrow molecular weight standards.
|
|||||||
| PMA | 10 | DMSO | 50 | 99 | 4400 | 5000 | 1.11 |
| P(PEGA480) | 10 | DMSO | 20 | 99 | 9500 | 9300 | 1.18 |
| P(EGA) | 10 | DMSO | 50 | 99 | 6700 | 7300 | 1.17 |
| PLA | 100 | Tol-MeOH | 50 | 75 | 9200 | 9400 | 1.19 |
| P(t-BA) | 100 | Tol-MeOH | 50 | 97 | 6400 | 7000 | 1.2 |
| PHA | 100 | TFE | 50 | 93 | 7400 | 7600 | 1.19 |
| PTFEA | 100 | TFE | 50 | 99 | 7900 | 8900 | 1.08 |
| PTFEMA | 10 | TFE | 50 | 93 | 8100 | 8400 | 1.15 |
Owing to the diverse properties of hydrophobic54–57 and semi-fluorinated58,59 materials we were then interested in polymerizing tert-butyl (t-BA), hexyl (HA, C6), lauryl (LA, C12) and trifluoroethyl (meth)acrylates (TFEA and TFEMA). However, DMSO has been reported as an unsuitable solvent leading to insoluble final polymeric materials and subsequent loss of control.60,61 As an alternative, the polymerization of t-BA and LA was attempted in mixtures of toluene/MeOH (4:1), where a small amount of MeOH is necessary to facilitate the complete solubilization of the catalyst, while toluene is needed to dissolve the monomers and the resulting polymers. Unfortunately, in the presence of a large headspace (10 μL reaction scale in a 200 μL vial insert) no polymerization was observed within 24 h for either t-BA or LA. Moreover, when the polymerization of MA was conducted in the same solvent system, similarly to the other monomers, no monomer conversion occurred. However, when the identical experiments were performed upon elimination of the headspace to almost zero, the polymerization of t-BA (Fig. 4c, Fig. S7† and Table 3), LA (Fig. 4d and Table 3) and MA (Fig. S8 and Table S1†) occurred in a controlled manner, exhibiting narrow molecular weight distributions. In a similar vein, the polymerization of HA and TFEA, as well as MA in the presence of a large headspace and in trifluroethanol (TFE) was unsuccessful, and no polymerization was observed. On the contrary, when the low scale polymerizations took place in full vial inserts, control over the polymerization was maintained leading to PHA50 (Fig. S9a, b† and Table 3), PMA50 (Fig. S10 and Table S1†) and PTFEA50 (Fig. 4f, Fig. S11† and Table 3) with low dispersities. Interestingly, although the polymerization of TFEA was unsuccessful in the presence of headspace, the methacrylate analogue (TFEMA) was polymerized with ∼90 μL headspace, yielding a well-defined PTFEMA50 with Mn,SEC = 8400 and Đ ∼ 1.15 (Fig. 4e, Fig. S12† and Table 3).
The ability of the semi-fluorinated methacrylate to undergo polymerization even in the presence of significant headspace, was attributed to the higher degree of oxygen tolerance for the methacrylates compared to acrylates.62 Overall, these experiments suggest that both toluene/MeOH mixture and TFE possess limited headspace tolerance. The limited headspace tolerance of TFE might not be surprising given the capability of fluorinated and semi-fluorinated solvents to act as oxygen carriers.63 Nevertheless, by eliminating the headspace, the polymerizations proceeded in a controlled manner in all attempted solvents allowing for the low volume polymerization of a wide range of materials at high conversions.
To investigate the extent of end-group fidelity for the photoinduced low volume RDRP experiments, matrix-assisted laser desorption/ionization time of flight (MALDI-ToF) mass spectrometry was employed to analyze PMA with targeted DP = 25. A predominant polymer peak distribution was identified corresponding to polymer chains initiated by EBiB and terminated by the desired bromine end-group (Fig. 5a).
