
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A simple and versatile route to amphiphilic polymethacrylates: catalytic chain transfer polymerisation (CCTP) coupled with post-polymerisation modifications†
Christophe J.
Atkins
 ,
Georgios
Patias
,
James S.
Town
,
Alan M.
Wemyss
,
Ahmed M.
Eissa
,
Ataulla
Shegiwal
and
David M.
Haddleton
*
,
Georgios
Patias
,
James S.
Town
,
Alan M.
Wemyss
,
Ahmed M.
Eissa
,
Ataulla
Shegiwal
and
David M.
Haddleton
*
Department of Chemistry, University of Warwick, Gibbet Hill, Coventry, CV4 7AL, UK. E-mail: d.m.haddleton@warwick.ac.uk
First published on 3rd January 2019
Abstract
Amphiphilic polymers have become key figures in the fields of pharmacology, medicine, agriculture and cosmetics. The use of reversible deactivation radical polymerisation (RDRP) techniques has allowed advances in the synthesis of amphiphilic polymers. However, the high price to performance ratio of these methods can limit their industrial application. Herein, poly(glycidyl methacrylate) polymers of varying molecular weights were first synthesised by catalytic chain transfer polymerisation (CCTP). Amphiphilic polymers were then prepared using a simple one-pot, post-polymerisation modification process involving Michael-thiol addition in the presence of a range of hydrophobic mercaptans, followed by ring-opening of the epoxide groups with ethanolamine using microwave-assisted synthesis. This procedure allows for the synthesis of fully functional polymers within 3 hours. A range of well-defined materials are prepared and characterised by GPC, NMR, FTIR, DLS, TGA, and TEM.
Introduction
The synthesis and behaviour of amphiphiles in solution has been the focus of numerous experimental1–4 and theoretical5–7 studies over the past few decades. Self-assembly, exemplified in nature with molecules such as phospholipids, cholesterol, or bile acids, occurs in order to reduce the overall free energy of a system by minimising interactions between water and hydrophobic moieties,8 resulting in the formation of supra-molecular structures of varying morphologies, such as spherical micelles,9,10 worm-like micelles,11 and vesicles.12Which of these morphologies is adopted can be generally predicted using the packing parameter, p,13 as defined in eqn (1):
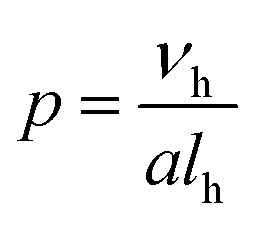 | (1) |
Developments in reversible-deactivation radical polymerisation (RDRP)14–19 techniques have allowed progress in the synthesis of functional amphiphilic polymers. For instance, Ning et al.20 have described the synthesis of biodegradable amphiphilic triblock copolymers using a combination of ring-opening polymerisation of ε-caprolactone and reversible addition–fragmentation chain transfer (RAFT) polymerisation of 2-(2-methoxyethoxy)ethyl methacrylate and ethylene glycol methacrylate. Research into the self-assembly of synthetic macromolecules often focusses on their potential use in drug delivery systems.21–23 Amphiphilic polymers have also found applications in bio-imaging,24 food processing,25 in the plastic industry as plasticisers,26 and in cleaning products as anti-redeposition agents, etc.27
Despite the academic interest in the synthesis and self-assembly of amphiphilic polymers, the implementation of RDRP within industrial settings has been somewhat limited. This is a consequence of RDRP techniques often being difficult to scale up due to a high oxygen sensitivity, being limited in their monomer compatibility, or producing polymers with chain transfer agent or halide end-groups, which are often difficult to remove from the final product (Fig. 1).28,29
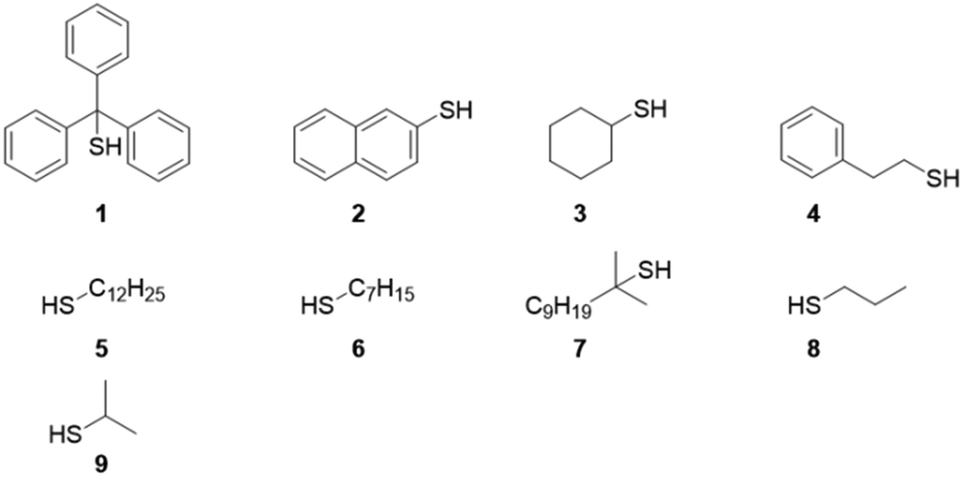 | ||
| Fig. 1 Commercially available thiols employed in this study: triphenylmethanethiol (1), 2-naphthalenethiol (2), cyclohexanethiol (3), phenylethylthiol (4), dodecanethiol (5), heptanethiol (6), tert-dodecanethiol (7), propanethiol (8), 2-propanethiol (9). | ||
Alternatively, the use of industrially proven free-radical polymerisation techniques such as catalytic chain transfer polymerisation (CCTP), which allows the synthesis of low molecular weight macromonomers offers a potentially interesting investigation avenue.30,31 CCTP yields polymers terminated with vinyl ω-end groups. Typically, low spin cobalt(II) complexes are used as catalytic chain transfer agents, and are only required in ppm amounts relative to the concentration of monomer due to extremely high chain transfer constants when compared with other chain transfer agents, such as thiols or halocarbons.32–35 The generally accepted mechanism for the CCTP of methacrylates proceeds via a two-step reaction, Scheme 1.36 The process is highly adaptable, and polymers produced by CCTP have found applications within hair care,37 as toner for printing applications,38 and have also been used for the synthesis of low-VOC (Volatile Organic Compound) high solid coatings.39 Products of CCTP have often been used alongside click-chemistry strategies due to their high levels of vinyl functionality. Several click reactions40 have been reported in the past decade and have since become workhorses in the field. Amongst these reactions can be found hydroamination,41 1,3-dipolar cycloadditions,42–45 Diels–Alder reactions,46,47 and particularly Michael additions (Fig. 2).48–51
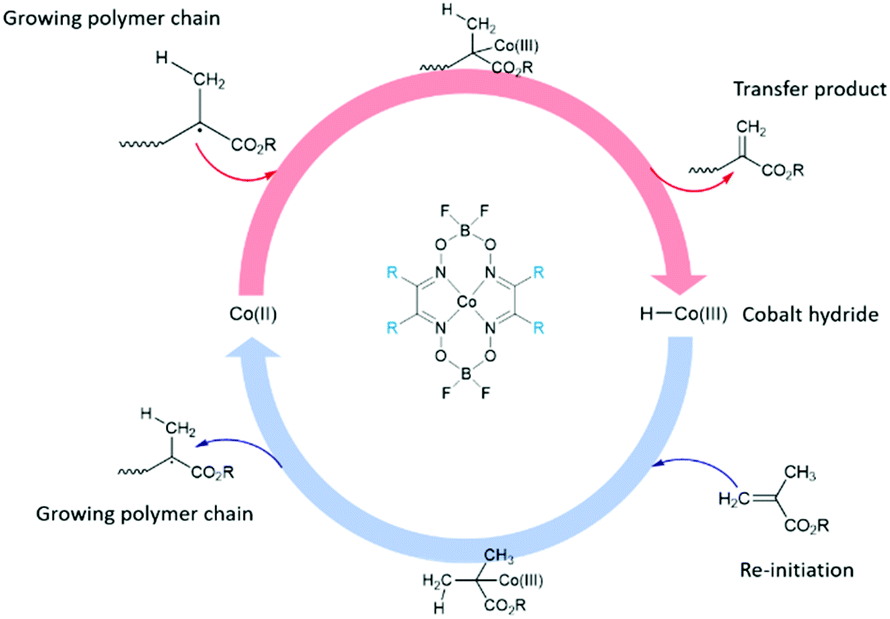 | ||
| Scheme 1 Polymerisation mechanism for methacrylic monomers utilising a cobaloxime (CoBF: R = Me) as chain transfer agent. | ||
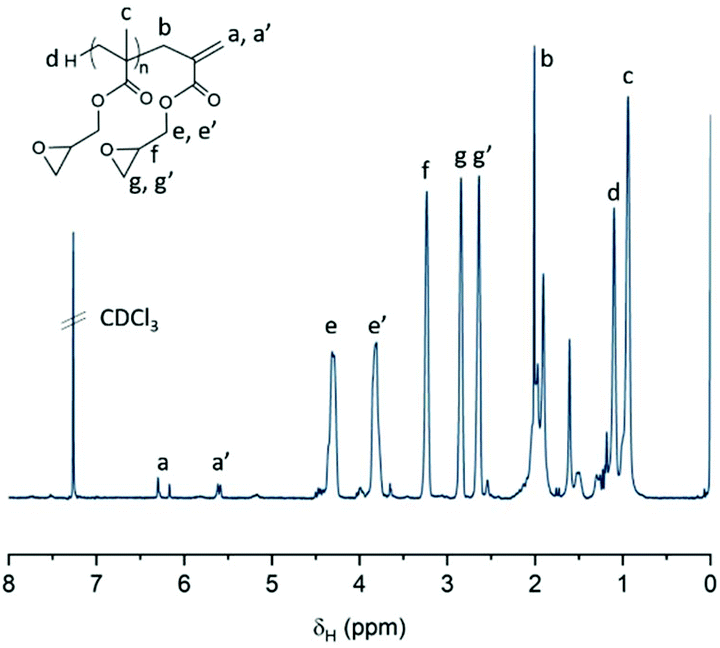 | ||
| Fig. 2 Typical CDCl31H-NMR spectrum of p(GMA) prepared by CCTP. | ||
In an earlier work from our group,34 we demonstrated the versatility afforded by the use of CCTP by polymerising an epoxide containing monomer, glycidyl methacrylate. Using the terminal vinyl bond, and the reactive side chains, the authors obtained functional material by combining hydroamination and epoxide ring-opening in a step-wise fashion.
