
Nanoporous silica nanoparticles functionalized with a fluorescent turn-on spirorhodamineamide as pH indicators†
M.
Di Paolo
ab,
M. J.
Roberti
a,
A. V.
Bordoni
 c,
P. F.
Aramendía
bd,
A.
Wolosiuk
bc and
M. L.
Bossi‡
*ab
c,
P. F.
Aramendía
bd,
A.
Wolosiuk
bc and
M. L.
Bossi‡
*ab
aINQUIMAE, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Argentina. E-mail: mariano.bossi@gmail.com
bDepartamento de Química Inorgánica, Analítica y Química Física. Facultad de Ciencias Exactas y Naturales. Universidad de Buenos Aires, Pabellón 2. Ciudad Universitaria, 1428 Ciudad de Buenos Aires, Argentina
cGerencia Química – Centro Atómico Constituyentes, Comisión Nacional de Energía Atómica, Av. Gral. Paz 1499, B1650KNA San Martín, Buenos Aires, Argentina
dCentro de Investigaciones en Bionanociencias “Elizabeth Jares-Erijman” CIBION-CONICET, Godoy Cruz 2390, 1425 Ciudad de Buenos Aires, Argentina
First published on 11th October 2018
Abstract
We prepared water soluble, biocompatible fluorescent turn-on pH nanosensors and characterized their behavior as a function of changes in pH. The response relies on a halochromic reaction of a spirorhodamineamide derived from the bright and highly chemically and photo-stable rhodamine 6G, encapsulated in core/nanoporous shell silica nanoparticles. The fluorescent sensors displayed a fast response in the pH range of intracellular compartments. The encapsulation conferred solubility in aqueous environments and biocompatibility. We assessed the two main properties of the sensor, namely the useful pH range and the kinetics of the response, and compared them to those of the free probe. We found that such properties are strongly dependent on the functionalization and position in the silica matrix relative to the core/shell structure. Finally, we demonstrated the cellular uptake of the nanosensors, and their localization in lysosomes of living cells, by fluorescence confocal microscopy.
Introduction
Xanthene dyes have been widely used as fluorescent probes for bioimaging applications due to their favorable photophysical and spectroscopic properties, namely excellent photostability, high brightness (due to a high absorption coefficient and emission quantum yield), and relatively long emission wavelength. While both fluorescein and rhodamine xanthene dyes have been used,1 or even diads containing both,2,3 rhodamines are more popular due to their red-shifted emission and higher photostability compared to fluoresceins4,5 that make them suitable to operate at the high intensities required at the focal plane of an epifluorescence microscope.6–8 In particular, the striking spectral changes induced by the ring-opening reaction of rhodamine spirolactam derivatives,9 eliciting the turn-on (photo- or chemically induced) of a fluorescence signal, have been extensively exploited for breaking the diffraction barrier in single molecule localization based far-field microscopy (photo-triggered),10–14 as well as for the fabrication of fluorescence sensors for different analytes (chemically triggered) such as pH, metal cations,15 and other small biologically relevant molecules.16–19 Moreover, some of these probes work well for intracellular measurements, particularly as pH sensors.20–22 In a cellular context, pH plays a crucial role in protein function regulation through proton-linked structural transitions,23 such as cell growth,24 apoptosis,25 and even the onset of severe degenerative diseases.26 Moreover, the pH is distinctive for each subcellular compartment; for instance, lysosomes are acidic (pH ∼ 4.7–6.5), the cytoplasm is close to neutral (pH ∼ 7.2–7.4), and mitochondria are slightly basic (pH ∼ 8.0).27 Thus, monitoring subcellular pH and its real-time fluctuations is of paramount relevance in living cells.One of the main advantages of spirorhodamineamide (SRA) sensors is their rapid response, reversibility and high sensitivity and selectivity. In addition, combined with far-field microscopy, they provide a non-invasive observation of the target analyte with high spatial resolution. The equilibrium of the thermally induced reaction of the spirolactam ring-opening reaction (Scheme 1) between the closed form (SRA-CF) and the protonated open form (SRA-OFH+) has a pKa value of typically 4 to 6,12,16,20 making them particularly suitable for imaging application of acidic organelles and cellular compartments, in particular lysosomes.22,28–30 Relevant processes have been related to changes in the lysosomal pH; for instance cell maturation resulted in a decrease of lysosomal pH from 5.4 to 4.6 in dendritic cells,31 while an increase in the pH of the lysosomes from 4.3 in healthy cells to 5.2 in apoptotic ones was observed in U937 cells.32 Tuning of the pH response range of a SRA is possible by tailoring the substituent of the lactame nitrogen or the fluorophore,12,33i.e. by changing R3 or R1/R2/R4 groups in Scheme 1.
 | ||
| Scheme 1 Thermal equilibrium of the spirorhodamineamides (SRAs) responsible for the absorption and emission changes as a function of pH. | ||
Although there are many reports in the literature of sensors with good performance in cuvette experiments, few examples are found in intracellular environments.2,20–22 Possible reasons are the potential toxicity of the probe, its solubility in aqueous media, and its cell impermeability. Nanoparticle (NP) encapsulation presents an interesting alternative as fluorescent sensor carriers and has been used extensively to overcome most of the mentioned problems.34,35 In addition, they also provide signal amplification,36 protection from photobleaching,37 easy signal-multiplexing by co-incorporation of several chromophores or sensors,38,39 and the possibility of surface modification with one or multiple groups to specifically target a desired structure.40,41 In particular, silica nanoparticles are an extremely popular biocompatible platform42 with several advantages: high physical and chemical stability, simple and well-established preparation, easy surface post-modification chemistry, and straightforward size tuning by selection of the synthesis conditions.43,44 Common approaches use core–shell structures, where the (fluorescence) sensor is incorporated either into the core, the shell or the interphase.45 Moreover, nanoporous shells are commonly used because they present a much higher surface area/volume ratio, tunable pore size and surface reactivity, and interconnected pores.46,47 The surface of the porous shell is an additional interface for locating (luminescent) sensors. Incorporation of the labels can be performed by different strategies, such as addition of chemical precursors during the NP synthesis or post-modification schemes, each method having their own advantages. Although a large number of fluorescent sensors based on silica NPs have been described,32,33,48–50 usually poor attention is paid to the effect of important variables, in particular the sensor position and structure of the silica matrix, to the response of the sensor, more specifically to the responsive pH range and to the response time. Sensor response kinetics is a particular feature of the nanosensors often disregarded.
In the current study, we present a fluorescent turn-on pH sensor based on a spirorhodamineamide incorporated into nanostructured silica NPs, at three different positions in core/nanoporous shell structures. The performance of the probe as a function of its location on the NP was studied and compared with that of the free probe (i.e. in solution), focusing on how the transition pH range and the reaction rate are affected by the local environment. Finally, we demonstrate the utility of such NP-sensors for assessing pH in intracellular acidic organelles.
Experimental section
Materials and general procedures
Rhodamine 6G (Rh6G, dye content ∼95%), triethylamine (TEA, ≥99%), 4-(N-maleimidomethyl)cyclohexanecarboxylic acid N-hydroxysuccinimide ester (SMCC, ≥98%, power), tetraethylorthosilicate (TEOS, 98%,), (3-mercaptopropyl)trimethoxysilane (MPTMS, 95%), cetyltrimethylammonium bromide anhydrous (CTAB), dimethylformamide (99.8%, DMF), diisopropylethylamine (DIPEA, 99%), and ethylenediamine (EN, ≥99%) were purchased from Sigma–Aldrich and used as received. Hydrochloric acid 37%, ammonium hydroxide (28%), absolute ethanol, hexane and ethyl acetate were purchased from Merck. Cell culture media, high-glucose DMEM (Dulbecco's Modified Eagle Medium), fetal bovine serum (FBS), and LysoTracker Red DND-99 were purchased from Thermo Fisher Scientific.Absorption spectra were recorded on a Shimadzu UV-3600 Plus (Shimadzu Scientific Instruments) UV-Vis-NIR Spectrophotometer, and fluorescence spectra were recorded on a PTI QuantaMaster 400 (Photon Technology International) steady-state spectrofluorometer, with 1 cm path quartz cuvettes. Solvents were of analytical grade (Sigma Aldrich, Merck) and used as received. 1H-NMR spectra were acquired on a Bruker Avance II 500 MHz multinuclear spectrometer, and 13C-NMR spectra in a Bruker Biospin AVIII600. Fluorescence confocal microscopy was performed with a LSM510 Meta (Carl Zeiss microscope).