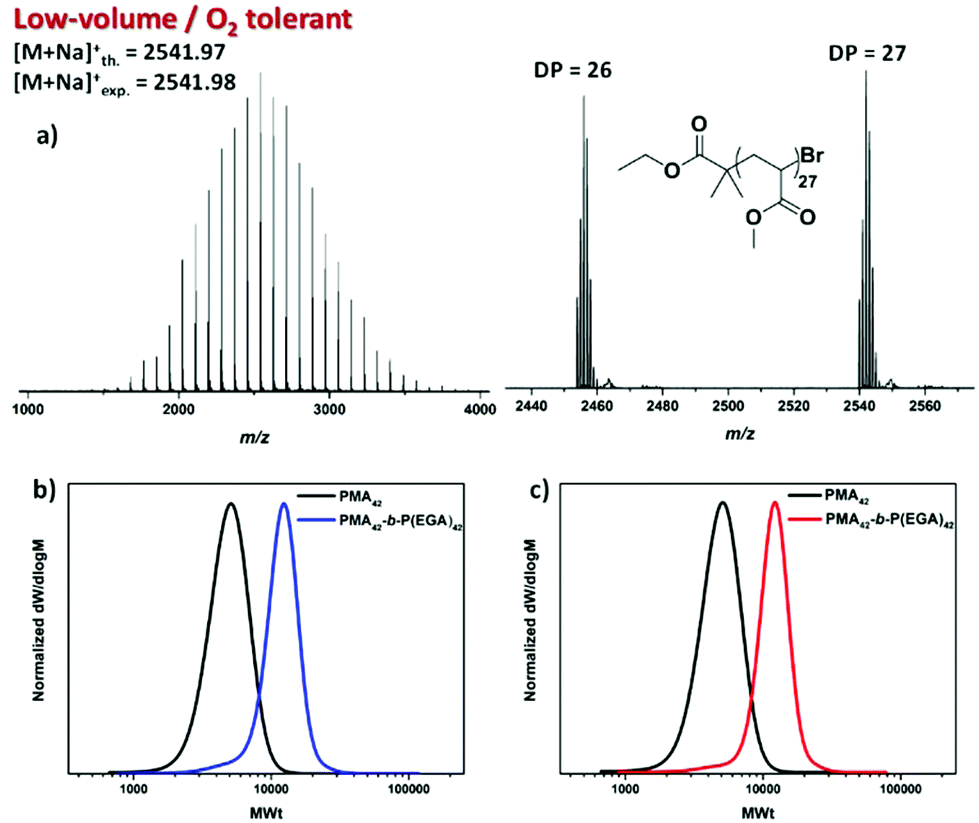 | ||
| Fig. 5 (a) MALDI-ToF spectrum for the deoxygenation-free 10 μL PMA25 revealing the predominant single peak distribution and SEC traces for the diblocks P(MA)42-b-P(EGA)42 on a (b) 200 μL scale (absence of headspace) and (c) 20 μL scale (∼180 μL of headspace) obtained after chain extension of the isolated PMA42 macroinitiator via oxygen tolerant photoinduced Cu-RDRP. | ||
This suggests that active end-groups can be maintained during polymerization which prompted us to attempt in situ chain extensions. However, upon chain extending PMA with an aliquot of EGA, inconsistent results were obtained. In particular, the conversion of the second block was either minimal, if any, (0–10%) or exhibited a significant tailing to low molecular weights indicating severe termination events (data not shown). This was rather surprising since both MALDI-ToF and 1H NMR showed bromine terminated polymer chains prior to chain extension. It was thus hypothesized that the additional oxygen (either dissolved in the second aliquot of monomer and/or added upon removal of the lid) introduced to the system via the addition of the second monomer was detrimental for preserving high end-group fidelity. To confirm whether this is the case, PMA with DP = 42 was synthesized and isolated prior to chain extension (Fig. S13†). Upon re-subjecting the PMA42 macroinitiator to irradiation in a fully filled vial insert, in the presence of EGA, well-defined block copolymers of P(MA)42-b-P(EGA)42 could be obtained with the molecular weight distribution shifting to higher molecular weights and negligible tailing observed. Importantly, the final dispersity was ∼1.15 and the control over the molecular weight distributions was not compromised (Mn,SEC = 10200) even at near quantitative conversions (99%) (Fig. 5b and Fig. S14†). An identical chain extension experiment was also performed in the presence of significant headspace (20 μL per 200 μL). Despite the extent of the headspace, a complete shift of the macroinitiator was evident through SEC analysis (Mn,SEC = 10900) yielding diblock copolymers with low dispersity value (Đ ∼ 1.12) and high conversion (99%) (Fig. 5c). Thus, in DMSO, successful chain extensions with or without headspace can be reproducibly achieved by isolating the macroinitiator. These results indicate that, indeed a high proportion of ω-bromo functionality can be maintained and that the unsuccessful in situ chain extensions can be explained by the inclusion of additional oxygen through the addition of the second monomer.
