
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThree-state switching in a double-pole change-over nanoswitch controlled by redox-dependent self-sorting†
Sudhakar
Gaikwad
,
Merve Sinem
Özer
,
Susnata
Pramanik
and
Michael
Schmittel
 *
*
Center of Micro-and Nanochemistry and Engineering, Organische Chemie I, Universität Siegen Adolf-Reichwein-Strasse-2, 57068 Siegen, Germany. E-mail: schmittel@chemie.uni-siegen.de
First published on 13th August 2019
Abstract
The four-arm nanomechanical switch 1 with four different terminals exhibits two switching arms (contacts A and D) and two distinct stations for binding (contacts B and C). In switching State I, the azaterpyridine arm is intramolecularly coordinated to a zinc(II) porphyrin station (connection A ↔ B) while contact D (a ferrocenylbipyridine unit) and contact C (phenanthroline) remain disconnected. After addition of copper(I) ions (State II) both connections A ↔ B and C ↔ D are established. Upon one-electron oxidation, double-pole change-over switching cleaves both connections A ↔ B & C ↔ D and establishes the new connection A ↔ C (State III). Fully reversible three-state switching (State I → State II → State III → State II → State I) was achieved by adding appropriate chemical and redox stimuli.
Introduction
In the quest for technological applications, a good variety of molecular switches have been reported that undergo nanomechanical toggling upon addition and/or removal of electrons.1,2 Despite spectacular cases of two-state switches,1,2 examples of multi-state toggling (three and more states) including at least one redox-driven switching event remain scarce.3 Actually, full reversibility in molecular switches with three and more states remains a general challenge toward the development of multi-functional devices.4Further challenges encompass the development of switching protocols that involve entangled nanomechanical motions, for instance when a toggling arm is replaced by another arm in a single step giving rise to a double-pole change-over switching. Such an entangled switching event is valuable because electronic and steric changes effectively happen at four sites simultaneously and may ignite a number of follow-up processes. Herein, we would like to report on a case of double-pole change-over switching by presenting nanoswitch 1 and its three-state toggling. In the key switching step, it involves self-sorting5 triggered by oxidation/reduction of a ferrocene-appended bipyridine ligand. Reliable redox-state dependent self-sorting has been demonstrated earlier in rotors,1a in a two-state nanoswitch,2d in chemical communication between up to three nanoswitches,6 and in switchable catalysis.2k
To accomplish three-state switching using chemical and electrochemical stimuli, we chose to integrate two distinct switching arms and stations into switch 1. As a result, four different terminals are attached at the central tetraphenylmethane core of 1, i.e. a (i) sterically shielded diarylphenanthroline (phenAr2) station, (ii) zinc(II) porphyrin (ZnPor) station, (iii) ferrocene-appended bipyridine (fcbipy) arm, and (iv) an azaterpyridine (azatpy) arm. For three-state switching altogether three orthogonal coordination binding motifs are needed, which are the (i) HETPHEN7 (HETeroleptic PHENanthroline) complexation in presence of copper(I) ions, (ii) HETTAP8 (HETeroleptic Terpyridine And Phenanthroline) complexation after oxidation and (iii) Nazatpy → ZnPor interaction.9
In State I (= nanoswitch 1) the azatpy arm is intramolecularly locked at the ZnPor station. After addition of copper(I) ions, we would expect that the presence of the fcbipy arm with its electron-rich ferrocene-appended bipyridine in 1 leads to the selective formation of the intramolecular HETPHEN-type7 complex [Cu(fcbipy)(phenAr2)]+ which should be favored over the HETTAP complex8 [Cu(azatpy)(phenAr2)]+. This binding preference in [Cu(1)]+ (State II) is warranted due to (i) a stronger binding of the electron-rich fcbipy unit to the copper(I)-loaded phenanthroline and (ii) the additional stabilization gained from maintaining the Nazatpy → ZnPor interaction. Upon oxidation at ferrocene, the binding strength of the fcbipy unit should be sufficiently reduced to allow toggling of the azatpy rotary arm in [Cu(1)]2+ from the ZnPor station to the copper(I) phenanthroline site thus affording a HETTAP complex (State III). Upon reduction of the ferrocenium unit, the binding strength of the fcbipy unit should be restored to regenerate State II. Finally, removal of copper(I) ions with 2-ferrocenyl-9-mesityl-[1,10]-phenanthroline is expected to reset the original locked State I (Fig. 1). Interconversion of States II and III fulfills the criteria for a double-pole change-over switching process (Fig. 2).
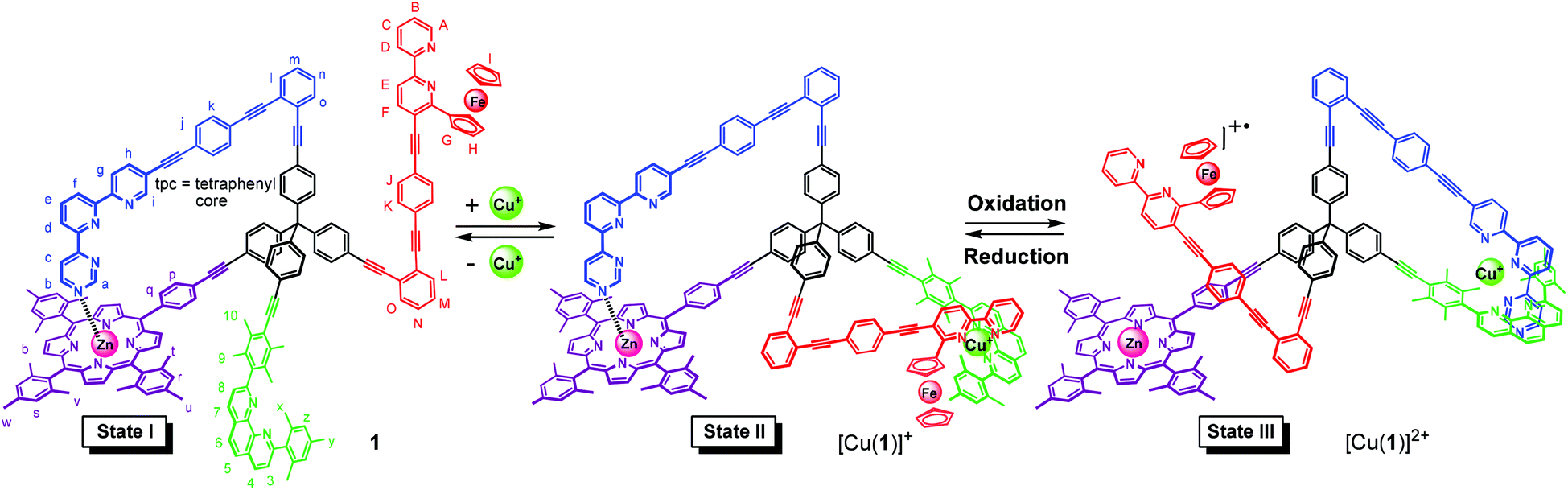 | ||
| Fig. 1 Three-state switching in nanoswitch 1. | ||
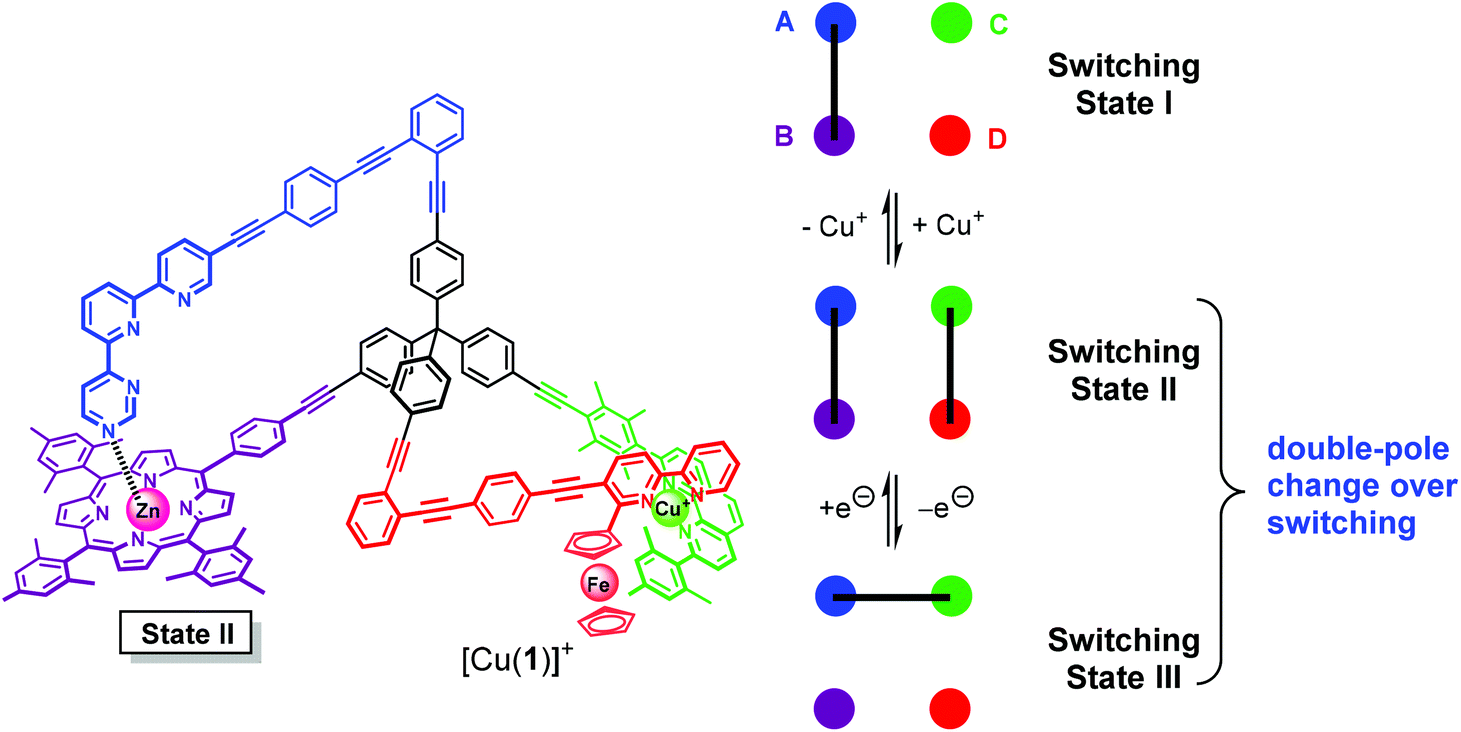 | ||
| Fig. 2 Nanoswitch 1 and the connections in all switching states. | ||
Results and discussion
Synthesis
Nanoswitch 1 was prepared through a series of Sonogashira cross-coupling reactions as shown in Scheme 1. First, 6-ferrocenyl-5-((trimethylsilyl)ethynyl)-2,2′-bipyridine10 (2) was deprotected in aq. K2CO3, THF and methanol yielding compound 3 in 74% yield. The Sonogashira reaction of compound 3 with ((4-bromophenyl)ethynyl)trimethylsilane11 in a mixture of THF/triethylamine in presence of Pd(PPh3)4 as a catalyst furnished compound 4 in good yield. Compound 4 was converted to the terminal ethynyl derivative 5 by treatment with aq. K2CO3 in THF and methanol. A follow-up Sonogashira coupling of compound 5 with 1,2-diiodobenzene using Pd(0) catalyst in anhydrous DMF and triethylamine provided the rotary arm 6 in 63% yield. Finally, Sonogashira coupling of compound 6 with the known scaffold 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4c using Pd(0) as catalyst in anhydrous DMF and triethylamine furnished nanoswitch 1 in 22% yield.