Synthesis of the amino terminated intermediate (2)
Compound 2 (Fig. 1) was first synthesized using commercial Rh6G as the starting product, and performing an amidation reaction5 with EN. A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 Rh6G:EN molar ratio (500 mg Rh6G, 1 mmol) was dissolved in DMF. The presence of excess EN avoids the reversibility of the reaction. The reaction mixture was stirred at room temperature for 24 h followed by thin-layer chromatography until disappearance of the reactant signals. The reaction crude product was placed in a water/ice bath where the formation of a red precipitate was almost immediately observed. The first purification was carried out by dissolution and re-precipitation cycles with methanol in a water/ice bath. Finally, the purification was performed by silica column chromatography, with hexane/ethyl acetate (1/99) as the elution solvent. Yield 402 mg (88%). The purity of the products obtained was finally assessed by TLC and NMR. Spectra shown in Fig. S9.†1H NMR (500 MHz, CDCl3) δ = 7.91–7.93 (1H, dd), 7.43–7.48 (2H, m), 7.04–7.06 (1H, dd), 6.34 (2H, s), 6.22 (2H, s), 3.19–3.23 (4H, m), 3.15–3.18 (2H, t), 2.36–2.39 (2H, t), 1.90 (6H, s), 1.31–1.34 (6H, t). 13C NMR (151 MHz, CDCl3) δ = 168.64, 162.18, 153.51, 151.69, 147.45, 132.48, 131.19, 128.30, 128.07, 123.84, 122.77, 118.01, 106.13, 96.53, 65.06, 43.92, 42.02, 41.16, 40.73, 40.52, 39.17, 38.67, 38.36, 29.70, 16.70, 14.74, 1.02.
:3 Rh6G:EN molar ratio (500 mg Rh6G, 1 mmol) was dissolved in DMF. The presence of excess EN avoids the reversibility of the reaction. The reaction mixture was stirred at room temperature for 24 h followed by thin-layer chromatography until disappearance of the reactant signals. The reaction crude product was placed in a water/ice bath where the formation of a red precipitate was almost immediately observed. The first purification was carried out by dissolution and re-precipitation cycles with methanol in a water/ice bath. Finally, the purification was performed by silica column chromatography, with hexane/ethyl acetate (1/99) as the elution solvent. Yield 402 mg (88%). The purity of the products obtained was finally assessed by TLC and NMR. Spectra shown in Fig. S9.†1H NMR (500 MHz, CDCl3) δ = 7.91–7.93 (1H, dd), 7.43–7.48 (2H, m), 7.04–7.06 (1H, dd), 6.34 (2H, s), 6.22 (2H, s), 3.19–3.23 (4H, m), 3.15–3.18 (2H, t), 2.36–2.39 (2H, t), 1.90 (6H, s), 1.31–1.34 (6H, t). 13C NMR (151 MHz, CDCl3) δ = 168.64, 162.18, 153.51, 151.69, 147.45, 132.48, 131.19, 128.30, 128.07, 123.84, 122.77, 118.01, 106.13, 96.53, 65.06, 43.92, 42.02, 41.16, 40.73, 40.52, 39.17, 38.67, 38.36, 29.70, 16.70, 14.74, 1.02.
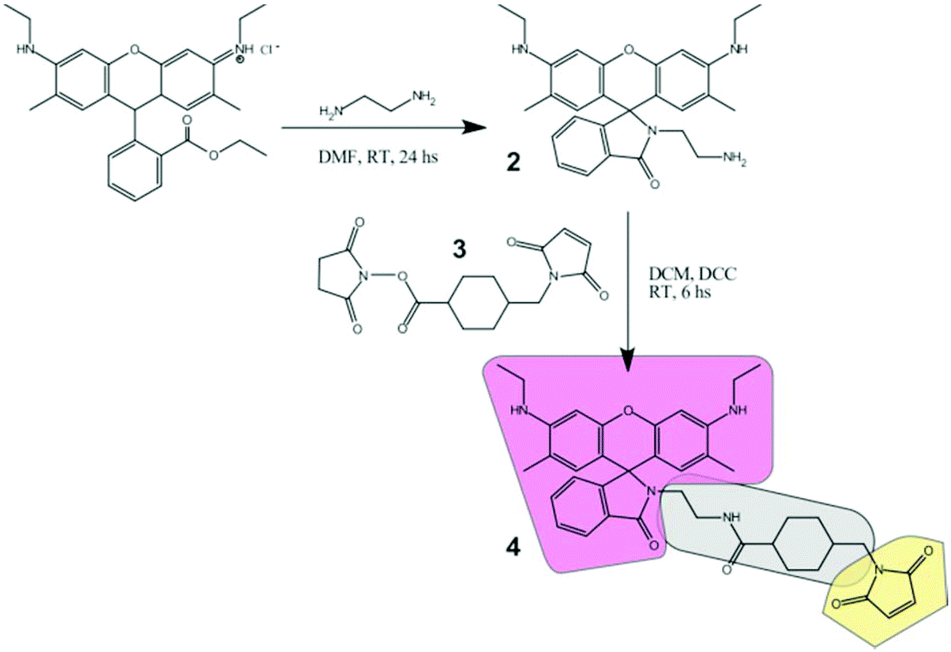 | ||
| Fig. 1 Preparation route of the molecular sensor (4) for modification of silica nanostructures. The building blocks and structural parts are highlighted with different colors (the core fluorescent probe in pink, the linker in gray, and the thiol-reactive group in yellow). | ||
Synthesis of the free marker (4)
The thiol reactive marker was obtained from the reaction of 2 (15 mg, 0.033 mmol) and SMCC (3) (12 mg, 0.036 mmol) in DMF, in the presence of catalytic amounts of TEA. The reaction mixture was stirred at room temperature for 24 h, followed by thin-layer chromatography until disappearance of the reactant signals. The product was purified by silica column chromatography, with pure ethyl acetate as the elution solvent. Yield 14 mg (62%). The purity of the products obtained was finally assessed by TLC and NMR. Spectra shown in Fig. S9.†1H NMR (500 MHz, CDCl3): δ = 7.92–7.93 (1H, m), 7.46–7.47 (2H, m), 7.04–7.05 (1H, m), 6.70 (2H, s), 6.34 (2H, s), 6.19 (2H, s), 3.36–3.37 (2H, d), 3.22–3.28 (2H, t), 3.18–3.21 (4H, q), 2.93–2.96 (2H, q), 1.90–1.93 (2H, m), 1.89 (6H, s), 1.83–1.86 (2H, m), 1.67–1.73 (4H, m), 1.37–1.39 (2H, m), 1.31–1.34 (6H, t). 13C NMR (151 MHz, CDCl3) δ = 175.4, 171.0, 169.9, 153.9, 151.7, 147.6, 134.0, 132.8, 130.4, 128.2, 127.9, 123.9, 122.9, 118.2, 105.3, 96.6, 65.7, 59.5, 45.0, 44.9, 44.5, 43.8, 40.4, 39.9, 38.4, 38.2, 36.4, 31.9, 31.2, 30.0, 29.7, 28.8, 28.7, 25.0, 16.8, 14.7, 14.1, 1.0.Preparation of SiO2 cores (B-NPs)
Monodisperse solid cores were synthesized by the Stöber method from a mixture of ammonia, water and TEOS.37 Typically, we prepared these particles by mixing 71.2 ml of absolute ethanol, 2.8 ml of aqueous ammonia, and 14.6 ml of H2O, and then 4.56 ml of TEOS was added under vigorous stirring. The reaction was carried out at room temperature (25 °C) for 18 hours, and the resulting product was used without any treatment as seeds for the core–shell particle synthesis.Preparation of thiol functionalized SiO2 cores (S-SH-NPs)
They were prepared by adding drop by drop a solution of 0.02 ml of MPTMS in 0.4 ml of pure ethanol to 15 ml of B-NPs under vigorous stirring at room temperature. After 18 h, the S-SH-NPs were centrifuged at 6000 rpm and washed twice in ethanol and several times in deionized water. Finally, they were dried overnight at 60 °C.Preparation of thiol functionalized mesoporous core–shell colloids (CS-SH NPs)
Core–shell particles were obtained using a suspension of B-NPs as solid cores. A mesoporous SiO2 layer was deposited using a combination of 0.5:0.5 TEOS and MPTMS with CTAB as the porogen, following reported procedures.51 To form the functionalized mesoporous shell, 6.25 ml of the B-NP suspension was added slowly to a mixture of 82.45 g of deionized H2O, 23.54 g of ethanol, 0.44 g of aqueous ammonia and 0.48 g of CTAB. After stirring for 30 min, 2.1 mmol of the precursors (TEOS:MPTMS 0.5:0.5 molar ratio) were added under vigorous stirring. Typically, the MPTMS/TEOS mixture was added simultaneously after 30 min stirring drop by drop. The reaction mixture was stirred at room temperature for 24 h. The core–shell nanoparticles were centrifuged and redispersed in a 0.1 M HCl ethanolic solution in an ultrasonic bath to remove the surfactant. This process was repeated at least five times.
