
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the role of N-vinylpyrrolidone in the aqueous radical-initiated copolymerization with PEGDA mediated by eosin Y in the presence of O2†
Alan
Aguirre-Soto
 a,
Seunghyeon
Kim
a,
Kaja
Kaastrup
a and
Hadley D.
Sikes
*ab
a,
Seunghyeon
Kim
a,
Kaja
Kaastrup
a and
Hadley D.
Sikes
*ab
aDepartment of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: sikes@mit.edu
bProgram in Polymers and Soft Matter, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA
First published on 19th December 2018
Abstract
The photochemistry of eosin Y has attracted attention for its role in visible-light induced polymerization reactions that proceed in the presence of ambient oxygen to form various macromolecular architectures that are useful for a wide range of applications, including biosensing and drug delivery. N-Vinylpyrrolidone (NVP) has been employed as a comonomer in the eosin-mediated synthesis of hydrogels with polyethylene glycol (PEG) based multifunctional monomers to aid in reducing oxygen inhibition and enhancing the rate of radical polymerization and the final conversion. However, the mechanism by which NVP reduces the oxygen inhibition time (tinh) remains unclear. Additionally, no investigations were found on the integration of NVP into the radical-generating photocatalytic mechanism of eosin Y. Towards a better understanding of eosin-mediated synthesis of PEG-based hydrogels, we analyzed the effect of NVP on the steady-state kinetics of the aqueous NVP/PEG-diacrylate copolymerization reaction. In this case, the reduction in tinh is lower than that reported for copolymerization with neat (meth)acrylate monomers. We propose the formation of a ground-state complex between eosin and NVP as the main reason for the reduction in oxygen inhibition and contrast it with previous theories. In addition, we discuss the role of this eosin/NVP complex and cross-propagation kinetics to explain the ∼70% increase in the initial rate of polymerization upon addition of NVP. The effect of cross-propagation kinetics is enhanced at the later stages, leading to a 10% increase in final vinyl conversion in this relatively mobile network. By analyzing the change in the scaling of the eosin decay coefficient as a function of light intensity during and after oxygen inhibition, we then link eosin inactivation to radical termination kinetics. Finally, we discuss the role of radical recombination between semireduced eosin and the propagating radicals as an additional eosin inactivation route by which leuco-eosin ends tethered to the network. These insights contribute to a thorough understanding of visible-light activated polymerization in the presence of oxygen and of the role of NVP in eosin-mediated radical production.
Introduction
Hydrogels have led to important advances in biomaterials,1 biomedicine2 and bioengineering,3 including contact lenses,4 drug delivery,5 cell encapsulation,6 3D printing,7 tissue engineering,8 soft robotics,9 biosensing,10 and regenerative medicine.11 A practical and versatile method to synthesize hydrogels is through radical-initiated crosslinking of multifunctional monomers, such as acrylates.1,12 However, translation into practice can be hindered by the challenge of forming hydrogels in situ against the inhibitory effect of physiological oxygen on the radical initiation process.13,14 Although several chemistries have been successfully implemented to overcome oxygen inhibition, many of them require reagents, concentration regimes, or energy doses that may reasonably be expected to introduce unwanted perturbations to proteins, cells, tissues and organisms.15Organic photoredox catalysis has garnered attention as a means to initiate radical polymerizations in the presence of naturally-occurring oxygen concentrations under low-intensity visible radiation with water-soluble biocompatible photocatalysts at significantly lower concentrations than those typically required by other radical initiation methods.16 Eosin Y (eosin, EY) is a particularly exceptional organic photocatalyst useful in forming hydrogels under atmospheric conditions and low-intensity visible light with sub-micromolar concentrations.6,17,18
In addition to the benefits provided by eosin photocatalysis to counteract the inhibitory effect of oxygen, N-vinylpyrrolidone (NVP) has been repeatedly employed as a water-soluble comonomer to further reduce the oxygen inhibition time (tinh), increase the rate of polymerization and the final conversion for the synthesis of hydrogels with various crosslinker chemistries.19,20 Generally, NVP has found application as a reactive diluent and comonomer in several formulations, especially when its hydrophilicity is acceptable or preferred.21–23 However, the mechanism by which NVP reduces oxygen inhibition when copolymerized with other monomers remains elusive. Furthermore, no reports were found that provide mechanistic insights into the role of NVP in the eosin-mediated initiation process.
Hoyle and coworkers studied the copolymerization of various N-vinyl amides with diacrylate monomers to explain the reductions in tinh.24 Three theories emerged from this work in neat monomer systems (Scheme 1): (1) NVP can potentially enhance the scavenging of molecular oxygen via an irreversible photo-oxidation reaction; (2) NVP could form a charge-transfer complex (CTC) or exciplex with molecular oxygen, thus accelerating the rate of de-oxygenation, while producing initiating radicals; and (3) NVP may form a complex with certain functional groups, which would increase the reaction rate constant for propagation and/or reduce the reaction rate for oxygen inhibition. In more recent studies, these theories were re-examined, revealing that they appear insufficient to explain the observed results.14,22,25 An important remark in this work was that there is still limited evidence favoring either one of these theories.14,22
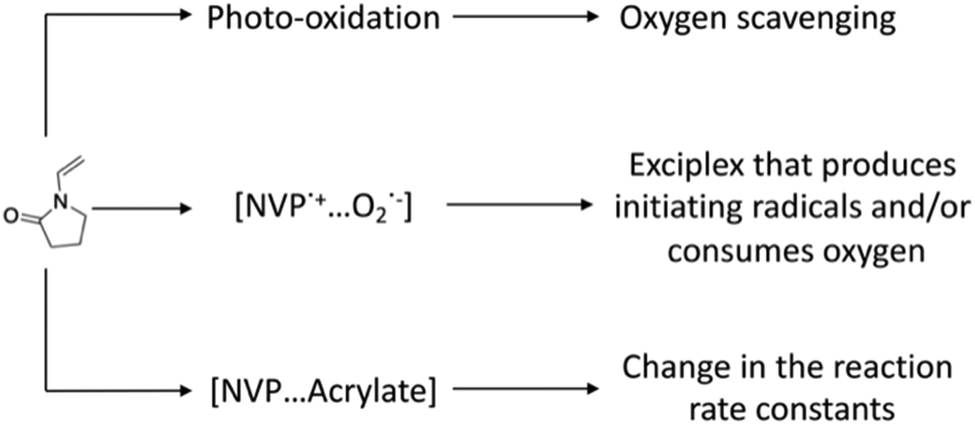 | ||
| Scheme 1 Previously proposed mechanisms for the reduction of oxygen inhibition times upon addition of N-vinylpyrrolidone to (meth)acrylate monomers. | ||
In contrast, the role of NVP in the rate of polymerization and final conversion has been more extensively researched. Seemingly conclusive theories are available to explain the observed increases in the rate of polymerization and final conversion upon addition of NVP to several multifunctional monomers.25,26 At first, however, the issue was that the initial observations were counterintuitive to what would be expected from monofunctional monomers functioning as reactive diluents, i.e. these are expected to lower the concentration of the vinyl groups and the crosslink density, leading to higher final conversion from the delayed onset of reaction–diffusion control kinetics and vitrification. However, this increase in mobility is also expected to decrease the rate of polymerization by increasing radical termination in the initial stages. Two alternative theories arose to explain the enhanced effects of NVP that could not be explained by the reactive plasticizer effect: (1) cross-propagation may lead to an increase in rates and final conversions, and (2) a ground state complex between NVP and certain monomers may increase the reaction rate constant for propagation of the macroradicals.
The theories of cross-propagation and of NVP/acrylate ground-state complexation are difficult to extrapolate from observations in neat monomer formulations to the present NVP/poly-ethylene glycol diacrylate (PEGDA) copolymerization in dilute aqueous solutions, where the reactive diluent contributions to the rate of polymerization and final conversion are limited, if present. However, previous studies in neat monomers served as the starting point for our studies. White and Guymon analyzed the effects of NVP concentration on the copolymerization kinetics and the mechanical properties of the polymers with various mono- to penta-functional acrylate monomers using phosphine oxide photocleavable initiators.25 They concluded that NVP homopolymerization only occurs at high NVP concentrations, that NVP increases network mobility at low concentrations, and thus delays the onset of reaction–diffusion termination and that cross-propagation is most likely responsible for the enhanced effects on the rate of polymerization over what is expected for a typical reactive diluent, like hexyl acrylate. In our highly dilute diacrylate system, however, mobility is considerably greater, so the delayed onset in the reaction–diffusion kinetics may not account for the higher rates of polymerization observed with NVP.