In this current work, a highly efficient one-pot dual post-polymerisation strategy is developed to produce amphiphilic polymers within 3 hours using thiol–ene chemistry and microwave-assisted epoxide ring-opening. A comparison of the influence of molecular weights of the polymers and clicked thiols on the thermal properties is provided, with an initial investigation into self-assembly behaviours. This study was designed to provide a streamlined, potentially scalable method that allows the synthesis of amphiphilic polymers in the shortest amount of time without the need for intermediate purification steps.
Results and discussion
Synthesis of poly(glycidyl methacrylate)
Three different batches of p(GMA), were prepared with bis[(difluoroboryl)dimethylglyoximato]cobalt(II), CoBF (Scheme 1), as catalytic chain transfer agent. All polymerisations were carried out on a 70 g scale, consistently achieving conversions of ≥95% (Table 1). Moreover, good control over molecular weights was observed by GPC with varying concentrations of catalyst (2, 5 and 8 ppm) (Fig. 3). As expected, molecular weights were found to decrease with increasing concentrations of CoBF. None of the polymerisation products showed signs of side-reactions of the epoxide side chains, as evidenced by FT-IR spectroscopy (ESI, Fig. S1†).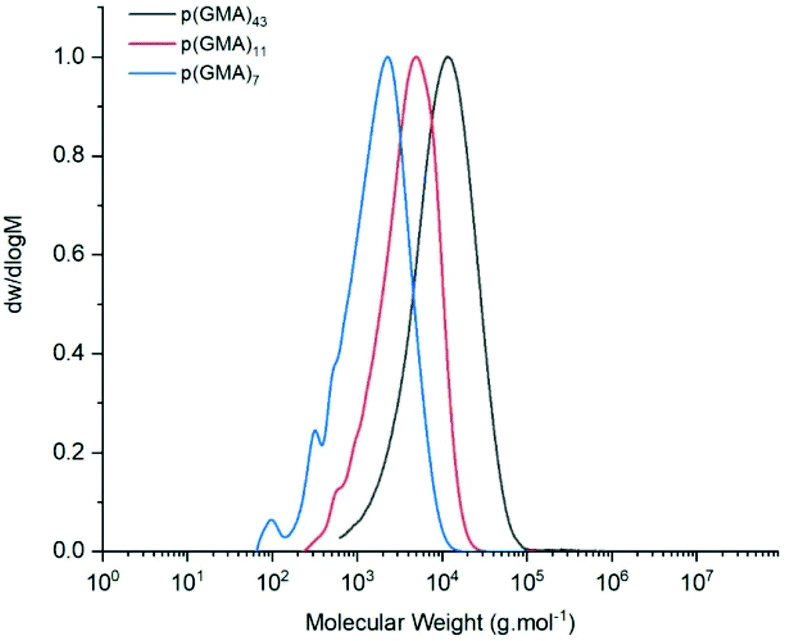 | ||
| Fig. 3 Normalised GPC (DMF, 50 °C) traces comparison of p(GMA) batches A, B and C (2, 5 and 8 ppm CoBF respectively). | ||
| Entry | CoBF (ppm) | Conversion (%) | DPNMR | M n,GPC (g mol−1) | M w,GPC (g mol−1) | Đ |
|---|---|---|---|---|---|---|
| A | 2 | 98 | 43 | 6670 | 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 500 |
2.17 |
| B | 5 | 96 | 11 | 2600 | 4920 | 1.87 |
| C | 8 | 95 | 7 | 2170 | 3380 | 1.55 |
As a catalyst, CoBF can be somewhat susceptible to hydrolysis or oxidation.35 In order to reduce the impact of this, our group previously described a protocol where a solution of CoBF in monomer is fed into the reaction mixture in order to maintain a relatively constant concentration of active catalyst.52 Our results further demonstrate that a feeding of CoBF allows the reactions to achieve high conversions and prevents an uncontrolled increase in dispersity following catalyst destruction.
MALDI-ToF analysis (Fig. 4) of the products confirmed the presence of a polymer with a repeating unit of 142 Da, as expected from GMA. A −14 Da impurity was also observed. Electrospray ionisation mass spectrometry of glycidyl methacrylate monomer confirmed the presence of an unknown compound, which was hypothesised to stem from the monomer purity stated by the supplier (97%) (ESI, Fig. S2†).