Covalent attachment of the maleimide marker to S-SH-NPs
Dry S-SH-NPs (20 mg) were dispersed in DMF (2 ml) by sonication for 30 min and 0.02 ml of DIPEA was added to this suspension. After 10 min of stirring, 0.1 ml of a 1.04 mM solution of 4 in DMF was added, and the mixture was left to react for 16 h at room temperature. The reaction mixture was centrifuged at 14000g for 15 min and the supernatant was discarded. The dye-labelled nanoparticles (S-NPs) were redispersed in ethanol by sonication and washed three times with ethanol and twice with water in order to remove traces of non-reacted dye.
Covalent attachment of maleimide dye to CS-SH NPs
Dry CS-SH NPs (20 mg) were dispersed in 2 ml of DMF by sonication for at least 30 min; later, 0.04 ml of DIPEA was added under stirring. After 15 min, 0.05 ml of a 1.04 mM solution of 4 in DMF was added. The reaction was carried out at room temperature for 16 h. The resulting labeled nanoparticles (PG-NPs) were isolated by centrifugation. Unreacted dye was washed by several cycles of redispersion and centrifugation in ethanol and water.Synthesis of the 4-MPTMS conjugate
The conjugate was prepared by a similar protocol to the one described by Herz et al.52 Briefly, MPTMS (0.28 mmol, 59 μl) was added to a solution containing the dye (4.3 mg, 6.36 μmol) and DIPEA (2 μl) in DMF. The reaction mixture was stirred for 16 h at room temperature and was used for the co-condensation core shell synthesis without any further treatment.Co-condensed NPs (CC-NPs)
Core–shell CC-NPs were synthesized using 2.0 ml of the suspension of the B-NPs that were added slowly to a mixture of 27.7 g of deionized H2O, 7.84 g of ethanol, 0.16 g of aqueous ammonia and 0.16 g of CTAB as the porogen. After stirring for 30 min, a mesoporous SiO2 layer was deposited from a previously mixed solution of TEOS (0.16 ml, 0.72 mmol) and 10 μl of the 4-MPTMS conjugate solution. The reaction mixture was stirred at room temperature for another 24 h. The resulting NPs (CC-NPs) were centrifuged and redispersed by sonication in a 0.1 M HCl ethanolic solution in order to remove the surfactant. This process was repeated at least five times.Single particle kinetics
8-Well chambered slides (Lab-Tek) were activated with NaOH (0.1 M) for 5 min and washed thoroughly with water. Then a 0.1% poly-L-lysine solution was applied for 1 min and washed again with water. Finally, a diluted suspension of the NPs in ethanol was added, which was allowed to stand for 5 min. The sample was kept in a saturated alcoholic environment to prevent evaporation. The sample was rinsed with water and placed in PBS (phosphate buffered saline) pH 7.4 for ca. 2 h for equilibration before beginning the imaging experiment. The PBS was then replaced for a buffered solution at the final pH to start the kinetic measurements.Cell imaging experiments
Vero cells were plated in 8-well chambered slides coated with poly-L-lysine (Lab-Tek) in culture media (high-glucose DMEM with 10% FBS), 24 h before the experiment. Then, the cells were starved for 10 min at 37 °C by replacing the culture media with PBS (phosphate-buffered saline). The cells were then incubated with a diluted suspension of the corresponding nanoparticles in the culture media (the suspension was previously sonicated for 5 min) and incubated for 6 hours at 37 °C (control cells were incubated only with culture media at this step). After this step, the cells were rinsed with PBS (pH 7.4), and mounted in PBS for imaging in a confocal microscope. Excitation was performed with a 514 nm argon laser, and detection was limited between 525 nm and 595 nm, both separated by a 488/514 nm dichroic mirror. Image acquisition was performed at 37 °C. For co-localization experiments, LysoTracker Red was used according to the manufacturer's protocol. Briefly, cells were incubated for 30 min with a 50 nM concentration of the dye in culture media, after the incubation step with the nanoparticles.Results and discussion
Fluorescent marker design and synthesis
Rh6G (1) was selected as the starting base fluorophore due to its excellent photophysical properties: it displays one of the highest fluorescent quantum yields (0.95), a high absorption coefficient at the maximum (∼105 M−1 cm−1), absorption and emission in the center of the visible range where excitation sources are available (in particular for high-end microscopies), and very high photostability, allowing its detection at the single molecule level. Derivatization with ethylenediamine renders the lactam 2 that is halochromic, working as a pH sensor in ethanolic/aqueous mixtures (see below). Further reaction with cross-linker 3 (SMCC) provides a thiol reactive group (maleimide) at the extreme of the molecule as well as a flexible linker that allows a fair separation (ca. 8 Å) between the chromophore and the target structure. The structure of compound 4 is analyzed in Fig. 1: it comprises a pH- and light-sensitive chromophore (pink), a reactive group (yellow), and a connecting spacer (gray). Chemical attachment via thiol groups is a facile and commonly used route for silica gel modification. Most importantly, this covalent linkage precludes any possible leaking of the dye.Labeled silica nanoparticles
For the current study, we selected three types of nanostructured SiO2 architectures with the goal of providing different locations and environments for the fluorescent molecular marker (Fig. 2). The first one consists of a SiO2 dense core surface modified with the marker (S-NPs). This configuration has the advantage of being compact and leaving a well exposed probe, limited only by the nanosphere surface geometrical area. However, core/shell structures incorporate the marker into a porous shell, providing better chemical protection and accessibility to analytes, via a connected net of nanopores.41,45 Co-condensed NPs (CC-NPs) were prepared by incorporating the labeled silica precursor during the growth of the shell, and thus the marker can be localized either within the bulk of the SiO2 structure (major amount) and/or on the pore surface (minor amount). Finally, in the post-grafted NPs (PG-NPs) the mesoporous shell was grown in a first stage and subsequently the pore surface was functionalized with the fluorescent probe. The CC approach has the possibility to incorporate a larger density of markers per particle, while the PG strategy has the advantage of exposing them better to the environment. Moreover, the larger surface to volume ratio provided by both mesoporous shells represents an advantage in these two architectures. We anticipate an increased density of fluorescent markers compared to S-NPs. Altogether, these structures represent three different ways of NP functionalization with a pH responsive probe. The different environments in each case modulate the fluorescent sensor performance in terms of the turning point (pKa) and the kinetic response to pH changes.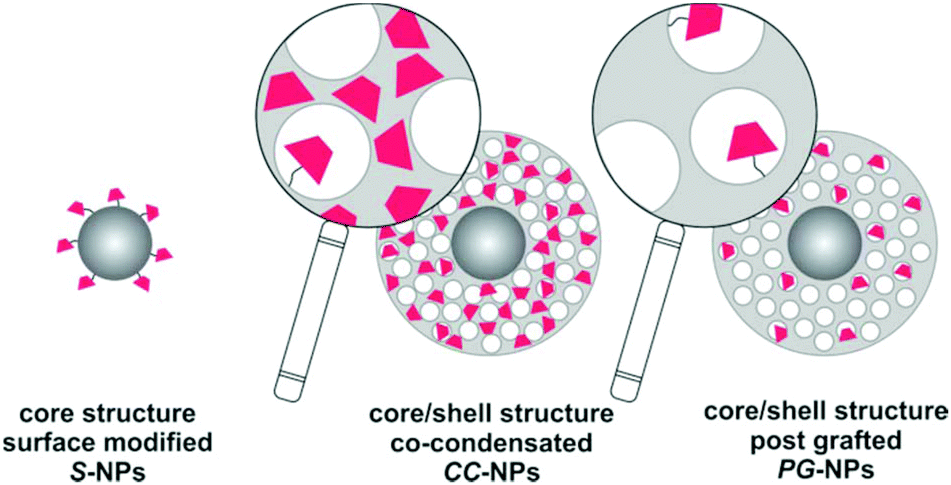 | ||
| Fig. 2 Schematic structure of the three types of labeled silica nanoparticles (S-NPs, CC-NPs and PG-NPs) prepared in this work. Nanopores are represented as white circles and the fluorescent marker as pink trapezoids, on top of the gray silica matrix. | ||
The three types of NPs present a spherical shape with a defined size and narrow polydispersity. SEM images are presented in Fig. 3. Visual inspection of the three architectures revealed uniform spheres with average sizes of 61 ± 7 nm (S-NPs), 403 ± 15 nm (CC-NPs) and 380 ± 31 nm (PG-NPs). This indicates shell thicknesses of 171 and 160 nm, respectively. The mesoporous shells on the synthesized particles have pores that lie in the 2.5 to 2.0 nm range in diameter, a size consistent with the CTAB micelle size. Moreover, the BET area of these kinds of materials is around 600–800 m2 g−1 with pores in the shell having a hexagonal order46 (not shown).