To explore the robustness of our oxygen tolerant photoinduced Cu-RDRP for the synthesis of well-defined materials on a high-multigram scale, we initially attempted to scale up the polymerization of MA in DMSO (100 ml scale, 50% solids) utilizing a custom made UV box equipped with light bulbs with λmax ∼ 360 nm (Fig. S15†). However, in all our attempts, the septa/lid “popped off” leading to poor monomer conversions and a slightly brown colour attributed to the oxidized catalyst, as a result of the continuous exposure to oxygen. Due to this exothermic nature of the reaction, an exit needle was employed to release the increase in pressure. Although the monomer conversion increased, very high conversions were not achieved and the dispersity was significantly higher (Đ ∼ 1.3) when compared to identical experiments at lower volumes. We then envisaged that TFE might be a better alternative given the high-end-group fidelity of polymers synthesized in TFE as well as the significant thermal stability provided by this solvent.60,61 As a result, the polymerization was successfully conducted at 100 mL (Fig. 6c and Table S2†) and 250 mL (Fig. S16†) at high conversions (91–94%), exhibiting similar initiator efficiency with the lower volume polymerizations and low dispersity values (Đ ∼ 1.12). We also managed to perform the polymerization on a 0.5 L scale yielding well-defined PMA with narrow molecular weight distributions (Đ ∼ 1.19) and high conversions (91%), thus further highlighting the versatility of the reported approach (Fig. 6d).
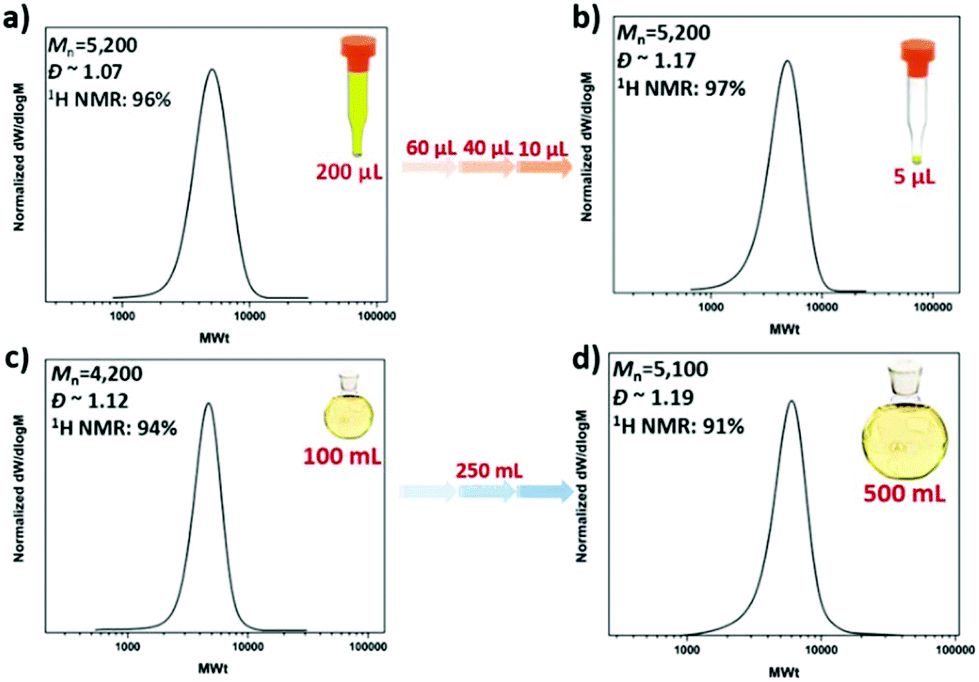 | ||
| Fig. 6 SEC traces for (a),( b) the low volume PMA50 and (c),( d) the high scale PMA50 synthesized via oxygen tolerant photoinduced Cu-RDRP under a UV lamp with broad band λmax ∼ 360 nm. | ||
Preliminary insights into the oxygen consumption mechanism
