
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTopology effects on protein–polymer block copolymer self-assembly†
Takuya
Suguri
ab and
Bradley D.
Olsen
 *a
*a
aDepartment of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Ave, Cambridge, MA 02139, USA. E-mail: bdolsen@mit.edu; Tel: +1 (617) 715-4548
bYokkaichi Research Center, JSR Corporation, 100 Kawajiricho, Yokkaichi, Mie, Japan
First published on 5th March 2019
Abstract
Bioconjugates made of the model red fluorescent protein mCherry and synthetic polymer blocks show that topology, i.e. the BA, BA2, ABA and ABC chain structure of the block copolymers, where B represents the protein and A and C represent polymers, has a significant effect on ordering transitions and the type and size of nanostructures formed during microphase separation. ABA and ABC type block copolymers were synthesized by using two site-specific bioconjugation reactions: the thiol–ene reaction with a cysteine on mCherry and maleimide functionalized polymers, and the sortase A ligation reaction with an LPETG sequence at the C-terminus on mCherry and a triglycine functionalized polymer. The phase behaviors of mCherry–poly(N-isopropylacrylamide) (PNIPAM) and mCherry–(PNIPAM)2 show that the shapes of the phase diagrams are similar overall, but mCherry–(PNIPAM)2, i.e. BA2 type, yields a narrower domain spacing than mCherry–PNIPAM, i.e. BA type. PNIPAM–mCherry–PNIPAM (ABA type) shows only lamellar phases in the range of conditions under which ordered structures appear. PDMAPS–mCherry–PNIPAM (ABC type) shows an ordered structure across the widest range of conditions in the four bioconjugates and also the widest range of different nanodomain structures. The phase behavior of the ABC type implies that the repulsive interaction between two water-soluble coil polymers can be a key factor in enhancing the self-assembly of globular protein–polymer block copolymers.
Introduction
Protein bioconjugates1 provide a great method to immobilize2 or stabilize proteins,3,4 construct nanoscale architectures5 and control protein activity.6 These materials are useful for a variety of applications such as biocatalysts,7 biosensors,8 biofuel cells,9 and bioelectronic devices.10 For many of these applications, the materials must maintain or enhance the stability of the protein fold, achieve a high areal density of active sites, control orientation for recognition,11 and provide structured channels for transport of reactants/products.12 To achieve all of these design criteria, material nanostructure must be carefully controlled. To control the position, orientation, and density of proteins, three broad approaches to nanopatterning have been demonstrated. First, top-down approaches that combine lithographic techniques including photolithography,13 electron beam lithography,14,15 dip-pen nanolithography,16 and nanocontact printing17 with protein immobilization chemistry can form protein nanopatterns on substrates. Second, a template can guide protein nanostructure formation. For example, premade 2D or 3D structures such as block copolymer films18 and nanoporous materials19 can be used as a template. Also, some fabrication methods such as layer-by-layer deposition20 can control the nanostructure of protein-based materials. The final approach is self-assembly of protein–polymer block copolymers, which are created by bioconjugation of a globular protein and a coil polymer block21 or by fusion of a functional and a non-functional protein block.22,23 This technique can achieve high three-dimensional protein density, connectivity of domains, and control over nanoscale morphology.12,24Because the self-assembly of protein–polymer block copolymers is very different from traditional block copolymer systems, understanding the physics of this self-assembly process remains a challenge.25–27 One important parameter that has an impact on block copolymer self-assembly is the topology of the block, for example changes between BA, ABA, and BA2 copolymers. Although there are many reports about topology effects on polymer–polymer block copolymers including branch and triblock types of coil–coil28–32 and rod–coil block copolymers,33–38 the effect on protein–polymer block copolymers has not been clarified. Recently, we have reported the topological effects on the self-assembly of globular protein–coil protein fusion proteins22,39 and protein polymer block copolymers.40 In this work, changing the elastin-like polypeptide (ELP) tails from the N to C termini of mCherry has no impact on the ability to form well-ordered nanostructures.39 On the other hand, a bola fusion protein, which has two ELP tails on both N and C termini of mCherry, showed a lower order–disorder transition concentration than the linear fusion protein, which has one EPL tail on the N terminus of mCherry.22 This work provided important new insight into the effect of chain topology in the self-assembly of block copolymers including globular proteins. However, fusion proteins have some limitations of topological and functional designs because globular and coil proteins can be connected to each other only at the N and C termini, and only linear primary chain structure is possible. Furthermore, new genes are necessary for each design to modify or introduce functions derived from the coil protein, such as stimuli responsiveness. In addition to these fusions, changing the bioconjugation site in mCherry–PNIPAM bioconjugates has been explored. Changing the bioconjugation site showed qualitatively similar phase diagrams, indicating that self-assembly is robust with respect to changes in the conjugation site. However, differences in domain spacing were observed, suggesting changes in protein orientation within the lamellar phase.40
The protein–polymer block copolymer approach is a promising approach to expand the capability to obtain topological designs and functional designs. To obtain well-defined bioconjugates, site-specific modification is one of the most important tools. To control topologies of polymers, living polymerizations such as RAFT polymerization are also necessary.41 Two or more orthogonal site-specific reactions provide the ability to access a wider variety of bioconjugates; however, this remains challenging.42
In this study, the effect of topology on the self-assembly of protein–polymer bioconjugates is investigated by comparing four types of protein–polymer bioconjugates, namely BA, BA2, ABA and ABC types, where B represents the protein and A and C represent polymers. In order to examine the effect of branching architecture, 1 and 2-arm PNIPAMs are respectively introduced on mCherry, i.e. mCherry–PNIPAM and mCherry–(PNIPAM)2. In addition, in order to examine the effect of ABA and ABC triblock types, PNIPAM–mCherry–PNIPAM and PDMAPS–mCherry–PNIPAM are also studied. The use of multiple conjugations necessitated the use of two orthogonal site-specific bioconjugations, motivating the development of RAFT polymers functionalized for sortase ligation. Finally, the microphase separation behaviour of each bioconjugate is investigated using small-angle X-ray scattering (SAXS) and depolarized light scattering (DPLS).