In contrast, the role of NVP in copolymerization reactions in highly dilute monomer solutions has been relatively less studied. Hubbell and coworkers analyzed the effects of NVP concentration on the structure of a polymer network and obtained kinetic parameters from a polymer release model after lysis of the polymer network.25,26 These studies confirmed that NVP incorporates in a semi-alternating fashion into the PEG-based hydrogel, as had been proposed for neat monomers, with the extent of homopolymerization varying with NVP concentration. However, the kinetics of oxygen inhibition and the initiation process were not discussed.
Eosin and triethanolamine (TEOA) have been most often employed as a visible-light-sensitive, radical production system for the synthesis of various hydrogels based on acrylate, thiol–vinyl and thiol–ene monomer chemistries copolymerized with NVP.27 Notably, no studies were found linking the radical production mechanism by eosin photocatalysis to the copolymerization reaction of the NVP/PEGDA monomers. While numerous investigations have focused on the photochemistry of eosin,28–35 few have attempted to propose a comprehensive mechanism that integrates the photochemical reactions of eosin with the non-photochemical reactions leading to the radical copolymerization of NVP and PEGDA in the presence of excess oxygen.18,36–38 Oster and Delzenne studied the regeneration of eosin by oxygen in the solution polymerization of acrylamide.39,40 Fouassier and coworkers later studied the regeneration of eosin in neat (meth)acrylate monomers without NVP.32 Later, Avens et al. analyzed the initiation kinetics of the copolymerization of PEGDA with NVP to form hydrogels with the goal of analyzing eosin regeneration as an explanation for the shorter oxygen inhibition times.18 Recently, we used kinetic modeling to discuss the link between eosin regeneration and oxygen consumption.36,37 We suggested that regeneration and resilience to O2 inhibition appeared to be associated with a primary photochemical process from the excited triplet state of eosin.38 This led us to propose a process that involves the formation of a semireduced radical trianion (EY˙3−) that can be directly oxidized back to an eosin dianion (EY2−) via oxidation by molecular oxygen, where excitation of the metastable intermediate further improves EY2− regeneration.41 However, in none of these studies was the role of NVP integrated into the proposed eosin-mediated radical production mechanism.
Here, we elucidate the role of NVP in the radical production mechanism responsible for consuming molecular oxygen and subsequently initiating the aqueous copolymerization of NVP and PEGDA monomers with eosin. We provide a clearer picture of the synergistic contribution of eosin photocatalysis and the NVP comonomer in enabling practically relevant rates of hydrogel formation in the presence of oxygen. This work bridges between eosin photocatalysis and the NVP/PEGDA copolymerization to provide additional insights into a comprehensive mechanism. We examine the kinetics of eosin and acrylate group consumption during and after oxygen inhibition to connect the photocatalytic radical production to the observed effects on the polymerization kinetics upon addition of NVP. A steady-state kinetics study of the vinyl group and eosin consumption in aqueous solution under various irradiation regimes was performed in the presence and absence of oxygen. Analysis of the initial rate of polymerization (Rp0) and the oxygen inhibition time (tinh) was employed to investigate the radical generation efficiency of the photoredox cycle. Absorbance and fluorescence spectra were then investigated to support the formation of a ground-state complex between eosin and NVP. From these studies, we discuss the applicability, or lack thereof, of the previous theories (Scheme 1) for the aqueous copolymerization of NVP and PEGDA using eosin and TEOA under exposure to green light (λmax = 500 nm) at low monomer concentrations. Most importantly, the formation of a ground-state complex between NVP and eosin is introduced as a new theory to explain the observed increase in the rates of oxygen and acrylate group consumption by the addition of NVP. Finally, we propose a previously-missed eosin inactivation pathway by radical recombination of the semireduced eosin intermediate with the propagating radicals. Our overarching goal is to provide guidelines to reduce tinh and increase the rate of polymerization at low photocatalyst concentrations.
Experimental
Materials
Poly(ethylene glycol) diacrylate (average Mn 575), triethanolamine (TEOA), 1-vinyl-2-pyrrolidone (NVP), ethyl pyrrolidone (EP), and 2′,4′,5′,7′-tetrabromofluorescein disodium salt (eosin Y) were purchased from Sigma Aldrich and used as received. Distilled water was used for the preparation of the solutions.Preparation of monomer solutions
Monomer solutions were prepared containing combinations of 420 mM PEGDA, 35 mM NVP, 210 mM TEOA, and 5 μM eosin in DI water (equivalent to 21.6% PEGDA, 2.8% TEOA, 0.4% NVP, and 75.2% water by volume). While sub-micromolar concentrations of eosin have been used, micromolar concentrations provide better UV-Vis signals for the steady-state kinetics experiments. In the case of the purged samples, argon or nitrogen gas was bubbled through the solution for 5 minutes prior to transfer to the cuvettes for polymerization. The reaction chamber was de-oxygenated for ∼10 min before the start of irradiation. Then, the samples were continuously purged during the experiments under a nitrogen or argon flow (∼8 psi) to test the effect of O2 on the reactions. An average of four replicates was performed for every set of conditions.Reaction chamber
As previously described,42 we utilized a coupled FT-NIR/UV-vis set-up. Dual pathlength (10 × 2 mm) PMMA cuvettes (UVette, Eppendorf, Hauppauge, NY) with transmission in the 220–1600 nm range were used inside a modified cuvette adapter (UVette, Eppendorf, Hauppauge, NY) with custom optical apertures. The 10 mm pathlength was used for UV-Vis probing based on the molar absorptivity of eosin and the vinyl groups, while the 2 mm pathlength was used for NIR probing, i.e. to allow detection of the low vinyl group concentration from the background associated with the broad –OH bands. The sample volume was 50 μL, which results in sample dimensions of 2 mm × 10 mm × 2.5 mm, where the latter is the thickness in the direction of the excitation light from the LED. At the initial eosin concentrations used, the 2.5 mm depth ensures operation within the thin-film approximation. Cuvettes were placed inside a CUV-ALL-UV 4-way cuvette holder (Ocean Optics, Dunedin, FL) with SMA connectors for fiber integration. Fiber optic cables were connected perpendicularly for UV-Vis and FT-NIR analyses at the same z-plane of ∼1.25 mm (half the sample depth).Steady-state kinetic monitoring of eosin decay
A fiber optic coupled UV-Vis spectrophotometer (USB4000-FL Miniature Fiber Optic Spectrometer, Ocean Optics, Dunedin, FL) was used to monitor absorbance within the 350–1000 nm range. A UV-Vis-NIR light source was used to emit the probing beam (DH-Mini, Ocean Optics, Dunedin, FL) which contains two bulbs: a deuterium and a halogen bulb. The capability of utilizing these light sources independently allowed us to eliminate the violet light from the probing beam in the kinetic experiments. The UV-Vis probing light was fed into the cuvette holder via a 600 μm solarization resistant fiber optic cable (QP-600-1-SR, Ocean Optics, Dunedin, FL), and a 50 μm receiving fiber optic cable (P50-1-Vis-NIR, Ocean Optics, Dunedin, FL) was connected to the UV-Vis spectrometer. The collimating lens in the cuvette holder and the ≲1 mm diameter pinhole limit the set of incidence angles of the UV-Vis probing beam in this set-up. The acquisition time for the UV-Vis spectrometer was set to ∼0.5 s (50 ms integration time, 10 scans to average) and a boxcar width of 4 was used. A (background) reference spectrum was collected prior to every experiment. Eosin-free solutions of PEGDA, NVP, TEA or combinations thereof showed no absorption in the visible region, but NVP and TEOA absorbed strongly in the ultraviolet region. Only the water peaks overlap slightly with the R–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H overtone. The UV-Vis probing light was adjusted to the same initial threshold intensity (∼5000 photon counts) before every experiment, as recommended by the manufacturer of the spectrophotometer. The UV-Vis probing beams in the span of hours triggered no reaction.
C–H overtone. The UV-Vis probing light was adjusted to the same initial threshold intensity (∼5000 photon counts) before every experiment, as recommended by the manufacturer of the spectrophotometer. The UV-Vis probing beams in the span of hours triggered no reaction.