 | ||
| Fig. 3 SEM images of the labeled silica nanoparticles. (A) Surface modified (S-NPs); (B) co-condensed NPs (CC-NPs); (C) post-grafted NPs (PG-NPs). | ||
Sensor pH response
The emission spectrum of the fluorescent form of the marker has a small but noticeable dependence on the environment (Fig. S1†). The emission maximum is at 553 nm for S-NPs, very similar to the one observed for the free dye in ethanol (550 nm) and is shifted to 547 nm and 556 nm for CC-NPs and PG-NPs, respectively. As expected, rhodamine dyes show low dependence on the emission properties with the polarity of the environment. Due to the high dilution of the samples, changes in the absorption spectra were not observed, because the absorption signal was smaller than the scattered light. However, when more concentrated dispersions were prepared, the absorption of the open isomer of the SRA clearly appears over the scattered background (Fig. S2†).The steady-state response of the three NPs was characterized and compared to the response of the free marker 2.53 To this aim, aliquots of each sample were mixed with buffered solutions and allowed to react at room temperature in the dark for 24 h, to ensure complete stabilization of all conditions, and the emission spectra were recorded (Fig. S3†). Compound 2 was characterized in (1:1) methanol/buffer mixtures, due to its poor solubility in aqueous solution. All the observed changes were reversible: upon pH increase to pH = 8.0, the emission of all solutions/suspensions disappears.
The apparent or operative pKa values (IF = 0.5) found for the NPs (Fig. 4) are considerably shifted (1–3 pH units) with respect to the free compound 2 and strongly depend on the environment or position of the probe in the structure. Assuming that there is a simple equilibrium between two forms (SRA-CF and SRA-OFH+ in Scheme 1), we calculated the pKa (ref. 33) in the three different environments and for the free probe (dashed lines in Fig. 4). Except for the free dye, the fits to a single pKa are of poor quality (see Table 1), pointing to a microheterogeneous environment in the silica host. A simple two-site model (solid lines in Fig. 4) shows improved fits (Table 1). Similar behavior has been reported for other pH sensitive chromogenic probes in nanospheres.54 Nevertheless, independent of the model used to adjust the pH response, incorporation of the dye into the NPs shifts the response range to higher pH values in all cases. For in-cell applications, this is a positive feature. In particular, co-condensation provides a response to a more acidic range, while post-grafting allows the use of the sensor up to pH ∼ 7.0, a biologically relevant value for most applications. In addition, these results demonstrate that one of the most important properties of the sensor depends on the environment or more precisely, on the location of the probe in the host silica structure. The turning point of the pH indicator can be tuned in the range of pH from around 2 in the free dye to 5 in PG-NPs.
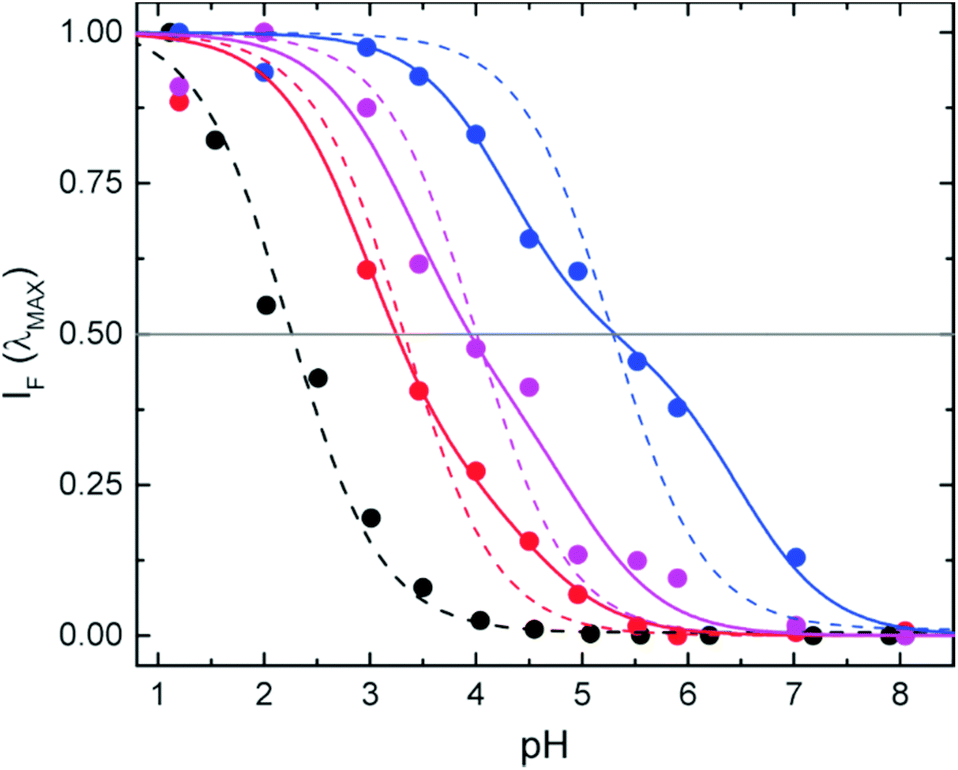 | ||
| Fig. 4 Normalized fluorescence intensity at the emission maximum as a function of pH for CC-NPs (red), PG-NPs (blue), S-NPs (purple), and the free dye 2 (black), stabilized in different aqueous buffers. Operative pKa values (IF = 0.5) are approximately 3.3 (CC-NPs), 5.3 (PG-NPs), 4.0 (S-NPs), and 2.2 (free dye). The latter was characterized in 1:1 methanol/buffer mixtures. Dotted lines correspond to fits to a single pKa value (homogeneous distribution of markers), and solid lines to a two pKa model (a two sites approximation). | ||
| One-site model | Two-site model | |||||
|---|---|---|---|---|---|---|
| pKa | R 2 | F | pKa1 | pKa2 | R 2 | |
| Free dye | 2.3 | 0.988 | — | — | — | — |
| CC-NPs | 3.3 | 0.969 | 0.75 | 3.0 | 4.5 | 0.989 |
| PG-NPs | 5.3 | 0.900 | 0.51 | 4.2 | 6.5 | 0.994 |
| S-NPs | 4.0 | 0.943 | 0.59 | 3.4 | 5.0 | 0.980 |
To gain a better insight into the sensor performance, we studied the kinetics of the response to pH changes. The NPs were first stabilized in aqueous solutions at pH 7.4 (by dilution of the stock solutions in methanol), where all the markers are at the non-emissive form SRA-CF (Scheme 1). This ensures solvent stabilization of the NPs in the buffer, as their response is probably different to that in methanol, and avoids the latter having an effect on the kinetic response. Then, a pH jump was induced (final pH < 7.4) and the time course of the changes was followed by fluorescence emission. Some examples are shown at two different pH jumps (final pH values of 2 and 4) in Fig. S4.† Under these conditions, the NPs tend to aggregate and precipitate at low pH. The effect was more drastic for S-NPs. The incorporation of positive charges through the halochromic ring opening of the probe reduces the negative charge of the NPs,55 thus enabling its flocculation at higher pH values. That is, the typical isoelectric point of the silica around pH ∼ 2–356 might be shifted by the presence of SRA-OFH+. This effect may not be important in CC-NPs and should play no role in the PG-NPs. We did not observe a significant downward curvature of the kinetics at long times in these two types of NPs, in the timespan studied.