Results and discussion
Design and approach to synthesize diblock and triblock type protein–polymer block copolymers
To explore the effects of architecture on self-assembly in globular protein–polymer block copolymers, four types of architectures were chosen: BA, BA2, ABA, ABC. The protein mCherry was selected as the model protein21,43–46 because it has a well-defined structure, color may be used as an indicator that it maintains its folded structure, and it has been extensively studied previously as a model in globular protein–polymer self-assembly. Water-soluble but neither cationic nor anionic polymers are suitable to investigate microphase separation behaviors in aqueous solution because protein charge may cause aggregation with cationic or anionic polymers.47,48 Because it is well-studied and shows a wide region for phase separation from proteins in simple diblocks, PNIPAM was selected as the first polymer for the construction of different topologies. For ABC triblocks, it may be favourable that the two synthetic polymers are immiscible to generate phase separation. Preliminary experiments showed that the mixture of PNIPAM and PDMAPS was immiscible at higher concentrations in aqueous solution at room temperature (Fig. S14†); therefore, these two polymers were selected as model polymers. The different phase behaviors of these two polymers in solution, i.e. lower critical solution temperature (LCST) of PNIPAM and upper critical solution temperature (UCST) of PDMAPS, may also enhance self-assembly over a wide range of temperatures.49To obtain BA and BA2 type copolymers, a site-specific thiol–maleimide coupling between a cysteine on the mutated mCherry (mCherryS131C) and maleimide–PNIPAM was used (Scheme 1). To obtain one and two arm PNIPAMs, two types of reversible addition fragmentation chain transfer (RAFT) agents containing a protected maleimide group (pMI-EMP and pMI-(EMP)2) were used. After polymerization, the maleimide groups were thermally deprotected to yield maleimide end-functionalized 1-arm and 2-arm PNIPAMs. Each conjugate was designed to have an approximately equal volume fraction of mCherry and PNIPAM, because previous results suggested that an approximately equal volume fraction of each polymer would enhance self-assembly and show a minimum order–disorder transition concentration.46 Bioconjugation reactions were performed under mild conditions similar to literature procedures.21
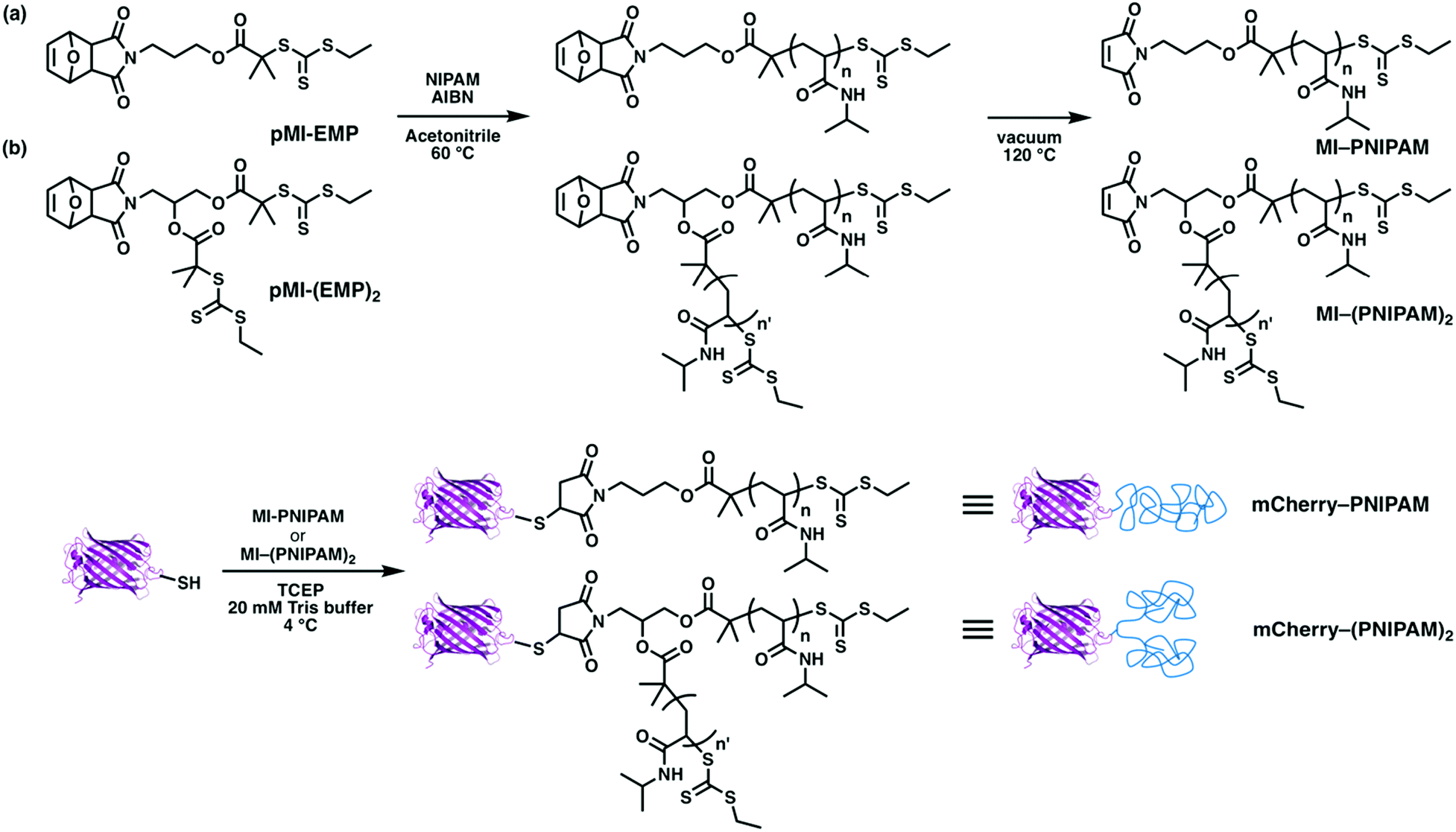 | ||
| Scheme 1 Synthesis of (a) mCherry–(PNIPAM) and (b) mCherry–(PNIPAM)2. | ||
For ABA and ABC copolymers, two distinct bioconjugation sites are necessary, and ABC type copolymers need two orthogonal site-specific chemistries to attach the two different polymers. Cysteine is selected as the first reacting group for thiol–ene reactions, which is on the one side of the β-barrel structure; the mutation site is the same as mCherryS131C. The second group is the LPETG sequence, installed at the C-terminus on the other side of the β-barrel structure for Sortase A mediated ligation (SrtA ligation). Sortase A from Staphylococcus aureus catalyzes the cleavage reaction between threonine and glycine at an LPXTG recognition sequence on the first molecule, where the variable residue X is most commonly E, followed by the formation of an amide bond between the carboxyl group of threonine and a second molecule with an N-terminal nucleophilic group, typically an oligoglycine motif. SrtA ligation was selected for the second bioconjugation reaction because of its high specificity, mild reaction conditions, ease of introducing the LPETG sequence into recombinant proteins, and compatibility with synthetic polymers. In addition, a histidine tag (His-tag) could be introduced in mCherry downstream from the LPETG sequence to facilitate purification. To introduce N-terminal oligoglycine in polymers, a new RAFT agent with the tert-butyloxycarbonyl (Boc) protected triglycine (Boc-GGG-EMP) was synthesized (Scheme 2). HO-EMP was synthesized according to previously reported procedure (details in ESI†). HO-EMP was reacted with Boc-GGG-OH in the presence of N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride and 4-dimethylaminopyridine to give Boc-GGG-EMP. The Boc moiety was removed after RAFT polymerization.
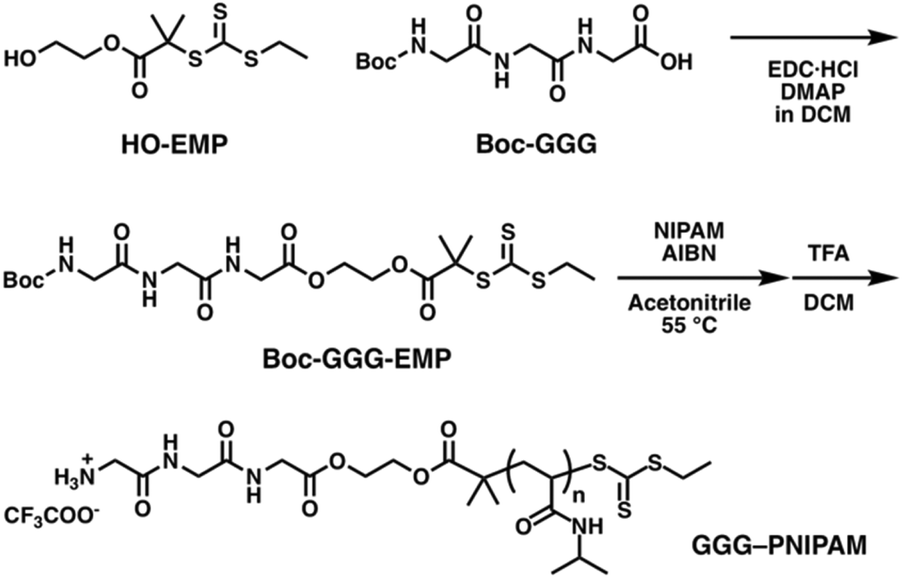 | ||
| Scheme 2 Synthesis of triglycine functionalized PNIPAM. | ||
ABA and ABC type triblock copolymers were synthesized in two steps (Scheme 3). First, PNIPAM and PDMAPS with maleimide end groups were reacted with a cysteine on mCherryS131C–LPETGG–His6 to give PNIPAM–mCherry–LPETGG–His6 and PDMAPS–mCherry–LPETGG–His6. After both bioconjugates were purified, they were reacted with triglycine–PNIPAM in the presence of sortase A. Both polymer chains for each triblock were designed to have approximately equal volume fraction in the final block copolymer. Unreacted bioconjugates (PNIPAM–mCherry–LPETGG–His6 and PDMAPS–mCherry–LPETGG–His6) and sortase A which has a His-tag were removed using Ni-NTA. Bioconjugates (PNIPAM–mCherry–PNIPAM and PDMAPS–mCherry–PNIPAM) and excess polymer (GGG-PNIPAM) were collected as flow-through. Then, FPLC purification was used to isolate PNIPAM–mCherry–PNIPAM (ABA) and PDMAPS–mCherry–PNIPAM (ABC), respectively. The mCherry function after purification of the triblock type was relatively lower than that of the diblock type: BA = 94%, BA2 = 90%, ABA = 74%, and ABC = 85% based on UV-vis absorbance measurements (Fig. S17, S20 and S22†). These differences suggest that sortase ligation or the longer two step conjugation and purification process results in some small loss of protein function. The SDS-PAGE shows that the bioconjugates are pure, indicating that the self-assembly behavior is due to the bioconjugates, not due to a mix of native or partially modified and fully modified proteins. The molecular properties of all four protein–polymer block copolymers are summarized in Table 1.