Steady-state kinetics of vinyl group consumption
A fiber optic coupled FT-NIR spectrometer (Nicolet Magna-IR Series II, Thermo Scientific, West Palm Beach, FL) was used to track the vinyl group concentration. FT-NIR spectra were collected with a resolution of 8, a gain of 1, and an optical aperture of 10 with 4 scans to average for every time point. Acquisition time for the FT-NIR spectroscopy was between 0.5 and 3 s. Two 1000 μm fibers were used to feed the NIR probing light from the spectrometer to the sample and from the sample back into the InGaAs detector. The FT-NIR spectrometer has a built-in white lamp as the probing light source. No reaction was observed in the negative controls from exposure to the IR probing or reference beams.Excitation sources
A high-power fiber coupled multi-wavelength light-emitting diode (LED) light source including a green 500 nm LED (FC8-LED, Prizmatix, Southfield, MI) was used to excite eosin and initiate the polymerization from above the sample. Irradiance (power density) was controlled with a built-in potentiometer and measured with a radiometer (6253, International Light Technologies, Peabody, MA) within the 400–700 nm range. The fiber optic cable was connected to a collimating (focusing) lens. A 3D printed cap was placed on top of the cuvette to reduce the amount of noise read by the UV-Vis spectrophotometer from the LED. The irradiance values from the radiometer were corrected for the presence of the smaller cross-section area provided by the 3D printed cap.A custom-made polycarbonate transparent box was built to enclose the CUV-ALL-UV 4-way cuvette holder for oxygen-free experiments. The enclosure has fittings for every fiber optic cable described above, as well as gas fittings to allow the flow of nitrogen or argon during the experiments. The enclosure was sealed after being assembled and before every experiment to ensure that a positive pressure is built inside the reaction chamber.
Initial experiments were performed in water without a hydrogel precursor, e.g. PEGDA. However, coupled UV-Vis/FT-NIR allowed us to replicate these experiments with PEGDA to validate the proposed mechanism in polymerizing media, focusing on the O2 inhibition.
UV-Vis spectroscopy
A Cary 50 Bio UV-Vis spectrophotometer (Varian Inc.-Agilent Technologies, Santa Clara, CA) was used to study the photoreduction of eosin Y in the presence of TEOA and NVP. A custom-made green LED (530 nm) source was utilized to irradiate 3 ml aqueous solutions inside glass cuvettes. Discrete absorbance scans (<1 min) were taken at 20, 30 and 60 s intervals. The absorbance was also monitored after >95% of the eosin Y was consumed. The irradiance of the green LED was estimated to be ∼35 mW cm−2 using an Advanced Light Meter (SPER Scientific, Scottsdale, AZ).Aqueous solutions including 0.035 mM NVP, 0.035 mM PEGDA, or 1.5 mM TEOA were prepared. Each solution was purged in a UV cuvette for 30 minutes under pressurized nitrogen gas with a flow rate of 5 LPM.
Steady-state fluorescence
Aqueous solutions including 5 μM eosin Y, 100 mM NVP, 210 mM TEOA, or 420 mM PEGDA were prepared. A Tecan Infinite 200 (Tecan Austria GmbH, 5082 Grödig, Austria) was used to obtain the absorbance and fluorescence spectra of 200 μl of each prepared solution. For fluorescence measurements, eosin Y in each solution was excited at 480 nm.LC-MSD
Liquid chromatography (LC), Agilent 1100 LC, coupled and a single quadrupole mass spectrometer spectrometry (LC–MS), Agilent.Results and discussion
Reduction of the oxygen inhibition time by NVP
From a linear regression of the acrylate group conversion data between 2 and 12% conversion we obtain values for tinh as the intercept when conversion approaches zero (Fig. 1). Despite the limitations of analyzing conversion with FT-NIR, other methods, like photo-DSC, are not suitable for highly diluted polymerizations in water. Useful data were acquired under every set of conditions from at least 5 replicates. From the analysis of the data in Fig. 1B, tinh = 41.81 ± 1.24 s for the copolymerization of NVP and PEGDA equilibrated with atmospheric oxygen. It should be noted that when the solutions were purged with argon or nitrogen, a shorter tinh was obtained from residual oxygen, 2 < tinh < 10 s, omitted from the analysis (Fig. 1A). When NVP is removed, the inhibition time increases to tinh = 48.33 ± 4.25 s (Fig. 1B). Hence, NVP reduced tinh by 6.5 s when copolymerized with PEGDA. This reduction in tinh is lower than that reported for the solvent-less copolymerization with acrylate monomers,22,24,25 which could be explained by the elimination of the reactive diluent effects in our water-based systems. It is worth noting that all photopolymerizations were run under quasi-laminated conditions since the cuvettes were capped with a custom-made plastic slit and inside a closed reaction chamber.33 Hence, it can be expected that the rate of replenishment of molecular oxygen is significantly slower as compared to surface polymerization completely exposed to ambient oxygen through a much wider area, which is the case for previous thin-film experiments run in the FT-NIR spectroscopy.25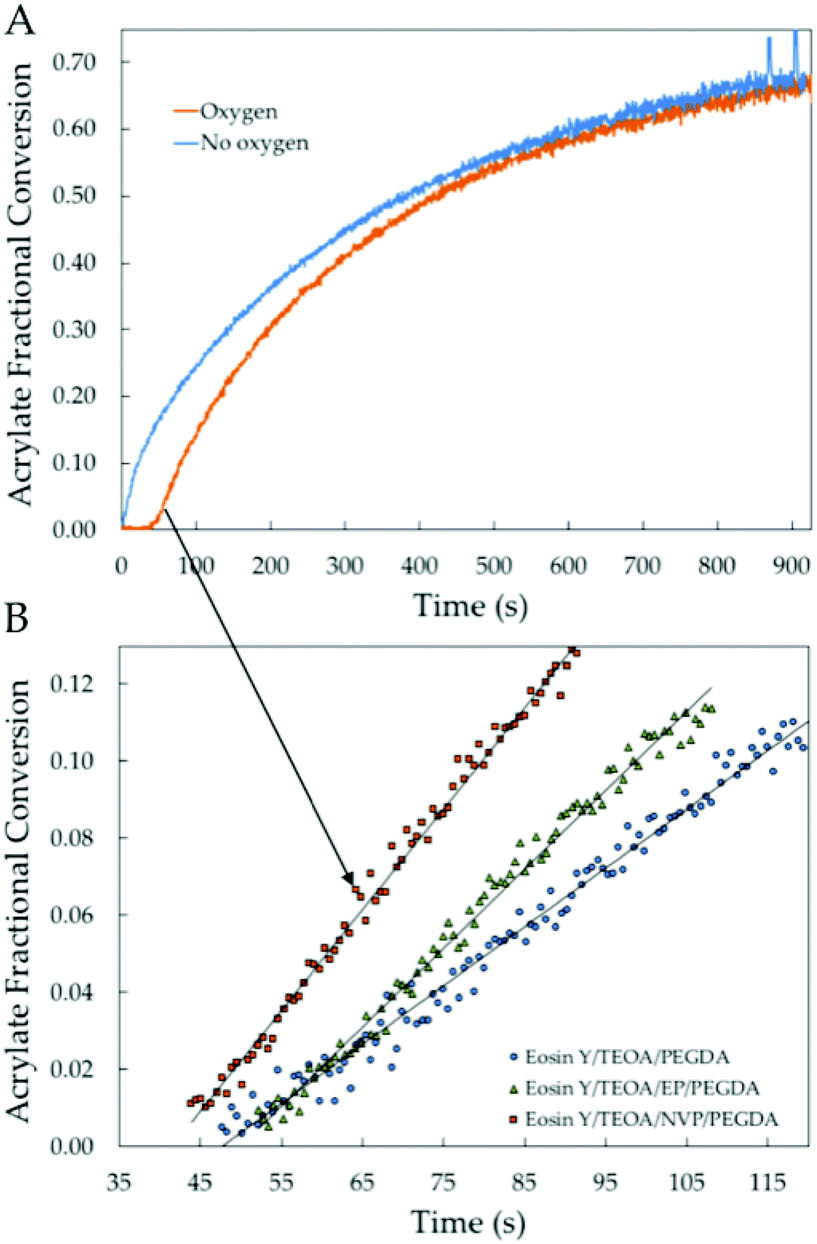 | ||
| Fig. 1 (A) Averaged fractional vinyl conversion related to the acrylate groups as a function of time for the polymerization of PEGDA in the presence and absence of oxygen. (B) From the slopes of the averaged vinyl fractional conversion results between 0 and 12% conversion we extract the initial rate of polymerization Rp0 in order to analyze the relative rates of initiation (Ri) from the expected scaling. | ||
To analyze the effect of the vinyl group of NVP on the reduction of tinh we substituted NVP for ethylpyrrolidone (EP) as its non-vinyl analog. The calculated value for the polymerization of PEGDA in the presence of EP was tinh = 50.95 ± 4.73 s. The oxygen inhibition time does not appear to change upon addition of EP to PEGDA solutions (Fig. 1B). While determining the relevance of the vinyl group is helpful, there are many ways in which the vinyl group can participate in the oxygen consumption pathway. Husár et al. compared N-methyl-pyrrolidone with NVP and determined that both had a positive effect.22 However, their analysis was based on the final double bond conversion, and not on the oxygen inhibition time. No values were found for tinh obtained for the copolymerization of different N-vinyl amides with multifunctional acrylates.
