
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dimeric benzoboroxoles for targeted activity against Mycobacterium tuberculosis†
Collette S.
Guy
 ab,
Kathryn
Murray
b,
Matthew I.
Gibson
*bc and
Elizabeth
Fullam
*a
ab,
Kathryn
Murray
b,
Matthew I.
Gibson
*bc and
Elizabeth
Fullam
*a
aSchool of Life Sciences, University of Warwick, CV4 7AL, UK. E-mail: e.fullam@warwick.ac.uk
bDepartment of Chemistry, University of Warwick, CV4 7AL, UK. E-mail: m.i.gibson@warwick.ac.uk
cWarwick Medical School, University of Warwick, CV4 7AL, UK
First published on 29th October 2019
Abstract
Dimeric benzoboroxoles that are covalently linked by a short scaffold enhance selective anti-tubercular activity. These multimeric benzoboroxole compounds are capable of engaging the specific extracellular Mycobacterium tuberculosis glycans, do not lead to the evolution of resistance and bypass the need to cross the impermeable mycobacterial cell envelope barrier.
Introduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis, is a major global health problem with over 10 million new cases of TB and 1.6 million deaths reported by the World Health Organisation in 2017.1 The rapid emergence of drug resistance has led to an alarming increase in both multidrug-resistant (MDR)-TB and extensively drug-resistant (XDR)-TB rendering the current frontline TB therapeutic regimens ineffective.1 Clearly there is an urgent need to develop innovative therapeutic interventions that have distinct mechanisms of action to reduce the global TB burden.The Mtb cell envelope is distinct from other microorganisms and contributes to the survival and virulence of this pathogen.2–4 The complex lipids and glycans within the cell envelope provide a formidable, impermeable barrier to antibiotics that further complicates efforts to eradicate TB.2–5 The architecture of the Mtb cell envelope comprises a complex macromolecular core consisting of peptidoglycan linked to arabinogalactan that in turn is covalently attached to long-chain mycolic acids (mAGP complex).3 Additional non-covalently linked lipids and lipoglycans are interspersed within the mycolate layer of the mAGP complex to form an outer, waxy, ‘mycomembrane’.2–4
The current anti-tubercular agents are lethal to Mtb through modulation of well-defined intracellular pathways.6 In particular, targeting key enzymes that are involved in the synthesis and assembly of the Mtb cell envelope has proved to be a highly effective strategy for the development of new TB therapeutics,5,6 highlighted by the clinical use of the front-line antitubercular agents isoniazid and ethambutol and several second-line drugs that include cycloserine and ethionamide.7–9
Recent studies have shown that an alternative anti-tubercular approach is possible through the direct capture of the structurally unique mycobacterial extracellular glycans with multimeric boronic acids.10 Specifically, two boronic acid units separated by a short poly(ethylene glycol) (PEG) linker were found to be optimal for antimycobacterial selectivity and potency.10 This concept exploits the vulnerability of the essential mycobacterial cell wall with the advantage that the antimycobacterial agent does not need to cross the Mtb cell envelope and avoids one of the major obstacles in the development of new TB therapeutics.
Multimeric boronic acids have emerged as synthetic receptors for the specific detection of glycans.11 The nature of both the boronic acid and the scaffold linker can be tuned and modulated to enable selective glycan recognition.11 Benzoboroxoles are water soluble heterocyclic-modified boronic acids,12,13 and in addition to their role as glycan sensors have also emerged as an interesting class of biomolecules that have a range of potent biological activities against a range of diseases.14–16 This includes potent activity and selectivity against Mtb, and the GlaxoSmithKline (GSK) aminobenzoboroxole analogue (GSK656) that targets leucyl-tRNA synthetase is a promising anti-tubercular candidate that has been progressed into phase II clinical trials.17,18
Here, we therefore replaced the boronic acid units to generate a panel of dimeric benzoboroxoles to promote the interaction with the unique extracellular mycobacterial glycans. These compounds have excellent, selective activity for mycobacteria, including the Mtb pathogen, are capable of complexing to Mtb glycans without resulting in the evolution of resistance and have a distinct mode of action to monomeric benzoboroxole analogues.