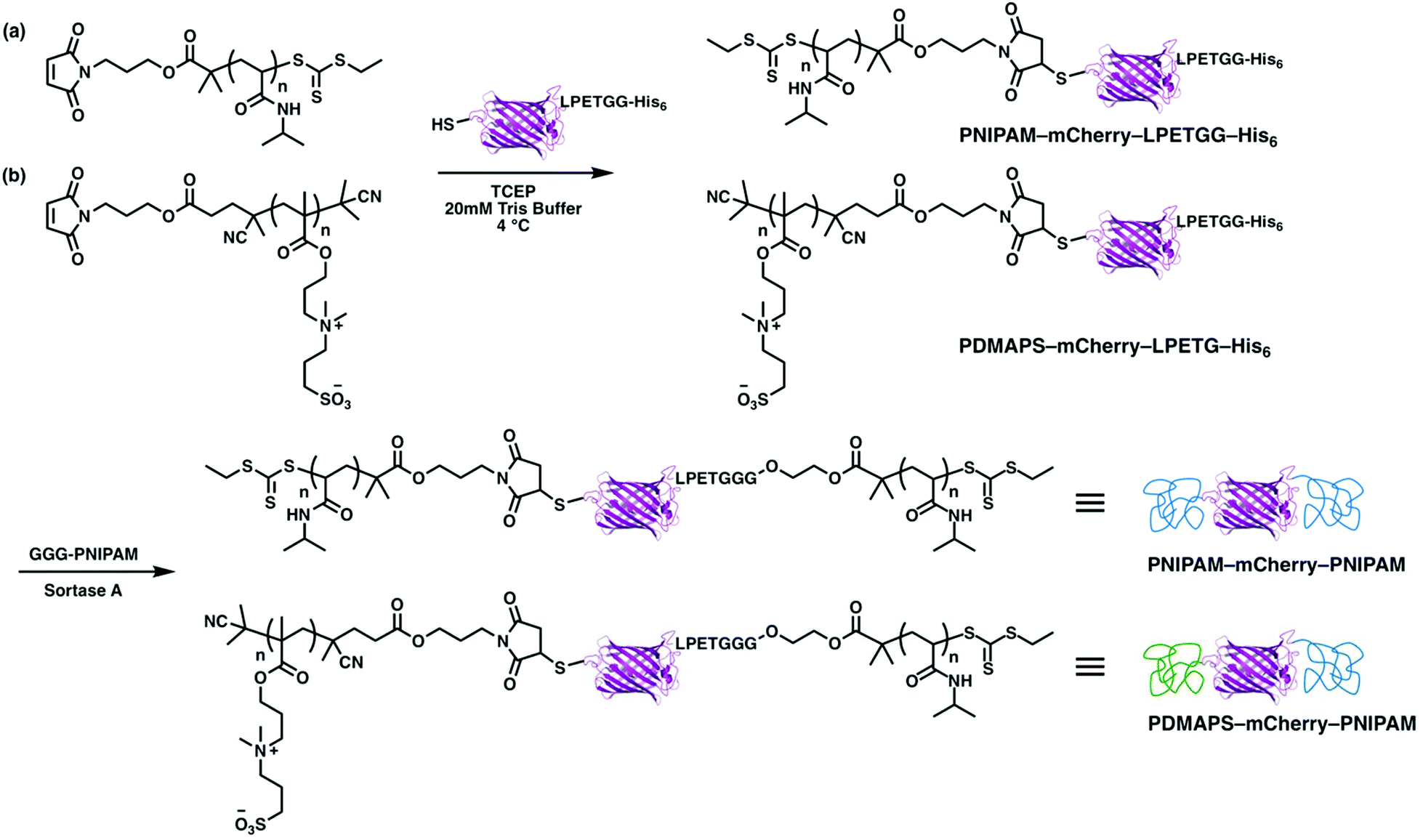 | ||
| Scheme 3 Synthesis of (a) PNIPAM–mCherry–PNIPAM and (b) PDMAPS–mCherry–PNIPAM by thiol–ene bioconjugation reaction and sortase A ligation reaction. | ||
| Bioconjugates | Type | mCherry | Maleimide–PNIPAM | Maleimide–PDMAPS | Triglycine–PNIPAM | Polymer volume fractiona | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Reaction site | M n | PDI | M n | PDI | M n | PDI | ϕ maleimide | ϕ triglycine | ϕ total | ||
| a Density of PNIPAM 1.05, PDMAPS 1.37 and mCherry 1.35 g cm−3 and a molecular weight of mCherry 28.2 kDa were used to calculate the volume fraction of polymers in bioconjugates. | |||||||||||
| mCherry–PNIPAM | BA | Cys | 29.1 | 1.16 | — | — | — | — | 0.606 | — | 0.606 |
| mCherry–(PNIPAM)2 | BA2 | Cys | 28.6 | 1.15 | — | — | — | — | 0.599 | — | 0.599 |
| PNIPAM–mCherry–PNIPAM | ABA | Cys/LPETG | 17.8 | 1.05 | — | — | 17.4 | 1.07 | 0.313 | 0.318 | 0.631 |
| PDMAPS–mCherry–PNIPAM | ABC | Cys/LPETG | — | — | 19.2 | 1.20 | 17.4 | 1.07 | 0.303 | 0.322 | 0.625 |
Phase behavior in concentrated aqueous solution
When self-assembled in concentrated solution above an order–disorder transition concentration, all four bioconjugates form ordered nanostructures reminiscent of traditional block copolymers. To investigate self-assembly in concentrated solution, the purified bioconjugates were spin-concentrated and dried under vacuum to give pellets, and then the collected pellets were re-dissolved to prepare concentrated solution samples in the range of 30–60 wt%. The microphase separated nanostructures of samples are examined with small-angle X-ray scattering (SAXS) over a range of temperatures between 10–40 °C (all data are shown in Fig. S24 and S27†).Comparison of mCherry–PNIPAM (BA type) and mCherry–(PNIPAM)2 (BA2 type) block copolymer self-assemblies in concentrated solution shows that both materials exhibit a similar phase behavior. Phase identifications were performed using small-angle X-ray scattering (SAXS) and depolarized light scattering (DPLS), and each identified phase is summarized in phase diagrams (Fig. 1a and b). The phase diagrams of both mCherry–PNIPAM and mCherry–(PNIPAM)2 showed a quite similar general shape (Fig. 1c and d). In both bioconjugates, disordered phases are observed at concentrations below 40 wt% at low temperature. Increasing temperature above 35 °C yields a disordered micellar phase due to the LCST behavior of PNIPAM46 which causes desolvation of this block in the copolymer upon heating. Increasing concentration to 40 wt% yields a lamellar phase with peaks at q* and 2q*. At 50 wt%, both bioconjugates show well-ordered lamellar phases, and the peaks broaden with further increasing concentration to 60 wt%. This suggests that the degree of order decreases. Only in mCherry–PNIPAM, lamellar–lamellar coexistence is observed at 45 and 50 wt% (see also Fig. S25†). Increasing the temperature at concentrations above 40 wt% yielded a hexagonal phase with peaks at q*, √3q* and √4q* for a narrow range of temperature and concentration. In addition, in both bioconjugates, birefringent lamellae are observed at temperatures below 25 °C (mCherry–PNIPAM) or 30 °C (mCherry–(PNIPAM)2). In the case of mCherry–(PNIPAM)2, a birefringent lamellar phase is observed from 50 wt% to 60 wt%, while mCherry–PNIPAM shows birefringent lamellae from 40 wt% to 60 wt%. While these results might imply that the BA2 type shows relatively weaker ordered structure than the BA type, it has also previously been shown that liquid crystalline ordering can emerge in protein–polymer conjugates at high concentrations,43 and it is possible that the propensity for such ordering is stronger in the linear BA conjugate.