4c using Pd(0) as catalyst in anhydrous DMF and triethylamine furnished nanoswitch 1 in 22% yield.
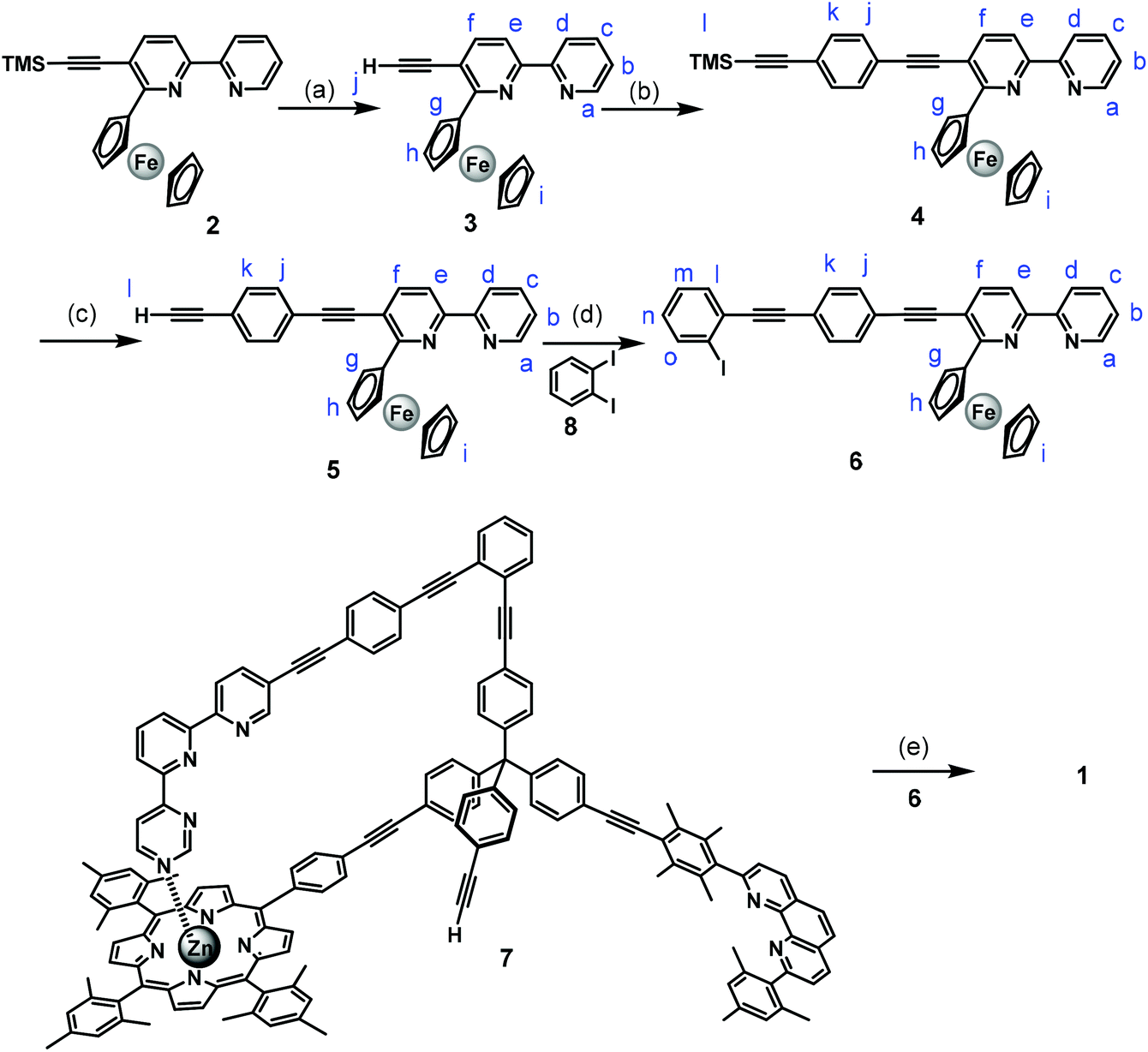 | ||
| Scheme 1 Synthesis of nanoswitch 1. (a) aq. K2CO3, MeOH–THF, 74%; (b) Pd(PPh3)4, THF–Et3N, 60 °C, 18 h, 98%; (c) aq. K2CO3, MeOH–THF, 98%; (d) Pd(PPh3)4, THF–Et3N, 60 °C, 18 h, 63%; (e) Pd(PPh3)4, DMF–Et3N, 60 °C, 24 h, 22%. | ||
Nanoswitch 1 was fully characterized using NMR, UV-vis spectroscopy, ESI-MS, and elemental analysis. The ESI-MS displays a molecular ion peak at m/z = 1309.7 that is diagnostic for the doubly protonated [1·2H]2+ with the experimental isotopic splitting pattern matching the computed one (ESI, Fig. S32†). In the 1H NMR spectrum of 1, pyrimidine protons a-H and b-H of the azatpy arm are located in the aliphatic region at 3.76 and 2.73 ppm, respectively, which confirms the immersion of the terminal pyrimidine (pym) ring into the ZnPor's shielding zone suggesting a Nazatpy → ZnPor interaction (ESI, Fig. S13†). The sharp signals of protons a-H and b-H are concentration independent (c = 0.55 mM to 2.9 mM, in CD2Cl2, ESI, Fig. S17†) precluding intermolecular coordination. Equally, the Q-band absorption of 1 at 561 nm remained constant when the concentration was varied from c = 10−6 to 10−4 M (in DCM, ESI, Fig. S24†). Moreover, the Soret band absorption of 1 at 429 nm (c = 10−6 M, in DCM) supports the coordination of the pyrimidine 4N nitrogen to the ZnPor station. The 8 nm bathochromic shift of the Soret band in 1 may be compared to that of an uncoordinated tetraphenyl zinc porphyrin (ZnTPP) at 421 nm (ESI, Fig. S23†).9,12 In summary, combined 1H NMR and UV-vis data corroborate intramolecular coordination of the pyrimidine ring to the zinc porphyrin unit in 1.
Three-state switching in nanoswitch 1
At first, we investigated reversible switching between States I and II by addition and removal of copper(I) ions. Addition of 1.0 equiv. of [Cu(CH3CN)4][B(C6F5)4] to a solution of 1 in dichloromethane-d2 resulted in the formation of the intramolecular HETPHEN complex [Cu(1)]+ (= State II) in 96% yield. The assignment was corroborated by 1H, 1H–1H COSY, ESI-MS and UV-vis data. The 1H NMR was informative in various ways: signals of phenanthroline protons z-H at δ = 6.95 ppm shifted and split into four sets at δ = 6.97, 6.72 and 6.96, 6.70 ppm as a result of breaking the local symmetry and formation of two diastereomers in 55:45 ratio. In addition, bipyridine protons A-H and E-H were upfield shifted from δ = 8.57 and 8.22 ppm to δ = 7.56 and 7.77 ppm, respectively, due to ring currents of the phenanthroline aryl groups (Fig. 3B). These NMR observations thus unambiguously confirmed formation of [Cu(1)]+ (= State II). The ESI-MS analysis of the resultant complex exhibited a molecular ion peak at m/z = 2741.1 (i.e. [Cu(1)·MeCN·H2O]+) supporting formation of State II (ESI, Fig. S33†). The complexation constant of [Cu(1)]+ was determined using a UV-vis titration as logK = 9.39 (ESI, Fig. S42†).