In order to investigate the fate of the dissolved oxygen in the photoinduced Cu-RDRP system an oxygen probe was employed for the in situ [O2] monitoring. Under conditions identical to our polymerization ([M]:[I]:[Cu(II)Br2]:[L] = [50]:[1]:[0.02]:[0.12]) and upon UV irradiation (λmax ∼ 360 nm), complete oxygen consumption was observed in ∼5 min (Solution 1) (Fig. 7a). This rapid oxygen consumption can be potentially attributed to the reduction of Cu(II)Br2 (by an excess of free amine) to active species (Cu(I) and/or Cu(0)). The active species can then consume oxygen via two different pathways. In particular, the active species can either react directly with oxygen or abstract the bromine from the initiator leading to the generation of initiating radicals which can then react with oxygen. To investigate this hypothesis, we investigated the role of each component including the copper source (Cu(II)Br2), the ligand (Me6Tren) and the initiator (EBiB). Initially, the same polymerization mixture (Solution 1) was investigated in the absence of Cu(II)Br2. Interestingly, when only initiator and ligand were present, the oxygen consumption was decelerated to ∼45 min, thus verifying the importance of Cu(II)Br2 to enhance the rate of oxygen consumption. In addition, experiments where the concentration of Cu(II)Br2 was altered were also performed (Fig. 7b). At very low Cu(II)Br2 concentration (0.001 equiv. with respect to initiator), the oxygen consumption was completed after ∼20 min. This is attributed to the slow generation of active species which can then lead to oxygen consumption. However, at higher amounts of Cu(II)Br2 (0.005–0.05 equiv.) little, if any, differences in the rate of oxygen consumption were observed (∼6 min). This suggests that upon sufficient generation of active species, the oxygen consumption can proceed at the maximum rate. It should also be noted that when the concentration of Cu(II)Br2 either exceeded or equalled the ligand concentration, no oxygen consumption was observed. This is to be expected as according to the literature, excess of free amine is required to mediate the reduction of the copper complex.64,65
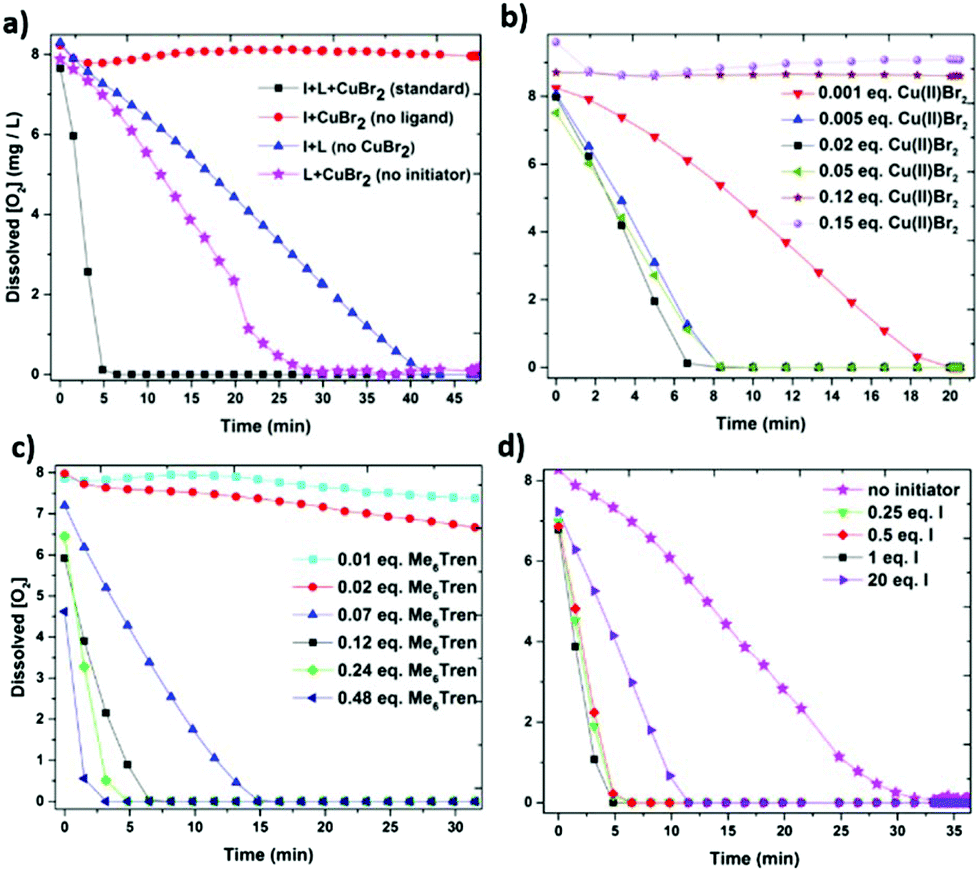 | ||
| Fig. 7 Graphical illustration of the dissolved oxygen consumption for (a) the standard system (solution 1) and the role of the polymerization components, and the effect of different equivalents of (b) the copper source (Cu(II)Br2), (c) the ligand (Me6Tren) and (d) the initiator (EBiB) on oxygen consumption. All the solutions were exposed under a UV lamp with broad band λmax ∼ 360 nm. | ||