In general, few reports have reported the quantitative values of the oxygen inhibition time. Inhibition times are often difficult to accurately measure due to the limitations in measuring small changes in the FT-IR peaks at the start of the polymerization process. In these studies, the irradiation intensity and the FT-NIR parameters were selected to minimize the experimental errors. Boyer and coworkers have studied oxygen inhibition in controlled/living radical polymerization with various monomers and reported that NVP showed no inhibition time in DMSO when exposed to visible-light in the presence of Ir[ppy]3, as compared to other unconjugated and conjugated monomers that show inhibition times of up to 5 hours.21 While eosin has been the preferred photocatalyst for metal-free ATRP, we could not find studies on the effect of the photocatalyst on the inhibition period for these systems. Molecular oxygen was not integrated as part of the initiation mechanism, thus an explanation of the absence of an inhibition period was not discussed. It appears possible that regardless of the oxygen scavenging that may occur as part of iridium photocatalysis, the NVP monomer exhibits some unusual resilience to oxygen inhibition, perhaps by a lower reactivity of the NVP propagating radicals towards the reaction with molecular oxygen. In our case, however, we aim at integrating NVP into the radical production mechanism to propose an explanation for the observed reduction in oxygen inhibition.
Oxygen scavenging by NVP photo-oxidation does not appear to explain the results
It is widely known that amines reduce oxygen inhibition through a chain peroxidation reaction, where oxygen quenches the aminoalkyl radicals from the photo-oxidation of TEOA to form peroxyl radicals that abstract a hydrogen from an unreacted amine to form a new aminoalkyl radical and scavenge molecular oxygen in the process.14,22 While chain peroxidation certainly takes place in most neat (meth)acrylate monomers and resin formulations, it has been reported that amino peroxyl radicals are quite unstable in basic aqueous solutions.43 For instance, triethylamine does not appear to significantly form the expected peroxyl radicals upon radiolysis, and instead decays to a stable iminium species depending on the pH of the aqueous solution.Here, we propose that molecular oxygen is primarily scavenged by a non-chain peroxidation of TEOA that culminates in the production of stable iminium cations and superoxide. The superoxide can subsequently react to form relatively stable hydroperoxide by disproportionation41 and also alkyl peroxides. If this is the case, it implies that every aminoalkyl radical scavenges one oxygen molecule.
In fact, we previously documented that the inhibition time (tinh) scales linearly with incident irradiance (I0). From kinetic simulations, we had proposed that a chain peroxidation seemed insufficient to account for the oxygen inhibition times that were obtained experimentally.36 Here, we confirmed that the linear scaling is preserved for a wider range of eosin concentrations and irradiance levels, expressed in terms of the number of moles of photons (Einsteins) absorbed per time per volume (Fig. 2). This would be expected of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 O2 per aminoalkyl radical ratio, where chain peroxidation does not occur, or is not the rate limiting process.30,31
:1 O2 per aminoalkyl radical ratio, where chain peroxidation does not occur, or is not the rate limiting process.30,31
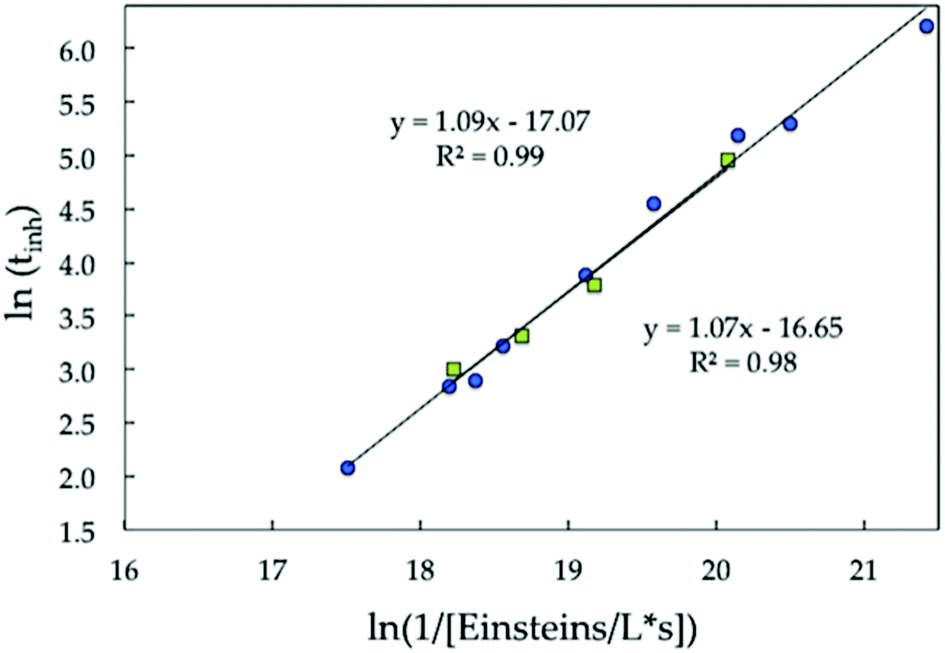 | ||
| Fig. 2 Scaling of the oxygen inhibition time (tinh) with the number of photons absorbed obtained at irradiance values between 1 and 10 mW cm−2 with emission at λmax = 500 nm (green squares) and eosin concentrations varying between 0.5 and 20 micromolar (blue dots). | ||
We then proceeded to verify if a ratio of one oxygen molecule consumed per aminoalkyl radical could account for the observed rates of oxygen consumption. The experimentally measured tinh for an incident irradiation of ∼1 mW cm−2 was found to be on average 50 seconds. Considering a quantum yield of photoreduction by TEOA of 0.76 and an extinction coefficient of 5.26 × 104 M−1 cm−1 at λex = 500 nm, and a pathlength of 0.5 cm, we calculated a tinh between 30 and 50 s, depending on the rate of photobleaching by the optically thick samples, which indicates that an oxygen concentration higher than the initial can be readily consumed at this photon absorption rate in about the same tinh that was observed experimentally. This supports the idea that a chain peroxidation is not required to explain the oxygen inhibition times reported for the eosin/TEOA/NVP/PEGDA system.
Additionally, we detect the formation of some of the expected photodecomposition products when irradiating aqueous solutions containing eosin/TEOA/NVP in the presence of oxygen. LC-MS with a UV-Vis detector shows a complex mixture of low and high molecular weight products from the irradiation of TEOA/NVP aqueous solutions in the presence of oxygen (Fig. S1 ESI†).
If chain peroxidation is precluded by the lack of formation of amino peroxy radicals in water, it is reasonable to expect that NVP does not participate as a hydrogen donor in the oxygen scavenging mechanism. In consequence, it appears unlikely that neither the photo-oxidation nor the ground-state oxidation of NVP can explain the small albeit noticeable reduction in oxygen inhibition. No studies were found on the photochemical oxidation of NVP that showed a unique photo-oxidative process that could explain the observed reductions in tinh.
No evidence of the formation of a ground-state charge-transfer complex or an exciplex between NVP and oxygen was obtained
The formation of a sufficiently strong ground-state charge-transfer complex can be typically evidenced in the UV-Vis spectrum of either the donor or acceptor molecules. If the absorbance peak for the acceptor molecule is analyzed, the shift is expected to occur to lower energy frequencies due to the increase in electronic resonance. We analyzed the UV-Vis spectra of all binary combinations: eosin/TEOA, eosin/NVP, eosin/PEGDA, TEOA/NVP, TEOA/PEGDA, NVP/PEGDA and NVP/O2 to investigate if ground-state complexes were formed. For the case of weaker EDA complexes, other techniques have been used: EPR, NMR, polarography and fluorescence spectroscopy. We analyzed changes in the fluorescence emission for cases where the UV-Vis spectra indicated that a weak complex was possible.Takeishi and Tao reported a reversible bathochromic shift in the absorbance spectrum of NVP upon addition and removal of molecular oxygen.44 Their results were later validated by Hoyle and coworkers, adding that the vinyl group in NVP did not have a significant effect on the formation of the CTC.24 We replicated their experiments in our aqueous NVP solutions and found no evidence of the formation of a CTC between NVP and O2 (Fig. S2 ESI†). The latter indicates that while the NVP/O2 CTC may form in neat monomers, it does not seem to be significant in water. Furthermore, for the formation of a complex with oxygen to have an effect in tinh, one must have evidence of NVP oxidation products. Since the participation of NVP in the oxygen scavenging mechanism appears to be unlikely in this case, as discussed above, one can safely assume that any change in the reaction kinetics or production of an additional initiating radical from an NVP/O2 complex cannot explain the present results. Thus, it seems that the formation of a CTC or exciplex between NVP and oxygen is also not significantly occurring in the present formulation.