Results and discussion
Dimeric benzoboroxoles were synthesised such that the inter-boroxole distance was systematically varied by the use of various oligo(ethylene glycol) diamine linkers (Fig. 1). Dimeric benzoboroxoles were obtained in one step by reaction of benzoboroxole-carboxylic acid (1-hydroxy-1,3-dihydro-2,1-benzoxaborole-6-carboxylic acid) 6 with the diamines using 1.1 equiv. PyBOP as the coupling reagent (Scheme S1, ESI†). Following purification all compounds were characterised by 1H and 13C NMR and mass spectrometry confirming successful coupling and high purity of 2–5 (Fig. S3–12, ESI†). An Alizarin red based assay confirmed carbohydrate-benzoboroxole binding with similar trends and affinities observed for the carbohydrates with the monomeric benzoboroxole 1 and the dimeric analogues 2–4 (Fig. S1, ESI†).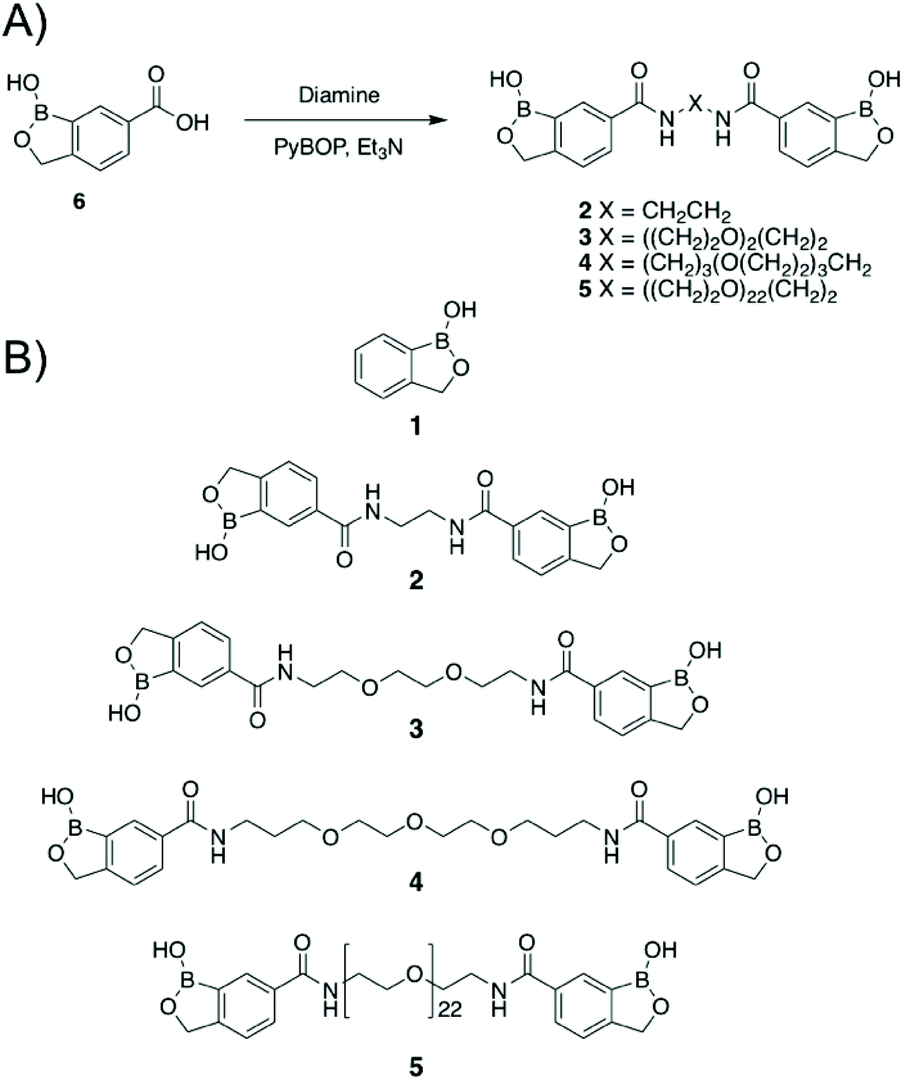 | ||
| Fig. 1 Synthesis of dimeric benzoboroxoles (2–5). (A) Synthetic strategy (Et3N: triethylamine; PyBOP: benzotriazol-1-yl-oxytripyrrol-idinophosphonium hexafluorophosphate); (B) compounds used in this study. | ||
Next, the panel of dimeric benzoboroxole compounds (2–5) was evaluated for antibacterial activity using the resazurin reduction assay19 to determine the minimum inhibitory concentrations (MIC) against Mtb, Mycobacterium bovis BCG and Mycobacterium smegmatis and the Gram-negative strains Escherichia coli and Pseudomonas putida. The monomeric benzoboroxole compound 1, which is known to target leucyl-tRNA synthetase (LeuRS),18,20 had micromolar potencies with similar MIC values for all of the mycobacterial and Gram-negative strains evaluated (Table 1, Fig. 2). However, by changing the benzoboroxole to a dimeric unit (2–4) a large increase in selectivity for mycobacteria was observed (Table 1, Fig. 