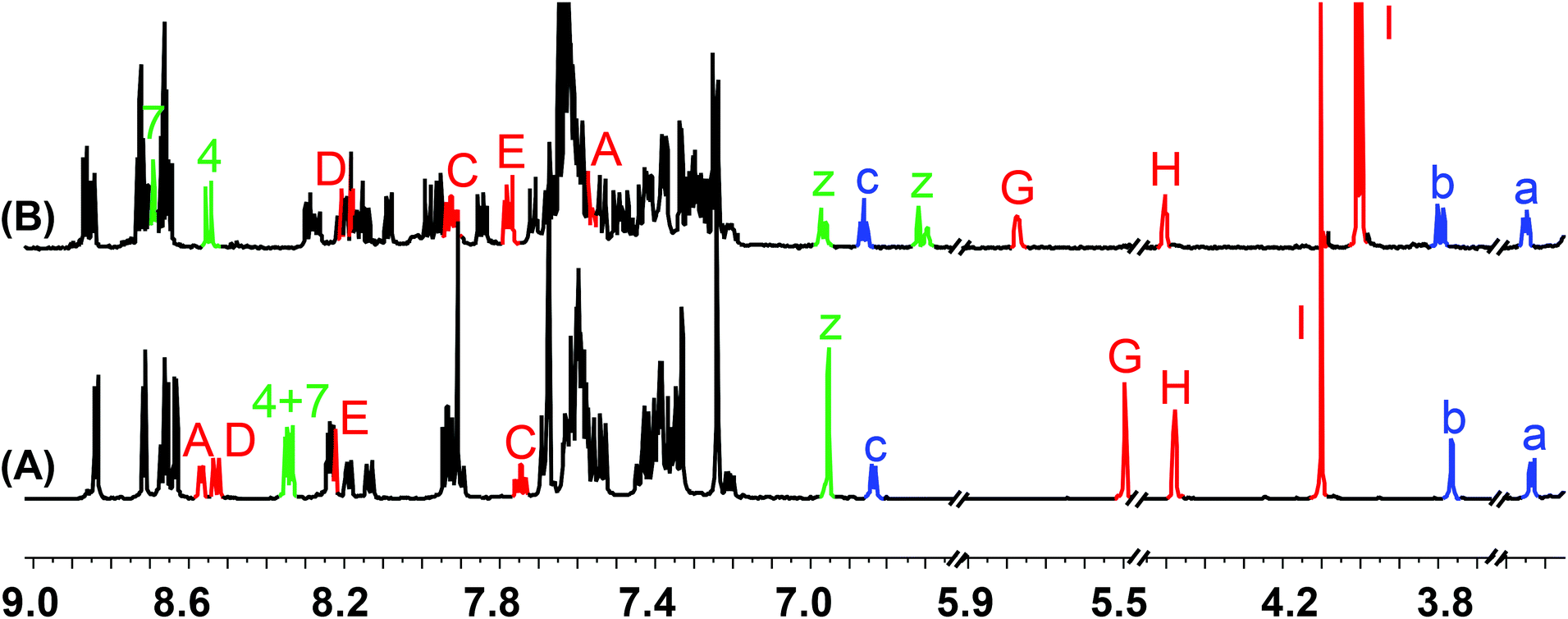 | ||
| Fig. 3 Comparison of partial 1H NMR spectra (600 MHz, CD2Cl2, 298 K) of (A) nanoswitch 1 = Switching State I and (B) complex [Cu(1)]+ = Switching State II. | ||
In addition, a UV-vis study of [Cu(1)]+ showing the Q-band absorption at 561 nm proved the existence of Nazatpy → ZnPor coordination in State II (ESI, Fig. S25†). Addition of 2.0 equiv. of 2-ferrocenyl-9-mesityl-[1,10]-phenanthroline regenerated 1, i.e. the original locked State I. Reversible switching between States I and II was checked for two cycles by addition and removal of copper(I) ions (ESI, Fig. S20†).
Thereafter, we investigated reversible switching between States II and III by redox input. A combined cyclic voltammetry (CV) and differential pulse voltammetry (DPV) study of [Cu(1)]+ in dichloromethane displayed E1/2 = 0.74 and 0.78 VSCE for Fc/Fc+ and ZnPor0/+ transitions (Fig. S38 & 39†). In contrast, with 1 vol% of acetonitrile, the complex [Cu(1)]+ revealed two well separated oxidation steps (Fig. 4), i.e. Fc/Fc+ at E1/2 = 0.47 VSCE and ZnPor0/+ at E1/2 = 0.67 VSCE, while the third wave at E1/2 = 1.11 VSCE is attributed to the Cu+/2+ transition. The data of 1 with E1/2 = 0.45 VSCE for Fc/Fc+ and ZnPor0/+ at E1/2 = 0.66 VSCE as a reference (Fig. S36 & S37†) suggest that in acetonitrile the ferrocenyl arm upon oxidation readily detaches from the copper(I) site.
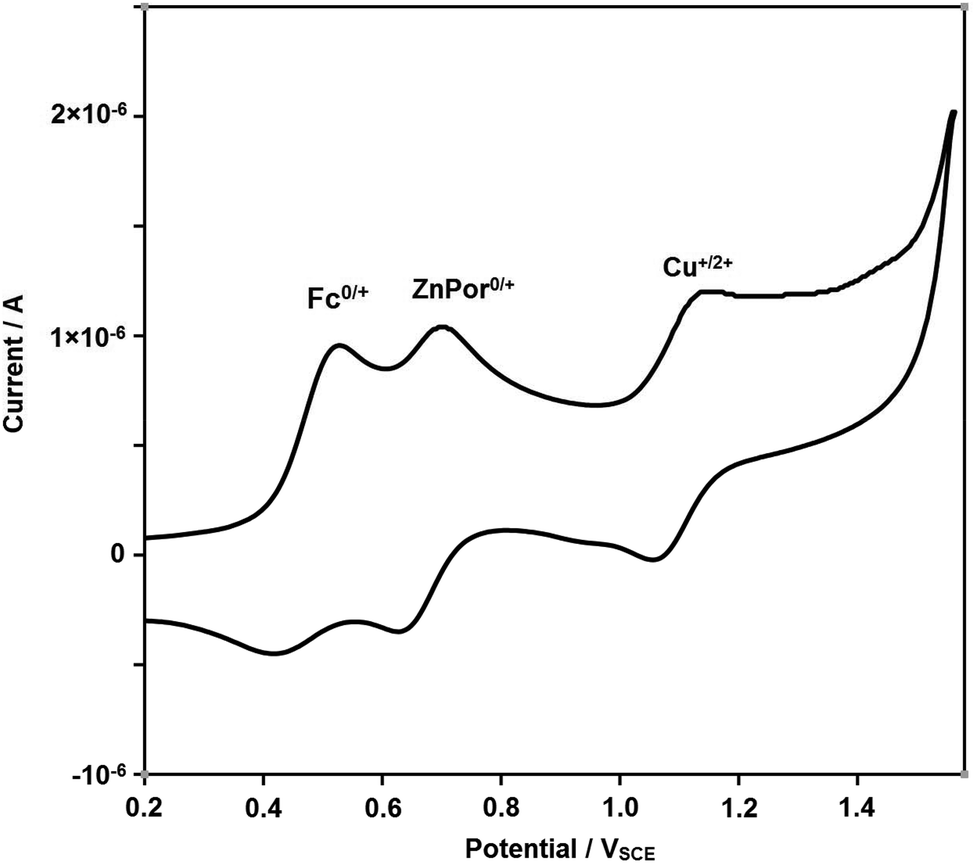 | ||
| Fig. 4 CV of complex [Cu(1)]+ (scan rate of 100 mV s−1) in dichloromethane + 1 vol% dry acetonitrile: Fc0/+ at E1/2 = 0.47 VSCE, ZnPor0/+ at E1/2 = 0.67 VSCE and Cu+/2+ at E1/2 = 1.11 VSCE. | ||
Even at a low scan rate of 50 mV s−1 and in presence of 1 vol% of acetonitrile for accelerating metal–ligand dissociation, the CV (ESI, Fig. 4†) did not show any indication of redox toggling of the azatpy rotary arm. Because switching of the azatpy arm could not be detected due to the inadequate timescale of CV, we decided to utilize chemical redox reagents.13 For chemical oxidation, we used tris(4-bromophenyl)aminium hexachloroantimonate (TBA+˙SbCl6−) or tris(4-bromophenyl)aminium tetrafluoroborate (TBA+˙BF4−), and for reduction13 decamethylferrocene (dmfc) or 3-(11-bromoundecyl)-1,1′-biferrocenylene (BFD).14
At first, 1.0 equiv. of TBA+˙SbCl6− was added to a solution of [Cu(1)]+ in dichloromethane, then after 5 min an oxidative scan was started that showed a new oxidation peak at 0.69 VSCE (ESI, Fig. S40†) which indicated the presence of a copper HETTAP complex and thus formation of State III (= [Cu(1)]2+). Then the resultant solution of State III was treated with 1.0 equiv. of dmfc. Two min after mixing a scan showed the CV signature of State II, i.e. oxidation waves at 0.59 VSCE (merged wave for Fc0/+ and ZnPor0/+) and at 1.16 VSCE (for Cu+/Cu2+) (see ESI, Fig. S41†). Apparently, the reduction of the ferrocenium unit in [Cu(1)]2+ regenerated the electron-donating fcbipy unit thus triggering formation of the HETPHEN complex of the fcbipy unit with the copper(I)-phenanthroline station. At the same time the azatpy arm returned to the ZnPor station.
The equilibrium position in State III was assessed by a thermochemical cycle (ESI, Fig. S43†) based on logK = 9.39 determined for [Cu(1)]+ (ESI, Fig. S42†). In dichloromethane, the change of the ferrocene potential in the metal-free nanoswitch 1 (Fc0/+, E1/2 = 450 mVSCE) and in its Cu+ complex (Fc0/+, E1/2 = 740 mVSCE, ESI, Fig. S39†) show a reduced binding in the [Cu(fcbipy+˙)(phenAr2)]+ complex of logK = 4.5 (ESI, Fig. S43†).6b In contrast, the [Cu(azatpy)(phenAr2)]+ complex in State III is known from earlier work on [Cu(7)]+ to correspond to logK = 7.42, a rather low value for a HETTAP complex since it incorporates the parallel cleavage of the N → ZnPor interaction. Switching the azatpy arm in the process of State II → III thus involves a stronger binding of the [Cu(azatpy)(phenAr2)]+ unit by ΔlogK = 7.4–4.5 = 2.9. This data suggests a 99.9% preference of the [Cu(azatpy)(phenAr2)]+ over the [Cu(fcbipy+˙)(phenAr2)]+ complexation site in State III (ESI, Fig. S44†).
