
Acidity-triggered zwitterionic prodrug nano-carriers with AIE properties and amplification of oxidative stress for mitochondria-targeted cancer theranostics†
 a
Haibo
Wang
*a
and
a
Haibo
Wang
*a
and
Abstract
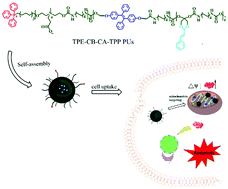
Although biodegradable polymer–drug conjugate delivery systems exhibit great promise toward many types of cancers, several challenges still remain, associated with specific targeting of organelles as well as real-time tracking and observing. Herein, an innovative pH-responsive zwitterionic prodrug is prepared that can easily self-assemble into stable micelles with high drug loading. This new nanocarrier is composed of the following vital factors: (i) a zwitterionic outer shell to prolong the blood circulation and aggregate at the tumor site; (ii) surface-encoded triphenyl-phosphonium groups to enhance organelle targeting; (iii) ketal bonds of nanomicelles to realize the controlled release of therapeutic molecules; (iv) aggregation-induced emission (AIE) luminogen tetraphenylethene (TPE) to realize labelling and real-time monitoring of the mitochondria in living cells. This targeted pH-responsive polyprodrug can effectively release therapeutic molecules to activate the generation of the local ROS level and enable real-time monitoring of the status of mitochondria, which presents a practicable strategy for simultaneous cancer diagnostics and therapeutics.
 Please wait while we load your content...
Please wait while we load your content...