We subsequently sought to study the importance of ligand (Fig. 7c). In the absence of ligand, no oxygen consumption was evident within a 60 minutes scale, suggesting lack of generated radicals under these conditions. A similar trend was observed when less ligand equivalents than Cu(II)Br2 (0.01 and 0.02 with respect to initiator) were employed validating our previous results, in which an excess of ligand is essential to consume oxygen. However, at higher ligand loadings (0.07 w.r.t initiator), oxygen was fully consumed in ∼15 min. A further gradual increase of the ligand concentration led to even faster oxygen consumption (as fast as ∼3 min). It can thus be concluded that (i) an excess of ligand is necessary to consume the oxygen and (ii) more ligand leads to the generation of more active species which can then directly or indirectly consume the oxygen. Finally, in situ [O2] monitoring of Solution 1 in the absence of initiator was also conducted. When only Cu(II)Br2 and ligand were present, the second fastest oxygen consumption rate (with the first one being the Solution 1 with all the components included) was monitored at ∼27 min (Fig. 7a). This observation verifies our initial hypothesis that the copper complex is primarily responsible for the oxygen consumption. Moreover, by altering the initiator equivalents (Fig. 7d), it can be concluded that when sufficient amount is present, the oxygen consumption remains equally fast (∼5 min) regardless of the initiator concentration (0.25, 0.5 and 1 equivalents of initiator). This is reasonable as the complex is the main factor that determines the oxygen consumption and as a result, the same amount of active species generated will only react with a constant amount of initiator, even if further excess of initiator is available. Interestingly, at extremely high initiator loadings (20 equiv. or ∼25% v/v), slower oxygen consumption was observed (∼12 min) which is likely due to the change of the reaction medium.