Switching between States II and III was extensively studied by UV-vis and ESI-MS spectroscopy. When 1.0 equiv. of TBA+˙SbCl6− was added to a solution of [Cu(1)]+ in dichloromethane (c = 2 × 10−4 M), the Q-band absorption at 561 nm shifted to 553 nm within 12 min at room temperature (Fig. 5, blue trace and ESI, Fig. S27†). This 8 nm shift is in perfect agreement with a swing of the azatpy rotary arm from the ZnPor station to the copper(I)-phenanthroline binding site and concomitant formation of the HETTAP complex. After oxidation the ESI-MS spectrum displayed a molecular ion peak at m/z = 1340.9 Da that corresponds to [Cu(1)]2+ with the experimental isotopic splitting pattern matching with the calculated one (Fig. S34†). State III was then treated with one equiv. of dmfc as reducing agent. The Q-band of the resultant solution fully shifted from 553 to 561 nm within 1 min at room temperature suggesting intramolecular return of the azatpy arm to the ZnPor station (Fig. 5, red trace and ESI, Fig. S27†). The ESI-MS spectrum of the resultant solution shows a molecular ion peak at m/z = 2738.4 Da for [Cu(1)·CH3CN·H2O]+ that is assigned to State II (ESI, Fig. S35†).
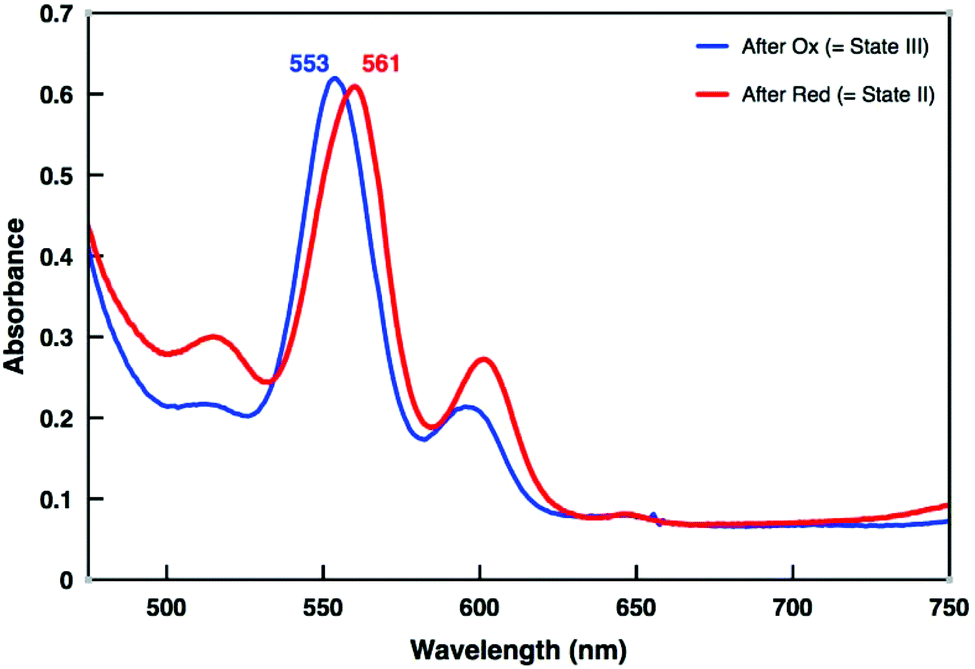 | ||
| Fig. 5 UV-vis spectra: Blue trace: Oxidation of State II by TBA+˙SbCl6− furnishes State III (Fig. 1). Red trace: Reduction of State III by dmfc generates State II. | ||
The switching process from State III → II was also studied by 1H NMR now using 3-(11-bromoundecyl)-1,1′-biferrocenylene (BFD) as a reducing agent because its oxidation produces a diamagnetic species. Hereunto, the solution of State II was first oxidized by addition of one equiv. of TBA+˙BF4−. Subsequent treatment with BFD provided [Cu(1)]+ in 95:5 selectivity of [Cu(fcbipy)(phenAr2)]+:[Cu(azatpy)(phenAr2)]+ (ESI, Fig. S21†), slightly different from the thermodynamically expected 99:1 ratio derived from the known logK values.
Next we examined the switching between all three states by addition of appropriate chemical inputs: State I → State II → State III → State II → State I. Addition of 1.0 equiv. of [Cu(CH3CN)4][B(C6F5)4] to a solution of 1 (State I) in dichloromethane-d2 provided the HETTAP complex [Cu(1)]+ (= State II). The complex [Cu(1)]+ was then treated with 1.0 equiv. of TBA+˙BF4− to afford [Cu(1)]2+ (= State III) which was subsequently reduced by addition of 3-(11-bromoundecyl)-1,1′-biferrocenylene to afford State II in 94% yield. Finally, 2.0 equiv. of 2-ferrocenyl-9-mesityl-[1,10]-phenanthroline was added to reset to the original locked State I (ESI, Fig. S22†).
The reversible double-pole change-over switching process as demonstrated in State II → State III → State II is readily understood since the binding strength of the fcbipy arm is notably deteriorated by oxidation of the ferrocenyl unit by ΔlogK = 4.9 (vide supra). As a result, the azatpy arm detaches from the ZnPor site and associates with the copper(I) phenanthroline site.
Conclusion
We describe the synthesis of the nanomechanical switch 1 in which four distinct terminals are mounted on a tetraphenylmethane scaffold. Bidirectional switching of nanoswitch 1 between three different states is revealed upon addition of chemical and redox inputs. A key step is the reversible double-pole change-over switching process that was ignited by one-electron oxidation and reduction of the ferrocenyl subunit and that involves electronic and steric changes at all four terminals. In detail, upon oxidation/reduction two toggling arms, an azaterpyridine and a ferrocenylbipyridine arm, dissociate from their binding sites and reversibly exchange at the copper(I) phenanthroline station.Experimental
General information
All reagents were obtained from commercial providers and used without further purification. Technical grade solvents were distilled prior to use. Tetrahydrofuran (THF) was predried over basic alumina and then distilled over potassium. Dimethylformamide (DMF) and triethylamine were distilled on calcium hydride. Melting points of compounds were measured using a Büchi SMP-11 instrument. 1H, 13C, and 1H–1H COSY NMR spectra were recorded on Bruker Avance 400 and Varian (600 MHz) spectrometers. Chemical shifts refer to the residual protiated fraction of the NMR solvent (CHCl3: δH = 7.26 ppm, δC = 77.0 ppm; CHDCl2: δH = 5.32 ppm, δC = 53.8 ppm). Abbreviations were used in 1H NMR assignments to describe splitting patterns (s: singlet, d: doublet, t: triplet, dd: doublet of doublet, ddd: doublet of doublet of doublet, bs: broad singlet, td: triplet of doublets, quint: quintet, m: multiplet), the value of coupling constant(s) is reported in Hertz (Hz) and the number of protons are implied. Numbering of the carbon atoms is not in accordance with IUPAC nomenclature. UV-vis spectra were measured on a Cary Win 50 spectrometer. Electrospray ionization mass spectra (ESI-MS) were recorded on a Thermo-Quest LCQ Deca instrument. Infrared (IR) spectra were recorded using a PerkinElmer Spectrum-Two FT-IR spectrometer. Column chromatography was performed on silica gel 60 (60–230 mesh) or on neutral alumina (0.05–0.15 mm, Brockmann Activity 1). Thin layer chromatography (TLC) was performed using Merck silica gel (60 F254) or on neutral Al2O3 (150 F254) sheets. Size exclusion chromatography was performed on BioRads Biobeads-SX3 using toluene or tetrahydrofuran as an eluents.The cyclic voltammetry (CV) and DPV experiments were carried out by using a standard three-electrode setup (1.0 mm Pt working electrode, Pt auxiliary electrode, and a silver wire as the reference electrode) connected to a Princeton Applied Research PARSTAT 2273 Advanced Electrochemical System. Calibration was done with 2,4,6-triphenylpyrylium tetrafluoroborate (E1/2 = −0.39 V vs. SCE) or dmfc as an internal standard (E1/2 = −0.14 V vs. SCE).
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 3272, 3013, 2241, 1710, 1586, 1574, 1544, 1474, 1436, 1391, 1361, 1230, 1103, 1094, 1077, 991, 856, 831, 818, 798, 748, 721, 670, 616, 516 cm−1. 1H NMR (CDCl3, 400 MHz): δ = 8.69 (ddd, 3J = 4.8 Hz, 4J = 1.8 Hz, 5J = 0.9 Hz, 1H, a-H), 8.57 (ddd, 3J = 7.6 Hz, 4J = 1.2 Hz, 5J = 0.9 Hz, 1H, d-H), 8.21 (d, 3J = 8.1 Hz, 1H, f-H), 7.88 (d, 3J = 8.1 Hz, 1H, e-H), 7.86 (td, 3J = 7.6 Hz, 4J = 1.8 Hz, 1H, c-H), 7.32 (ddd, 3J = 7.6 Hz, 3J = 4.8 Hz, 4J = 1.2 Hz, 1H, b-H), 5.49 (t, 3J = 2.0 Hz, 2H, g-H), 4.44 (t, 3J = 2.0 Hz, 2H, h-H), 4.11 (s, 5H, i-H), 3.60 (s, 1H, j-H). 13C NMR (CDCl3, 100 MHz): δ = 159.3, 155.8, 154.4, 149.1, 143.2, 136.8, 123.9, 121.5, 116.7, 114.7, 84.4, 83.5, 82.9, 69.8, 69.7 (2C). Elemental analysis: Calcd for C22H16FeN2: C, 72.55; H, 4.43; N, 7.69; found: C, 72.22; H, 4.41; N, 7.45. ESI-MS: Calcd for [C22H16FeN2·H]+ = [3·H]+, m/z = 365.1; found: [3·H]+, m/z = 365.2.