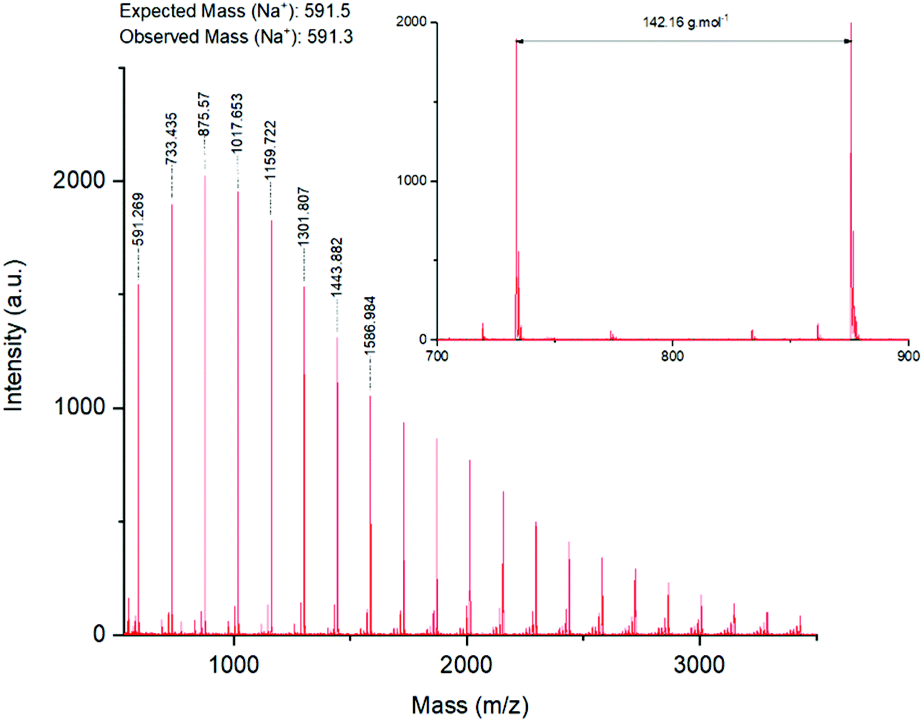 | ||
| Fig. 4 MALDI-ToF spectra of A with detailed zoom between 600 and 1000 Da. | ||
Michael-thiol addition of hydrophobic thiols
Michael-thiol additions allow the formation of alkyl sulphides in the presence of an amine (base-catalysed Michael-thiol addition) or a phosphine (nucleophilic-catalysed Michael-thiol addition) catalyst. Here, reactions were carried out according to a literature protocol in the presence of 0.8 equivalent of dimethylphenylphosphine (DMPP) to double-bonds (1 per polymer chain).53 All reactions were carried out in acetonitrile on a 0.10 g scale of macromonomer under ambient temperature without prior de-oxygenation, utilising 1.2 equivalents of thiols. Initial experiments were conducted using triphenylmethanethiol (1), 2-naphthalenethiol (2) or dodecanethiol (5). Reactions were monitored by 1H-NMR and, within 3 hours, only the reaction of p(GMA) and dodecanethiol showed complete disappearance of vinyl peaks (Fig. 5). Lower yields observed with triphenylmethanethiol and naphthalenethiol were attributed to increased steric hindrance and electronic stabilisation of the thiolate anion intermediates respectively. The reaction of dodecanethiol with p(GMA) was repeated several times and showed some inconsistencies with several reactions reaching noticeably lower functionalisation levels of 60 to 75%. It is known in the literature that DMPP is oxygen sensitive and must be stored under inert gas and this sensitivity, combined with the low quantities used, could play a role in the observed variations. The issue was avoided by purging the oxygen from all reaction vessels used thereafter. Further MALDI-ToF analysis showed that side-reactions between epoxide side-chains and thiols were negligible within the period of the reaction. For instance, with the propanethiol terminated p(GMA) (Fig. 6), we find a series matching the expected structure adducted with a single sodium (c + Na). This series, however, is not the series which has the highest intensity in the spectra. The mains series of peaks suggest that DMPP is capable of ionising the sample (c + DMPP). We therefore propose a mechanism of adduction onto the carbonyl group as shown in the mechanism of DMPP catalysis.53 Although notable, proving this mechanism does not fall within the scope of this work, and should be examined in the future to confirm the validity of the assignment. Other minute impurities include evidence of a structure with a single ring-opened repeat unit due to thiol activation (b + Na, b + DMPP), and a very small peak related to the unreacted p(GMA) which maintained its unsaturated vinyl end group (a + DMPP). If stored unpurified, a full consumption of the epoxides from side-reactions can be observed after twelve hours. This is easily seen through NMR, with the disappearance of the three membered ring's peaks (ESI, Fig. S3†), which suggests a preferential reactivity of the thiols toward the olefinic bonds. To investigate the reaction further, a screening of commercially available hydrophobic thiols was carried out (Fig. 7). Most investigated compounds reached yields ≥98% within 3 hours. Differences in reactivity were also observed between iso-propylthiol and propanethiol, as well as between dodecanethiol and tert-dodecanethiol. Although each set composed of isomers, differences were attributed to stereochemistry, the presence of iso-propyl groups likely slowing down the addition reaction (Scheme 2).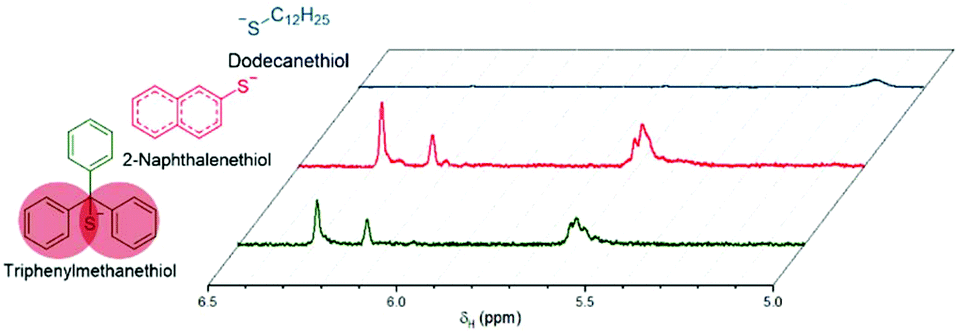 | ||
| Fig. 5 Comparison of vinyl peaks between 7 and 4.5 ppm observed by 1H-NMR (CDCl3) after Michael-thiol addition after 180 min of, from top to bottom: dodecanethiol, 2-naphthalenethiol and triphenylmethanethiol onto p(GMA). | ||