In summary, from these preliminary experiments it can be inferred that the combination of Cu(II)Br2, ligand and initiator synergistically contribute to the oxygen consumption (∼5 min). Upon exclusion of initiator, the second fastest oxygen consumption is being monitored (∼27 min) which can be predominantly attributed to the reduction of the copper complex into active species. Therefore, the presence of initiator is important to accelerate the rate of consumption suggesting that the initiating radicals react with oxygen more rapidly than the active species. At the same time, in the absence of Cu(II)Br2, an even slower oxygen consumption is observed (∼45 min) which implies that the initiator and the ligand in the absence of copper, are less significant than the complex for the process of oxygen consumption. Although slower, this oxygen consumption can be attributed to either the light-induced C–Br bond scission of the initiator (generating initiating/propagating radicals) or by the formation of a radical cation from the ligand upon irradiation.65 Finally, since no oxygen consumption is evident in the absence of ligand, it is hypothesized that either the C–Br cleavage does not occur at large extent or that the presence of the deactivator is somehow hindering the cleavage even in the absence of ligand (i.e. by delivering the bromine back to the initiator). However, further experiments are required to verify this hypothesis and a more detailed analysis of the mechanism will be the subject of a forthcoming publication.
Conclusions
In summary, we report the first low volume oxygen tolerant photoinduced Cu-RDRP method independent of complex oxygen scavengers or external deoxygenation methods. Good control over the polymerization and high-end group fidelity were maintained, yielding well-defined homo- and block co-polymers with a range of monomers with different hydrophobicity, as well as, semi-fluorinated (meth)acrylates having been successfully polymerized. The facile and efficient nature of this methodology is also applicable to high scale polymerizations (up to 0.5 L). Furthermore, the employment of an oxygen probe enabled us to investigate the role of the polymerization components into oxygen consumption. The proposed methodology renders the oxygen-tolerant photoinduced Cu-RDRP a multi-applicable strategy for the synthesis of a range of materials, on different scales with undemanding setup.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support from the University of Warwick (E. L., R. W., C. E. I., G. P., N. G. E., A. M., G. R. J.), Syngenta (A. M.) and Lubrizol (G. P.) is gratefully acknowledged. R. W. acknowledges the Institute of Advanced Study (University of Warwick) for postdoctoral funding. A.A. acknowledges the Global Marie Curie Fellowship (BINAMA 705041) for financial support. We thank Dr David Fox for his valuable input. We are also grateful for the Polymer Characterization RTP and Dr Daniel Lester (University of Warwick) for providing use of SEC equipment.References
- A. Anastasaki, V. Nikolaou and D. M. Haddleton, Polym. Chem., 2016, 7, 1002–1026 RSC.
- A. Anastasaki, J. Willenbacher, C. Fleischmann, W. R. Gutekunst and C. J. Hawker, Polym. Chem., 2017, 8, 689–697 RSC.
- C. Boyer, N. A. Corrigan, K. Jung, D. Nguyen, T.-K. Nguyen, N. N. M. Adnan, S. Oliver, S. Shanmugam and J. Yeow, Chem. Rev., 2016, 116, 1803–1949 CrossRef CAS PubMed.
- G. R. Jones, A. Anastasaki, R. Whitfield, N. Engelis, E. Liarou and D. M. Haddleton, Angew. Chem., Int. Ed. Engl., 2018, 57, 10468–10482 CrossRef CAS PubMed.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- K. Matyjaszewski, Adv. Mater., 2018, 30, 1706441 CrossRef PubMed.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069–5119 CrossRef CAS PubMed.
- E. Baeten, J. J. Haven and T. Junkers, Polym. Chem., 2017, 8, 3815–3824 RSC.
- J. De Neve, J. J. Haven, L. Maes and T. Junkers, Polym. Chem., 2018, 9, 4692–4705 RSC.
- R. Aksakal, M. Resmini and C. R. Becer, Polym. Chem., 2016, 7, 171–175 RSC.
- Y. Abdouni, G. Yilmaz and C. R. Becer, Macromol. Rapid Commun., 2017, 38, 1700212 CrossRef PubMed.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS PubMed.
- M. R. Hill, R. N. Carmean and B. S. Sumerlin, Macromolecules, 2015, 48, 5459–5469 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- L. Barner, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2007, 28, 539–559 CrossRef CAS.