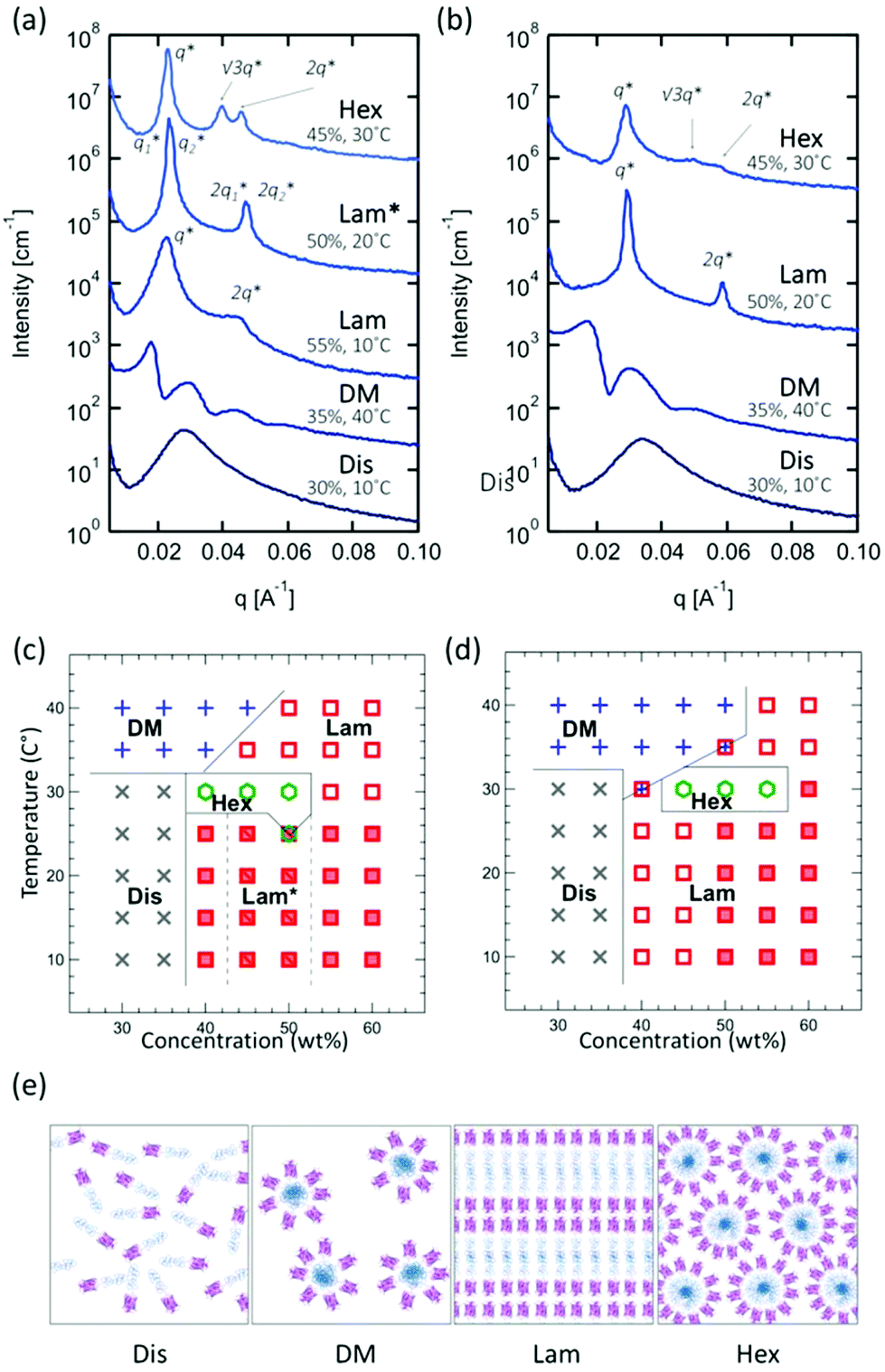 | ||
| Fig. 1 Representative SAXS curves for (a) mCherry–PNIPAM and (b) mCherry–(PNIPAM)2, and phase diagrams for (c) mCherry–PNIPAM and (d) mCherry–(PNIPAM)2. (e) Cartoon schematics of each identified phase. Phases are labeled as disordered (Dis), disordered micellar (DM), lamellar (Lam), lamellar–lamellar coexistence (Lam*) and hexagonally packed cylindrical (Hex). Open symbols represent non-birefringent structures and solid symbols represent birefringence structures. | ||
In a previous comparison of bola and linear block copolymers using mCherry and ELP fusion proteins,22 the bola type which has two ELP tails showed a much wider concentration range for ordering than the linear fusion copolymer. While the linear mCherry–ELP fusion protein self-assembled over a similar concentration range to the linear mCherry–PNIPAM bioconjugate (above 40%), ordered structures were observed for the bola fusion for concentrations above 30 wt%. Comparing the BA type copolymer in this work and linear fusion in the previous work, both block copolymers have similar structures, which are globular protein–coil block copolymers, despite differences in the chemistry of the coil block. Furthermore, both phase diagrams suggest a similar phase behavior. In other words, these results imply that the difference between PNIPAM and ELP has only a minor impact on the phase behavior of these two polymers. In contrast, BA2 type and bola type show different behaviors for the ELP. Because ELP and PNIPAM behave similarly in the linear system, this difference in the effect of topology between PNIPAM and ELP may arise from the disparities in the way the individual chains are connected to mCherry: there is a single branch point for PNIPAM, while the ELP is connected in two different locations on the same side of the protein.
A theoretical study of the BAn Gaussian coil block copolymer systems50,51 and rod–coil block copolymer systems including coil branching52,53 predicts that phases could be shifted by the molecular architectural change; however, this is not observed with these protein copolymers. Recent experimental results show a topology effect on self-assembly of polyhedral oligomeric silsesquioxane (POSS)–polystyrene (PS) conjugates, which suggests that introduction of two PS tails with half-length of one tail affects the boundaries between different ordered phases, order–disorder transition temperatures and packing configurations of the functional POSS cages.54 Although both BA and BA2 in this study show similar boundaries and order–disorder transition temperatures, the difference between topologies affected the packing configurations, namely domain spacings and cross-sectional areas.
The domain spacings of both mCherry–PNIPAM and mCherry–(PNIAPM)2 vary with temperature and concentration over the range of 24.8–30.8 nm in the case of mCherry–PNIPAM and 20.2–24.2 nm in the case of mCherry–(PNIAPM)2 (Fig. 2a and b). In mCherry–PNIPAM at 50 wt% and 10 °C, the domain spacing is 27.4 nm, which is nearly two times as large as the calculated domain spacing (ESI†).
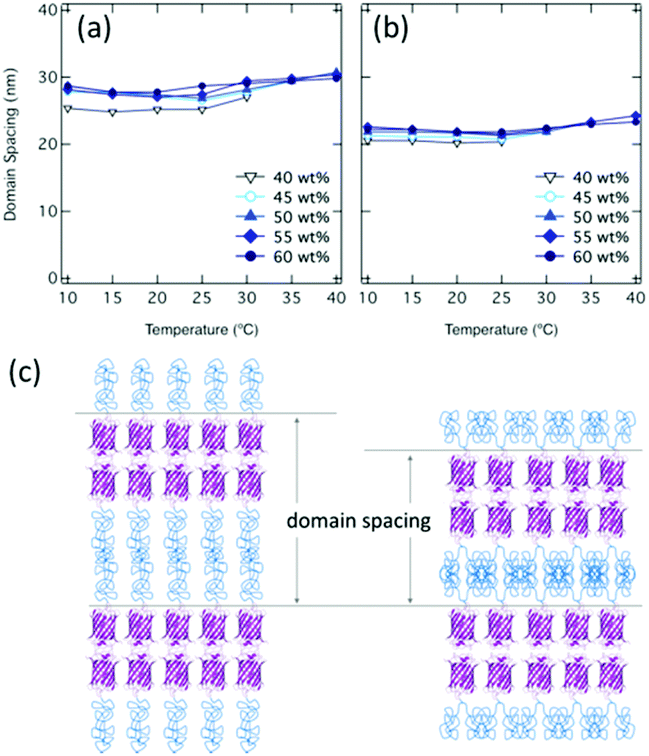 | ||
| Fig. 2 Domain spacing calculated by primary peaks of SAXS for (a) mCherry–PNIPAM and (b) mCherry–(PNIPAM)2 and (c) cartoon schematics of lamellar phase. | ||
This value suggests bilayer lamellar morphologies, as previously reported.45 In mCherry–(PNIPAM)2, the smaller domain spacing (e.g. 21.7 nm at 50 wt% 10 °C) suggests that branched architectures result in narrower polymer domain morphologies (Fig. 2c) because of the lower hydrodynamic volumes, as observed in other studies.37,38 For this to occur while conserving the volume of each component, there must be a larger average lateral spacing between proteins. To estimate lateral spacing, cross-section area54 can be calculated using domain spacings based on SAXS curves (ESI†). The cross-section area corresponding one bioconjugate of mCherry–PNIPAM and mCherry–(PNIAPM)2 at 50 wt%, 10 °C is calculated to be 6.43 nm2 and 7.97 nm2 based upon SAXS curves, which suggest that diameters of them are 2.90 nm and 3.22 nm, respectively. These estimated diameters are reasonable considering the previous study54 and the calculated diameter at 50 wt% based upon mCherry β-barrel structure of 3.15 nm (ESI, Fig. S29†).