= 2999, 2952, 2209, 2150, 1585, 1571, 1541, 1502, 1473, 1431, 1386, 1247, 1218, 1105, 1030, 1001, 861, 814, 772, 740 cm−1. 1H NMR (CDCl3, 400 MHz): δ = 8.70 (ddd, 3J = 4.7 Hz, 4J = 1.8 Hz, 5J = 0.9 Hz, 1H, a-H), 8.59 (ddd, 3J = 7.6 Hz, 4J = 1.2 Hz, 5J = 0.9 Hz, 1H, d-H), 8.25 (d, 3J = 8.1 Hz, 1H, f-H), 7.91 (d, 3J = 8.1 Hz, 1H, e-H), 7.86 (td, 3J = 7.6 Hz, 4J = 1.8 Hz, 1H, c-H), 7.57 (d, 3J = 8.8 Hz, 2H, j-H), 7.51 (d, 3J = 8.8 Hz, 2H, k-H), 7.33 (ddd, 3J = 7.6 Hz, 3J = 4.7 Hz, 4J = 1.2 Hz, 1H, b-H), 5.50 (t, 3J = 1.9 Hz, 2H, g-H), 4.47 (t, 3J = 1.9 Hz, 2H, h-H), 4.11 (s, 5H, i-H), 0.28 (s, 9H, l-H). 13C NMR (CDCl3, 100 MHz): δ = 158.8, 155.8, 154.1, 149.1, 142.2, 136.8, 132.2, 131.0, 123.8, 123.3 (2C), 121.4, 116.8, 115.7, 104.5, 96.7, 95.6, 90.8, 83.7, 69.9, 69.7, 69.6, −0.1. Elemental analysis: Calcd for C33H28FeN2Si: C, 73.87; H, 5.26; N, 5.22; found: C, 73.50; H, 5.21; N, 5.17. ESI-MS: Calcd for [C33H28FeN2Si·H]+ = [4·H]+, m/z = 537.1; found: [4·H]+, m/z = 537.2.
= 3279, 3073, 2921, 2852, 2336, 1586, 1542, 1508, 1477, 1441, 1430, 1383, 1361, 1242, 1103, 1075, 1034, 993, 837, 824, 772, 741, 663, 621, 492 cm−1. 1H NMR (CDCl3, 400 MHz): δ = 8.70 (ddd, 3J = 4.8 Hz, 4J = 1.9 Hz, 5J = 0.9 Hz, 1H, a-H), 8.59 (ddd, 3J = 7.8 Hz, 4J = 1.2 Hz, 5J = 0.9 Hz, 1H, d-H), 8.25 (d, 3J = 8.1 Hz, 1H, f-H), 7.91 (d, 3J = 8.1 Hz, 1H, e-H), 7.87 (td, 3J = 7.8 Hz, 4J = 1.9 Hz, 1H, c-H), 7.60 (d, 3J = 8.5 Hz, 2H, j-H), 7.55 (d, 3J = 8.5 Hz, 2H, k-H), 7.33 (ddd, 3J = 7.8 Hz, 3J = 4.8 Hz, 4J = 1.2 Hz, 1H, b-H), 5.50 (t, 3J = 1.9 Hz, 2H, g-H), 4.48 (t, 3J = 1.9 Hz, 2H, h-H), 4.15 (s, 5H, i-H), 3.22 (s, 1H, l-H). 13C NMR (CDCl3, 100 MHz): δ = 158.8, 155.8, 154.2, 149.1, 142.3, 136.8, 132.3, 131.1, 123.9, 123.7, 122.3, 121.5, 116.8, 115.6, 95.3, 90.9, 83.7, 83.2, 79.2, 69.9, 69.7, 69.6. Elemental analysis: Calcd for C30H20FeN2: C, 77.60; H, 4.34; N, 6.03; found: C, 77.96; H, 4.49; N, 5.68. ESI-MS: Calcd for [C30H20FeN2·H]+ = [5·H]+, m/z = 465.1; found: [5·H]+, m/z = 465.2.
= 3035, 2957, 2925, 2851, 2216, 1898, 1737, 1571, 1511, 1474, 1431, 1385, 1361, 1106, 1093, 1015, 1007, 826, 818, 770, 744, 737, 497 cm−1. 1HNMR (CDCl3, 400 MHz): δ = 8.70 (ddd, 3J = 4.8 Hz, 4J = 1.8 Hz, 5J = 0.8 Hz, 1H, a-H), 8.60 (ddd, 3J = 7.6 Hz, 4J = 1.0 Hz, 5J = 0.8 Hz, 1H, d-H), 8.26 (d, 3J = 8.1 Hz, 1H, f-H), 7.93 (d, 3J = 8.1 Hz, 1H, e-H), 7.89 (dt, 3J = 7.8 Hz, 4J = 1.6 Hz, 1H, n-H), 7.87 (td, 3J = 7.6 Hz, 4J = 1.8 Hz, 1H, c-H), 7.66 (d, 3J = 8.4 Hz, 2H, j-H), 7.65 (d, 3J = 8.4 Hz, 2H, k-H), 7.56 (dd, 3J = 7.8 Hz, 4J = 1.6 Hz, 1H, l-H), 7.32–7.36 (m, 2H, m-, b-H), 7.04 (td, 3J = 7.8 Hz, 4J = 1.6 Hz, 1H, o-H), 5.52 (t, 3J = 2.0 Hz, 2H, g-H), 4.49 (t, 3J = 2.0 Hz, 2H, h-H), 4.13 (s, 5H, i-H). 13C NMR (CDCl3, 100 MHz): δ = 158.8, 155.8, 154.1, 149.1, 142.2, 138.8, 136.8, 132.5, 131.8, 131.2, 129.6, 129.5, 127.9, 123.9, 123.5, 123.1, 121.5, 116.8, 115.7, 101.2, 95.6, 93.7, 92.6, 91.0, 83.7, 69.9, 69.7 (2C). Elemental analysis: Calcd for C36H23FeIN2: C, 64.89; H, 3.48; N, 4.20; found: C, 64.62; H, 3.41; N, 4.12. ESI-MS: Calcd for [C36H23FeIN2·H]+ = [6·H]+, m/z = 667.0; found: [6·H]+, m/z = 667.1.
= 3031, 2918, 2854, 2737, 2210, 1916, 1732, 1586, 1541, 1506, 1477, 1443, 1378, 1242, 1203, 1100, 1061, 996, 798, 756, 721, 542 cm−1. 1H NMR (CD2Cl2, 600 MHz): δ = 8.83 (d, 3J = 4.5 Hz, 2H, β-H), 8.71 (d, 3J = 4.5 Hz, 2H, β-H), 8.67 (dd, 4J = 2.1 Hz, 5J = 0.8 Hz, 1H, i-H), 8.65 (d, 3J = 4.5 Hz, 2H, β-H), 8.63 (d, 3J = 4.5 Hz, 2H, β-H), 8.57 (ddd, 3J = 4.8 Hz, 4J = 1.8 Hz, 5J = 0.8 Hz, 1H, A-H), 8.53 (ddd, 3J = 7.6 Hz, 3J = 1.2 Hz, 4J = 0.8 Hz, 1H, D-H), 8.34 (d, 3J = 8.2 Hz, 1H, 4-H), 8.33 (d, 3J = 8.2 Hz, 1H, 7-H), 8.23 (d, 3J = 7.9 Hz, 2H, q-H), 8.22 (d, 3J = 8.1 Hz, 1H, E-H), 8.18 (t, 3J = 7.7 Hz, 1H, e-H), 8.13 (d, 3J = 8.2 Hz, 1H, g-H), 7.93 (d, 3J = 7.9 Hz, 2H, p-H), 7.92 (d, 3J = 8.1 Hz, 1H, F-H), 7.90 (2 s, merged, 2H, 5-, 6-H), 7.89 (dd, 3J = 8.2 Hz, 4J = 2.1 Hz, 1H, h-H), 7.74 (td, 3J = 7.6 Hz, 4J = 1.8 Hz, 1H, C-H), 7.68 (d, 3J = 8.5 Hz, 2H, j/k-H), 7.67 (2 d, merged, 3J = 8.2 Hz, 4H, J-, K-H), 7.57–7.63 (m, 16H, d-, f-, l-, o-, L-, O-, tpc-H), 7.56 (d, 3J = 8.2 Hz, 1H, 3-H), 7.53 (d, 3J = 8.2 Hz, 1H, 8-H), 7.34–7.44 (m, 12H, k/j-, m-, n-, M-, N-, tpc-H), 7.33 (s, 2H, s-H), 7.24 (s, 4H, r-H), 7.20 (ddd, 3J = 7.6 Hz, 3J = 4.8 Hz, 4J = 1.2 Hz, 1H, B-H), 6.95 (s, 2H, z-H), 6.83 (d, 3J = 5.8 Hz, 1H, c-H), 5.49 (t, 3J = 1.8 Hz, 2H, G-H), 4.48 (t, 3J = 1.8 Hz, 2H, H-H), 4.10 (s, 5H, I-H), 3.76 (s, 1H, a-H), 2.73 (d, 3J = 5.8 Hz, 1H, b-H), 2.64 (s, 3H, w-H), 2.58 (s, 6H, u-H), 2.52 (s, 6H, 9-H), 2.32 (s, 3H, y-H), 2.04 (s, 6H, x-H), 1.94 (s, 6H, v-H), 1.93 (s, 6H, 10-H), 1.74 (s, 12H, t-H). 13C NMR (CD2Cl2, 100 MHz): δ = 162.6, 161.4, 160.5, 159.2, 156.0, 155.0, 154.6, 154.3, 152.9, 151.5, 151.4, 151.2 (2C), 150.2 (2C), 149.9, 149.7, 149.4, 146.9, 146.8, 146.6, 146.5, 146.1, 144.1, 142.6, 141.9, 140.3, 139.8 (2C), 139.6, 139.5, 138.5, 138.2, 137.8, 137.7, 137.6, 137.0, 136.6, 136.4 (2C), 136.1, 135.1, 132.6 (2C), 132.4, 132.3, 132.2 (2C), 132.1, 132.0, 131.7, 131.6 (3C), 131.5 (2C), 131.3 (3C), 131.2, 130.9, 130.1, 128.8 (2C), 128.6 (2C), 127.9, 127.8, 127.6 (2C), 126.6, 126.5, 126.1, 125.7, 125.4, 124.9, 124.8, 124.1 (2C), 123.8 (2C), 123.1, 123.0, 122.9, 122.6, 122.3, 121.8, 121.6 (2C), 121.5, 121.3, 120.7, 118.9 (3C), 118.8, 117.1, 116.0, 96.8, 95.9, 93.9, 93.8, 93.6, 93.5, 93.3, 91.4, 90.8, 90.7, 90.3, 90.1, 89.8, 88.9, 88.8, 88.6, 84.1, 70.3, 70.1, 65.5, 22.0, 21.8, 21.6, 21.5, 21.2, 20.4, 18.6, 17.7. Elemental analysis: Calcd for C183H130FeN12Zn·CH2Cl2·MeOH: C, 81.23; H, 5.01; N, 6.14; found: C, 81.07; H, 5.40; N, 5.81. ESI-MS: Calcd for [C183H130FeN12Zn·2H]2+ = [1·2H]2+, m/z = 1309.5; [C183H130FeN12Zn·3H]3+ = [1·3H]3+, m/z = 873.3; found: [1·2H]2+, m/z = 1309.7; [1·3H]3+, m/z = 873.5.