- X. Pan, M. A. Tasdelen, J. Laun, T. Junkers, Y. Yagci and K. Matyjaszewski, Prog. Polym. Sci., 2016, 62, 73–125 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed.
- V. A. Bhanu and K. Kishore, Chem. Rev., 1991, 91, 99–117 CrossRef CAS.
- S. C. Ligon, B. Husár, H. Wutzel, R. Holman and R. Liska, Chem. Rev., 2014, 114, 557–589 CrossRef CAS PubMed.
- R. Shenoy and C. N. Bowman, Macromolecules, 2010, 43, 7964–7970 CrossRef CAS.
- J. Yeow, R. Chapman, A. J. Gormley and C. Boyer, Chem. Soc. Rev., 2018, 47, 4357–4387 RSC.
- T. Arakawa, S. J. Prestrelski, W. C. Kenney and J. F. Carpenter, Adv. Drug Delivery Rev., 2001, 46, 307–326 CrossRef CAS PubMed.
- Y. Wang, L. Fu and K. Matyjaszewski, ACS Macro Lett., 2018, 7, 1317–1321 CrossRef CAS.
- J. Tan, D. Liu, Y. Bai, C. Huang, X. Li, J. He, Q. Xu and L. Zhang, Macromolecules, 2017, 50, 5798–5806 CrossRef CAS.
- A. E. Enciso, L. Fu, S. Lathwal, M. Olszewski, Z. Wang, S. R. Das, A. J. Russell and K. Matyjaszewski, Angew. Chem., Int. Ed., 2018, 57, 16157–16161 CrossRef CAS PubMed.
- A. E. Enciso, L. Fu, A. J. Russell and K. Matyjaszewski, Angew. Chem., Int. Ed., 2017, 57, 933–936 CrossRef PubMed.
- Y. Lv, Z. Liu, A. Zhu and Z. An, J. Polym. Sci., Part A: Polym. Chem., 2016, 55, 164–174 CrossRef.
- R. Chapman, A. J. Gormley, K.-L. Herpoldt and M. M. Stevens, Macromolecules, 2014, 47, 8541–8547 CrossRef CAS.
- R. Chapman, A. J. Gormley, M. H. Stenzel and M. M. Stevens, Angew. Chem., Int. Ed. Engl., 2016, 55, 4500–4503 CrossRef CAS PubMed.
- Q. Yang, J. Lalevée and J. Poly, Macromolecules, 2016, 49, 7653–7666 CrossRef CAS.
- I.-H. Lee, E. H. Discekici, A. Anastasaki, J. R. de Alaniz and C. J. Hawker, Polym. Chem., 2017, 8, 3351–3356 RSC.
- S. Fleischmann, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1190–1196 CrossRef CAS.
- S. Fleischmann and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2243–2250 CrossRef CAS.
- G. Ng, J. Yeow, J. Xu and C. Boyer, Polym. Chem., 2017, 8, 2841–2851 RSC.
- J. Yeow, R. Chapman, J. Xu and C. Boyer, Polym. Chem., 2017, 8, 5012–5022 RSC.
- G. Ng, J. Yeow, R. Chapman, N. Isahak, E. Wolvetang, J. J. Cooper-White and C. Boyer, Macromolecules, 2018, 51, 7600–7607 CrossRef CAS.
- A. Anastasaki, V. Nikolaou, A. Simula, J. Godfrey, M. Li, G. Nurumbetov, P. Wilson and D. M. Haddleton, Macromolecules, 2014, 47, 3852–3859 CrossRef CAS.
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed.
- N. Corrigan, J. Yeow, P. Judzewitsch, J. Xu and C. Boyer, Angew. Chem., Int. Ed., 2018 DOI:10.1002/anie.201805473.
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed.
- J. Yeow, S. Joshi, R. Chapman and C. Boyer, Angew. Chem., Int. Ed. Engl., 2018, 57, 10102–10106 CrossRef CAS PubMed.