Alternatively, we used another strategy to measure the time evolution of the ring opening that on the one side is not affected by floculation and on the other side discards photobleaching as a reason for the signal decrease at long times. NPs were cast onto microscopy cover slides, and the emission intensity changes in a confocal microscope were studied after a pH-jump (Fig. 5A–C). After the frames were registered (to compensate for small in-plane drifts of the sample), the signal of each NP (or groups of few NPs) was plotted as a function of time. Kinetic curves are noisier (Fig. 5E), due to the lower signal of individual nanostructures as compared with ensemble measurements, but nevertheless no downward curvature was observed for any of the structures in the whole pH range studied (2–6). Transient monoexponential fits of the time dependent signals yield first order rate constants in agreement with those obtained in ensemble experiments. Considering that the excitation intensity in a confocal microscope is several orders of magnitude higher than that in a cuvette experiment, we can confirm that the decrease of the fluorescence signal after long irradiation time (ca. 1–2 hours) is due to partial flocculation of the colloids. The kinetic studies in a microscope show the high photostability of the fluorescent form of the probes, allowing the observation of even single particles for up to hours, with no evidence of photobleaching. The global kinetic constants for each condition and for the three types of NPs were calculated from an average of ca. 20 NPs in each case. Results are presented in Fig. 6. Surprisingly, despite the relative complex stationary behavior found requiring a two-site model, single exponential decays yielded reasonable fits for all conditions tested. This apparent contradiction can be explained if we consider that kinetics is dominated by the approach of the proton from solution to the dye location in the NP, and further protonation. This was demonstrated for the dye in solution.33 This diffusion is not selective to the dye environment. On the other side, the equilibrium constant is the ratio of the rate constant for the deprotonation of OFH and the protonation rate constant. The former is dependent on the dye location.
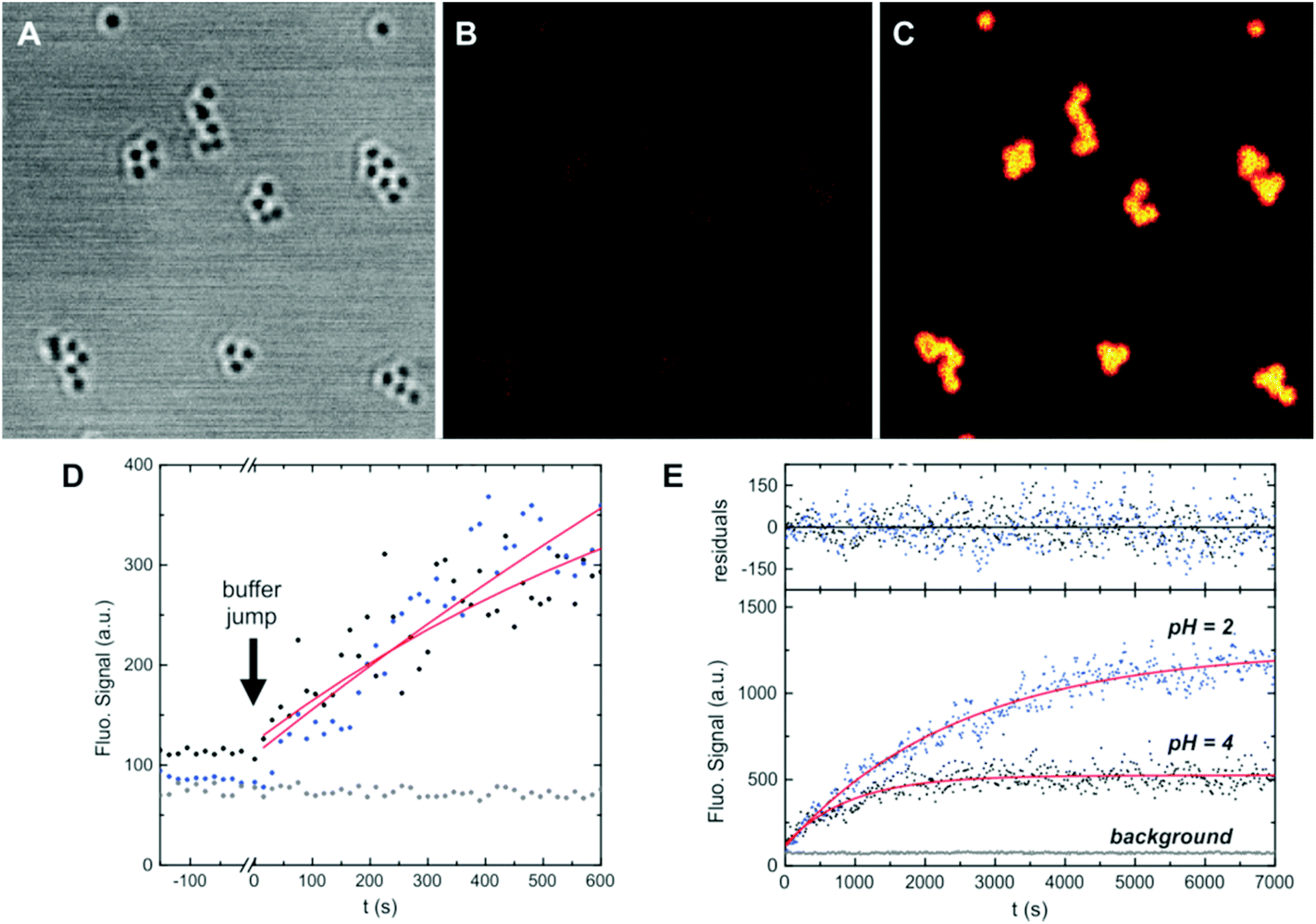 | ||
| Fig. 5 Single particle kinetics experiments with CC-NPs. Transmission (A), and fluorescence (B–C) images. The first frame (t = −150 s) before a buffer pH jump from 7.4 to 2 is presented in A–B (transmission and emission), and the last frame (t = 7000 s) is presented in C (emission). Kinetics for two pH jumps (7.4 → 4 and 7.4 → 2) is presented in E. Symbols represent the total fluorescence signal over a single NP (frames were recorded every 15 s), the lines are the best monoexponential growth fit to the data (from t = 0), and the corresponding residuals are plotted in the upper panel. A zoom to the first frames is presented in (D), where the change in the buffer conditions is highlighted. Gray symbols in (D–E) correspond to the background signal, recorded from an area with no NPs (same total area). | ||
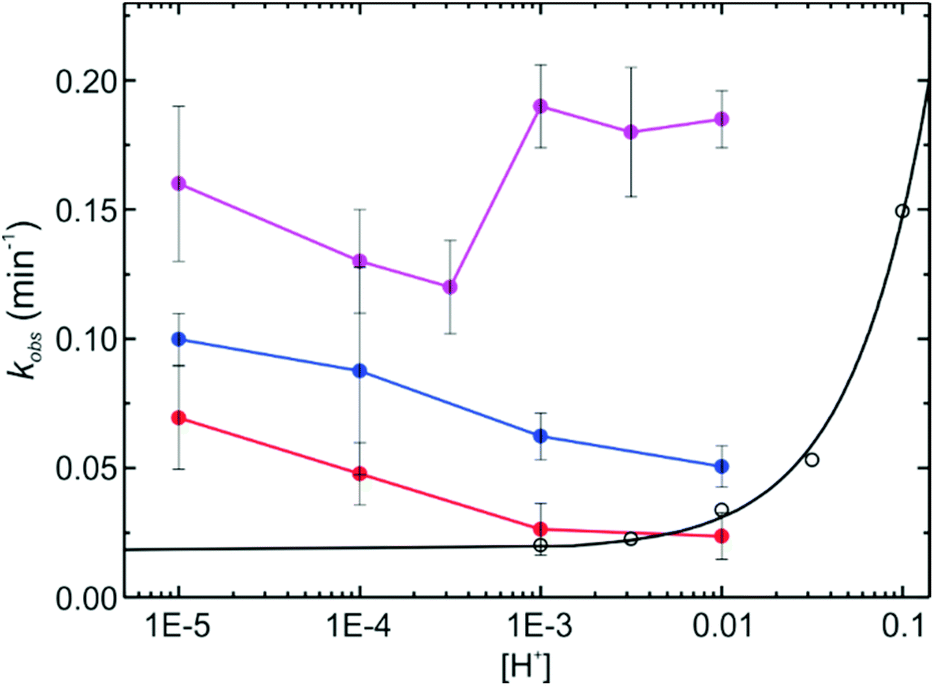 | ||
| Fig. 6 Global first order rate constants of the thermally (proton) induced isomerization of SRAs in the different NPs, after pH jumps from 7.4 to the indicated value controlled by buffered solution. S-NPs (purple), CC-NPs (red), and PG-NPs (blue). The kinetics of the free dye 2 (hollow black symbols) in methanol/buffer mixtures (proton concentration was regulated by TFA additions), and a linear regression for the latter (logkobsvs. pH) is also shown (solid black line). | ||
The kinetics of the halochromic transformation of the SRA in silica NPs shows a great influence of the NP environment. On the one side, at the same bulk pH, the kinetics of the ring opening is always faster in the NPs than in solution in the order: S-NP > PG-NPs > CC-NPs > free dye in solution (Fig. 6). On the other side, there is an inverse pH dependence of the observed first order reaction rate constant (kobs) in the NPs compared to the free dye in solution. Furthermore, the order of the rate constant increase does not correlate with the acidity, as measured by the pKa (Fig. 4). The latter shows an acidity in the order: free dye > CC-NPs > S-NPs > PG-NPs. The increase in kobs in the different types of NPs can be well explained by the accessibility of the proton to the dye location. Considering that the reaction rate is in all cases faster in the NPs than in solution, it is possible to conclude that chemical transformation when the proton reaches the dye location is much faster than proton diffusion to the dye and therefore, accessibility of the dye, i.e. diffusion of the proton within the NP to the dye location, is the rate determining step.