2). Whilst the MIC values of the dimeric benzoboroxoles 2–4 increased compared to mono-benzoboroxole 1 for all strains (Table 1, Fig. 2), there was a marked increase in the MICs of 2–4 against the Gram-negative strains that was not observed for mycobacteria. The observed change in the MIC profile of the dimeric benzoboroxoles 2–4 compared to the monomeric benzoboroxole unit 1 confirms that selectivity for mycobacteria is controlled by the dimeric presentation. Dimeric benzoboroxole 5, which contains a longer scaffold linker length (PEG22) between the boroxole pharmacophores, resulted in a much higher MIC (25 mM) than those with 0–3 PEG units (0.39–6.25 mM) (2–4) against mycobacteria and loss of specificity with similar potencies observed for the Gram-negative organisms E. coli and P. putida (Table 1, Fig. 2). These observations agree with our previous study using dimeric boronic acids which also found that short linker lengths were crucial for optimising anti-mycobacterial activity and selectivity.10
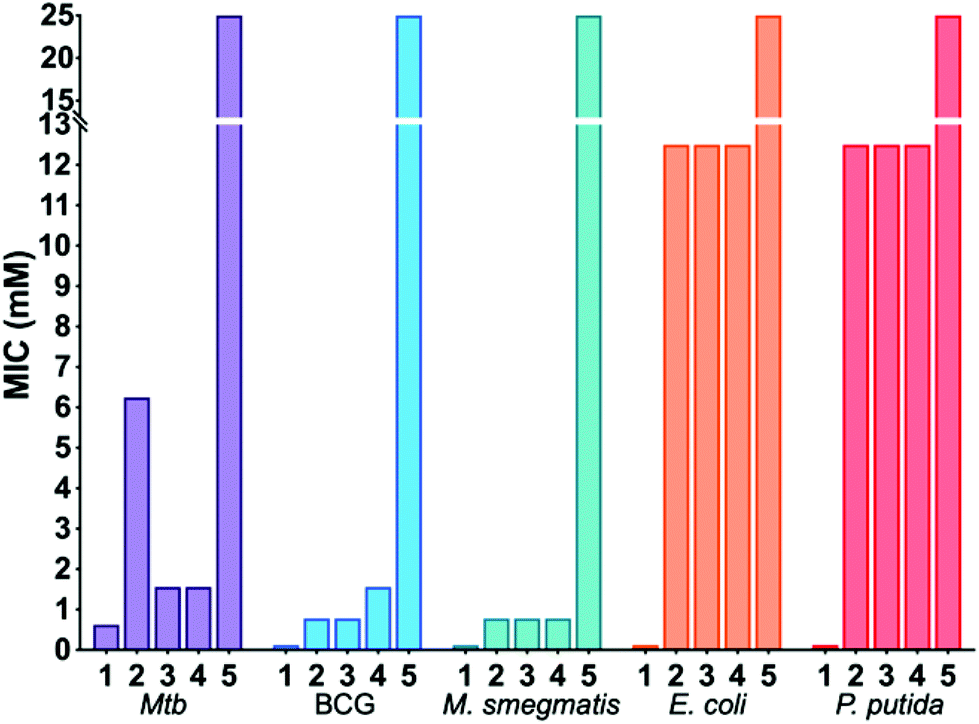 | ||
| Fig. 2 Antimicrobial activities of benzoboroxoles 1–5. | ||
| MIC mycobacteria (mM) | MIC Gram-negative (mM) | ||||
|---|---|---|---|---|---|
| Mtb | M. bovis BCG | M. smegmatis | E. coli | P. putida | |
| 1 | 0.625 | 0.125 | 0.078–0.125 | 0.125 | 0.125 |
| 2 | 6.25 | 0.78 | 0.78 | >12.5 | >12.5 |
| 3 | 1.56 | 0.78 | 0.39–0.78 | >12.5 | >12.5 |
| 4 | 1.56 | 1.56 | 0.39–0.78 | >12.5 | >12.5 |
| 5 | >25 | >25 | 25 | >25 | >25 |
Cytotoxicity evaluation of dimeric benzoboroxoles 2–4 against human lung epithelial A549 cells showed that compounds 2–4 were not toxic at concentrations >10 mM (3) and >5 mM (2, 4), which were the highest concentrations possible to test due to solubility limits and toxicity levels of DMSO (Table 2). Importantly, evaluation of compounds 2–4 against erythrocytes did not result in haemolysis or agglutination at concentrations above 10 mM (Table 2). Combined, these results demonstrate that dimeric benzoboroxoles 2–4 have a higher selectivity for mycobacteria than mammalian cells.
| Compound | A549 | Red blood cells | |
|---|---|---|---|
| MIC99 (mM) | Haemolysis (%) | Agglutination | |
| ND – no agglutination detected. | |||
| 2 | >5 | 1.1 | ND |
| 3 | >10 | 0.75 | ND |
| 4 | >5 | 1.25 | ND |
Boron containing molecules are known to inhibit β-lactamases that otherwise degrade β-lactam antibiotics and their combined use has proved to be a useful strategy to restore the activity of this important class of antibiotics.21,22 To determine whether dimeric benzoboroxoles also function in this manner we used a checkerboard assay to evaluate the potential interactions of compounds 3 and 4 with the β-lactam meropenem on Mtb. To quantify the interactions between these compounds the sum of the fractional inhibitory index (∑FIC) was calculated for each combination (Table 3).23 For meropenem, the ∑FIC was found to be 2 and 1.5 for compounds 3 and 4 respectively indicating no synergistic or antagonistic action with the β-lactam, in contrast to the calculated ∑FIC of 0.3 for meropenem in combination with the β-lactamase inhibitor sulbactam. These results provide evidence that the multimeric benzoboroxoles 3 and 4 are not recognised by β-lactamase targets in Mtb suggesting that these compounds have a unique mechanism of action. In addition, the interaction of benzoboroxoles 3 and 4 with the front-line TB drug rifampicin was also evaluated and again no synergistic nor antagonistic behaviour was observed with the ∑FIC calculated as 2 for both compounds (Table 3).