No evidence of the formation of a charge-transfer complex between monomers was found
We found no evidence for the formation of a CTC between NVP and PEGDA (Fig. S2†). While the formation of donor/acceptor complexes between NVP and other monomers, e.g. maleimides, has been reported, we observed no variations in the absorbance or fluorescence spectra upon addition of PEGDA to an NVP aqueous solution. The absence of changes in the spectra does not completely eliminate the possibility of a weak complex between the electron acceptor PEGDA and the electron donor NVP. However, the theory of CTC between NVP and acrylates has been deemed the least plausible theory to explain the observed results in neat monomer formulations.14Ground-state complex between eosin and NVP may explain the reduction in tinh
While the eosin spectrum remained constant upon addition of TEOA, we observed a bathochromic shift of Δλmax = +2 nm in the eosin spectrum upon addition of NVP (Fig. S3 and S4 ESI†). This shift is close to the instrumental error, but we hypothesized that it may indicate the formation of a weak ground-state complex. Hence, we looked for additional evidence by analyzing variations in the fluorescence spectra for all solutions excited at 480 nm (Fig. 3). We obtained the same +2 nm shift in the fluorescence maximum upon addition of NVP to the eosin solutions. | ||
| Fig. 3 Fluorescence emission spectra of eosin excited at 480 nm in water and the effect of NVP, TEOA, and PEGDA. | ||
Additionally, we observed that the fluorescence intensity decreased upon addition of NVP (Fig. 3). This is interesting, because NVP does not lead to eosin decomposition based on our steady-state kinetic studies (Fig. 4A). Hence, we know that the photoinduced electron transfer from NVP is not significant. These results strongly suggest that a ground-state eosin/NVP complex may be formed, and that it may change the excited state dynamics favoring photoinduced electron transfer. In contrast, we know that TEOA quenches the singlet and triplet states of eosin via photoinduced electron transfer, thus leading to a drastic decay in the fluorescence intensity.
 | ||
| Fig. 4 (A) Normalized eosin concentration as a function of irradiation time (s) at 1.25 mW cm−2 for solutions containing eosin, eosin/NVP, eosin/TEOA, eosin/TEOA/NVP and eosin/TEOA/NVP/PEGDA. (B) Electronic spectra of eosin in solutions upon addition of NVP, TEOA and PEGDA. | ||
We propose that NVP forms a weak eosin/NVP ground state complex that may sufficiently alter the electronic density of the excited state eosin and slightly increase the rate of photoinduced electron transfer to TEOA (Scheme 2). Since the rate of PET from TEOA is already close to diffusion control,44 it can be reasonably expected that any contribution of NVP to the rate of PET will be translated into a small improvement in the oxygen scavenging rate, where the formation of excited state eosin is rate-limiting at the relatively low irradiance and eosin concentrations used. It is likely that the ground-state complex is excited into an exciplex that then reacts with TEOA. NVP would then be unreacted after PET. This would be possible if NVP is complexed with the xanthene core and the excited-state dynamics allowed electron density to move to the benzoic moiety before reaction with TEOA. Quantum chemical calculations in the excited state and transient spectroscopy experiments could further aid in supporting the viability of this unexpected reaction step.
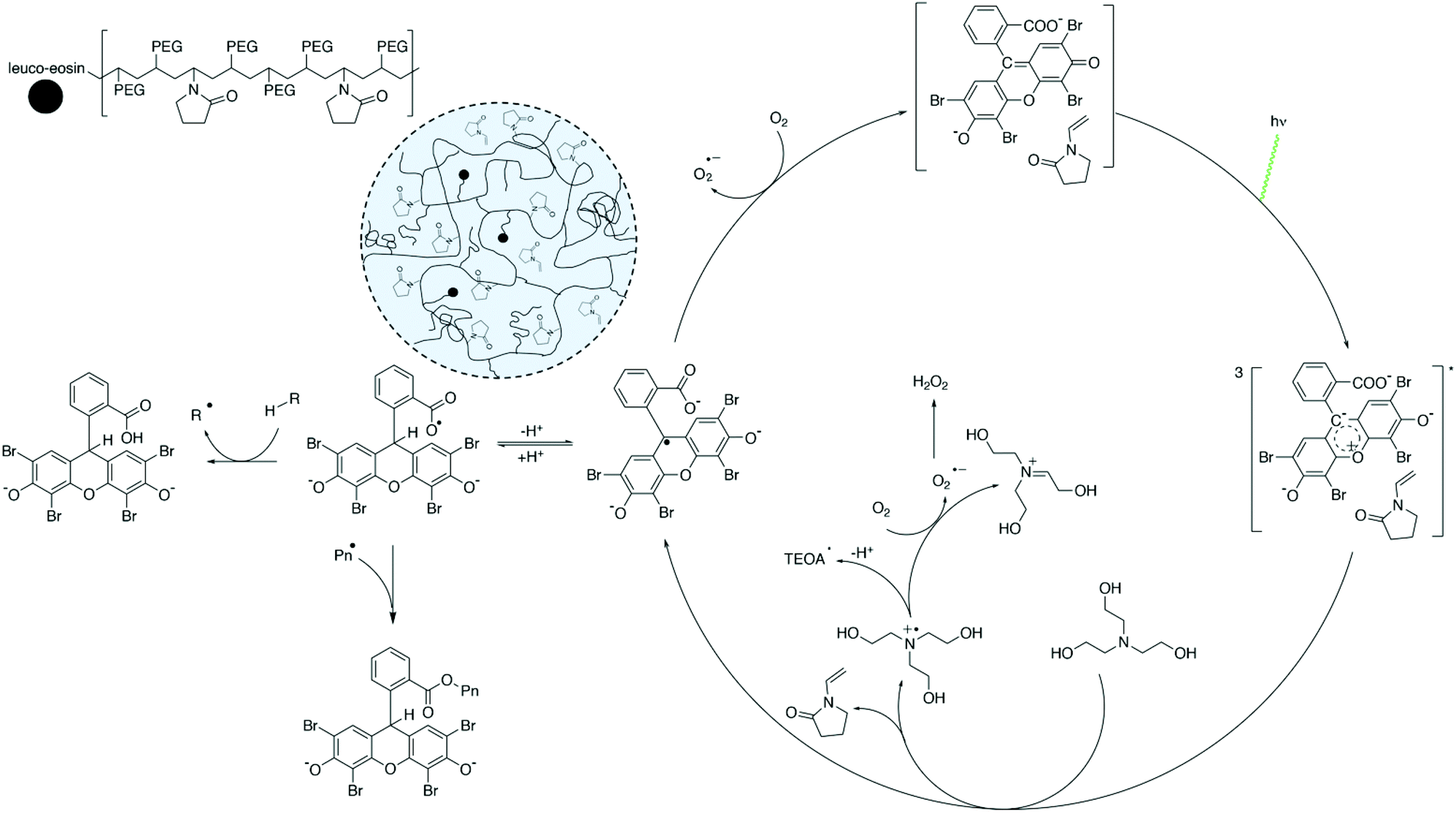 | ||
| Scheme 2 Proposed mechanism for the copolymerization of NVP and PEGDA in water induced by eosin including the proposed formation of an eosin/NVP CTC to enhance the rate of consumption of molecular oxygen. | ||
The formation of the ground-state complex between eosin/NVP is a significant finding because it supports the idea that eosin photocatalysis and NVP have a synergistic contribution towards oxygen scavenging (Scheme 2). Furthermore, the possibility of forming a ground-state complex with the photoinitiator or photosensitizer has not been considered as a likely explanation of the increase in the rates of oxygen scavenging. On one hand, a first oxygen molecule is converted into hydro or alkyl peroxides via the non-chain peroxidation of TEOA after photoinduced electron transfer to eosin with the help of NVP to slightly change the rate of photo-oxidation of TEOA. On the other hand, a second oxygen molecule is converted into hydrogen peroxide via the photocatalytic regeneration of eosin (Scheme 2).