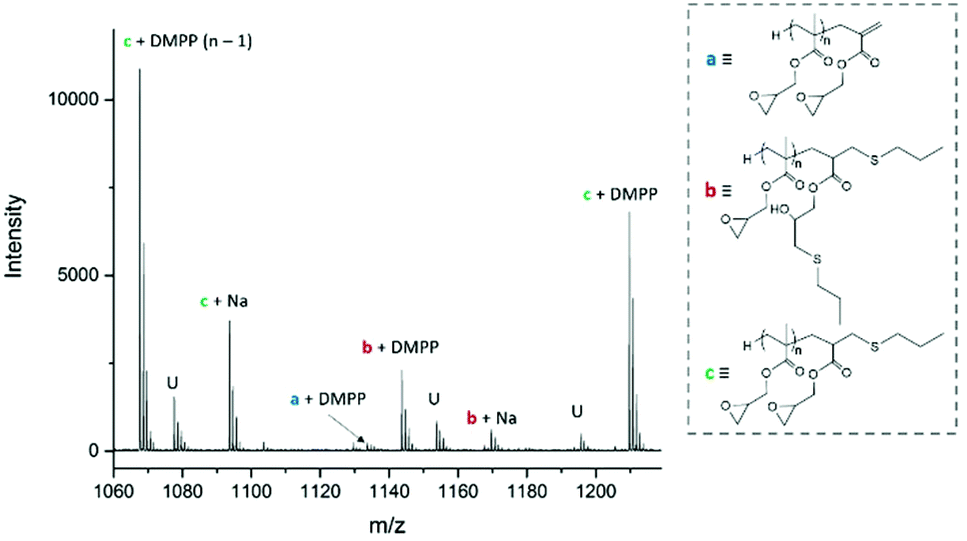 | ||
| Fig. 6 MALDI-ToF spectra of propanethiol functionalised p(GMA)11. | ||
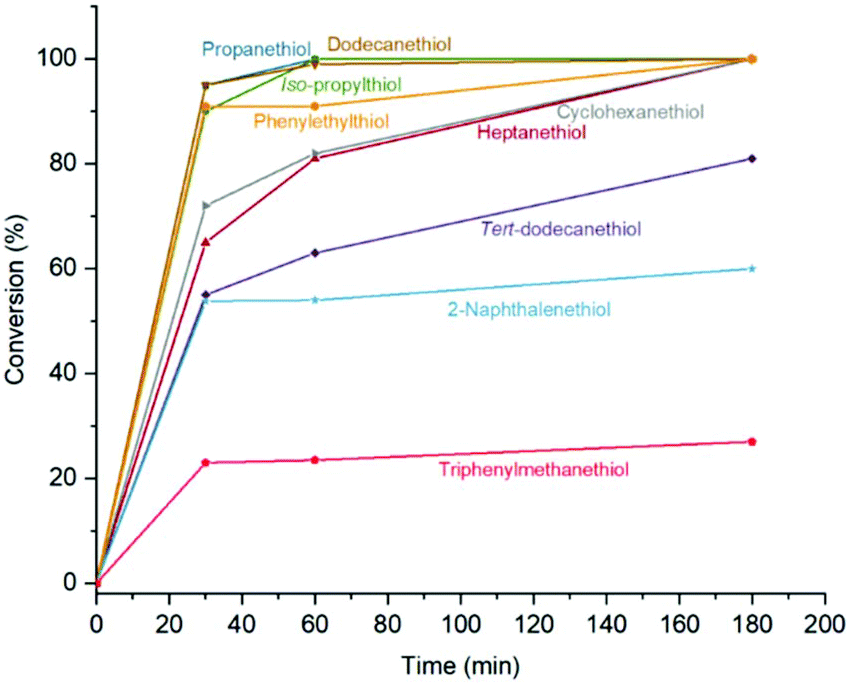 | ||
| Fig. 7 NMR conversion data from vinyl peaks disappearance for the Michael-thiol addition of various hydrophobic thiols. | ||
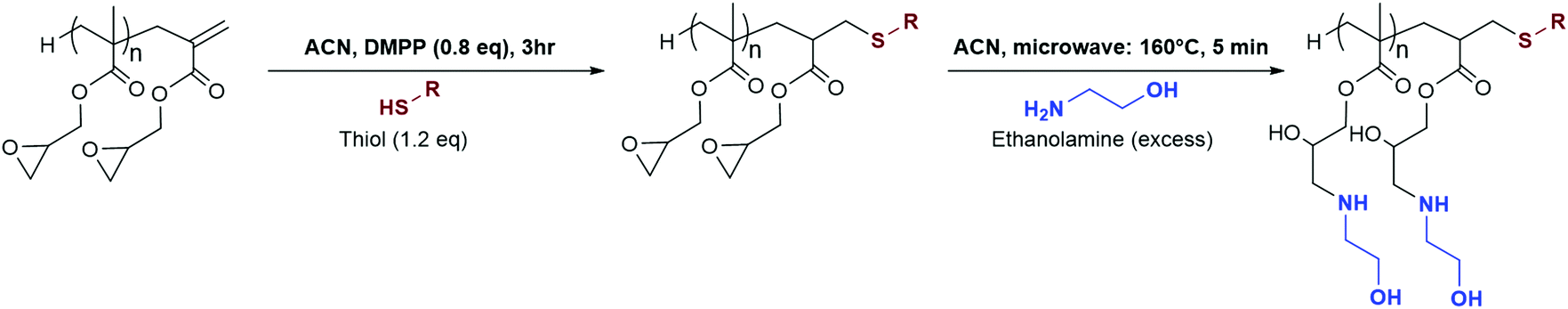 | ||
| Scheme 2 Synthetic pathway investigated in this study. | ||
Epoxide ring-opening using primary amines
p(GMA) polymers are susceptible to base or acid catalysed ring-opening reactions to give access to functional polymers. Epoxide ring-opening can be implemented as a way to introduce secondary reactive functionalities such as azide or carboxylic acid functional groups.54,55 Typically, these reactions are carried out with amines, during quite long reflux reactions which can potentially last up to 24 hours in order to reach high degrees of functionalisation under inert conditions. Microwave-assisted synthesis offers an elegant and efficient alternative, allowing for both a reduction in reaction times and for reactions to be carried out under aerobic conditions.The reaction of ethylamine with p(GMA)11 polymers at 160 °C for 5 minutes was therefore studied as model reaction. This was done in view of having no interference from other hydroxyl groups of hydrophilic amines during FT-IR studies, with ethanolamine being used later on in the study.
The instrument was set to apply energy at lower rates to achieve a more controlled temperature increase. 1, 1.2, 3 and 5 equivalents of ethylamine to epoxy group were used separately. All led to the appearance of an insoluble, white solid, likely the result of intermolecular cross-linking. The issue was described by different research groups,34,56,57 and is the result of the formed secondary amines undergoing another addition reaction to form a tertiary amine, Fig. 8. This issue can be prevented by either lowering reaction temperatures or utilising large excesses of amine. Consequently, reactions with 9-fold excess of amine were attempted (Fig. 8, image 5), and showed no visible sign of cross-linking. Ring-opening was observed with the appearance, on the FT-IR spectra, of characteristic –OH and –NH peaks at around 3400 and 1620 cm−1, respectively (Fig. 9). When cross-linking occurred, the latter peak remained negligible, albeit visible; because of the conversion of primary amines into tertiary amines. However, with the 9-fold excess both peaks became clearly visible.
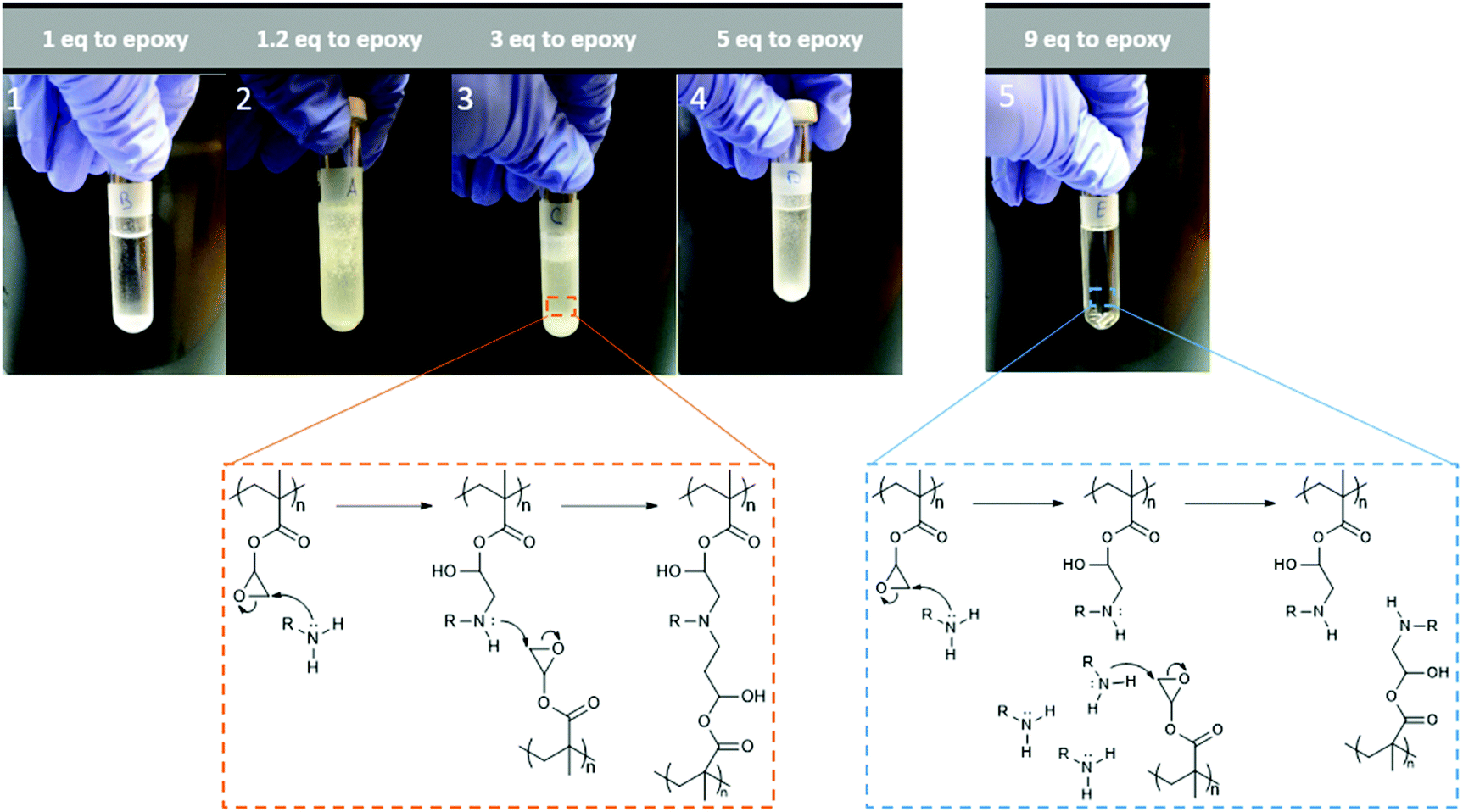 | ||
| Fig. 8 Example of observed cross-linking and ring-opening reactions of p(GMA) with varying ratios of amine to epoxy groups with corresponding reaction pathways. | ||
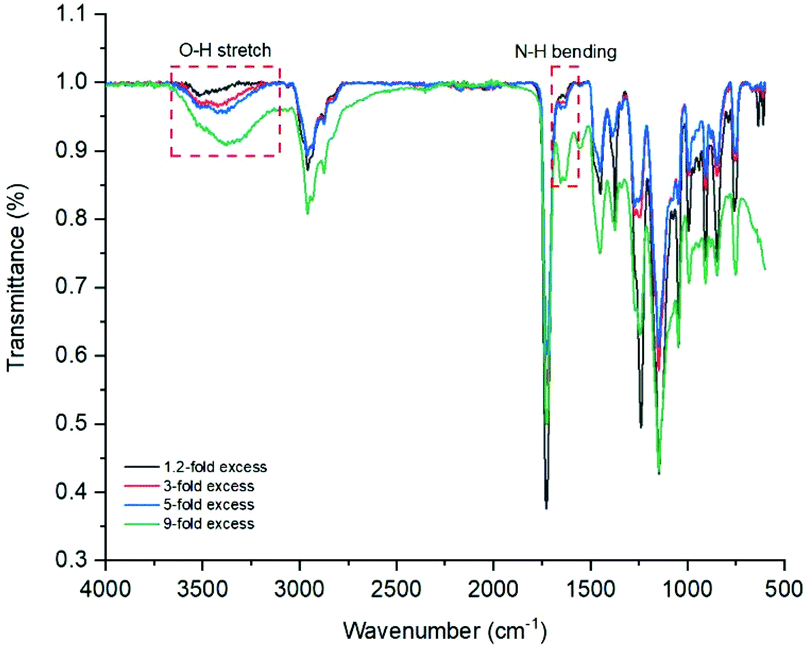 | ||
| Fig. 9 Overlay of FT-IR spectra of p(GMA) polymers ring-opened with varying quantities of ethanolamine. | ||
Dual functionalisation: one-pot Michael-thiol addition followed by epoxide ring-opening
In order to test the robustness of the formulated protocol, four thiols were selected: one aromatic (phenylethylthiol; PET), one containing an aliphatic ring (cyclohexanethiol; CHT), as well as both a long and a short acyclic thiol (dodecanethiol, DDT and iso-propylthiol, i-PT). A library of twelve different polymers was subsequently produced, Table 2. All experiments were carried out in a similar fashion as previously described, but in a sequential manner as one-pot: each thiol was first reacted with p(GMA) polymers from batches A, B and C, and then ring-opened with ethanolamine. Reactions were conducted on larger, 5 g scales of macromonomer.| Entry | Polymer | MT-Add conversion (%) | M n,GPC (g mol−1) | Đ | Average size (d nm) | PDi |
|---|---|---|---|---|---|---|
| P1 | p(GMA)7(DDT)(ETA) | ≥99 | 4650 | 2.87 | 7.48 | 0.0673 |