| Compound | MIC (mM) | Interaction profile with meropenem | Interaction profile with rifampicin | ||
|---|---|---|---|---|---|
| ∑FICa | Outcome | ∑FICa | Outcome | ||
| a Fractional inhibitory concentrations (FICs) were calculated by use of the formula as described previously.24–26 | |||||
| 3 | 1.56 | 2 | Additive | 2 | Additive |
| 4 | 1.56 | 1.5 | Additive | 2 | Additive |
| Sulbactam | 0.134 | 0.313 | Synergistic | — | — |
To provide more information of the mechanism of action and attempt to elucidate the target of the dimeric benzoboroxoles, we attempted to generate spontaneous resistant mutants of M. bovis BCG at 5× MIC of either the monomer 1 or the dimeric compound 3. Interestingly, we were unable to generate resistant mutants against the dibenzoboroxole (3) which further supports the hypothesis that these molecules have a distinct mode of action. In contrast, resistant mutants were derived against mono-benzoboroxole (1) with a frequency of five resistant mutants (BRX1–BRX5) generated from 1 × 108 cells. Whole genome sequencing of each resistant strain (BRX1–BRX5) revealed non-synonymous mutations in 5 gene loci compared to the wild-type M. bovis BCG reference strain (Table 4). In particular, we identified non-synonymous polymorphisms in the leuS gene (Table 4) in each resistant strain (BRX1–5), which encodes for the leucine-tRNA ligase (LeuS) and is reported to be a target of monomeric benzoboroxoles, including GSK656.18 To establish whether there is cross-resistance of the five mono-benzoboroxole M. bovis BCG mutants (BRX1–5) to the dimeric benzoboroxoles 2–4 we re-evaluated the MICs of 1–4 against these mutant strains. As expected, the MIC of mono-benzoboroxole 1 increased by 10–20 fold for each mutant (Table 5), whereas the MIC of the dimeric benzoboroxoles (2–4) were found to be comparable with the MICs that had been determined previously for wild-type M. bovis BCG (Table 5). These results provide robust evidence that the dimeric benzoboroxoles have a distinct molecular target compared to monomeric benzoboroxoles and that this does not involve interaction with LeuS.
| M. bovis BCG chromosome positiona | Mutation-type | Codon change | Amino acid change | Gene | Strainb |
|---|---|---|---|---|---|
| a Genomic positions are relevant to M. bovis BCG Pasteur 1173P2 strain (NC_008769). b Five resistant mutants of M. bovis BCG were generated against benzoboroxole 1 and referred to as BRX1, BRX2, BRX3, BRX4 and BRX5. | |||||
| 74515 | Non-synonymous | tAc/tGc | Y435C | leuS | BRX3 |
| 74521 | Non-synonymous | aCc/aTc | T437I | leuS | BRX5 |
| 74527 | Non-synonymous | gCa/gAa | A439E | leuS | BRX2 |
| BRX4 | |||||
| 74757 | Non-synonymous | Aaa/Gaa | K516E | leuS | BRX1 |
| 2434500 | Codon change plus codon insertion | acg/acCACg | T258TT | trpD | BRX5 |
| 3627670 | Non-synonymous | aCc/aAc | T245N | BCG_3323c | BRX2 |
| BRX3 | |||||
| BRX4 | |||||
| 3663833 | Non-synonymous | aCc/aAc | T245N | BCG_3359c | BRX2 |
| BRX3 | |||||
| BRX4 | |||||
| 4112296 | Frame shift | ggt/ggGt | G192G | glpK | BRX2 |
| BRX3 | |||||
| BRX4 | |||||
| BRX5 | |||||
| MIC (mM) | ||||||
|---|---|---|---|---|---|---|
| WT | BRX1 | BRX2 | BRX3 | BRX4 | BRX5 | |
| 1 | 0.078–0.125 | 1.25–2.5 | 1.25 | 1.25–2.5 | 1.25 | 1.25–2.5 |
| 2 | 0.78 | 0.78 | 0.78 | 0.78 | 1.56 | 1.56 |
| 3 | 0.78 | 1.56 | 1.56 | 1.56 | 1.56 | 1.56 |
| 4 | 1.56 | 3.13 | 3.13 | 3.13 | 1.56 | 1.56 |
Finally, we used biolayer interferometry (BLI) to probe whether the dimeric benzoboroxoles are able to specifically recognise glycans that are located in the mycobacterial cell envelope (Fig. S2 ESI†). A biotinylated benzoboronic acid (7) was synthesised (Scheme S2, ESI† and Fig. 3) and immobilised onto streptavidin-coated BLI sensors to ensure multivalent presentation of the glycan recognition units. Isolated components of the mycobacterial cell envelope were assessed for binding to the benzoboroxole sensors (Fig. 3). Of the Mtb glycans that we evaluated the highest affinity was observed with the benzoboroxole sensor for the trehalose containing glycolipids with a higher selectivity for trehalose dimycolate (TDM) over trehalose monomycolate (TMM) (Fig. 3). Benzoboroxoles are known to complex with hexopyranosides and nonreducing glycopyranosides and our findings are consistent with these results.12 Weak binding affinity towards arabinogalactan (AG) was observed and no specific binding to other mycobacterial cell envelope components was detected. This is in contrast to our previous studies which found that a boronic acid functionalised BLI sensor bound to TDM and TMM equally well but also had high affinity for AG as well as lipoarabinomannan (LAM) and peptidoglycan (PG) components.10 These data confirm that benzoboroxoles can engage with several components of the mycobacterial cell wall and that this has a fatal impact on mycobacterial cells.
