
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reaction mechanism of nucleoside 2′-deoxyribosyltransferases: free-energy landscape supports an oxocarbenium ion as the reaction intermediate†‡
Jon
del Arco
a,
Almudena
Perona
 a,
Leticia
González
b,
Jesús
Fernández-Lucas
ac,
Federico
Gago
d and
Pedro A.
Sánchez-Murcia
*b
a,
Leticia
González
b,
Jesús
Fernández-Lucas
ac,
Federico
Gago
d and
Pedro A.
Sánchez-Murcia
*b
aApplied Biotechnology Group, European University of Madrid, Villaviciosa de Odón, Spain
bInstitute of Theoretical Chemistry, Faculty of Chemistry, Währinger Str. 17, A-1090 University of Vienna, Vienna, Austria. E-mail: pedro.murcia@univie.ac.at
cGrupo de Investigación en Ciencias Naturales y Exactas, GICNEX, Universidad de la Costa, CUC, Calle 58 # 55–66, Barranquilla, Colombia
dDepartment of Biomedical Sciences and “Unidad Asociada IQM-CSIC”, School of Medicine and Health Sciences, University of Alcalá, Alcalá de Henares, Spain
First published on 31st July 2019
Abstract
Insight into the catalytic mechanism of Lactobacillus leichmannii nucleoside 2′-deoxyribosyltransferase (LlNDT) has been gained by calculating a quantum mechanics–molecular mechanics (QM/MM) free-energy landscape of the reaction within the enzyme active site. Our results support an oxocarbenium species as the reaction intermediate and thus an SN1 reaction mechanism in this family of bacterial enzymes. Our mechanistic proposal is validated by comparing experimental kinetic data on the impact of the single amino acid replacements Tyr7, Glu98 and Met125 with Ala, Asp and Ala/norLeu, respectively, and accounts for the specificity shown by this enzyme on a non-natural substrate. This work broadens our understanding of enzymatic C–N bond cleavage and C–N bond formation.
Introduction
Breakage/formation of C–N glycosidic bonds in nature is catalyzed by glycosyltransferases, a family of enzymes that catalyze the transfer of glycosyl moieties among a manifold of molecules.1 Increasingly widespread access to techniques such as directed evolution2 or computer-aided protein engineering3 in the last few years, amongst others, has helped to enrich our understanding of these remarkable proteins and their applications in biotechnology. In this context, we have recently reported the characterization and use of several nucleoside 2′-deoxyribosyltransferases (NDTs) isolated from different microorganisms for the industrial synthesis of nucleoside analogues.4–6 This family of enzymes has emerged in the last few years as a one-pot, selective and metal-free option for the catalysis of C–N bond formation. However, although the promiscuity of these enzymes allows them to recognize a broad range of non-natural substrates,7 there is still room for protein engineering on NDTs to expand their product repertoire and enhance the overall reaction yields. As illustrative examples, Kaminski and colleagues identified Gly9Ser and Ala15Thr variants of Lactobacillus leichmannii and Lactobacillus fermentum NDTs, respectively, which are suitable for the synthesis of 2′,3′-dideoxynucleosides8 and created a chimera composed of an NDT and a 5′-monophosphate-2′-deoxyribonucleoside hydrolase that can exchange deoxyribose 5′-(mono, di, and tri)-phosphate between natural bases and analogues.9The rational design of NDTs requires a deep understanding of the structural and mechanistic features of the catalytic mechanism carried out by these enzymes. Since the resolution of the first crystal structure of Lactobacillus leichmannii NDT (LlNDT) in atomic detail by Armstrong and colleagues,10 several experimental models for isoenzymes from other microbial species have been deposited in the Protein Data Bank (Table S1‡). NDTs (EC 2.4.2.6) catalyze the direct transfer of the 2′-deoxyribosyl moiety from a 2′-deoxynucleoside donor to a nucleobase acceptor11,12 (Scheme 1). The currently accepted mechanism for the exchange of a 2′-deoxyribosylribosyl moiety between two nucleobases catalyzed by NDTs involves two ‘half-reactions’ and a common covalent enzyme intermediate (Scheme 1). In the first half-reaction, the carboxylate of the catalytic glutamic acid (Glu98 in LlNDT) attacks the anomeric C1′ atom of 2′-deoxyribonucleoside in the precovalent complex I (e.g. 2′-deoxyinosine, dIno) to yield the covalent intermediate III and a free nucleobase (Hyp in this example). Thereafter, the sugar in III can be transferred to a different entering nucleobase – acting as an acceptor – following the same reaction mechanism but in the reverse direction. Since two inversions of configuration at the deoxyribose C1′ atom take place, its absolute stereochemistry in the leaving nucleoside is retained.