:45 ratio (due to the stereogenic center and axis). IR (KBr): = 3034, 2955, 2916, 2856, 2210, 1916, 1720, 1641, 1582, 1549, 1510, 1461, 1389, 1273, 1203, 1086, 979, 821, 800, 774, 755, 683 cm−1. 1H NMR (CD2Cl2, 600 MHz): δ = 8.86 (d, 3J = 4.5 Hz, 1H, β-H), 8.84 (d, 3J = 4.5 Hz, 1H, β-H), 8.72 (2 d, 3J = 4.6 Hz, 2H, β-H), 8.68–8.70 (m, 2H, 7/4-, i-H), 8.67 (3 d, 3J = 4.5 Hz, 3H, β-H), 8.64 (d, 3J = 4.5 Hz, 1H, β-H), 8.55 (d, 3J = 8.2 Hz, 1H, 4/7-H), 8.29 (d, 3J = 8.2 Hz, 1.1H, q-H), 8.27 (d, 3J = 8.2 Hz, 0.9H, q-H), 8.13–8.22 (m, 5H, D-, 5-, 6-, e-, g-H), 8.09 (d, 3J = 8.4 Hz, 0.45H, F-H), 8.08 (d, 3J = 8.4 Hz, 0.55H, F-H), 8.01 (dd, 3J = 8.2 Hz, 4J = 2.1 Hz, 1H, h-H), 7.98 (d, 3J = 8.2 Hz, 0.9H, p-H), 7.95 (d, 3J = 8.2 Hz, 1.1H, p-H), 7.92 (td, 3J = 7.6 Hz, 4J = 1.6 Hz, 1H, C-H), 7.85 (d, 3J = 8.2 Hz, 0.55H, 3/8-H), 7.84 (d, 3J = 8.4 Hz, 0.45H, 3/8-H), 7.76–7.78 (m, 1H, E-H), 7.71 (d, 3J = 8.4 Hz, 1H, tpc-H), 7.56–7.68 (m, 20H, A-, d-, f-, l-, o-, L-, O-, 8/3-, j/k-, J/K-, tpc-H), 7.53 (d, 3J = 8.4 Hz, 1H, tpc-H), 7.40–7.50 (m, 6H, tpc-H), 7.36–7.38 (m, 4H, k/j-, K/J-H), 7.331 (s, 1.1H, s-H), 7.327 (s, 0.9H, s-H), 7.26–7.32 (m, 4H, M-, N-, m-, n-H), 7.25 (s, 2.2H, r-H), 7.24 (s, 1.8H, r-H), 7.18–7.22 (m, 1H, B-H), 6.97 (s, 0.55 H, z-H), 6.96 (s, 0.45 H, z-H), 6.860 (d, 3J = 5.8 Hz, 0.55H, c-H), 6.858 (d, 3J = 5.8 Hz, 0.45H, c-H), 6.72 (s, 0.55 H, z-H), 6.70 (s, 0.45 H, z-H), 5.76–5.78 (m, 2H, G-H), 4.49–4.51 (m, 2H, H-H), 4.01 (s, 2.75 H, I-H), 3.99 (s, 2.25 H, I-H), 3.80 (s, 0.55H, a-H), 3.78 (s, 0.45H, a-H), 2.75 (d, 3J = 5.8 Hz, 0.45H, b-H), 2.74 (d, 3J = 5.8 Hz, 0.55H, b-H), 2.644 (s, 1.65H, w-H), 2.641 (s, 1.35H, w-H), 2.60 (s, 3.3H, u-H), 2.58 (s, 2.7H, u-H), 2.46 (s, 3H, 10-H), 2.24 (s, 1.65H, x-H), 2.22 (s, 1.35H, x-H), 2.13 (s, 1.65H, x-H), 2.12 (s, 1.35H, x-H), 1.95 (s, 3.3H, v-H), 1.94 (s, 2.7H, v-H), 1.89 (s, 3H, 10-H), 1.75 (s, 3.3H, t-H)*, 1.74 (s, 6H, t-H), 1.73 (s, 2.7H, t-H)*, 1.57 (s, 1.65H, y-H), 1.55 (s, 1.35H, y-H), 1.28 (s, 1.65H, 9-H), 1.27 (s, 1.35H, 9-H), 0.82 (s, 1.65H, 9-H), 0.81 (s, 1.35H, 9-H). Elemental analysis: Calcd for C207H130BCuF20FeN12Zn·2.5CH2Cl2: C, 70.42; H, 3.81; N, 4.70; found: C, 70.45; H, 3.48; N, 4.61. ESI-MS: Calcd for [C183CuH130FeN12Zn·MeCN·H2O]2+ = [Cu(1)·MeCN·H2O]+, m/z = 2740.9; found: [Cu(1)·MeCN·H2O]+, m/z = 2741.1.
= 3272, 3013, 2241, 1710, 1586, 1574, 1544, 1474, 1436, 1391, 1361, 1230, 1103, 1094, 1077, 991, 856, 831, 818, 798, 748, 721, 670, 616, 516 cm−1. 1H NMR (CDCl3, 400 MHz): δ = 8.69 (ddd, 3J = 4.8 Hz, 4J = 1.8 Hz, 5J = 0.9 Hz, 1H, a-H), 8.57 (ddd, 3J = 7.6 Hz, 4J = 1.2 Hz, 5J = 0.9 Hz, 1H, d-H), 8.21 (d, 3J = 8.1 Hz, 1H, f-H), 7.88 (d, 3J = 8.1 Hz, 1H, e-H), 7.86 (td, 3J = 7.6 Hz, 4J = 1.8 Hz, 1H, c-H), 7.32 (ddd, 3J = 7.6 Hz, 3J = 4.8 Hz, 4J = 1.2 Hz, 1H, b-H), 5.49 (t, 3J = 2.0 Hz, 2H, g-H), 4.44 (t, 3J = 2.0 Hz, 2H, h-H), 4.11 (s, 5H, i-H), 3.60 (s, 1H, j-H). 13C NMR (CDCl3, 100 MHz): δ = 159.3, 155.8, 154.4, 149.1, 143.2, 136.8, 123.9, 121.5, 116.7, 114.7, 84.4, 83.5, 82.9, 69.8, 69.7 (2C). Elemental analysis: Calcd for C22H16FeN2: C, 72.55; H, 4.43; N, 7.69; found: C, 72.22; H, 4.41; N, 7.45. ESI-MS: Calcd for [C22H16FeN2·H]+ = [3·H]+, m/z = 365.1; found: [3·H]+, m/z = 365.2.
= 2999, 2952, 2209, 2150, 1585, 1571, 1541, 1502, 1473, 1431, 1386, 1247, 1218, 1105, 1030, 1001, 861, 814, 772, 740 cm−1. 1H NMR (CDCl3, 400 MHz): δ = 8.70 (ddd, 3J = 4.7 Hz, 4J = 1.8 Hz, 5J = 0.9 Hz, 1H, a-H), 8.59 (ddd, 3J = 7.6 Hz, 4J = 1.2 Hz, 5J = 0.9 Hz, 1H, d-H), 8.25 (d, 3J = 8.1 Hz, 1H, f-H), 7.91 (d, 3J = 8.1 Hz, 1H, e-H), 7.86 (td, 3J = 7.6 Hz, 4J = 1.8 Hz, 1H, c-H), 7.57 (d, 3J = 8.8 Hz, 2H, j-H), 7.51 (d, 3J = 8.8 Hz, 2H, k-H), 7.33 (ddd, 3J = 7.6 Hz, 3J = 4.7 Hz, 4J = 1.2 Hz, 1H, b-H), 5.50 (t, 3J = 1.9 Hz, 2H, g-H), 4.47 (t, 3J = 1.9 Hz, 2H, h-H), 4.11 (s, 5H, i-H), 0.28 (s, 9H, l-H). 13C NMR (CDCl3, 100 MHz): δ = 158.8, 155.8, 154.1, 149.1, 142.2, 136.8, 132.2, 131.0, 123.8, 123.3 (2C), 121.4, 116.8, 115.7, 104.5, 96.7, 95.6, 90.8, 83.7, 69.9, 69.7, 69.6, −0.1. Elemental analysis: Calcd for C33H28FeN2Si: C, 73.87; H, 5.26; N, 5.22; found: C, 73.50; H, 5.21; N, 5.17. ESI-MS: Calcd for [C33H28FeN2Si·H]+ = [4·H]+, m/z = 537.1; found: [4·H]+, m/z = 537.2.
= 3279, 3073, 2921, 2852, 2336, 1586, 1542, 1508, 1477, 1441, 1430, 1383, 1361, 1242, 1103, 1075, 1034, 993, 837, 824, 772, 741, 663, 621, 492 cm−1. 1H NMR (CDCl3, 400 MHz): δ = 8.70 (ddd, 3J = 4.8 Hz, 4J = 1.9 Hz, 5J = 0.9 Hz, 1H, a-H), 8.59 (ddd, 3J = 7.8 Hz, 4J = 1.2 Hz, 5J = 0.9 Hz, 1H, d-H), 8.25 (d, 3J = 8.1 Hz, 1H, f-H), 7.91 (d, 3J = 8.1 Hz, 1H, e-H), 7.87 (td, 3J = 7.8 Hz, 4J = 1.9 Hz, 1H, c-H), 7.60 (d, 3J = 8.5 Hz, 2H, j-H), 7.55 (d, 3J = 8.5 Hz, 2H, k-H), 7.33 (ddd, 3J = 7.8 Hz, 3J = 4.8 Hz, 4J = 1.2 Hz, 1H, b-H), 5.50 (t, 3J = 1.9 Hz, 2H, g-H), 4.48 (t, 3J = 1.9 Hz, 2H, h-H), 4.15 (s, 5H, i-H), 3.22 (s, 1H, l-H). 13C NMR (CDCl3, 100 MHz): δ = 158.8, 155.8, 154.2, 149.1, 142.3, 136.8, 132.3, 131.1, 123.9, 123.7, 122.3, 121.5, 116.8, 115.6, 95.3, 90.9, 83.7, 83.2, 79.2, 69.9, 69.7, 69.6. Elemental analysis: Calcd for C30H20FeN2: C, 77.60; H, 4.34; N, 6.03; found: C, 77.96; H, 4.49; N, 5.68. ESI-MS: Calcd for [C30H20FeN2·H]+ = [5·H]+, m/z = 465.1; found: [5·H]+, m/z = 465.2.