| P2 | p(GMA)7(CHT)(ETA) | ≥99 | 4540 | 1.83 | 121.65 | 0.294 |
| P3 | p(GMA)7(PET)(ETA) | 97 | 5260 | 1.72 | 119.43 | 0.306 |
| P4 | p(GMA)7(i-PT)(ETA) | ≥97 | 4510 | 1.80 | 101.42 | 0.467 |
| P5 | p(GMA)11(DDT)(ETA) | ≥97 | 5850 | 2.00 | 8.60 | 0.889 |
| P6 | p(GMA)11(CHT)(ETA) | ≥97 | 2800 | 1.91 | 118.15 | 1.28 |
| P7 | p(GMA)11(PET)(ETA) | ≥97 | 3250 | 1.93 | 15.27 | 0.0851 |
| P8 | p(GMA)11(i-PT)(ETA) | ≥97 | 3200 | 1.85 | 13.53 | 0.0896 |
| P9 | p(GMA)43(DDT)(ETA) | 96.1 | 2420 | 1.67 | 41.01 | 0.0961 |
| P10 | p(GMA)43(CHT)(ETA) | 95.5 | 2560 | 1.97 | 11.36 | 0.0870 |
| P11 | p(GMA)43(PET)(ETA) | 96.8 | 3530 | 1.73 | 6.16 | 0.0663 |
| P12 | p(GMA)43(i-PT)(ETA) | 98.2 | 3000 | 1.35 | 9.66 | 0.0769 |
Due to the larger scale, reactions in the microwave had to be heated to 120 °C to avoid uncontrolled increase in internal pressure. However, both Michael-thiol addition and epoxide ring-opening produced polymers with high levels of thiol and amine functionalisation (Table 2). Functionalisation was first observed following GPC analysis, where a visible shift in molecular weight from approximately 2500 g mol−1 to 4500 g mol−1 could be observed, Fig. 10.
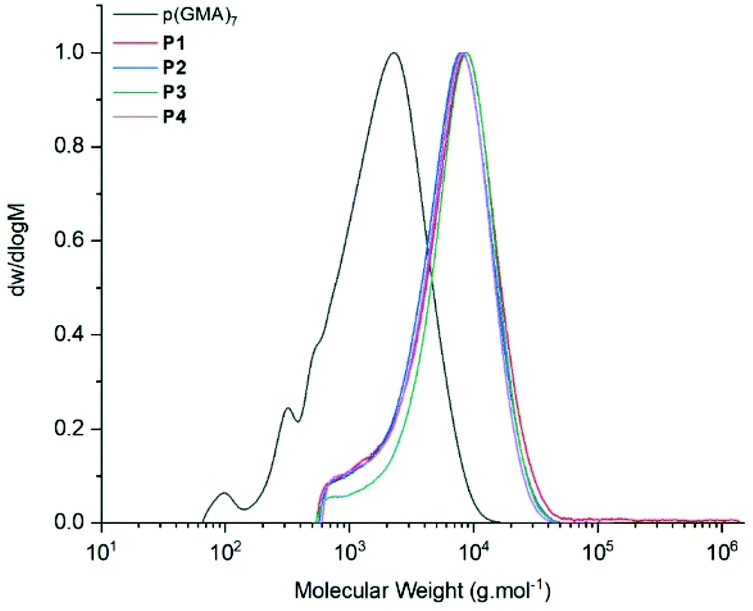 | ||
| Fig. 10 GPC (DMF, 50 °C) comparison of polymers before and after functionalisation of P1, P2, P3 and P4. | ||
Fig. 11 shows a comparison of the 1H-NMR spectra of P9 and P11, neither of which show any peaks corresponding to the vinyl peak or cyclic epoxide functionalities, confirming high levels of functionalisation. Hydroxyl and amine groups were, in both cases, visible at 4.5 and 5.0 ppm, respectively. Further indications of ring opening was noticed from the downfield shift of the OCH2 peak (c) from 3.25 ppm to 4 ppm, overlapping with the methylene peak (d) from the opened epoxide. Moreover, differences were also observed from the different thiols used. For instance, P9 showed dodecane methylene peaks at 1.25 ppm whilst P11, after react ion with phenylethylthiol, showed aromatic peaks at 7.30 ppm. Peaks from the hydrophobic segment showed aromatic peaks at 7.30 ppm. Peaks from the hydrophobic segments also disappeared when ran in D2O, a first indication of the self-assembly potential of the synthesised macromolecules (ESI, Fig. S4†). This self-assembly behaviour was later investigated using dynamic light scattering (DLS) and transmission electron microscopy (TEM).
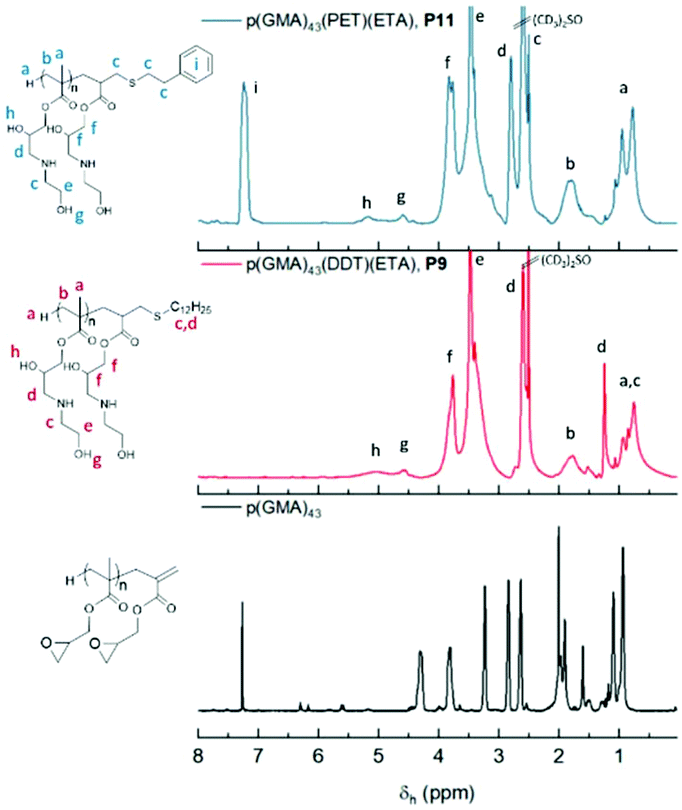 | ||
| Fig. 11 Comparison of 1H-NMR (DMSO-d6) of P9 (blue trace) and P11 (red trace) with DMSO-d6 as solvent. | ||
Most compounds self-assembled into polydisperse structures with sizes ranging from 10 to 30 nm in diameter, Fig. 12 and Table 2. TEM revealed structures to be swollen spherical micelles. The type of clicked thiol and the size of the polymer did not seem to bear any influence on the observed morphologies. From the DLS data, we see that P1, P5 and P9, which were functionalised with dodecanethiol, showed an expected increase in size with increasing hydrophilic block size. The opposite trend is however observed for all other polymers showed. The behaviour of amphiphilic polymers is expected to be intrinsically different when analysed under different conditions (in solution for DLS, and dried for TEM). In solution, the smaller polymers are thought to aggregate into undiscernible structures due to the small nature of both the clicked thiol and the polymer backbone (DP = 7), and progressively become more able to self-assemble in the case of bigger polymers or when solvent is removed for TEM. This is also supported by the fact that the polymers containing longer dodecane chains behave, and can be assimilated to di-block copolymers with a degree of polymerisation twelve polyethylene hydrophobic segments.