If the ring opening reaction is coupled to diffusion of the proton to the dye location in the NP, the following simple mechanism (eqn (1)) can be proposed:
 | (1) |
The resulting first order rate constant (kobs) for the decay of the closed isomer in the NPs (SRA-CF)NP is:
 | (2) |
If the reaction is faster than cage escape, i.e. kR ≫ k-diff, then, kobs ≈ kdiff·[H+]. The value of kdiff is influenced by the charge of the NP, which in turn depends on pH. Under this picture, kdiff is expected to decrease with the increase of [H+] due to the charge effect, compensating the accelerating effect of the concentration increase. All these influences result in a complex behavior of kobs with pH, as the one we have observed experimentally (Fig. 6), which is not a simple linear inverse plot as the one observed for the free dye, as well as for similar SRAs in solution.33
Intracellular pH sensing: acidic organelle imaging in living cells
The average response time of NP sensors is on the order of a few minutes (ca. 5–30 min) and thus compatible with the time scale of environment changes inside living cells. Therefore, we assessed their utility as pH sensors in such environments. To this end, the sensor-loaded NPs S-NPs and CC-NPs were incorporated into Vero cells by endocytosis and imaged with a confocal microscope (PP-NPs were not used because they are already fluorescent in the cell-culture medium, pH = 7.4 – see Fig. 4). The images (Fig. 7) clearly show several NPs incorporated within the cell, and some of them (marked with green arrows) display a high fluorescence emission. There must be also several NPs that are in the OFF state, presenting no signal, corresponding to endocyted nanosensors in different environments or cell compartments of higher pH (close to neutral). Although they cannot be distinguished from some high-contrast organelles (see the orange arrows in the control experiment in Fig. S6†), there is a clear difference in the amount of such objects between the endocyted and the control cells. Thus, it is reasonable to assume that a fair amount of these features (light blue arrows) are NPs in the OFF-state. To ensure that the signal observed is due to fluorescence and not to scattered light, NPs were also imaged under identical conditions, cast on the cover slide in PBS at pH 7.4 (Fig. S5†). Signal of less than 30 counts per frame in average were observed in contrast with values of up to 160–290 counts per frame (peak values) presented by bright (ON-state) NPs in the cells.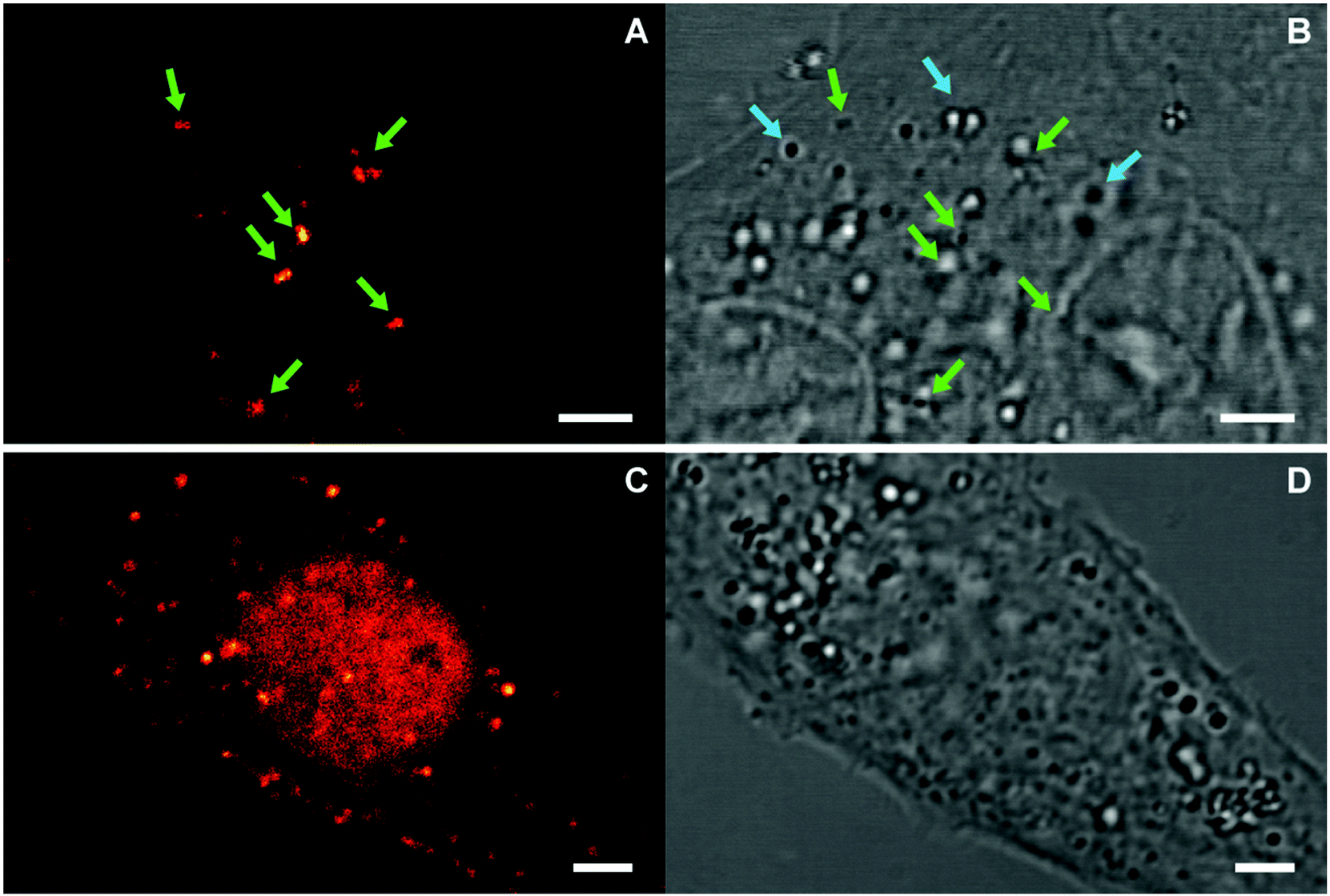 | ||
| Fig. 7 Confocal images of endocyted nanosensors CC-NPs (A–B), and S-NPs (C–D). Fluorescence (A and C) and transmission (B and D) images are presented. Green arrows highlight NPs in the ON-state, and light-blue arrows possible NPs in the OFF-state. The cells endocyted with S-NPs expressed GFP in the nucleus, whose signal is partially observed in the sensor channel (C). Scale bars: 3 μm. | ||
From the response curves (Fig. 4), we conclude that the nanosensors in the ON-state must be located in an acidic compartment. We hypothesize that they are probably lysosomes. To confirm this, co-staining with LysoTracker-Red was carried out (Fig. 8). Sequential two-channel imaging was performed with a rhodamine channel for our sensor (the same conditions as in Fig. 7, except that the detection bandwidth was narrowed to 525–578 nm to minimize cross-talk), and with a lysotracker channel with excitation performed with a 561 nm DPSS laser, detection bandwidth limited between 578 nm and 622 nm, and separated by a 488/561/633 nm dichroic mirror. The estimated colocalization value (Pearson's coefficient 0.79, Fig. S8†) confirms that the sensors are located in such organelles and the ring-opening necessary for the sensor response was completed within the timescale of the experiment. We also conclude that most lysosomes contain NPs, and from the size of the structures, each contains several ones.