= 3035, 2957, 2925, 2851, 2216, 1898, 1737, 1571, 1511, 1474, 1431, 1385, 1361, 1106, 1093, 1015, 1007, 826, 818, 770, 744, 737, 497 cm−1. 1HNMR (CDCl3, 400 MHz): δ = 8.70 (ddd, 3J = 4.8 Hz, 4J = 1.8 Hz, 5J = 0.8 Hz, 1H, a-H), 8.60 (ddd, 3J = 7.6 Hz, 4J = 1.0 Hz, 5J = 0.8 Hz, 1H, d-H), 8.26 (d, 3J = 8.1 Hz, 1H, f-H), 7.93 (d, 3J = 8.1 Hz, 1H, e-H), 7.89 (dt, 3J = 7.8 Hz, 4J = 1.6 Hz, 1H, n-H), 7.87 (td, 3J = 7.6 Hz, 4J = 1.8 Hz, 1H, c-H), 7.66 (d, 3J = 8.4 Hz, 2H, j-H), 7.65 (d, 3J = 8.4 Hz, 2H, k-H), 7.56 (dd, 3J = 7.8 Hz, 4J = 1.6 Hz, 1H, l-H), 7.32–7.36 (m, 2H, m-, b-H), 7.04 (td, 3J = 7.8 Hz, 4J = 1.6 Hz, 1H, o-H), 5.52 (t, 3J = 2.0 Hz, 2H, g-H), 4.49 (t, 3J = 2.0 Hz, 2H, h-H), 4.13 (s, 5H, i-H). 13C NMR (CDCl3, 100 MHz): δ = 158.8, 155.8, 154.1, 149.1, 142.2, 138.8, 136.8, 132.5, 131.8, 131.2, 129.6, 129.5, 127.9, 123.9, 123.5, 123.1, 121.5, 116.8, 115.7, 101.2, 95.6, 93.7, 92.6, 91.0, 83.7, 69.9, 69.7 (2C). Elemental analysis: Calcd for C36H23FeIN2: C, 64.89; H, 3.48; N, 4.20; found: C, 64.62; H, 3.41; N, 4.12. ESI-MS: Calcd for [C36H23FeIN2·H]+ = [6·H]+, m/z = 667.0; found: [6·H]+, m/z = 667.1.
= 3031, 2918, 2854, 2737, 2210, 1916, 1732, 1586, 1541, 1506, 1477, 1443, 1378, 1242, 1203, 1100, 1061, 996, 798, 756, 721, 542 cm−1. 1H NMR (CD2Cl2, 600 MHz): δ = 8.83 (d, 3J = 4.5 Hz, 2H, β-H), 8.71 (d, 3J = 4.5 Hz, 2H, β-H), 8.67 (dd, 4J = 2.1 Hz, 5J = 0.8 Hz, 1H, i-H), 8.65 (d, 3J = 4.5 Hz, 2H, β-H), 8.63 (d, 3J = 4.5 Hz, 2H, β-H), 8.57 (ddd, 3J = 4.8 Hz, 4J = 1.8 Hz, 5J = 0.8 Hz, 1H, A-H), 8.53 (ddd, 3J = 7.6 Hz, 3J = 1.2 Hz, 4J = 0.8 Hz, 1H, D-H), 8.34 (d, 3J = 8.2 Hz, 1H, 4-H), 8.33 (d, 3J = 8.2 Hz, 1H, 7-H), 8.23 (d, 3J = 7.9 Hz, 2H, q-H), 8.22 (d, 3J = 8.1 Hz, 1H, E-H), 8.18 (t, 3J = 7.7 Hz, 1H, e-H), 8.13 (d, 3J = 8.2 Hz, 1H, g-H), 7.93 (d, 3J = 7.9 Hz, 2H, p-H), 7.92 (d, 3J = 8.1 Hz, 1H, F-H), 7.90 (2 s, merged, 2H, 5-, 6-H), 7.89 (dd, 3J = 8.2 Hz, 4J = 2.1 Hz, 1H, h-H), 7.74 (td, 3J = 7.6 Hz, 4J = 1.8 Hz, 1H, C-H), 7.68 (d, 3J = 8.5 Hz, 2H, j/k-H), 7.67 (2 d, merged, 3J = 8.2 Hz, 4H, J-, K-H), 7.57–7.63 (m, 16H, d-, f-, l-, o-, L-, O-, tpc-H), 7.56 (d, 3J = 8.2 Hz, 1H, 3-H), 7.53 (d, 3J = 8.2 Hz, 1H, 8-H), 7.34–7.44 (m, 12H, k/j-, m-, n-, M-, N-, tpc-H), 7.33 (s, 2H, s-H), 7.24 (s, 4H, r-H), 7.20 (ddd, 3J = 7.6 Hz, 3J = 4.8 Hz, 4J = 1.2 Hz, 1H, B-H), 6.95 (s, 2H, z-H), 6.83 (d, 3J = 5.8 Hz, 1H, c-H), 5.49 (t, 3J = 1.8 Hz, 2H, G-H), 4.48 (t, 3J = 1.8 Hz, 2H, H-H), 4.10 (s, 5H, I-H), 3.76 (s, 1H, a-H), 2.73 (d, 3J = 5.8 Hz, 1H, b-H), 2.64 (s, 3H, w-H), 2.58 (s, 6H, u-H), 2.52 (s, 6H, 9-H), 2.32 (s, 3H, y-H), 2.04 (s, 6H, x-H), 1.94 (s, 6H, v-H), 1.93 (s, 6H, 10-H), 1.74 (s, 12H, t-H). 13C NMR (CD2Cl2, 100 MHz): δ = 162.6, 161.4, 160.5, 159.2, 156.0, 155.0, 154.6, 154.3, 152.9, 151.5, 151.4, 151.2 (2C), 150.2 (2C), 149.9, 149.7, 149.4, 146.9, 146.8, 146.6, 146.5, 146.1, 144.1, 142.6, 141.9, 140.3, 139.8 (2C), 139.6, 139.5, 138.5, 138.2, 137.8, 137.7, 137.6, 137.0, 136.6, 136.4 (2C), 136.1, 135.1, 132.6 (2C), 132.4, 132.3, 132.2 (2C), 132.1, 132.0, 131.7, 131.6 (3C), 131.5 (2C), 131.3 (3C), 131.2, 130.9, 130.1, 128.8 (2C), 128.6 (2C), 127.9, 127.8, 127.6 (2C), 126.6, 126.5, 126.1, 125.7, 125.4, 124.9, 124.8, 124.1 (2C), 123.8 (2C), 123.1, 123.0, 122.9, 122.6, 122.3, 121.8, 121.6 (2C), 121.5, 121.3, 120.7, 118.9 (3C), 118.8, 117.1, 116.0, 96.8, 95.9, 93.9, 93.8, 93.6, 93.5, 93.3, 91.4, 90.8, 90.7, 90.3, 90.1, 89.8, 88.9, 88.8, 88.6, 84.1, 70.3, 70.1, 65.5, 22.0, 21.8, 21.6, 21.5, 21.2, 20.4, 18.6, 17.7. Elemental analysis: Calcd for C183H130FeN12Zn·CH2Cl2·MeOH: C, 81.23; H, 5.01; N, 6.14; found: C, 81.07; H, 5.40; N, 5.81. ESI-MS: Calcd for [C183H130FeN12Zn·2H]2+ = [1·2H]2+, m/z = 1309.5; [C183H130FeN12Zn·3H]3+ = [1·3H]3+, m/z = 873.3; found: [1·2H]2+, m/z = 1309.7; [1·3H]3+, m/z = 873.5.