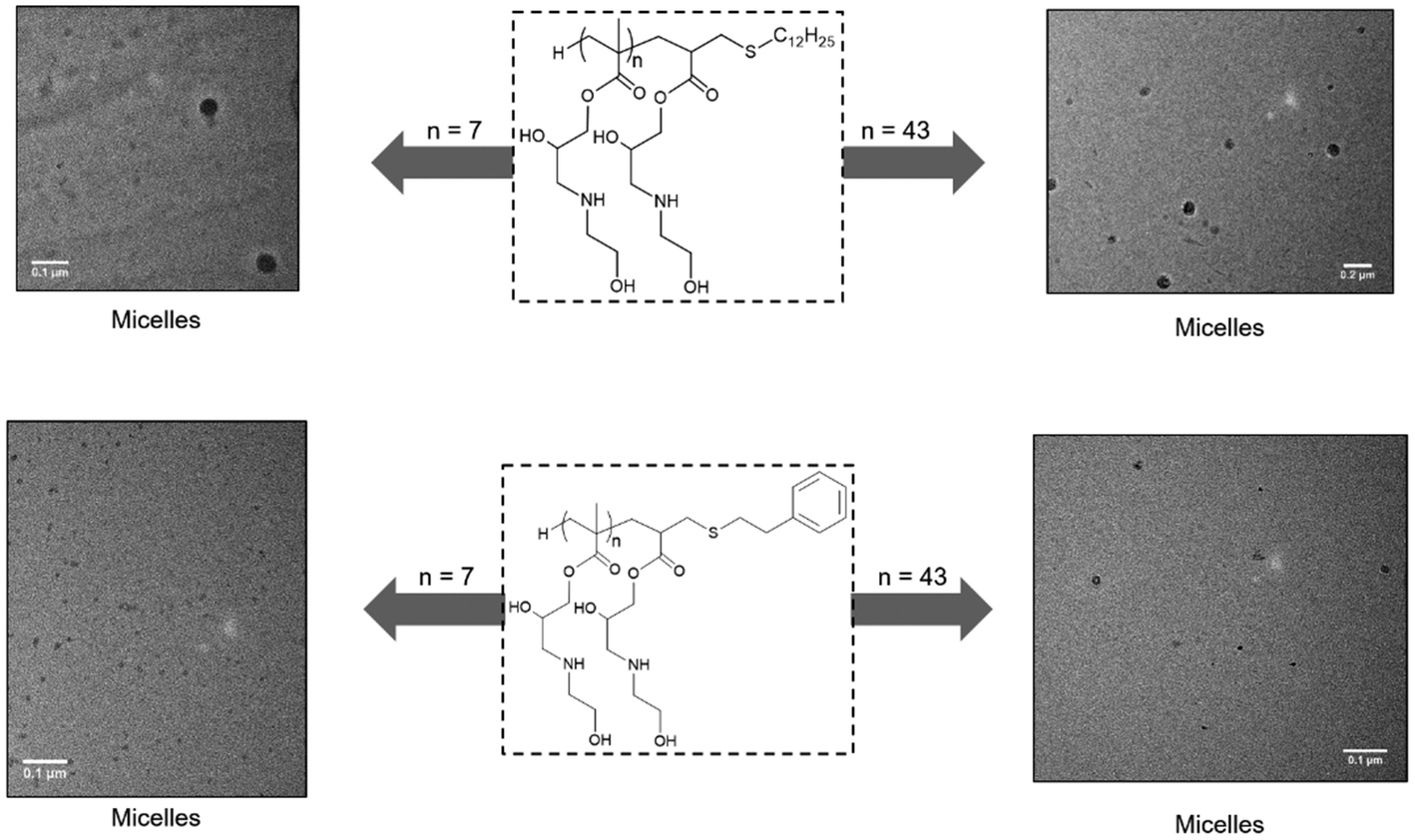 | ||
| Fig. 12 TEM images of P1, P3, P9 and P11 (respectively: top left, bottom left, top right and bottom right) showing self-assembly into spherical micelles. | ||
Neither the type of thiol, nor the size of the hydrophilic segment affected the thermal degradation properties of the polymers (ESI, Fig. S5†). There are two visible degradation steps at approximately 350 and 430 °C, amounting to around 10% residual product, which would involve two degradation mechanisms: depolymerisation to monomer initiated at random along the backbone of the polymer, and ester decomposition, which results in the elimination of different volatile compounds.58,59 Additionally we see, by derivative thermogravimetry (DTG), the appearance of a third degradation step at 260 °C, particularly visible with higher molecular weight polymers, which could correspond to degradation at initiation at unsaturated chain ends of polymers that were not functionalised during Michael-thiol addition, and is in accordance with conversion data reported in Table 2.
Conclusions
Through this work, multiple amphiphilic polymers were prepared using a potentially scalable combination of free-radical polymerisation and efficient post-polymerisation reactions.Molecular weights of p(GMA) were shown to be controllable depending on the concentration of catalyst CoBF, with no observable side-reaction. Amphiphilic polymers were obtained by dual post-polymerisation functionalisation on the macromonomers, which contained two potential reactive loci. Terminal vinyl bonds were reacted with commercially available hydrophobic thiols Michael-thiol addition and epoxide side chains were opened in the presence of ethanolamine, using microwave-assisted synthesis to reduce the reaction time by several orders of magnitude down to one hour.
This synthetic methodology circumvents all needs for protective chemistry, but also simplifies the process of obtaining amphiphilic polymers. With minimal work up, and the variety of available thiols, primary or secondary amine, our technique shows great potential for the synthesis of amphiphilic polymer of varied structure (branched, comb, star polymers etc.) and composition such as the introduction of carbohydrate, or other reactive functionalities.
Experimental
Materials
Dimethyl 2,2′-azobis(2-methylpropionate) (V-601) was obtained from Wako Chemicals, and bis[(difluoroboryl)dimethylglyoximato]cobalt(II) (CoBF) was synthesised according to the literature.60 All other chemicals were purchased from Sigma-Aldrich and used without further purification.Instrumentation
DMF Gel-Permeation Chromatography (GPC) was carried out on an Agilent Infinity II MDS instrument equipped with differential refractive index (DRI), viscometry (VS) and dual angle light scatter (LS) detectors. The system was equipped 2 × PLgel Mixed D columns (300 × 7.5 mm) and a PLgel 5 μm guard column. X12 poly(methyl methacrylate) standards (Agilent EasyVials) were used for calibration between 955000–550 g mol−1 fitted with an 3rd order polynomial. The eluent was DMF with 5 mmol NH4BF4 additive. Analyte samples were filtered through a nylon membrane with 0.22 μm pore size before injection, and samples were run at 1 ml min−1 at 50 °C. Number average molecular weight (Mn,GPC) and dispersity (Đ) values of synthesized polymers were determined by conventional calibration using Agilent GPC/SEC software.
1H-NMR spectra were recorded at room temperature on a Bruker Avance III HD-400 or 300 using either deuterated chloroform or deuterated dimethyl sulfoxide referenced against TMS.
Microwave-assisted reactions were performed in a Biotage initiator+. 15–20 mL Biotage vials were used.