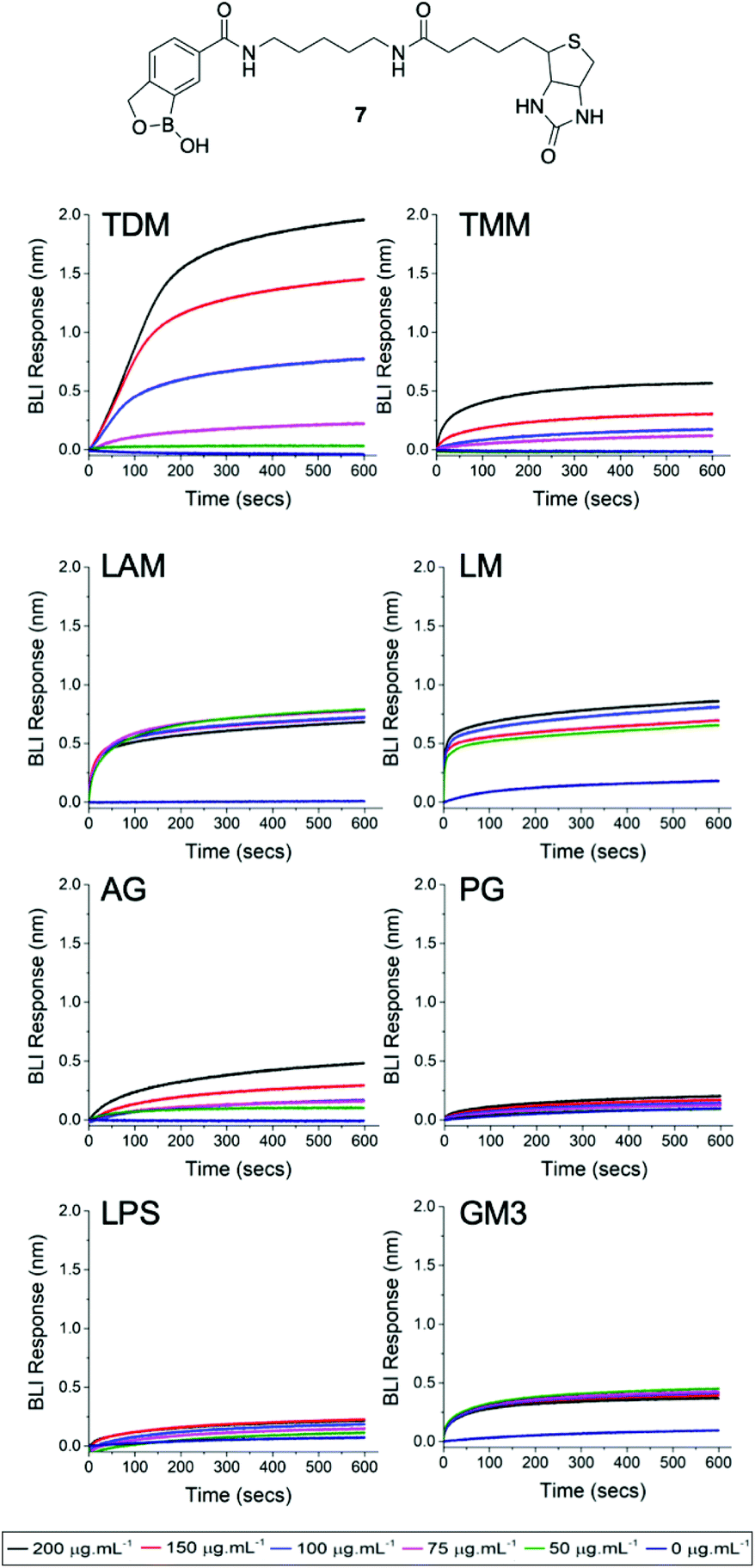 | ||
| Fig. 3 Biolayer interferometry analysis of 3-carboxy benzoboroxole functional sensor 7 against TDM (trehlaose dimycolate, TMM (trehalose monomycolate, LAM (lipoarabinomannan), LM (lipomannan), AG (arabinogalactan), PG (peptidoglycan), LPS (lipopolysaccharide), GM3 (glycan monosialodihexosylganglioside). | ||
Furthermore, we investigated whether the benzoboroxole unit is able to recognise glycans that are associated with either Gram-negative bacteria or mammalian cell surfaces. In these experiments, no specific binding was observed with the Gram-negative glycan lipopolysaccharide (LPS) or the mammalian specific glycan monosialodihexosylganglioside (GM3). Overall, these results may help to explain the apparent antimicrobial selectivity of the dimeric benzoboroxoles which may be due to the differences in how they target external mycobacterial specific glycans.