 | ||
| Fig. 8 Colocalization of endocyted nanosensors S-NPs with LysoTracker-Red in living cells. The two channels were sequentially imaged. Thus, small differences in the position are due to lysosome movement between frames. (A) Rhodamine channel (λEX = 514 nm; λEM = 525–578 nm); (B) LysoTracker-Red channel (λEX = 561 nm; λEM = 578–622 nm). (C) Overlay. The cells expressed GFP in the nucleus, whose signal partially bleeds through the sensor channel (A). Part of the nucleus is observed on the upper part of the image (see Fig. S7†). Scale bars: 2 μm. | ||
Conclusions
Biocompatible fluorescent pH nanostructured sensors were prepared and fully characterized from the spectroscopic and dynamic point of view. Their cellular uptake was demonstrated, as well as a potential application for the non-invasive observation of lysosomes in confocal far-field microscopy. A rational design of the nanosensor involved the preparation of the molecular probe with a response close to the desired pH range and its functionalization with the thiol reactive maleimide group to target the silica matrix at three different positions relative to the core/shell structure. The molecule proved to be stable under the chemical conditions used for the preparation of the silica through a basic TEOS condensation or the post-functionalization of their surface. The turn-on response at acidic pH values of the sensor arises from the halochromic reaction of a SRA, prepared from Rh6G. After the incorporation into the silica, the probe retained its responsivity. However, the two main properties, the useful pH range and the kinetics of the response, were considerably altered. The turning point was shifted from a pH of ∼2 for the free dye, to a pH of 3–5 depending on the location of the dye in the nanoparticle. The speed of the response was considerably increased, particularly at the biologically relevant pH values and around the turning point. The pH dependence of the time-response was opposed to the one presented by the free compound. This was interpreted as a change in the rate-determining step from the ring opening of the molecular probe to the proton diffusion through the silica, due to a change in the surface charge of the silica. These changes and in particular the tunability properties are remarkable and differ from similar reported probes.21,49 In fact, we were able to increase the turning point of the SRA up to three pH units, through a site-specific targeting of the structures, thus expanding the utility of the sensors to a pH range compatible with acidic subcellular compartments. The encapsulated probes were extremely photostable, and no sign of toxicity was detected during the time window of the experiments (ca. 2 h from the time of the NP incorporation in the medium to the end of the measurements).While this study focused mainly on the influence of the encapsulation on the properties on the molecular probe, and how this can be exploited to improve the performance of the marker, our nanoparticles still have plenty of room for improvement through the inclusion of further functionalization. For instance, a second pH-insensitive fluorescent dye can be incorporated for a more precise ratiometric determination of the intracellular pH, a strategy that has been successfully used in several publications.27,36,47–49 Also, the surface could be modified with an organelle-specific targeting moiety.27 Finally, the possibility to photo-activate the SRAs with UV light can be applied to the observation of the NP sensors located in neutral to basic subcellular compartments (i.e. at a pH ≥ pKa + 1) with modern super-resolution techniques based on the localization of single molecules.11–14
Conflicts of interest
There are no conflicts to declare.Acknowledgements
PFA, AW and AVB are staff members from CONICET (Consejo Nacional de Investigaciones Científicas y Técnicas, Argentina). MDP acknowledges a PhD fellowship and MJR a post-doctoral fellowship from CONICET. This work was performed under financial support from grants from UBA (UBACyT-20020110100203), CONICET (PIP11220130100795 & PIP 1220130100121) and ANPCyT (PICT 2012-2087 & PICT 2013-1931).References
- M. S. T. Gonçalves, Fluorescent Labeling of Biomolecules with Organic Probes, Chem. Rev., 2009, 109, 190–212 CrossRef PubMed.
- M. Lee, J. Han, J. Lee, N. Park, R. Kumar, C. Kang and J. Kim, Two-Color Probe to Monitor a Wide Range of pH Values in Cells, Angew. Chem., Int. Ed., 2013, 52, 6206–6209 CrossRef CAS PubMed.
- L. Li, C. Wang, J. Wu, Y. C. Tse, Y. Cai and K. M. Wong, A Molecular Chameleon with Fluorescein and Rhodamine Spectroscopic Behaviors, Inorg. Chem., 2016, 55, 205–213 CrossRef CAS PubMed.
- R. P. Haugland, Handbook of fluorescent probes and research chemicals, Molecular Probes Inc Eugene, OR. USA, 9th edn, 2002 Search PubMed.
- M. Beija, C. A. M. Afonso and J. M. G. Martinho, Synthesis and applications of Rhodamine derivatives as fluorescent probes, Chem. Soc. Rev., 2009, 38, 2410–2433 RSC.
- H. Giloh and J. W. Sedat, Fluorescence Microscopy: Reduced Photobleaching of Rhodamine and Fluorescein Protein Conjugates by n-Propyl Gallate, Science, 1982, 217, 1252–1255 CrossRef CAS PubMed.
- C. Eggeling, J. Widengren, R. Rigler and C. A. M. Seidel, Photobleaching of Fluorescent Dyes under Conditions Used for Single-Molecule Detection: Evidence of Two-Step Photolysis, Anal. Chem., 1998, 70, 2651–2659 CrossRef CAS PubMed.
- J. Widengren and R. Rigler, Mechanisms of photobleaching investigated by fluorescence correlation spectroscopy, Bioimaging, 1996, 4, 149–157 CrossRef CAS.
- K.-H. Knauer and R. Gleiter, Photochromism of Rhodarnine Derivatives, Angew. Chem., Int. Ed. Engl., 1977, 16, 113 CrossRef.
- J. Fölling, V. N. Belov, R. Kunetsky, R. Medda, A. Schönle, A. Egner, C. Eggeling, M. L. Bossi and S. W. Hell, Photochromic Rhodamines provide Nanoscopy with Optical Sectioning, Angew. Chem., Int. Ed., 2007, 46, 6266–6270 CrossRef PubMed.
- M. L. Bossi, J. Fölling, V. N. Belov, V. P. Boyarskiy, R. Medda, A. Egner, C. Eggeling, A. Schönle and S. W. Hell, Multicolor Far-Field Fluorescence Nanoscopy through Isolated Detection of Distinct Molecular Species, Nano Lett., 2008, 8, 2463–2468 CrossRef CAS PubMed.
- V. N. Belov, M. L. Bossi, J. Foelling, V. P. Boyarskiy and S. W. Hell, Rhodamine Spiroamides for Multicolor Single-Molecule Switching Fluorescent Nanoscopy, Chem. – Eur. J., 2009, 15, 10762–10776 CrossRef CAS PubMed.
- H. Aoki, K. Mori and S. Ito, Conformational analysis of single polymer chains in three dimensions by super-resolution fluorescence microscopy, Soft Matter, 2012, 8, 4390–4395 RSC.
- D. Aquino, A. Schönle, C. Geisler, C. v. Middendorff, C. A. Wurm, Y. Okamura, T. Lang, S. W. Hell and A. Egner, Two-color nanoscopy of three-dimensional volumes by 4Pi detection of stochastically switched fluorophores, Nat. Methods, 2011, 8, 353–359 CrossRef CAS PubMed.
- H. N. Kim, M. H. Lee, H. J. Kim, J. S. Kim and J. Yoon, A new trend in rhodamine-based chemosensors: application of spirolactam ring-opening to sensing ions, Chem. Soc. Rev., 2008, 37, 1465–1472 RSC.
- X. Chen, T. Pradhan, F. Wang, J. S. Kim and J. Yoon, Fluorescent Chemosensors Based on Spiroring-Opening of Xanthenes and Related Derivatives, Chem. Rev., 2012, 112, 1910–1956 CrossRef CAS PubMed.
- H. Zheng, X.-Q. Zhan, Q.-N. Bian and X.-J. Zhang, Advances in modifying fluorescein and rhodamine fluorophores as fluorescent chemosensors, Chem. Commun., 2013, 49, 429–447 RSC.
- D. T. Quang and J. S. Kim, Fluoro- and Chromogenic Chemodosimeters for Heavy Metal Ion Detection in Solution and Biospecimens, Chem. Rev., 2010, 110, 6280–6301 CrossRef CAS PubMed.
- Y. Yang, Q. Zhao, W. Feng and F. Li, Luminescent Chemodosimeters for Bioimaging, Chem. Rev., 2013, 113, 192–270 CrossRef CAS PubMed.
- W. Zhang, B. Tang, X. Liu, Y. Liu, K. Xu, J. Ma, L. Tong and G. Yang, A highly sensitive acidic pH fluorescent probe and its application to HepG2 cells, Analyst, 2009, 134, 367–371 RSC.
- A. Liu, M. Hong, W. Yang, S. Lu and D. Xu, One-pot synthesis of a new rhodamine-based dually-responsive pH sensor and its application to bioimaging, Tetrahedron, 2014, 70, 6974–6979 CrossRef CAS.
- Z. Li, S. Wu, J. Han and S. Han, Imaging of intracellular acidic compartments with a sensitive rhodamine based fluorogenic pH sensor, Analyst, 2011, 136, 3698–3706 RSC.
- K. Talley and E. Alexov, On the pH-optimum of activity and stability of proteins, Proteins, 2010, 78, 2699–2706 CAS.
- R. Martínez-Zaguilán, B. F. Chinnock, S. Wald-Hopkins, M. Bernas, D. Way, M. H. Witte and R. J. Gillies, [Ca2+]i and pHin homeostasis in kaposi sarcoma cells, Cell. Physiol. Biochem., 1996, 6, 169–148 CrossRef.