PEGDA may decrease the formation of the eosin/NVP complex
In addition to the observed changes in the absorbance spectra with the addition of NVP, we observed a greater bathochromic shift in the eosin electronic spectrum when PEGDA was added to the solutions from 517 nm to 525 nm (Fig. 4B). An isosbestic point was clearly identified around 440 nm. The red-shift in absorbance was accompanied by a shift in the fluorescence emission by about the same Δλ, along with a decrease in the fluorescence intensity. Variations in the absorbance and emission spectra of eosin as a function of the solvent polarity lead to exactly the same result.45 The Stokes shift appears to remain constant upon addition of both NVP and PEGDA (Fig. S5 ESI†).We propose that this shift is not due to the formation of a complex, but due to the change in the polarity of the medium where eosin resides and the associated consequences on the solvation, excited-state dipole moment, and solvent reorganization effects. However, it has proven difficult to establish a generalization of the trends in the effects of polarity on the Stokes shifts.45
Most importantly, these observations, along with the known hydrophobicity of eosin, allow us to propose that eosin is concentrated around PEGDA. Hydrophobic eosin may reasonably be expected to concentrate in the carbon-rich PEGDA domains, as evidenced by the SANS measurements of PEGDA solutions before photocrosslinking.46 In fact, we reported variations in light scattering associated with the formation of PEGDA clusters before and during irradiation. This is analogous to the well-documented nucleation of eosin, and other organic dyes, around surfactants and proteins.47,48 An important implication of this is that the local eosin concentration may be higher in these carbon-rich regions, thus affecting the quantum yields and reaction kinetics.
The effect of hydrogel formation on the oxygen inhibition
As we have previously reported, the regeneration of eosin can reach 100% under the appropriate oxygen concentration and irradiation regimes, e.g. when eosin/TEOA/NVP/PEGDA solutions are irradiated with 500 nm light.41 We confirmed that the eosin concentration is held almost constant for the initial ∼45 s of irradiation when PEGDA is present (Fig. 4B). The time to deplete O2 decreased from 250 s in plain water to 50 s with PEGDA, as extracted from the inflections in the eosin decay curves. After O2 is consumed, the rate of eosin reduction is slightly faster than the rate in the absence of PEGDA. This can be attributed to a lower O2 diffusion when a hydrogel is formed and/or an increase in the local concentration of eosin, as reported for PEG hydrogel microspheres.49Considering the initial oxygen concentration in the eosin/TEOA/NVP/PEGDA solution (∼0.58 mM),36 it is impressive that the oxygen inhibition times are shorter than in the absence of PEGDA, where the equilibrium oxygen concentration is ∼0.27 mM. These observations suggest that the aggregation of eosin in the PEGDA-rich regions accelerates O2 depletion, and eosin is then consumed more quickly in the presence of PEGDA because of a lower rate of O2 replenishment. It is reasonable to propose that during the PEGDA crosslinking process, oxygen replenishment into the eosin rich hydrophobic domains is hindered, where DO2–PEG ≈ 0.001 mm2 s−1 depending on the macromolecular architecture and the degree of crosslinking,49 as compared to that in plain water, DO2–H2O ≈ 0.002 mm2 s−1.50 Thus, eosin is consumed more rapidly after O2 depletion if PEGDA is present. Hence, we can conclude that PEGDA increases the initial oxygen solubility but reduces the rate of oxygen replenishment by diffusion once the hydrogel forms, which helps reduce the inhibitory effect.
Explanation for the increase in the initial rate of polymerization
After investigating the mechanism responsible for scavenging oxygen, we turn to analyze the effect of NVP addition on the rate of polymerization once oxygen is mostly depleted. As stated above, the initial rates of polymerization were obtained from the vinyl conversion data between 2 and 12%, typically, which fitted well a linear regression. In oxygenated solutions, the initial rate of polymerization with NVP, R′p0, gave a value of 0.0026 mol L−1 s−1, while the rate in the absence of NVP gave a value of Rp0 = 0.0015 mol L−1 s−1. This represents an ∼70% improvement in the rate of vinyl group consumption. Such an increase in Rp0 is considerable compared to the rate enhancements of up to 700% obtained for neat penta-acrylate monomers.25 In contrast, NVP increases the rate of polymerization of neat diacrylates only slightly. Hence, our observed rate increase falls closer to values reported for tri and tetra-functional neat monomers.
In contrast, when analyzing the effect of NVP in deoxygenated solutions we obtained an ∼40% improvement in the rate of vinyl group consumption. The smaller increase in Rp0 in deoxygenated solutions may be due to the effect that oxygen replenishment may have in the experiments that were not purged with argon or nitrogen during irradiation. It is important to note that a smaller increase in the rate of polymerization was observed when substituting NVP for EP. It appears that this increase falls within the instrumental error. However, an increase in the oxygen resilience and the final conversions has also been reported with N-alkyl amides in neat monomer formulations.14
From the analysis of the classical expression for the rate of polymerization we can deduce that the increase in the initial rate of polymerization after oxygen depletion cannot be solely explained by the formation of an eosin/NVP CTC. This stems from the fact that the rate of polymerization is proportional to the rate of initiation to the power of 0.5 for a model dominated by bimolecular termination. We obtained a scaling of 0.41 (Fig. S6 ESI†), which agrees well with the reported value of 0.46 by Avens and Bowman.18 This can be explained by a small contribution of the primary radical termination mechanism.

On the other hand, the local concentration of vinyl groups, [M], is not expected to be affected by the addition of such a small concentration of NVP, 35 mM, to an already highly dilute monomer solution at 75% water by volume. The double bond percent associated with NVP is calculated to be 4%.
In neat acrylate monomers of higher functionality, the rate enhancements have been associated partly with a plasticizing effect that aids in the propagation of the macroradicals.25 However, in this highly mobile system, these effects must be minimal, and a lower contribution of NVP can be expected. Furthermore, increasing the mobility would result in an increase in the rate of bimolecular termination, as was expected for conventional reactive diluents.25 However, the termination rate constant has been observed to remain relatively constant upon addition of NVP to acrylates. Hence, most of the improvements in the vinyl consumption rate have been proposed to be due to an increase in the kinetics of cross-propagation.25
NVP decreases the average functionality and it is expected that the crosslink density of the diacrylate network at a fixed double bond conversion is decreased by copolymerization with low concentrations of NVP (Scheme 2).25 A lower crosslink density means a more mobile network in which diffusional effects on the propagation and termination constants are delayed to higher conversions. The crosslinking density of the polymer network is expected to decrease upon incorporation of NVP into the network. The crosslinking of the network via the pendant cyclic amide groups appears to be negligible. While the addition of NVP does increase the acrylate conversion, which may lead to an increase in the crosslink density, this most likely has an effect at a later stage in the polymerization process. Therefore, we propose that the increase in the initial rate of polymerization is best explained by a reduction in the segmental mobility of the PEG segments upon addition of NVP units.
As a result, it appears that the increase in the rate of polymerization must be associated with an increase in the propagation rate constant, kp. The propagation rate constant is also expected to change due to cross-propagation. On the other hand, we hypothesize that the termination remains fairly constant due to two counter-acting effects: the decrease in cross-linking density due to NVP incorporation may favor bimolecular termination, while the increased stiffness of the polymer segments hinders macroradical motion. It is known that the glass transition temperature for the diacrylate is 100 °C, while the Tg for NVP is 180 °C.25 Given that poly-NVP has typically a low crosslink density, the higher Tg can be associated with the higher stiffness of the polymer backbone and the supramolecular interactions of the side groups. While these two counter-acting effects may keep the kt constant, the increased mobility of the hydrophilic NVP through the network and the cross-propagation could broaden the reaction–diffusion controlled kinetics region. Unsteady-state kinetic experiments are warranted in order to decouple kt and kp and confirm that in this formulation, kt also remains constant.
It has been reported that in most NVP/acrylate copolymerizations the acrylate groups are consumed considerably faster than the NVP vinyl groups, even reaching a ratio of 80% consumption of diacrylate to 20% consumption of the NVP vinyl group.24,25 The latter is due to the reactivity ratios, which are reported to be r1 ≈ 0.14 and r2 ≈ 0.09 for the acrylate/NVP copolymerization.26 As a result, and considering that in this formulation PEGDA and NVP appear to be concentrated in different regions, one can expect that the polymer network formed is rich in PEG. Hence, a significant fraction of NVP may be expected to remain unreacted once the plateau conversion is reached.