Both linear and branched copolymers show a similar dependence of the domain spacing on temperature and concentration. For temperatures below 30 °C, the nanostructures at 40 wt% show the smallest domain spacing; at higher temperatures, there is little dependence of the domain spacing on concentration. As temperature increases, domain spacing initially decreases as the solvent quality for the PNIPAM block grows worse and the PNIPAM chain contracts, and then increases when the solvent changes from good to poor for PNIPAM, presumably due to an increased strength of segregation. Over the range of conditions, mCherry–(PNIAPM)2 shows smaller domain spacings than mCherry–PNIPAM.
Compared to BA and BA2 diblock copolymers, ABA triblocks show a higher critical concentration for ordering. PNIPAM–mCherry–PNIPAM undergoes order–disorder transitions at 50 wt%, while diblock type bioconjugates undergo transitions at 40 wt% (Fig. 3a and b). The SAXS peaks of PNIPAM–mCherry–PNIPAM at concentrations above 50 wt% are broader than for diblock types. At concentrations below 45 wt%, increasing temperature yields a disordered micellar phase, which is similar to the behavior observed in BA and BA2 type copolymers. On the other hand, in the range of ordered concentration and temperature, only lamellae are observed, unlike mCherry–PNIPAM and mCherry–(PNIAPM)2 which also show hexagonal phases. In addition, unlike BA and BA2 type, PNIPAM–mCherry–PNIPAM never shows birefringent phases. Theoretical studies of ABA type Gaussian coil block copolymers55 and comparison between rod–coil and coil–rod–coil56 provide insights into the self-assembly behaviors of our bioconjugates, which suggested that the lamellar phase of a coil–rod–coil becomes more unstable than a coil–rod. Since mCherry has a large diameter compared to the rod, it is not straightforward to explain this self-assembly behavior using previous theory, but our results suggest similar behaviors.
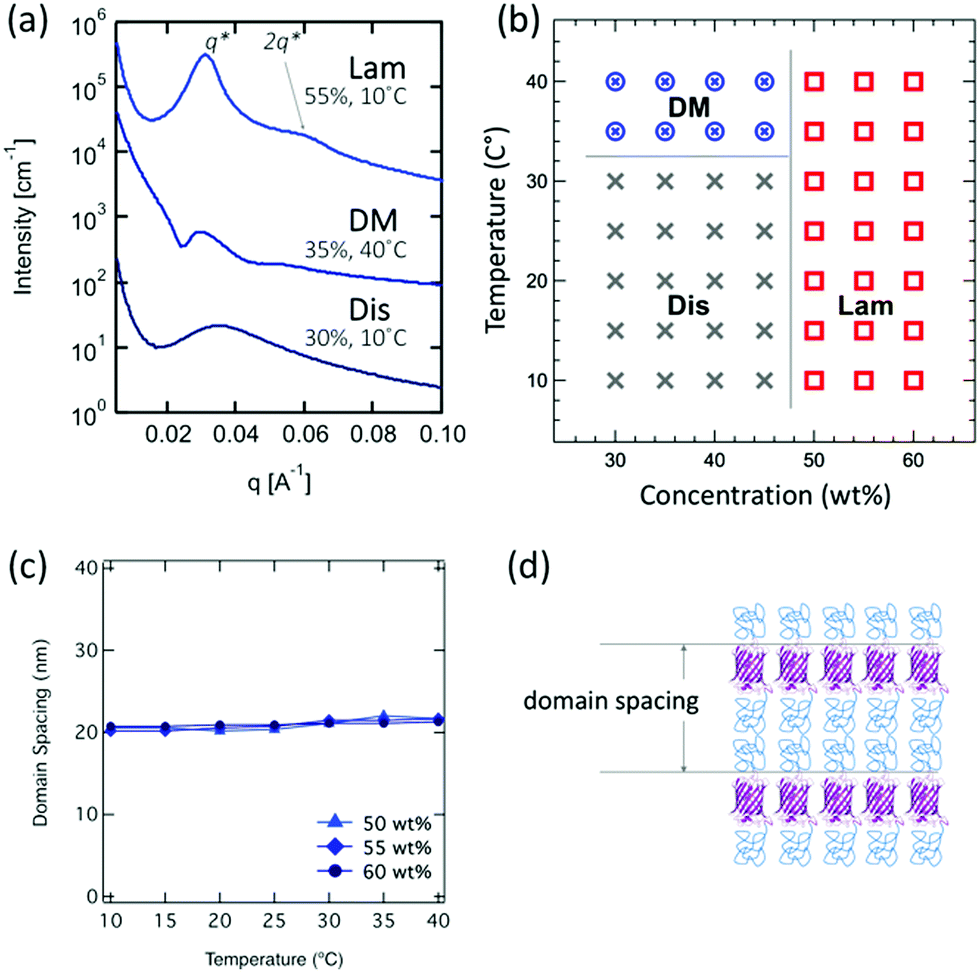 | ||
| Fig. 3 Representative pattern (a), phase diagram (b), domain spacing (c), and cartoon schematics (d) of PNIPAM–mCherry–PNIPAM. | ||
The relatively small domain spacings for PNIPAM–mCherry–PNIPAM change only slightly with varying concentration and temperature in the range 20.2–21.7 nm (Fig. 3c). The domain spacing of the lamellar morphology at 55 wt%, 10 °C is 20.2 nm, which is nearly 1.4 times larger than the calculated domain spacing for a rigid block copolymer. This value suggests a monolayer lamellar phase, unlike a bilayer lamellar phase in the case of the diblock type (Fig. 3d). This is consistent with the fact that the mCherry block has PNIPAM conjugated to both ends, preventing bilayer association.