- D. Perez-Sala, D. Collado-Escobar and D. F. Mollinedo, Intracellular alkalinization suppresses lovastatin-induced apoptosis in HL-60 cells through the inactivation of a pH-dependent endonuclease, J. Biol. Chem., 1995, 270, 6235–6242 CrossRef CAS PubMed.
- T. A. Davies, R. E. Fine, R. J. Johnson, C. A. Levesque, W. H. Rathbun, K. F. Seetoo, S. J. Smith, G. Strohmeier, L. Volicer, L. Delva and E. R. Simons, Non-age Related Differences in Thrombin Responses by Platelets from Male Patients with Advanced Alzheimer's Disease, Biochem. Biophys. Res. Commun., 1993, 194, 537–543 CrossRef CAS PubMed.
- W. Pan, H. Wang, L. Yang, Z. Yu, N. Li and B. Tang, Ratiometric Fluorescence Nanoprobes for Subcellular pH Imaging with a Single-Wavelength Excitation in Living Cells, Anal. Chem., 2016, 88, 6743–6748 CrossRef CAS PubMed.
- H. Li, H. Guan, X. Duan, J. Hu, G. Wang and Q. Wang, An acid catalyzed reversible ring-opening/ring-closure reaction involving a cyano-rhodamine spirolactam, Org. Biomol. Chem., 2013, 11, 1805–1809 RSC.
- K.-K. Yu, K. Li, J.-T. Hou, H.-H. Qin, Y.-M. Xie, C.-H. Qian and X.-Q. Yu, Rhodamine-based lysosome-targeted fluorescence probes: high pH sensitivity and their imaging application in living cells, RSC Adv., 2014, 4, 33975–33980 RSC.
- H. Li, C. Wang, M. She, Y. Zhu, J. Zhang, Z. Yang, P. Liu, Y. Wang and J. Li, Two rhodamine lactam modulated lysosome-targetable fluorescence probes for sensitively and selectively monitoring subcellular organelle pH change, Anal. Chim. Acta, 2015, 900, 97–102 CrossRef CAS PubMed.
- E. S. Trombetta, M. Ebersold, W. Garrett, M. Pypaert and I. Mellman, Activation of Lysosomal Function During Dendritic Cell Maturation, Science, 2003, 299, 1400–1403 CrossRef CAS PubMed.
- C. Nilsson, K. Kågedal, U. Johansson and K. Öllinger, Analysis of cytosolic and lysosomal pH in apoptotic cells by flow cytometry, Methods Cell Sci., 2003, 25, 185–194 CrossRef PubMed.
- H. Montenegro, M. Di Paolo, D. Capdevila, P. F. Aramendía and M. L. Bossi, The mechanism of the photochromic transformation of spirorhodamines, Photochem. Photobiol. Sci., 2012, 11, 1081–1086 RSC.
- R. V. Søndergaard, N. M. Christensen, J. R. Henriksen, E. K. Pramod Kumar, K. Almdal and T. L. Andresen, Facing the Design Challenges of Particle-Based Nanosensors for Metabolite Quantification in Living Cells, Chem. Rev., 2015, 115, 8344–8378 CrossRef PubMed.
- K. Wang, X. He, X. Yang and H. Shi, Functionalized Silica Nanoparticles: A Platform for Fluorescence Imaging at the Cell and Small Animal Levels, Acc. Chem. Res., 2013, 46, 1367–1376 CrossRef CAS PubMed.
- A. Burns, H. Ow and U. Wiesner, Fluorescent core–shell silica nanoparticles: towards “Lab on a Particle” architectures for nanobiotechnology, Chem. Soc. Rev., 2006, 35, 1028–1042 RSC.
- F. Wang, W. Tan, Y. Zhang, X. Fan and M. Wang, Luminescent nanomaterials for biological Labelling, Nanotechnology, 2006, 17, R1–R13 CrossRef CAS.
- L. Wang, C. Yang and W. Tan, Dual-Luminophore-Doped Silica Nanoparticles for Multiplexed Signaling, Nano Lett., 2005, 5, 37–43 CrossRef CAS PubMed.
- F. Gao, L. Tang, L. Dai and L. Wang, A fluorescence ratiometric nano-pH sensor based on dual-fluorophore-doped silica nanoparticles, Spectrochim. Acta, Part A, 2007, 67, 517–521 CrossRef PubMed.
- R. P. Bagwe, L. R. Hilliard and W. Tan, Surface Modification of Silica Nanoparticles to Reduce Aggregation and Nonspecific Binding, Langmuir, 2006, 22, 4357–4362 CrossRef CAS PubMed.
- J. M. Rosenholm, A. Meinander, E. Peuhu, R. Niemi, J. E. Eriksson, C. Sahlgren and M. Lindén, Targeting of Porous Hybrid Silica Nanoparticles to Cancer Cells, ACS Nano, 2009, 3, 197–206 CrossRef CAS PubMed.
- B. Korzeniowska, R. Nooney, D. Wencel and C. McDonagh, Silica nanoparticles for cell imaging and intracellular sensing, Nanotechnology, 2013, 24, 442002 CrossRef CAS PubMed , and references therein.
- W. Stöber, A. Fink and E. J. Bohn, Controlled growth of monodisperse silica spheres in the micron size range, Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- O. S. Wolfbeis, An overview of nanoparticles commonly used in fluorescent bioimaging, Chem. Soc. Rev., 2015, 44, 4743–4768 RSC.
- A. Van Blaaderen and A. Vrij, Synthesis and Characterization of Colloidal Dispersions of Fluorescent, Monodisperse Silica Spheres, Langmuir, 1992, 8, 2921–2931 CrossRef CAS.
- C. Argyo, V. Weiss, C. Bräuchle and T. Bein, Multifunctional Mesoporous Silica Nanoparticles as a Universal Platform for Drug Delivery, Chem. Mater., 2014, 26, 435–451 CrossRef CAS.
- V. Cauda, A. Schlossbauer, J. Kecht, A. Zürner and T. Bein, Multiple Core–Shell Functionalized Colloidal Mesoporous Silica Nanoparticles, J. Am. Chem. Soc., 2009, 131, 11361–11370 CrossRef CAS PubMed.
- S. Hornig, C. Biskup, A. Gräfe, J. Wotschadlo, T. Liebert, G. J. Mohr and T. Heinze, Biocompatible fluorescent nanoparticles for pH-sensoring, Soft Matter, 2008, 4, 1169–1172 RSC.
- J. Lei, L. Wang and J. Zhang, Ratiometric pH sensor based on mesoporous silica nanoparticles and Förster resonance energy transfer, Chem. Commun., 2010, 46, 8445–8447 RSC.
- S. Wu, Z. Li, J. Han and S. Han, Dual colored mesoporous silica nanoparticles with pH activable rhodamine-lactam for ratiometric sensing of lysosome acidity, Chem. Commun., 2011, 47, 11276–11278 RSC.
- M. H. Marchena, M. Granada, A. V. Bordoni, M. Joselevich, H. Troiani, F. J. Williams and A. Wolosiuk, Organized thiol functional groups in mesoporous core shell colloids, J. Solid State Chem., 2012, 187, 97–102 CrossRef CAS.
- E. Herz, H. Ow, D. Bonner, A. Burns and U. Wiesner, Dye structure-optical property correlations in near-infrared fluorescent core-shell silica nanoparticles, J. Mater. Chem., 2009, 19, 6341–6347 RSC.
- The properties of compounds 2 and 4 were found to be identical, within experimental errors. Thus, compound 2 was used as model compound, to avoid any issues with the stability of the maleimide group.
- X. Xie, J. Zhai, Z. Jarolímová and E. Bakker, Determination of pKa Values of Hydrophobic Colorimetric pH Sensitive Probes in Nanospheres, Anal. Chem., 2016, 88, 3015–3018 CrossRef CAS PubMed.
- K.-M. Kim, H. M. Kim, W.-J. Lee, C.-W. Lee, T. Kim, J.-K. Lee, J. Jeong, S.-M. Paek and J.-M. Oh, Surface treatment of silica nanoparticles for stable and charge-controlled colloidal silica, Int. J. Nanomed., 2014, 9(Suppl 2), 29–40 Search PubMed.
- A. Zane, C. McCracken, D. A. Knight, T. Young, A. D. Lutton, J. W. Olesik, W. J. Waldman and P. K. Dutta, Uptake of bright fluorophore core-silica shell nanoparticles by biological systems, Int. J. Nanomed., 2015, 10, 1547–1567 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8pp00133b |
| ‡ Current address: Max-Planck-Institute for Medical Research, Jahnstraße 29, 69120-Heidelberg, Germany. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2019 |