NVP increases the mobility of the polymer network
Now that we established that the cross propagation mediated by the mobile hydrophilic NVP is largely responsible for the increment in the initial rate of polymerization, it follows that it may also explain the higher final conversions observed upon addition of NVP. We observed that NVP increases the final overall conversion from 50 to 60% (Fig. 5). On the other hand, EP only increases the final conversion by 2%, which is essentially within the instrumental error (standard deviation of 1.8%). To put this into perspective, a 12% increase in the final double bond conversion has been observed upon addition of NVP to formulations for organic coatings.22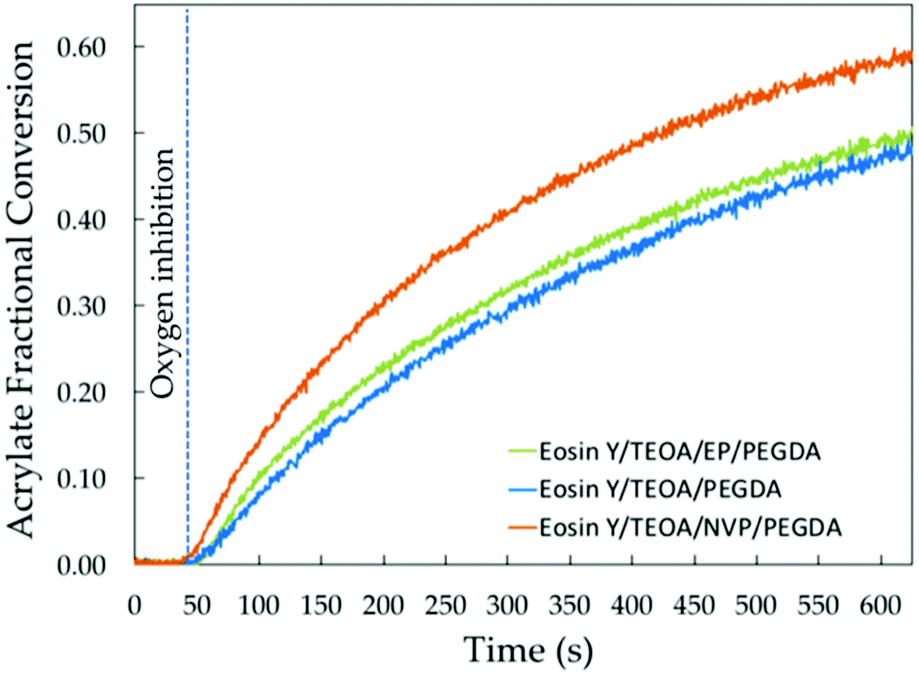 | ||
| Fig. 5 Vinyl fractional conversion from the acrylate groups as a function of irradiation time (s) for solutions containing PEGDA, EP/PEGDA and NVP/PEGDA. | ||
Others have decoupled the acrylate and NVP vinyl conversions using FT-IR to confirm that at high acrylate-to-NVP ratios, a significant amount of NVP remains unreacted by the time the final conversion in the acrylate polymerization is reached.25 Here, we examined aqueous NVP solutions in the absence of PEGDA with FT-NIR to test whether the conversion of the vinyl groups in NVP at the concentrations used could be quantified. However, the signal in the FT-NIR spectra was too low for conversion to be resolved. Nonetheless, the comparison with EP implies that the vinyl groups in NVP are being consumed as NVP copolymerizes into the hydrogel, but that a significant fraction may remain unreacted by the time the plateau conversion is reached. The overall fractional conversions of CC bonds reported herein are associated with the total concentration of double bonds from both acrylate and NVP monomers.
The increase in the final conversion can be explained by cross propagation. As previously reported, addition of NVP results in a higher final crosslink density due to higher double bond conversion.25,26 Additionally, increasing the NVP/acrylate ratio has been reported to result in an increase in the molecular weight of the copolymer backbone.26 Hence, the swelling rate is expected to decrease due to the additional mobility restrictions. This, however, may exacerbate the effect of cross-propagation on the polymerization kinetics.
Radical recombination between semireduced eosin and propagating radicals after oxygen depletion
From the analysis of the copolymerization kinetics, we wanted to explore the connection between the eosin initiation kinetics and the polymerization kinetics.41 The scaling of the initial rate of polymerization to the incident irradiance tells us that there must be some contribution of primary radical termination in the system. Considering the previously reported theories on eosin regeneration by peroxy radicals and molecular oxygen, we then analyzed the scaling of the eosin consumption coefficient as a function of incident irradiance. This had not been done, since the eosin concentration is not typically monitored during the photopolymerization reaction.We investigated the dependence of eosin consumption on intensity for the case of eosin/TEOA in the absence of oxygen. We indeed obtained a linear scaling in this case. Then, we studied the system in the presence of the PEGDA monomer and the NVP comonomer in the absence of oxygen. In this case, only three of the four experiment sets were useful due to the increase in the experimental error with the increasing extent of polymerization associated with light scattering. From the three data points, we were able to identify that the scaling in this case approximates to 0.73 (Fig. 6). This could be explained by a process in which the concentration of eosin is dependent on the concentration of propagating radicals (Scheme 2). In this case, carbon-centered radicals can be expected to react with eosin.
 | ||
| Fig. 6 (A) Correlation of the eosin decay coefficient with the incident irradiation intensity for the solutions with and without monomers in the absence of oxygen. (B) Correlation of the eosin decay coefficient with the incident irradiation intensity for eosin/TEOA/NVP/PEGDA solutions during and after oxygen inhibition. | ||
Then, we analyzed the scaling of eosin during and after oxygen inhibition by irradiating the samples. We confirmed a linear scaling during oxygen inhibition and a 0.6 scaling after oxygen inhibition (Fig. 4B). This further supports the idea that eosin and its intermediates may react with propagating radicals after oxygen is depleted.
We propose that the scaling must be linear in the presence of oxygen but between 0.5 and 1.0 after oxygen inhibition. The latter would be the result of eosin concentration being coupled to the radical propagation kinetics. This implies that while the semireduced eosin could abstract a hydrogen in the presence of good hydrogen donors, an additional eosin inactivation pathway is the radical recombination with the macroradicals from the copolymerization between NVP and PEGDA. If the semi-reduced eosin radicals react with the macroradicals that would lead to the covalent attachment of leuco-eosin to the polymer network (Scheme 2). In this case, we propose this reaction step as an additional eosin reduction pathway. We hypothesize that grafted leuco-eosin will not be active for further photoinitiation under exposure to green light. This differs from previous reports where eosin was grafted to surfaces and nanoparticles via peptide bond forming conjugation methods before polymerization.19,51 Kızılel and coworkers showed that eosin can initiate the interfacial polymerization of PEG/NVP hydrogels both when eosin is covalently attached to the surface and when it is simply adsorbed by electrostatic interactions. In our case, we did not aim to create a functional eosinylated polymer network, but rather to suggest a possible route for eosin decay that has not been noted previously.
Conclusions
Here, we investigated the role of NVP in the copolymerization with PEGDA in water to form hydrogels mediated by eosin. NVP reduces the oxygen inhibition time and increases the initial rate of polymerization and the final plateau conversion. We propose that the reduction in oxygen inhibition is due to the formation of a ground-state complex between NVP and eosin, which slightly enhances the rate of photoinduced electron transfer to TEOA, where two oxygen molecules are consumed per PET event. Complexation between NVP and the chromophore in photopolymerizations is an important new theory that may explain the reduction of oxygen inhibition in other formulations. Furthermore, we discuss the explanation of the increase in the rate of polymerization as a complex effect of the incorporation of NVP into the polymer network with unique cross-propagation kinetics in a system that transitions from diffusion-control to reaction–diffusion control. Finally, we introduce a previously missed eosin inactivation pathway whereby semireduced eosin reacts via radical recombination, explaining termination kinetics between primary radical and bimolecular termination.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
A CONACYT (Consejo Nacional de Ciencia y Tecnología Award No. I0010-2015-01-263622/I0010-2016-02-275449) Postdoctoral Research Award (to A. A.), Kwanjeong Educational Foundation Scholarship (to S. K.), a National Science Foundation Graduate Research Fellowship (to K. K.), a Burroughs Wellcome Fund Career Award at the Scientific Interface (to H. D. S.) and the Department of Defense (Congressionally Directed Medical Research Program, Prostate Cancer Research Program Award No. W81XWH-13-1-0272) supported this work. The opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense. We thank Prof. Jeffrey W. Stansbury for the use of the coupled UV-Vis/FT-NIR spectroscopy apparatus, developed with the support of the NSF Industry/University Cooperative Research Center for Fundamentals and Applications of Photopolymerization.Notes and references
- C. Wang, Y. Ye, Q. Hu, A. Bellotti and Z. Gu, Adv. Mater., 2017, 29, 1606036–1606024 CrossRef PubMed.
- J. E. V. Ramirez, L. A. Sharpe and N. A. Peppas, Adv. Drug Delivery Rev., 2017, 114, 116–131 CrossRef PubMed.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- G. Qin, Z. Zhu, S. Li, A. M. McDermott and C. Cai, Biomaterials, 2017, 124, 55–64 CrossRef CAS PubMed.