In contrast to PNIPAM–mCherry–PNIPAM, PDMAPS–mCherry–PNIPAM shows ordered structures at the lowest concentration of any of the four copolymers studied here. This ABC copolymer shows ordered structure at all concentrations studied above 30% (Fig. 4a and b). In the concentration range of 30–35 wt%, a lamellar morphology was observed at 10–25 °C. Increasing temperature in the concentration range of 30–35 wt% yields phase transitions to disordered and disordered micellar phases at temperatures above 35 °C due to the LCST behavior of PNIPAM. Increasing concentration to 40 wt%, hexagonal and lamellar ordering within a perforated lamellar structure are observed. The perforated lamellar phase is characterized by SAXS peaks at (100), (001), (110), (002), and (003) (e.g. 45 wt%, 10 °C) or (100), (001), (002), (210), (300), (310) and (003) (e.g. 50 wt%, 20 °C) (Fig. S26(b)†). Increasing concentration from 40 wt% to 60 wt% yields phase transitions from perforated lamellar to lamellar to hexagonal phases. Over the concentration range of 40–50 wt%, increasing temperature yields phase transitions from lamella to perforated lamellar to hexagonal to cubic phases including co-existence regions. Changes in the experimental conditions change the form factor of the cylinder nanodomains, resulting in changes in the relative peak intensities for the hexagonal phase: 1: 2q*: √7q* (e.g. 40 wt%, 30 °C), 1: √3q*: √7q* (e.g. 60 wt%, 10–40 °C), 1: √3q*: 2q*: √7q* (e.g. 45 wt%, 30 °C) with higher-order peaks (3q*, √12q*, v13q*) at some conditions (Fig. S26(a)†). The cubic phase is characterized by SAXS peaks at q*, √2q*, √3q* √5q* and √6q*, matching the pattern commonly observed for BCC sphere phases formed by coil–coil block copolymers. Phase transitions from lamellar to hexagonal to cubic are possible in theory,57,58 and experimental studies have been reported as a function of changing concentration, salt concentration, or polymer volume fraction.59–63 Therefore, there is precedence in the literature for such transitions, which can be driven in this system by changes in volume fraction and the LCST/UCST behaviors of PNIPAM and PDMAPS which changes polymer hydration as a function of temperature. PDMAPS–mCherry–PNIPAM shows ordered structures across the widest range of concentration in four bioconjugates in this study. In addition, PDMAPS–mCherry–PNIPAM shows a larger number of higher order peaks, q*: 2q*: 3 q*: 4 q* (lamellar) and q*: √3q*: 2q*: √7q*: 3q*: √12q*: v13q* (hexagonal), than the other three types of bioconjugates. Higher order peaks and ordered structures across the widest range of concentration suggest that ABC type block copolymers (e.g. PDMAPS–mCherry–PNIPAM) can achieve well-controlled nanostructured protein materials driven in part by repulsive interactions between two different water-soluble polymers.
 | ||
| Fig. 4 Representative patterns (a), phase diagram (b), domain spacing (c), and cartoon schematics (d) of PDMAPS–mCherry–PNIPAM. Open symbols represent non-birefringent structures and solid symbols represent birefringence structures. | ||
The domain spacings of PDMAPS–mCherry–PNIPAM change from 27.3 nm to 32.9 nm depending on the concentrations and temperatures (Fig. 4c). However, there is no obvious trend for the domain spacings depending on the concentrations or temperatures, potentially due to the fact that many phase boundaries are observed. However, the domain spacings are typically twice the calculated domain spacing, suggesting bilayer type arrangements (Fig. 4d), which is expected due to the asymmetry of the block copolymer.
In previous studies, it was revealed that electrostatic segregation of the protein block from a polymer block contributes significantly to the driving force for microphase separation, and the ionic polymer block results in much weaker segregation: mCherry–PNIPAM showed the lowest CODT but mCherry–PDMAPS showed the highest CODT.44 In this study, PDMAPS–mCherry–PNIPAM shows ordered structure across the widest range of concentration of the four bioconjugates (Fig. 5). Because the protein–polymer repulsion for PDMAPS and mCherry is relatively weak, this comparison implies that the repulsive interaction between two coil polymers is one of the key factors enhancing the self-assembly of ABC triblock copolymers (e.g. PDMAPS–mCherry–PNIPAM). The use of triblocks therefore provides a powerful method to induce ordering in protein–polymer bioconjugate systems that may be weakly ordered or disordered.
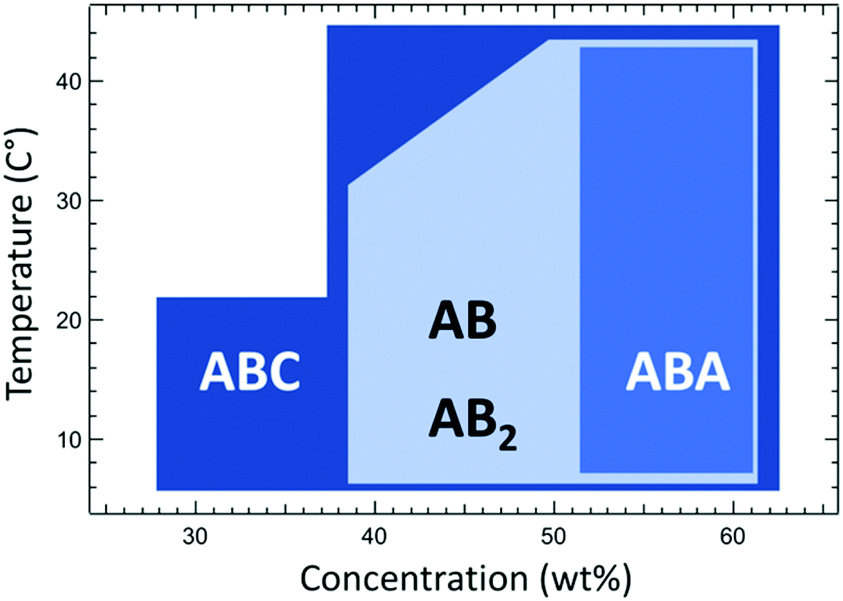 | ||
| Fig. 5 The range of concentration and temperature yielding ordered structures for the four types of bioconjugates. | ||
Conclusions
The self-assembly of four types of mCherry–polymer bioconjugates with different topologies, namely BA, BA2, ABA, and ABC type, was investigated in concentrated solutions. To obtain ABA and ABC type block copolymers, two different reaction sites were introduced on mCherry: cysteine for thiol–ene conjugation with maleimide functionalized polymers and a C-terminal LPETG sequence for sortase A ligation with a triglycine-functionalized polymer. Triglycine-functional polymer was produced using a novel RAFT agent, providing the first demonstration of sortase ligation to an end-functional RAFT polymer. The two step bioconjugation reaction successfully yielded ABA and ABC type block copolymers, which are PNIPAM–mCherry–PNIPAM and PDMAPS–mCherry–PNIPAM. This approach enables future work to target a wide variety of asymmetric ABC type copolymers to introduce functions based on polymer structure.Despite their different topologies, the phase diagrams of mCherry–PNIPAM (BA) and mCherry–(PNIPAM)2 (BA2) showed a quite similar general shape. However, the BA2 type showed smaller domain spacing than the BA type, suggesting that branching architectures result in narrower polymer domain morphologies and larger cross-section area. PNIPAM–mCherry–PNIPAM (ABA) showed weaker ordered structures than other bioconjugates: only lamellar ordered structures were observed at concentrations above 50 wt%. In contrast, PDMAPS–mCherry–PNIPAM (ABC) shows ordered structure across the widest range of conditions in the four bioconjugates. Our previous studies showed that (1) volume fraction of the polymer46 and (2) electrostatic segregation of the protein block from the polymer block44 contribute significantly to the driving force for microphase separation. This study further reveals that (3) topology is also a key factor in governing microphase separation and (4) the repulsive interaction between the two coil polymers of ABC type bioconjugates is the main factor enhancing the self-assembly of globular protein materials. Therefore, the ABC topology provides a practical approach to enhance microphase separation of a variety of proteins, allowing nanostructural control in protein materials.
Experimental details
Methods
NMR spectra were acquired on A Varian Mercury 300 NMR spectrometer. The residual undeuterated solvent peaks were used as references. The following abbreviations are used to denote the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, and br = broad.The molar masses and dispersities (Đ) of synthesized PNIPAMs were analyzed by gel permeation chromatography using a Waters GPC system equipped with a Waters 1515 Isocratic HPLC Pump, Waters 2487 Dual λ Absorbance Detector, and two ResiPore columns, 7.5–300 mm (Agilent Technologies) in N,N-dimethylformamide (DMF) with 0.02 M LiBr as the mobile phase. The signals were collected from a miniDAWN TREOS MALS detector and a Wyatt Optilab T-rEX refractometer. A dn/dc value of PNIPAM = 0.0761 mL g−1 was used to obtain absolute molar masses. The molar masses and dispersity of synthesized PDMAPS were analyzed by gel permeation chromatography using an Agilent Technologies 1260 Infinity system with two Aquagel columns in 0.5 M NaCl (aq) with 0.02% sodium azide as the mobile phase. The signals were collected from a Wyatt DAWN HELEOS II multi-angle light scattering detector and a Wyatt Optilab T-rEX refractometer. A dn/dc value of PDMAPS in 0.5 M NaCl solution of 0.1423 mL g−1 was used to obtain absolute molar masses.