Conclusions
In conclusion, we have demonstrated that dimeric benzoboroxoles are selective antimycobacterial agents and are lethal to Mtb via a different mode of action from the monomeric benzoboroxole analogue (1). Further evidence to support and validate our findings came from the resistance studies that confirms there is no cross-resistance between benzoboroxole 1 and the dimeric compounds 2–4. Importantly, we clearly identified that we were unable to evolve resistance against the dimeric benzoboroxole 3, which is advantageous in the ongoing battle to combat antibiotic resistance. This study has revealed that multimeric benzoboroxoles interact directly with the mycobacterial trehalose glycolipids that are absent in the human host. It is possible that the interaction of two benzoboroxoles, which are covalently linked via a short scaffold, with these essential components of the Mtb cell envelope impacts on the survival of mycobacteria. The ability to target surface-exposed glycans directly bypasses the obstacle of the mycobacterial cell envelope and the need to cross this impermeable barrier and may guide the design of an entire new class of highly selective anti-mycobacterial agents.Conflicts of interest
EF, MIG and CSG are named inventors on a patent application relating to this work.Acknowledgements
This work was supported by the Antimicrobial Resistance Cross Council Initiative supported by the seven Research Councils (MR/N006917/1), the EPSRC (EP/M027503/1), a Sir Henry Dale fellowship to EF jointly funded by the Wellcome Trust and the Royal Society (104193/Z/14/Z) and the European Research Council (789182). Genome sequencing was provided by MicrobesNG (http://www.microbesng.uk), which is supported by the BBSRC (BB/L024209/1).References
- WHO Global Tuberculosis Report, http://www.who.int/tb/publications/global_report/en/.
- P. J. Brennan, Tuberculosis, 2003, 83, 91–97 CrossRef CAS PubMed
.