- J. Liu, Y. Pang, S. Zhang, C. Cleveland, X. Yin, L. Booth, J. Lin, Y.-A. L. Lee, H. Mazdiyasni, S. Saxton, A. R. Kirtane, T. von Erlach, J. Rogner, R. Langer and G. Traverso, Nat. Commun., 2017, 8, 124 CrossRef PubMed.
- A. S. Sawhney, C. P. Pathak and J. A. Hubbell, Biomaterials, 1993, 14, 1008–1016 CrossRef CAS PubMed.
- L. A. Hockaday, K. H. Kang, N. W. Colangelo, P. Y. C. Cheung, B. Duan, E. Malone, J. Wu, L. N. Girardi, L. J. Bonassar, H. Lipson, C. C. Chu and J. T. Butcher, Biofabrication, 2012, 4, 035005 CrossRef CAS PubMed.
- C. A. DeForest and D. A. Tirrell, Nat. Mater., 2015, 14, 523–531 CrossRef CAS PubMed.
- S. A. Morin, R. F. Shepherd, S. W. Kwok, A. A. Stokes, A. Nemiroski and G. M. Whitesides, Science, 2012, 337, 828–832 CrossRef CAS PubMed.
- H. D. Sikes, R. R. Hansen, L. M. Johnson, R. Jenison, J. W. Birks, K. L. Rowlen and C. N. Bowman, Nat. Mater., 2007, 7, 52–56 CrossRef PubMed.
- B. V. Slaughter, S. S. Khurshid, O. Z. Fisher, A. Khademhosseini and N. A. Peppas, Adv. Mater., 2009, 21, 3307–3329 CrossRef CAS PubMed.
- W. E. Hennink and C. F. van Nostrum, Adv. Drug Delivery Rev., 2012, 64, 223–236 CrossRef.
- K. Sadtler, A. Singh, M. T. Wolf, X. Wang, D. M. Pardoll and J. H. Elisseeff, Nat. Rev. Mater., 2016, 1, 16040 CrossRef CAS.
- S. C. Ligon, B. Husár, H. Wutzel, R. Holman and R. Liska, Chem. Rev., 2014, 114, 557–589 CrossRef CAS PubMed.
- C. G. Williams, A. N. Malik, T. K. Kim, P. N. Manson and J. H. Elisseeff, Biomaterials, 2005, 26, 1211–1218 CrossRef CAS PubMed.
- P. Xiao, J. Zhang, F. Dumur, M.-A. Tehfe, F. Morlet-Savary, B. Graff, D. Gigmes, J.-P. Fouassier and J. Lalevée, Prog. Polym. Sci., 2015, 41, 32–66 CrossRef CAS.
- L. Kuck and A. Taylor, Biotechnology, 2008, 45, 179–186 CrossRef CAS PubMed.
- H. J. Avens and C. N. Bowman, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6083–6094 CrossRef CAS PubMed.
- S. Kızılel, V. H. Pérez-Luna and F. Teymour, Langmuir, 2004, 20, 8652–8658 CrossRef PubMed.
- K. S. Anseth, C. N. Bowman and L. Brannon-Peppas, Biomaterials, 1996, 17, 1647–1657 CrossRef CAS PubMed.
- (a) S. Shanmugam, J. Xu and C. Boyer, Macromolecules, 2014, 47, 4930–4942 CrossRef CAS; (b) J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed; (c) J. Yeow, R. Chapman, A. J. Gormley and C. Boyer, Chem. Soc. Rev., 2018, 47, 4357–4387 RSC.
- B. Husár, S. C. Ligon, H. Wutzel, H. Hoffmann and R. Liska, Prog. Org. Coat., 2014, 77, 1789–1798 CrossRef.
- Y. C. Lai and J. Appl, Polym. Sci., 1997, 66, 1475–1484 CAS.
- C. W. Miller, C. E. Hoyle, S. Jönsson, C. Nason, T. Y. Lee, W. F. Kuang and K. Viswanathan, in Polymerization Reactors and Processes, American Chemical Society, Washington, DC, 2009, vol. 847, pp. 2–14 Search PubMed.
- (a) T. J. White, W. B. Liechty, L. V. Natarajan, V. P. Tondiglia, T. J. Bunning and C. A. Guymon, Polymer, 2006, 47, 2289–2298 CrossRef CAS; (b) T. J. White, W. B. Liechty and C. A. Guymon, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4062–4073 CrossRef CAS.
- C.-Y. Lee, F. Teymour, H. Camastral, N. Tirelli, J. A. Hubbell, D. L. Elbert and G. Papavasiliou, Macromol. React. Eng., 2013, 8, 314–328 CrossRef.
- Y. Hao, H. Shih, Z. Muňoz, A. Kemp and C.-C. Lin, Acta Biomater., 2014, 10, 104–114 CrossRef CAS PubMed.
- G. Oster and A. H. Adelman, J. Am. Chem. Soc., 1956, 78, 913–916 CrossRef CAS.
- J. S. Bellin and G. Oster, J. Am. Chem. Soc., 1957, 79, 2461–2464 CrossRef CAS.
- Y. Usui, K. Itoh and M. Koizumi, Bull. Chem. Soc. Jpn., 1965, 38, 1015–1022 CrossRef CAS.
- V. Kasche and L. Lindqvist, Photochem. Photobiol., 1965, 4, 923–933 CrossRef CAS.
- E. Chesneau and J. P. Fouassier, Angew. Makromol. Chem., 1985, 135, 41–64 CrossRef CAS.
- J. P. Fouassier, E. Chesneau and M. LeBaccon, Makromol. Chem., Rapid Commun., 1988, 9, 223–227 CrossRef CAS.
- J. P. Fouassier and E. Chesneau, Makromol. Chem., 1991, 192, 1307–1315 CrossRef CAS.
- J. P. Fouassier and E. Chesneau, Makromol. Chem., 1991, 192, 245–260 CrossRef CAS.
- J. Wong, K. Kaastrup, A. Aguirre-Soto and H. D. Sikes, Polymer, 2015, 69, 169–177 CrossRef CAS.
- J. Wong and H. D. Sikes, Macromol. Theory Simul., 2016, 25, 229–237 CrossRef CAS.
- K. Kaastrup, A. Aguirre-Soto, C. Wang, C. N. Bowman, J. W. Stansbury and H. D. Sikes, Polym. Chem., 2016, 7, 592–602 RSC.
- G. Oster, Nature, 1954, 173, 300–301 CrossRef CAS.
- G. Delzenne, S. Toppet and G. Smets, J. Polym. Sci., 1960, 48, 347–355 CrossRef CAS.
- A. Aguirre-Soto, K. Kaastrup, S. Kim, K. Ugo-Beke and H. D. Sikes, ACS Catal., 2018, 8, 6394–6400 CrossRef CAS.
- A. Aguirre-Soto, A. T. Hwang, D. Glugla, J. W. Wydra, R. R. McLeod, C. N. Bowman and J. W. Stansbury, Macromolecules, 2015, 48, 6781–6790 CrossRef CAS.
- S. Das, M. N. Schuchmann, H. P. Schuchmann and C. Vonsonntag, Chem. Ber./Recl., 1987, 120, 319–323 CrossRef CAS.
- M. Takeishi and G. Y. Tao, J. Polym. Sci., Part C: Polym. Lett., 1989, 27, 301–305 CrossRef CAS.
- M. Chakraborty and A. K. Panda, Spectrochim. Acta, Part A, 2011, 81, 458–465 CrossRef CAS PubMed.
- S. Lin-Gibson, S. Bencherif, J. M. Antonucci, R. L. Jones and F. Horkay, Macromol. Symp., 2005, 227, 243–254 CrossRef CAS.
- E. I. Alarcon, H. Poblete, H. Roh, J.-F. Couture, J. Comer and I. E. Kochevar, ACS Omega, 2017, 2, 6646–6657 CrossRef CAS.
- N. R. Jana, Z. L. Wang and T. Pal, Langmuir, 2000, 16, 2457–2463 CrossRef CAS.
- K. Krutkramelis, B. Xia and J. Oakey, Lab Chip, 2016, 16, 1457–1465 RSC.
- C. R. Wilke and P. Chang, AIChE J., 1955, 1, 264–270 CrossRef CAS.
- M. Satoh, K. Shirai, H. Saitoh, T. Yamauchi and N. Tsubokawa, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 600–606 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8py01459k |
| This journal is © The Royal Society of Chemistry 2019 |