- J. Xu, S. Shanmugam, H. T. Duong and C. Boyer, Polym. Chem., 2015, 6, 5615–5624 RSC.
- S. Shanmugam, J. Xu and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9174–9185 CrossRef CAS PubMed.
- Q. Fu, K. Xie, T. G. McKenzie and G. G. Qiao, Polym. Chem., 2017, 8, 1519–1526 RSC.
- K. Borská, D. Moravčíková and J. Mosnáček, Macromol. Rapid Commun., 2017, 38, 1600639 CrossRef PubMed.
- Z. Huang, C. Feng, H. Guo and X. Huang, Polym. Chem., 2016, 7, 3034–3045 RSC.
- J. Mosnáček, A. Eckstein-Andicsová and K. Borská, Polym. Chem., 2015, 6, 2523–2530 RSC.
- X. Pan, S. Lathwal, S. Mack, J. Yan, S. R. Das and K. Matyjaszewski, Angew. Chem., Int. Ed., 2017, 56, 2740–2743 CrossRef CAS PubMed.
- E. Liarou, R. Whitfield, A. Anastasaki, N. G. Engelis, G. R. Jones, K. Velonia and D. M. Haddleton, Angew. Chem., Int. Ed., 2018, 57, 8998–9002 CrossRef CAS PubMed.
- G. R. Jones, R. Whitfield, A. Anastasaki, N. Risangud, A. Simula, D. J. Keddie and D. M. Haddleton, Polym. Chem., 2018, 9, 2382–2388 RSC.
- R. B. Smail, R. L. Jezorek, J. Lejnieks, M. Enayati, S. Grama, M. J. Monteiro and V. Percec, Polym. Chem., 2017, 8, 3102–3123 RSC.
- L. Voorhaar, S. Wallyn, F. E. Du Prez and R. Hoogenboom, Polym. Chem., 2014, 5, 4268–4276 RSC.
- M. Enayati, R. L. Jezorek, M. J. Monteiro and V. Percec, Polym. Chem., 2016, 7, 3608–3621 RSC.
- R. Whitfield, A. Anastasaki, G. R. Jones and D. M. Haddleton, Polym. Chem., 2018, 9, 4395–4403 RSC.
- E. H. Discekici, A. Anastasaki, R. Kaminker, J. Willenbacher, N. P. Truong, C. Fleischmann, B. Oschmann, D. J. Lunn, J. Read de Alaniz, T. P. Davis, C. M. Bates and C. J. Hawker, J. Am. Chem. Soc., 2017, 139, 5939–5945 CrossRef CAS PubMed.
- S. R. Samanta, R. Cai and V. Percec, Polym. Chem., 2014, 5, 5479–5491 RSC.
- V. Nikolaou, A. Anastasaki, F. Brandford-Adams, R. Whitfield, G. R. Jones, G. Nurumbetov and D. M. Haddleton, Polym. Chem., 2016, 7, 191–197 RSC.
- A. Anastasaki, B. Oschmann, J. Willenbacher, A. Melker, M. H. C. Van Son, N. P. Truong, M. W. Schulze, E. H. Discekici, A. J. McGrath, T. P. Davis, C. M. Bates and C. J. Hawker, Angew. Chem., Int. Ed., 2017, 56, 14483–14487 CrossRef CAS PubMed.
- T. Y. Lee, C. A. Guymon, E. S. Jönsson and C. E. Hoyle, Polymer, 2004, 45, 6155–6162 CrossRef CAS.
- J. G. Riess, Tetrahedron, 2002, 58, 4113–4131 CrossRef CAS.
- T. G. Ribelli, D. Konkolewicz, S. Bernhard and K. Matyjaszewski, J. Am. Chem. Soc., 2014, 136, 13303–13312 CrossRef CAS PubMed.
- E. Frick, A. Anastasaki, D. M. Haddleton and C. Barner-Kowollik, J. Am. Chem. Soc., 2015, 137, 6889–6896 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8py01720d |
| This journal is © The Royal Society of Chemistry 2019 |