:45 ratio (due to the stereogenic center and axis). IR (KBr): = 3034, 2955, 2916, 2856, 2210, 1916, 1720, 1641, 1582, 1549, 1510, 1461, 1389, 1273, 1203, 1086, 979, 821, 800, 774, 755, 683 cm−1. 1H NMR (CD2Cl2, 600 MHz): δ = 8.86 (d, 3J = 4.5 Hz, 1H, β-H), 8.84 (d, 3J = 4.5 Hz, 1H, β-H), 8.72 (2 d, 3J = 4.6 Hz, 2H, β-H), 8.68–8.70 (m, 2H, 7/4-, i-H), 8.67 (3 d, 3J = 4.5 Hz, 3H, β-H), 8.64 (d, 3J = 4.5 Hz, 1H, β-H), 8.55 (d, 3J = 8.2 Hz, 1H, 4/7-H), 8.29 (d, 3J = 8.2 Hz, 1.1H, q-H), 8.27 (d, 3J = 8.2 Hz, 0.9H, q-H), 8.13–8.22 (m, 5H, D-, 5-, 6-, e-, g-H), 8.09 (d, 3J = 8.4 Hz, 0.45H, F-H), 8.08 (d, 3J = 8.4 Hz, 0.55H, F-H), 8.01 (dd, 3J = 8.2 Hz, 4J = 2.1 Hz, 1H, h-H), 7.98 (d, 3J = 8.2 Hz, 0.9H, p-H), 7.95 (d, 3J = 8.2 Hz, 1.1H, p-H), 7.92 (td, 3J = 7.6 Hz, 4J = 1.6 Hz, 1H, C-H), 7.85 (d, 3J = 8.2 Hz, 0.55H, 3/8-H), 7.84 (d, 3J = 8.4 Hz, 0.45H, 3/8-H), 7.76–7.78 (m, 1H, E-H), 7.71 (d, 3J = 8.4 Hz, 1H, tpc-H), 7.56–7.68 (m, 20H, A-, d-, f-, l-, o-, L-, O-, 8/3-, j/k-, J/K-, tpc-H), 7.53 (d, 3J = 8.4 Hz, 1H, tpc-H), 7.40–7.50 (m, 6H, tpc-H), 7.36–7.38 (m, 4H, k/j-, K/J-H), 7.331 (s, 1.1H, s-H), 7.327 (s, 0.9H, s-H), 7.26–7.32 (m, 4H, M-, N-, m-, n-H), 7.25 (s, 2.2H, r-H), 7.24 (s, 1.8H, r-H), 7.18–7.22 (m, 1H, B-H), 6.97 (s, 0.55 H, z-H), 6.96 (s, 0.45 H, z-H), 6.860 (d, 3J = 5.8 Hz, 0.55H, c-H), 6.858 (d, 3J = 5.8 Hz, 0.45H, c-H), 6.72 (s, 0.55 H, z-H), 6.70 (s, 0.45 H, z-H), 5.76–5.78 (m, 2H, G-H), 4.49–4.51 (m, 2H, H-H), 4.01 (s, 2.75 H, I-H), 3.99 (s, 2.25 H, I-H), 3.80 (s, 0.55H, a-H), 3.78 (s, 0.45H, a-H), 2.75 (d, 3J = 5.8 Hz, 0.45H, b-H), 2.74 (d, 3J = 5.8 Hz, 0.55H, b-H), 2.644 (s, 1.65H, w-H), 2.641 (s, 1.35H, w-H), 2.60 (s, 3.3H, u-H), 2.58 (s, 2.7H, u-H), 2.46 (s, 3H, 10-H), 2.24 (s, 1.65H, x-H), 2.22 (s, 1.35H, x-H), 2.13 (s, 1.65H, x-H), 2.12 (s, 1.35H, x-H), 1.95 (s, 3.3H, v-H), 1.94 (s, 2.7H, v-H), 1.89 (s, 3H, 10-H), 1.75 (s, 3.3H, t-H)*, 1.74 (s, 6H, t-H), 1.73 (s, 2.7H, t-H)*, 1.57 (s, 1.65H, y-H), 1.55 (s, 1.35H, y-H), 1.28 (s, 1.65H, 9-H), 1.27 (s, 1.35H, 9-H), 0.82 (s, 1.65H, 9-H), 0.81 (s, 1.35H, 9-H). Elemental analysis: Calcd for C207H130BCuF20FeN12Zn·2.5CH2Cl2: C, 70.42; H, 3.81; N, 4.70; found: C, 70.45; H, 3.48; N, 4.61. ESI-MS: Calcd for [C183CuH130FeN12Zn·MeCN·H2O]2+ = [Cu(1)·MeCN·H2O]+, m/z = 2740.9; found: [Cu(1)·MeCN·H2O]+, m/z = 2741.1.
*The distinct shifts of t-H indicate a different angle at the Nazatpy → ZnPor unit as a result of diastereomer formation.
Conflicts of interest
There are not conflicts to declare.Acknowledgements
We are grateful to the DAAD (Deutscher Akademischer Austauschdienst), DFG (Deutsche Forschungsgemeinschaft, Schm-647/19-2), and the Universität Siegen for financial support. We thank Abir Goswami for determination of a binding constant.References
- Selected reviews: (a) M. Nishikawa, S. Kume and H. Nishihara, Phys. Chem. Chem. Phys., 2013, 15, 10549–10565 RSC; (b) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed; (c) A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729 CrossRef CAS PubMed; (d) N. Le Poul and B. Colasson, ChemElectroChem, 2015, 2, 475–496 CrossRef CAS; (e) S. Ø. Scottwell and J. D. Crowley, Chem. Commun., 2016, 52, 2451–2464 RSC; (f) Y. Wang, M. Frasconi and J. F. Stoddart, ACS Cent. Sci., 2017, 3, 927–935 CrossRef CAS PubMed; (g) T. Fukino, H. Yamagishi and T. Aida, Adv. Mater., 2017, 29, 1603888 CrossRef PubMed; (h) A. Goswami and M. Schmittel, Coord. Chem. Rev., 2018, 376, 478–505 CrossRef CAS.
- (a) F. Niess, V. Duplan and J.-P. Sauvage, J. Am. Chem. Soc., 2014, 136, 5876–5879 CrossRef CAS PubMed; (b) S. K. Samanta, A. Rana and M. Schmittel, Dalton Trans., 2014, 43, 9438–9447 RSC; (c) J. Sun, Y. Wu, Y. Wang, Z. Liu, C. Cheng, K. J. Hartlieb, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2015, 137, 13484–13487 CrossRef CAS PubMed; (d) M. Schmittel, S. De and S. Pramanik, Org. Biomol. Chem., 2015, 13, 8937–8944 RSC; (e) S. Ø. Scottwell, A. B. S. Elliott, K. J. Shaffer, A. Nafady, C. J. McAdam, K. C. Gordon and J. D. Crowley, Chem. Commun., 2015, 51, 8161–8164 RSC; (f) C. R. Benson, A. I. Share, M. G. Marzo and A. H. Flood, Inorg. Chem., 2016, 55, 3767–3776 CrossRef CAS PubMed; (g) H. V. Schröder, S. Sobottka, M. Nößler, H. Hupatz, M. Gaedke, B. Sarkar and C. A. Schalley, Chem. Sci., 2017, 8, 6300–6306 RSC; (h) C. Pezzato, M. T. Nguyen, C. Cheng, D. J. Kim, M. T. Otley and J. F. Stoddart, Tetrahedron, 2017, 73, 4849–4857 CrossRef CAS; (i) Y. Wang, J. Sun, Z. Liu, M. S. Nassar, Y. Y. Botros and J. F. Stoddart, Chem. Sci., 2017, 8, 2562–2568 RSC; (j) S. Gaikwad, S. Pramanik, S. De and M. Schmittel, Dalton Trans., 2018, 47, 1786–1789 RSC; (k) D. T. Payne, W. A. Webre, Y. Matsushita, N. Zhu, Z. Futera, J. Labuta, W. Jevasuwan, N. Fukata, J. S. Fossey, F. D'Souza, K. Ariga, W. Schmitt and J. P. Hill, Nat. Commun., 2019, 10, 1007 CrossRef PubMed.
- (a) B. S. L. Collins, J. C. M. Kistemaker, E. Otten and B. L. Feringa, Nat. Chem., 2016, 8, 860–866 CrossRef CAS; (b) B. Doistau, L. Benda, J.-L. Cantin, L.-M. Chamoreau, E. Ruiz, V. Marvaud, B. Hasenknopf and G. Vives, J. Am. Chem. Soc., 2017, 139, 9213–9220 CrossRef CAS PubMed.
- (a) H. Zhang, X.-X. Kou, Q. Zhang, D.-H. Qu and H. Tian, Org. Biomol. Chem., 2011, 9, 4051–4056 RSC; (b) F. Durola, V. Heitz, F. Reviriego, C. Roche, J.-P. Sauvage, A. Sour and Y. Trolez, Acc. Chem. Res., 2014, 47, 633–645 CrossRef CAS PubMed; (c) S. Gaikwad, A. Goswami, S. De and M. Schmittel, Angew. Chem., Int. Ed., 2016, 55, 10512–10517 CrossRef CAS PubMed; (d) I. Aprahamian, Chem. Commun., 2017, 53, 6674–6684 RSC; (e) M. Baroncini, M. Canton, L. Casimiro, S. Corra, J. Groppi, M. La Rosa, S. Silvi and A. Credi, Eur. J. Inorg. Chem., 2018, 4589–4603 CrossRef CAS PubMed; (f) B. Mondal, A. Kumar Ghosh and P. S. Mukherjee, J. Org. Chem., 2017, 82, 7783–7790 CrossRef CAS PubMed; (g) C. Burkhart and G. Haberhauer, Eur. J. Org. Chem., 2017, 1308–1317 CrossRef CAS; (h) S. Kassem, T. van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Chem. Soc. Rev., 2017, 46, 2592–2621 RSC; (i) S. Gaikwad and M. Schmittel, J. Org. Chem., 2017, 82, 343–352 CrossRef CAS PubMed; (j) H. V. Schröder, J. M. Wollschläger and C. A. Schalley, Chem. Commun., 2017, 53, 9218–9221 RSC.
- W. Jiang and C. A. Schalley, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10425–10429 CrossRef CAS PubMed.
- (a) S. Pramanik, S. De and M. Schmittel, Angew. Chem., Int. Ed., 2014, 53, 4709–4713 CrossRef CAS PubMed; (b) S. Pramanik, S. De and M. Schmittel, Chem. Commun., 2014, 50, 13254–13257 RSC.
- M. L. Saha, S. Neogi and M. Schmittel, Dalton Trans., 2014, 43, 3815–3834 RSC.
- M. Schmittel, V. Kalsani, P. Mal and J. W. Bats, Inorg. Chem., 2006, 45, 6370–6377 CrossRef CAS PubMed.
- M. Schmittel, S. De and S. Pramanik, Angew. Chem., Int. Ed., 2012, 51, 3832–3836 CrossRef CAS PubMed.
- F. L. Hedbern and H. Rosenberg, Tetrahedron Lett., 1969, 10, 4011–4012 CrossRef.
- A. G. Bonn and O. S. Wenger, J. Org. Chem., 2015, 80, 4097–4107 CrossRef CAS PubMed.
- M. Schmittel, S. Pramanik and S. De, Chem. Commun., 2012, 48, 11730–11732 RSC.
- F. A. Bell, A. Ledwith and D. C. Sherrington, J. Chem. Soc. C, 1969, 2719–2720 RSC.
- (a) R. Breuer and M. Schmittel, Organometallics, 2012, 31, 6642–6651 CrossRef CAS; (b) R. Breuer and M. Schmittel, Organometallics, 2013, 32, 5980–5987 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ob01456j |
| This journal is © The Royal Society of Chemistry 2019 |