Synthetic methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DCM = 5:1 for washing and DCM:methanol = 19:1 for elution.). The fractions containing Boc-GGG-EMP were collected, and the solvent was removed by rotary evaporation to yield Boc-GGG-EMP as a sticky yellow solid (1.96 g, 79%). 1H NMR (CDCl3, δ): 1.32 (t, 3H, –SCH2C
:DCM = 5:1 for washing and DCM:methanol = 19:1 for elution.). The fractions containing Boc-GGG-EMP were collected, and the solvent was removed by rotary evaporation to yield Boc-GGG-EMP as a sticky yellow solid (1.96 g, 79%). 1H NMR (CDCl3, δ): 1.32 (t, 3H, –SCH2C![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) 3), 1.44 (s, 9H, tert-Bu), 1.69 (s, 6H, –(C
3), 1.44 (s, 9H, tert-Bu), 1.69 (s, 6H, –(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)C(C3)2S–), 3.28 (q, 2H, –SC2CH3), 3.84 (d, 2H, –(NH)C2C(O)–), 4.03 (overlapped doublet, 4H, –(NH)C2C(O)–), 4.28–4.38 (m, 4H, –OC2C2O–), 5.28 (t, 1H, N), 6.89 (br, 1H, N), 6.94 (t, 1H, N). The 1H NMR spectrum is shown in Fig. S8.†
O)C(C3)2S–), 3.28 (q, 2H, –SC2CH3), 3.84 (d, 2H, –(NH)C2C(O)–), 4.03 (overlapped doublet, 4H, –(NH)C2C(O)–), 4.28–4.38 (m, 4H, –OC2C2O–), 5.28 (t, 1H, N), 6.89 (br, 1H, N), 6.94 (t, 1H, N). The 1H NMR spectrum is shown in Fig. S8.†
Protein expression
Bioconjugation
After bioconjugation with PNIPAM, the bioconjugates were purified by 1 M ammonium phosphate precipitation to remove unreacted mCherry (three times) and then by NiNTA chromatography to remove the excess PNIPAM.
After bioconjugation with PDMAPS, the bioconjugates were purified by Ni–NTA chromatography to remove the excess PDMAPS. The eluent containing free protein and bioconjugate was dialyzed into 10 mM Tris–Cl pH 7.0 buffer for FPLC purification. Bioconjugates were purified using a HiTrap Q HP 5 mL anionic exchange column, 10 mM Tris–Cl pH 7.0 as buffer A and 10 mM Tris–Cl 0.5 M NaCl pH 7.0 as buffer B in an AKTA pure FPLC system. The results of SDS-PAGE are shown in Fig. S16 and S19.†
Sample preparation
Each purified bioconjugate was dialyzed into deionized water. The bioconjugate solutions were concentrated using Millipore Ultra-15 centrifugal filters with a molecular weight cut-off (MWCO) of 30000 kDa, concentrated solutions were cast on Teflon sheets, and conjugates were dried under a controlled vacuum with a ramp rate of 50 Torr per hour until a final pressure of 10 Torr, followed by holding at this pressure overnight at room temperature. Solid pellets were weighed, and deionized water was added to hydrate the pellets at a desired concentration which is indicated as wt% = mass of pellets/mass of solution. Typically, the density of water was assumed to be 1 g cm−3 and the volume was measured when added to pellets. Protein folding and activity retention for mCherry conjugates after each step were evaluated by UV-vis spectrophotometry and CD spectroscopy (Fig. S17, S18 and S20–S23†).
SAXS characterization
SAXS samples were loaded into a 1 mm thick washer and sealed with Kapton tape. mCherry–PNIPAM and mCherry–(PNIPAM)2 were measured at the Argonne National Laboratory Advanced Photon Source at Beamline 8-ID-E. PNIPAM–mCherry–PNIPAM was measured at the Brookhaven National Laboratory NSLS-II 11-BL. PDMAPS–mCherry–PNIPAM was measured at the Argonne National Laboratory Advanced Photon Source at Beamline 12-ID-C,D. Samples were equilibrated for 10 minutes at all temperatures (10, 15, 20, 25, 30, 35, 40 °C) prior to data collection (Fig. S24†). Native mCherry, mCherryS131C, mCherryS131C-LPETG-His were also measured, but they did not show any peaks (Fig. S28†).DPLS characterization
Samples for turbidimetry and depolarized light scattering (DPLS) measurements were prepared by loading the solution into the center of 1 mm thick PTFE washers sandwiched between two quartz disks. A Coherent OBIS LX660 laser (λ = 662 nm) outputting 20 mW was used for the measurements. The data was collected while samples were heated from 10 °C to 40 °C at a rate of 1 °C min−1 and cooled back down from 40 °C to 10 °C at a rate of 1 °C min−1. This process was done with a rear polarizer for birefringence and without a rear polarizer for turbidimetry measurements. The birefringence was corrected for transmission and dark field background (Fig. S27†).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Department of Energy Office of Basic Energy Sciences (award number DE-SC0007106). We thank Dr Joseph Strzalka for experimental assistance with SAXS at the Argonne National Laboratory Advanced Photon Source at Beamline 8-ID-E. We thank Dr Masafumi Fukuto for experimental assistance with SAXS at the Brookhaven National Laboratory NSLS-II 11-BL. We thank Dr Soenke Seifert for experimental assistance with SAXS at the Argonne National Laboratory Advanced Photon Source at Beamline 12-ID-C,D.Notes and references
- I. Cobo, M. Li, B. S. Sumerlin and S. Perrier, Nat. Mater., 2015, 14, 143–159 CrossRef CAS PubMed.
- L. S. Wong, F. Khan and J. Micklefield, Chem. Rev., 2009, 109, 4025–4053 CrossRef CAS PubMed.
- R. J. Mancini, J. Lee and H. D. Maynard, J. Am. Chem. Soc., 2012, 134, 8474–8479 CrossRef CAS PubMed.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288–11289 CrossRef CAS PubMed.
- K. Velonia, Polym. Chem., 2010, 1, 944 RSC.
- T. Shimoboji, E. Larenas, T. Fowler, S. Kulkarni, A. S. Hoffman and P. S. Stayton, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16592–16596 CrossRef CAS PubMed.
- M. L. E. Gutarra, L. S. M. Miranda and R. O. M. A. de Souza, Enzyme Immobilization for Organic Synthesis, in Organic Synthesis Using Biocatalysis, ed. A. Goswami and J. D. Stewart, Elsevier, 2016, pp. 99–126 Search PubMed.
- Enzyme and Microbial biosensors, ed. A. Mulchandani and K. Rogers, Springer, New York, 1998, pp. 1–196 Search PubMed.
- M. Rasmussen, S. Abdellaoui and S. D. Minteer, Biosens. Bioelectron., 2016, 76, 91–102 CrossRef CAS PubMed.
- Biosensors and Biodetection: Methods and Protocols, ed. B. Prickril and A. Rasooly, Humana Press, New York, NY, 2017, vol. 1 Search PubMed.
- K. Yoichi, T. Yasuhide, I. Hiroyuki, I. Koreyoshi, S. Takaharu, K. Shigeo and N. Kazuhiro, Biotechnol. Prog., 2006, 22, 401–405 CrossRef PubMed.
- X. H. Dong, A. C. Obermeyer and B. D. Olsen, Angew. Chem., Int. Ed., 2017, 56, 1273–1277 CrossRef CAS PubMed.
- J. F. Mooney, A. J. Hunt, J. R. McIntosh, C. A. Liberko, D. M. Walba and C. T. Rogers, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 12287–12291 CrossRef CAS.
- U. Y. Lau, S. S. Saxer, J. Lee, E. Bat and H. D. Maynard, ACS Nano, 2016, 10, 723–729 CrossRef CAS PubMed.
- K. L. Christman, E. Schopf, R. M. Broyer, R. C. Li, Y. Chen and H. D. Maynard, J. Am. Chem. Soc., 2009, 131, 521–527 CrossRef CAS PubMed.
- K. B. Lee, S. J. Park, C. A. Mirkin, J. C. Smith and M. Mrksich, Science, 2002, 295, 1702–1705 CrossRef CAS PubMed.
- J. D. Hoff, L.-J. Cheng, E. Meyhöfer, L. J. Guo and A. J. Hunt, Nano Lett., 2004, 4, 853–857 CrossRef CAS.