MALDI-ToF spectra were collected using a Bruker Autoflex Speed, equipped with a 337 nm nitrogen laser, operating in reflectron positive mode with an ion source voltage of 19 kV. Results were accumulated in 10 readings of each spot with 500 laser shots, leading to a total of 5000 laser shots per spectra. Laser power was tuned to keep noise low while maintaining the signal as to not remove any trace peaks. The samples were dissolved into THF at concentrations of 10 mg ml−1, along with the cationizing agent sodium iodide (NaI) at 0.1 mg ml−1. A matrix solution was then made up of trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) in THF at a concentration of 40 mg ml−1, along with NaI at 0.1 mg ml−1. 10 μl of both the matrix and sample solutions were then mixed together, and 0.5 μl of the resulting solution was then spotted on an MTP 384 ground steel target plate.
FT-IR spectra were recorded on a Bruker Vector 22 FT–IR Spectrometer fitted with a diamond crystal plate and a pressure tower. The instrument was set to perform 64 scans per sample at a scan speed of 0.5 cm s−1.
Dynamic light scattering (DLS) analyses were carried out on a Malvern Instruments Zetasizer Nano-ZS in water at 25 °C fitted with a 4 mW He–Ne 633 nm laser set at a back-scattering angle of 173°. Samples were prepared by dissolving 1 mg of product in 20 mL of HPLC-grade water. All solutions were sonicated for 2 minutes at 50 °C and allowed to settle for a further 10 minutes Small aliquots were then pipetted into disposable DLS cuvettes. The equilibration time was set at 2 minutes and five measurements of eleven runs were conducted each time. Sizes are reported as averages of 5 runs and polydispersities were calculated using eqn (2):
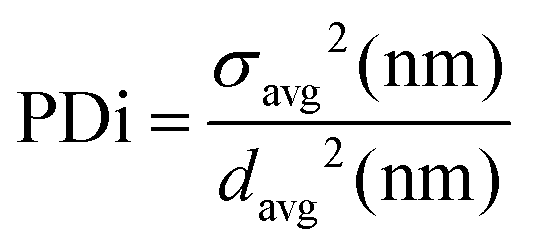 | (2) |
Thermo-gravimetric (TGA) data was obtained using a Mettler-Toledo TGA/DSC1 with autosampler. Measurements were carried out under a nitrogen atmosphere from 25 to 600 °C at a rate of 10 °C min−1 in a 40 μL aluminium crucible.
Transmission Electron Microscopy (TEM) images were obtained on a JEOL 2100 TEM fitted with a Gatan Ultrascan 1000 camera. Samples were diluted at 0.1% v/v and one drop was cast on a carbon coated TEM copped grid. After 2 minutes the drop was blotted off with filter paper. All samples were prepared without using a stain.
Synthesis of poly(glycidyl methacrylate)
0.25 mg mL−1 stock solutions of CoBF in GMA were prepared by separately de-oxygenating 5 mg of CoBF and 20 mg of GMA for 1 hour and 30 minutes respectively using nitrogen bubbling. GMA was then transferred, using degassed syringes, into the CoBF flask, previously equipped with a magnetic stirrer. The solution was then sonicated for 10 min and left to stir for up to 1 hour to ensure proper mixing. Stock solutions were stored at 4 °C for up to two months.All polymerisations were carried out in a similar fashion. VA-601 (552.62 mg, 2.4 mmol) was dissolved in acetonitrile (60 mL) and introduced into a 250 mL tri-neck round bottom flask (rbf 1) equipped with a stirrer bar. In parallel, another round bottom flask (rbf 2) was loaded with an appropriate volume of GMA, taking into consideration the volume of CoBF in GMA required from the stock solution to reach the required final CoBF concentration within the reaction mixture. The two flasks were then sealed with rubber septa and degassed for 30 minutes each. Once all components were purged of oxygen, a CoBF/GMA volume from the stock solution was transferred into rbf 2. The final CoBF in GMA solution was fed into rbf 1, which was placed in an oil bath pre-heated to 70 °C, over a 3 hour period. The reaction mixture was allowed to react for 24 hours. Samples for 1H-NMR, GPC, MALDI-ToF MS and IR were taken after termination of the reaction and removal of solvent using rotary evaporation.
Michael-thiol addition of hydrophobic thiols
Michael-thiol additions were typically carried out by introducing 100 mg of p(GMA) and 3 mL of acetonitrile into vials along with magnetic stirrers. Vials were sealed and de-oxygenated for 15 minutes. 1.2 equivalents of de-oxygenated thiol were subsequently added to the vials, as well as 0.8 eq. of dimethylphenylphosphine (DMPP) using de-oxygenated syringes. All reactions were then left to react for 3 hours under vigorous stirring (500 rpm). No workup was carried out between the termination of the reaction and MALDI-TOF analysis.Epoxide ring-opening using hydrophilic amines
Ring-opening of epoxide was conducted by adding an excess of amine (1, 1.2, 3, 5 or 9 equivalents to nepoxide) to 100 mg of p(GMA)11 dissolved using 3 mL of acetonitrile in a 2–5 mL microwave vial. The reaction mixtures were then mixed under vigorous stirring for 5 minutes, sealed using microwave caps, and placed in the microwave reactor to react for 5 minutes at 160 °C.Dual functionalisation: one-pot Michael-thiol addition followed by epoxide ring-opening
Dual functionalisation reactions of p(GMA) polymers were carried out on a 5 g scale of macromolecules (1 eq.). P(GMA) was first introduced – along with magnetic stirrers – to 10–20 mL microwave vials and dissolved with 8 mL of acetonitrile. Vials were then sealed and de-oxygenated using nitrogen bubbling for 30 minutes. 1.2 equivalents of thiols and DMPP (0.8 eq.), which were degassed in parallel, were then transferred into the vials with de-oxygenated syringes. The reaction was allowed to proceed, after 3 hours, the seal was removed, and 9 equivalents excess of ethanolamine to the number of moles of epoxide were added to the vials without intermediate purification step. The vials were sealed, keeping an ambient atmosphere, and placed in the microwave for 5 minutes reaction cycle at 120 °C. Samples were then taken out of the instrument, and acetonitrile evaporated by blowing oxygen through the clear solution for 12 hours. The resulting viscous solution was dissolved with 10 mL of methanol, and dialysed against methanol for 50 h to remove residual acetonitrile and impurities. Methanol was removed by a combination of rotary evaporation, freeze-drying on Schlenk lines. All products were finally placed in a vacuum oven overnight before carrying out product analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Equipment used was supported by the Warwick Polymer Characterisation Research and Technology Platform and help from Dr D. Lester is appreciated. Funding for studentships for GP and CJA from Lubrizol and the University of Warwick is appreciated.Notes and references
- V. P. Torchilin, V. S. Trubetskoy, K. R. Whiteman, P. Caliceti, P. Ferruti and F. M. Veronese, J. Pharm. Sci., 1995, 84, 1049–1053 CrossRef CAS.
- X. Qu, Y. Zou, C. He, Y. Zhou, Y. Jin, Y. Deng, Z. Wang, X. Li, Y. Zhou and Y. Liu, Drug Delivery, 2018, 25, 210–225 CrossRef CAS PubMed.
- W. W. Yu, E. Chang, J. C. Falkner, J. Zhang, A. M. Al-Somali, C. M. Sayes, J. Johns, R. Drezek and V. L. Colvin, J. Am. Chem. Soc., 2007, 129, 2871–2879 CrossRef CAS PubMed.
- H. S. Sundaram, Y. Cho, M. D. Dimitriou, J. A. Finlay, G. Cone, S. Williams, D. Handlin, J. Gatto, M. E. Callow, J. A. Callow, E. J. Kramer and C. K. Ober, ACS Appl. Mater. Interfaces, 2011, 3, 3366–3374 CrossRef CAS PubMed.
- V. V. Vasilevskaya, P. G. Khalatur and A. R. Khokhlov, Macromolecules, 2003, 36, 10103–10111 CrossRef CAS.
- A. F. Miller, M. R. Wilson, M. J. Cook and R. W. Richards, Mol. Phys., 2003, 101, 1131–1138 CrossRef CAS.
- K. Kempe, R. A. Wylie, M. D. Dimitriou, H. Tran, R. Hoogenboom, U. S. Schubert, C. J. Hawker, L. M. Campos and L. A. Connal, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 750–757 CrossRef CAS.
- M. Müller and K. C. Daoulas, J. Chem. Phys., 2008, 128, 32 Search PubMed.