- P. J. Brennan and H. Nikaido, Annu. Rev. Biochem., 1995, 64, 29–63 CrossRef CAS PubMed
- M. Jackson, Cold Spring Harb. Perspect Med., 2014, 4(10), a021105 CrossRef PubMed
- K. A. Abrahams and G. S. Besra, Parasitology, 2018, 145, 116–133 CrossRef PubMed
- S. Wellington and D. T. Hung, ACS Infect. Dis., 2018, 4, 696–714 CrossRef CAS PubMed
- A. Banerjee, E. Dubnau, A. Quemard, V. Balasubramanian, K. S. Um, T. Wilson, D. Collins, G. de Lisle and W. R. Jacobs Jr., Science, 1994, 263, 227–230 CrossRef CAS PubMed
- K. Mikusova, R. A. Slayden, G. S. Besra and P. J. Brennan, Antimicrob. Agents Chemother., 1995, 39, 2484–2489 CrossRef CAS PubMed
- G. A. Prosser and L. P. de Carvalho, ACS Med. Chem. Lett., 2013, 4, 1233–1237 CrossRef CAS PubMed
- C. S. Guy, M. I. Gibson and E. Fullam, Chem. Sci., 2019, 10, 5935–5942 RSC
- X. Wu, Z. Li, X. X. Chen, J. S. Fossey, T. D. James and Y. B. Jiang, Chem. Soc. Rev., 2013, 42, 8032–8048 RSC
- M. Berube, M. Dowlut and D. G. Hall, J. Org. Chem., 2008, 73, 6471–6479 CrossRef CAS PubMed
- M. Dowlut and D. G. Hall, J. Am. Chem. Soc., 2006, 128, 4226–4227 CrossRef CAS PubMed
- A. Larcher, A. Nocentini, C. T. Supuran, J. Y. Winum, A. van der Lee, J. J. Vasseur, D. Laurencin and M. Smietana, ACS Med. Chem. Lett., 2019, 10, 1205–1210 CrossRef CAS PubMed
- A. Adamczyk-Wozniak, K. M. Borys and A. Sporzynski, Chem. Rev., 2015, 115, 5224–5247 CrossRef CAS PubMed
- B. E. Elewski, R. Aly, S. L. Baldwin, R. F. Gonzalez Soto, P. Rich, M. Weisfeld, H. Wiltz, L. T. Zane and R. Pollak, J. Am. Acad. Dermatol., 2015, 73, 62–69 CrossRef CAS PubMed
- M. A. Alam, K. Arora, S. Gurrapu, S. K. Jonnalagadda, G. L. Nelson, P. Kiprof, S. C. Jonnalagadda and V. R. Mereddy, Tetrahedron, 2016, 72, 3795–3801 CrossRef CAS PubMed
- X. Li, V. Hernandez, F. L. Rock, W. Choi, Y. S. L. Mak, M. Mohan, W. Mao, Y. Zhou, E. E. Easom, J. J. Plattner, W. Zou, E. Perez-Herran, I. Giordano, A. Mendoza-Losana, C. Alemparte, J. Rullas, I. Angulo-Barturen, S. Crouch, F. Ortega, D. Barros and M. R. K. Alley, J. Med. Chem., 2017, 60, 8011–8026 CrossRef CAS PubMed
- J. C. Palomino, A. Martin, M. Camacho, H. Guerra, J. Swings and F. Portaels, Antimicrob. Agents Chemother., 2002, 46, 2720–2722 CrossRef CAS PubMed
- F. L. Rock, W. Mao, A. Yaremchuk, M. Tukalo, T. Crepin, H. Zhou, Y. K. Zhang, V. Hernandez, T. Akama, S. J. Baker, J. J. Plattner, L. Shapiro, S. A. Martinis, S. J. Benkovic, S. Cusack and M. R. Alley, Science, 2007, 316, 1759–1761 CrossRef CAS PubMed
- J. Brem, R. Cain, S. Cahill, M. A. McDonough, I. J. Clifton, J. C. Jimenez-Castellanos, M. B. Avison, J. Spencer, C. W. Fishwick and C. J. Schofield, Nat. Commun., 2016, 7, 12406 CrossRef CAS PubMed
- S. G. Kurz, S. Hazra, C. R. Bethel, C. Romagnoli, E. Caselli, F. Prati, J. S. Blanchard and R. A. Bonomo, ACS Infect. Dis., 2015, 1, 234–242 CrossRef CAS PubMed
- B. Lechartier, R. C. Hartkoorn and S. T. Cole, Antimicrob. Agents Chemother., 2012, 56, 5790–5793 CrossRef CAS PubMed
- B. Lechartier, R. C. Hartkoorn and S. T. Cole, Antimicrob. Agents Chemother., 2012, 56, 5790–5793 CrossRef CAS PubMed
- F. C. Odds, J. Antimicrob. Chemother., 2003, 52, 1 CrossRef CAS PubMed
- K. H. Rand, H. J. Houck, P. Brown and D. Bennett, Antimicrob. Agents Chemother., 1993, 37, 613–615 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic, characterization, sequencing and experimental details. See DOI: 10.1039/c9ob02222h |
| This journal is © The Royal Society of Chemistry 2019 |