- K. H. A. Lau, J. Bang, D. H. Kim and W. Knoll, Adv. Funct. Mater., 2008, 18, 3148–3157 CrossRef CAS.
- K. J. Stine, K. Jefferson and O. V. Shulga, Methods Mol. Biol., 2011, 679, 67–83 CrossRef CAS PubMed.
- Y. Lvov, K. Ariga, I. Ichinose and T. Kunitake, J. Am. Chem. Soc., 1995, 117, 6117–6123 CrossRef CAS.
- C. S. Thomas, M. J. Glassman and B. D. Olsen, ACS Nano, 2011, 5, 5697–5707 CrossRef CAS PubMed.
- G. Qin, M. J. Glassman, C. N. Lam, D. Chang, E. Schaible, A. Hexemer and B. D. Olsen, Adv. Funct. Mater., 2015, 25, 729–738 CrossRef CAS.
- O. S. Rabotyagova, P. Cebe and D. L. Kaplan, Biomacromolecules, 2011, 12, 269–289 CrossRef CAS PubMed.
- A. Huang, G. Qin and B. D. Olsen, ACS Appl. Mater. Interfaces, 2015, 7, 14660–14669 CrossRef CAS PubMed.
- C. S. Thomas and B. D. Olsen, Soft Matter, 2014, 10, 3093–3102 RSC.
- C. N. Lam, H. Yao and B. D. Olsen, Biomacromolecules, 2016, 17, 2820–2829 CrossRef CAS PubMed.
- D. Chang, A. Huang and B. D. Olsen, Macromol. Rapid Commun., 2017, 38, 1600449 CrossRef PubMed.
- N. Hadjichristidis, H. Iatrou, S. K. Behal, J. J. Chludzinski, M. M. Disko, R. T. Garner, K. S. Liang, D. J. Lohse and S. T. Milner, Macromolecules, 1993, 26, 5812–5815 CrossRef CAS.
- D. J. Pochan, S. P. Gido, S. Pispas, J. W. Mays, A. J. Ryan, J. P. A. Fairclough, I. W. Hamley and N. J. Terrill, Macromolecules, 1996, 29, 5091–5098 CrossRef CAS.
- W. Shi, Y. Tateishi, W. Li, C. J. Hawker, G. H. Fredrickson and E. J. Kramer, ACS Macro Lett., 2015, 4, 1287–1292 CrossRef CAS.
- H. Kim, B.-G. Kang, J. Choi, Z. Sun, D. M. Yu, J. Mays and T. P. Russell, Macromolecules, 2018, 51, 1181–1188 CrossRef CAS.
- H. Minehara, L. M. Pitet, S. Kim, R. H. Zha, E. W. Meijer and C. J. Hawker, Macromolecules, 2016, 49, 2318–2326 CrossRef CAS.
- J. Park, H. C. Moon, C. Choi and J. K. Kim, Macromolecules, 2015, 48, 3523–3530 CrossRef CAS.
- J. Babin, D. Taton, M. Brinkmann and S. Lecommandoux, Macromolecules, 2008, 41, 1384–1392 CrossRef CAS.
- S. Junnila, N. Houbenov, A. Karatzas, N. Hadjichristidis, A. Hirao, H. Iatrou and O. Ikkala, Macromolecules, 2012, 45, 2850–2856 CrossRef CAS.
- R. Goseki, A. Hirao, M.-A. Kakimoto and T. Hayakawa, ACS Macro Lett., 2013, 2, 625–629 CrossRef CAS.
- T. Isono, I. Otsuka, Y. Kondo, S. Halila, S. Fort, C. Rochas, T. Satoh, R. Borsali and T. Kakuchi, Macromolecules, 2013, 46, 1461–1469 CrossRef CAS.
- T. Isono, I. Otsuka, D. Suemasa, C. Rochas, T. Satoh, R. Borsali and T. Kakuchi, Macromolecules, 2013, 46, 8932–8940 CrossRef CAS.
- G. Qin, P. M. Perez, C. E. Mills and B. D. Olsen, Biomacromolecules, 2016, 17, 928–934 CrossRef CAS PubMed.
- A. Huang and B. D. Olsen, Macromol. Rapid Commun., 2016, 37, 1268–1274 CrossRef CAS PubMed.
- A. Gregory and M. H. Stenzel, Prog. Polym. Sci., 2012, 37, 38–105 CrossRef CAS.
- Y. Hou, J. Yuan, Y. Zhou, J. Yu and H. Lu, J. Am. Chem. Soc., 2016, 138, 10995–11000 CrossRef CAS PubMed.
- D. Chang, C. N. Lam, S. Tang and B. D. Olsen, Polym. Chem., 2014, 5, 4884–4895 RSC.
- D. Chang and B. D. Olsen, Polym. Chem., 2016, 7, 2410–2418 RSC.
- C. N. Lam, M. Kim, C. S. Thomas, D. Chang, G. E. Sanoja, C. U. Okwara and B. D. Olsen, Biomacromolecules, 2014, 15, 1248–1258 CrossRef CAS PubMed.
- C. N. Lam and B. D. Olsen, Soft Matter, 2013, 9, 2393 RSC.
- C. L. Cooper, P. L. Dubin, A. B. Kayitmazer and S. Turksen, Curr. Opin. Colloid Interface Sci., 2005, 10, 52–78 CrossRef CAS.
- P. Dubin, J. Bock, R. Davis, D. N. Schulz and C. Thies, Macromolecular complexes in chemistry and biology, Springer Science & Business Media, 1994 Search PubMed.
- Y. J. Shih, Y. Chang, A. Deratani and D. Quemener, Biomacromolecules, 2012, 13, 2849–2858 CrossRef CAS PubMed.
- S. T. Milner, Macromolecules, 1994, 27, 2333–2335 CrossRef CAS.
- G. M. Grason and R. D. Kamien, Macromolecules, 2004, 37, 7371–7380 CrossRef CAS.
- J.-Z. Chen, C.-X. Zhang, Z.-Y. Sun, Y.-S. Zheng and L.-J. An, J. Chem. Phys., 2006, 124, 104907 CrossRef PubMed.
- Y. Xia, Z. Sun, T. Shi, J. Chen, L. An and Y. Jia, Polymer, 2008, 49, 5596–5601 CrossRef CAS.
- K. Yue, C. Liu, M. Huang, J. Huang, Z. Zhou, K. Wu, H. Liu, Z. Lin, A.-C. Shi, W.-B. Zhang and S. Z. D. Cheng, Macromolecules, 2017, 50, 303–314 CrossRef CAS.
- A. M. Mayes and M. Olvera de la Cruz, J. Chem. Phys., 1989, 91, 7228–7235 CrossRef CAS.
- J.-Z. Chen, C.-X. Zhang, Z.-Y. Sun, L.-J. An and Z. Tong, J. Chem. Phys., 2007, 127, 024105 CrossRef PubMed.
- L. Leibler, Macromolecules, 1980, 13, 1602–1617 CrossRef CAS.
- C.-I. Huang and T. P. Lodge, Macromolecules, 1998, 31, 3556–3565 CrossRef CAS.
- S. Naidu, H. Ahn, J. Gong, B. Kim and D. Y. Ryu, Macromolecules, 2011, 44, 6085–6093 CrossRef CAS.
- L.-Y. Shi, Y. Zhou, X.-H. Fan and Z. Shen, Macromolecules, 2013, 46, 5308–5316 CrossRef CAS.
- X. Yu, K. Yue, I. F. Hsieh, Y. Li, X.-H. Dong, C. Liu, Y. Xin, H.-F. Wang, A.-C. Shi, G. R. Newkome, R.-M. Ho, E.-Q. Chen, W.-B. Zhang and S. Z. D. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10078 CrossRef CAS PubMed.
- S.-H. Choi, F. S. Bates and T. P. Lodge, Macromolecules, 2014, 47, 7978–7986 CrossRef CAS.
- H.-S. Jang, T.-H. Kim, C. Do, M.-J. Lee and S.-M. Choi, Soft Matter, 2013, 9, 3050–3056 RSC.
- S. Perrier, P. Takolpuckdee and C. A. Mars, Macromolecules, 2005, 38, 2033–2036 CrossRef CAS.
- I. Chen, B. M. Dorr and D. R. Liu, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11399–11404 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8py01228h |
| This journal is © The Royal Society of Chemistry 2019 |