- G. Kwon, M. Naito, M. Yokoyama, T. Okano, Y. Sakurai and K. Kataoka, Langmuir, 1993, 9, 945–949 CrossRef CAS.
- D. Kiserow, C. Ramireddy, P. Munk, S. E. Webber, K. Prochazka and Z. Tuzar, Macromolecules, 1992, 25, 461–469 CrossRef CAS.
- S. Cai, K. Vijayan, D. Cheng, E. M. Lima and D. E. Discher, Pharm. Res., 2007, 24, 2099–2109 CrossRef CAS PubMed.
- A. Choucair, C. Lavigueur and A. Eisenberg, Langmuir, 2004, 20, 3894–3900 CrossRef CAS.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- A. Mühlebach, S. G. Gaynor and K. Matyjaszewski, Macromolecules, 1998, 31, 6046–6052 CrossRef.
- J. Li, H. Chen, G. Mu, J. Sun, Y. Sun, C. Wang, Q. Ren and J. Ji, React. Funct. Polym., 2013, 73, 1517–1522 CrossRef CAS.
- M. Mertoglu, S. Garnier, A. Laschewsky, K. Skrabania and J. Storsberg, in Polymer, Elsevier, 2005, vol. 46, pp. 7726–7740 Search PubMed.
- H. Götz, E. Harth, S. M. Schiller, C. W. Frank, W. Knoll and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3379–3391 CrossRef.
- G. R. Jones, R. Whitfield, A. Anastasaki, N. Risangud, A. Simula, D. J. Keddie and D. M. Haddleton, Polym. Chem., 2018, 9, 2382–2388 RSC.
- R. Whitfield, A. Anastasaki, G. R. Jones and D. M. Haddleton, Polym. Chem., 2018, 9, 4395–4403 RSC.
- W. Ning, P. Shang, J. Wu, X. Shi and S. Liu, Polymers, 2018, 10, 214 CrossRef.
- F. Suriano, R. Pratt, J. P. K. Tan, N. Wiradharma, A. Nelson, Y. Y. Yang, P. Dubois and J. L. Hedrick, Biomaterials, 2010, 31, 2637–2645 CrossRef CAS PubMed.
- P. S. Stayton, A. S. Hoffman, N. Murthy, C. Lackey, C. Cheung, P. Tan, L. A. Klumb, A. Chilkoti, F. S. Wilbur and O. W. Press, J. Controlled Release, 2000, 65, 203–220 CrossRef CAS PubMed.
- F. Wang, T. K. Bronich, A. V. Kabanov, R. D. Rauh and J. Roovers, Bioconjugate Chem., 2005, 16, 397–405 CrossRef CAS PubMed.
- Z. Huang, Q. Chen, Q. Wan, K. Wang, J. Yuan, X. Zhang, L. Tao and Y. Wei, Polym. Chem., 2017, 8, 4805–4810 RSC.
- E. Dickinson and D. Lorient, Food Macromolecules and Colloids, Royal Society Of Chemistry, Cambridge, United Kingdom, 1995 Search PubMed.
- I. Ahmed, M. B. K. Niazi, A. Hussain and Z. Jahan, Polym.-Plast. Technol. Eng., 2018, 57, 17–27 CrossRef CAS.
- I. Sroková, V. Tomanová, A. Ebringerová, A. Malovíková and T. Heinze, Macromol. Mater. Eng., 2004, 289, 63–69 CrossRef.
- M. Destarac, Macromol. React. Eng., 2010, 4, 165–179 CrossRef CAS.
- M. Destarac, Polym. Chem., 2018, 9, 4947–4967 RSC.
- N. G. Engelis, A. Anastasaki, G. Nurumbetov, N. P. Truong, V. Nikolaou, A. Shegiwal, M. R. Whittaker, T. P. Davis and D. M. Haddleton, Nat. Chem., 2017, 9, 171–178 CrossRef CAS PubMed.
- N. G. Engelis, A. Anastasaki, R. Whitfield, G. R. Jones, E. Liarou, V. Nikolaou, G. Nurumbetov and D. M. Haddleton, Macromolecules, 2018, 51, 336–342 CrossRef CAS.
- A. Gridnev, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1753–1766 CrossRef CAS.
- A. A. Gridnev and S. D. Ittel, Chem. Rev., 2001, 101, 3611–3659 CrossRef CAS PubMed.
- K. a. McEwan, S. Slavin, E. Tunnah and D. M. Haddleton, Polym. Chem., 2013, 4, 2608 RSC.
- J. P. A. Heuts and N. M. B. Smeets, Polym. Chem., 2011, 2, 2407 RSC.
- D. Kukulj and T. P. Davis, Macromol. Chem. Phys., 1998, 199, 1697–1708 CrossRef CAS.
- S. Slavin, E. Khoshdel and D. M. Haddleton, Polym. Chem., 2012, 3, 1461–1466 RSC.
- C.-M. Cheng and D. J. Tshudy, US Pat, 5928829, 1998 Search PubMed.
- K. Adamsons, G. Blackman, B. Gregorovich, L. Lin and R. Matheson, Prog. Org. Coat., 1998, 34, 64–74 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- A. L. Reznichenko and K. C. Hultzsch, in Organic Reactions, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2015, vol. 88, pp. 1–554 Search PubMed.
- J. A. Opsteen and J. C. M. Van Hest, Chem. Commun., 2005, 57–59 RSC.
- W. H. Binder and C. Kluger, Macromolecules, 2004, 37, 9321–9330 CrossRef CAS.
- B. Helms, J. L. Mynar, C. J. Hawker and J. M. J. Fréchet, J. Am. Chem. Soc., 2004, 126, 15020–15021 CrossRef CAS PubMed.
- G. Mantovani, V. Ladmiral, L. Tao and D. M. Haddleton, Chem. Commun., 2005, 2089–2091 RSC.
- B. Gacal, H. Durmaz, M. A. Tasdelen, G. Hizal, U. Tunca, Y. Yagci and A. L. Demirel, Macromolecules, 2006, 39, 5330–5336 CrossRef CAS.
- H. Durmaz, B. Colakoglu, U. Tunca and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1667–1675 CrossRef CAS.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- M. Uygun, M. A. Tasdelen and Y. Yagci, Macromol. Chem. Phys., 2010, 211, 103–110 CrossRef CAS.
- J. P. Menzel, D. M. Haddleton and E. Khoshdel, Polym. Prepr., 2010, 51, 721–722 Search PubMed.
- A. A. Aimetti, A. J. Machen and K. S. Anseth, Biomaterials, 2009, 30, 6048–6054 CrossRef CAS PubMed.
- D. M. Haddleton, E. Depaquis, E. J. Kelly, D. Kukulj, S. R. Morsley, S. A. F. Bon, M. D. Eason and A. G. Steward, Polym. Chem., 2001, 39, 2378–2384 CrossRef CAS.
- G.-Z. Li, R. K. Randev, A. H. Soeriyadi, G. Rees, C. Boyer, Z. Tong, T. P. Davis, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1196 RSC.
- Q. Zhang, S. Slavin, M. W. Jones, A. J. Haddleton and D. M. Haddleton, Polym. Chem., 2012, 3, 1016–1023 RSC.
- S. C. Chang, S. J. Chiu, C. Y. Hsu, Y. Chang and Y. L. Liu, Polym., 2012, 53, 4399–4406 CrossRef CAS.
- H. Gao, X. Lu, Y. Ma, Y. Yang, J. Li, G. Wu, Y. Wang, Y. Fan and J. Ma, Soft Matter, 2011, 7, 9239–9247 RSC.
- F. J. Xu, M. Y. Chai, W. B. Li, Y. Ping, G. P. Tang, W. T. Yang, J. Ma and F. S. Liu, Biomacromolecules, 2010, 11, 1437–1442 CrossRef CAS PubMed.
- S. Zulfiqar, M. Zulfiqar and M. N, Polym. Degrad., 1990, 30, 195–203 CrossRef CAS.
- M. Ferriol, A. Gentilhomme, M. Cochez, N. Oget and J. L. Mieloszynski, Polym. Degrad. Stab., 2003, 79, 271–281 CrossRef CAS.
- A. Bakac and J. H. Espenson, J. Am. Chem. Soc., 1984, 106, 5197–5202 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Complementary figures and schemes. See DOI: 10.1039/c8py01641k |
| This journal is © The Royal Society of Chemistry 2019 |