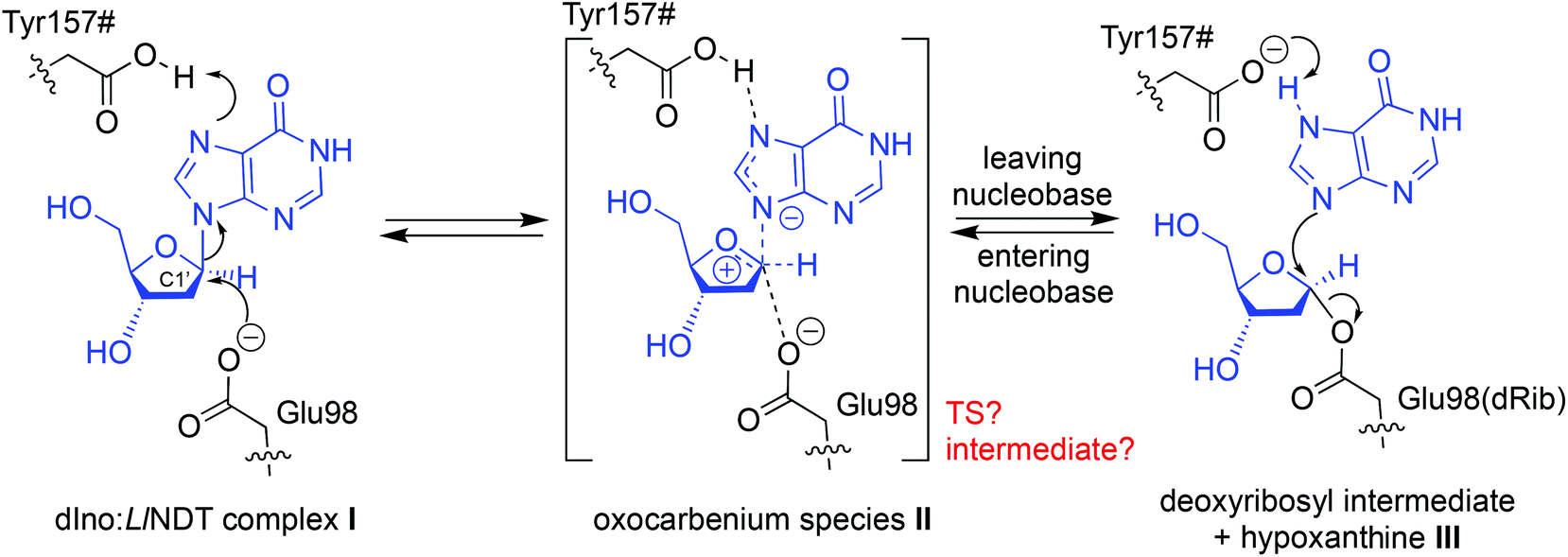 | ||
| Scheme 1 Proposed reaction mechanism for the LlNDT-catalyzed C–N bond cleavage/formation using 2′-deoxyinosine (dIno) and hypoxanthine (Hyp) as the nucleoside donor and nucleobase acceptor, respectively. | ||
This proposal for the reaction mechanism and the catalytic residues involved arises from structural11 and kinetic studies,13,14 with some of them supported by the results from site-directed mutagenesis.14 The role of Glu98 as the nucleophile in the trans-glycosylation reaction is clear.13 However, the nature of the oxocarbenium species II (an oxocarbenium ion-like transition state (TS) or an oxocarbenium intermediate) in this family of transglycosidases is still under debate.12 Depending on the temporal ordering of the breaking and generation of the glycosidic bond in II, this oxocarbenium could be a TS (SN2-like) – if the two processes occur at the same time – or an intermediate (SN1-like) if one of them antedates the other one.15 Besides, although the C-terminal carboxylic acid of Tyr157# from another subunit of the enzyme (dimer, tetramer or hexamer) has been suggested to protonate the leaving nucleobase,11 this event is still not completely understood. By addressing all of these questions, in this work we propose a unifying mechanism for the reaction catalyzed by LlNDT involving a nucleoside and a nucleobase. QM/MM MD simulations have allowed us to compute the reaction pathway within the active site while simultaneously considering the flexibility of the enzyme.16 Our mechanistic proposal is corroborated by crossmatching with the experimental data pertaining to LlNDT.
Results and discussion
We captured an ensemble of dIno binding geometries in the Michaelis complex (complex I, Fig. 1A) by means of a combination of MD and QM/MM MD simulations, during which all the relevant residues of the active site were included into the QM region (see Computational methods). This approach allowed us to efficiently sample the conformational space of the Michaelis complex17 and to capture the optimal orientation of the reactive atoms shown in Scheme 1 (Fig. S1‡). In this complex, the substrate is tightly bound to the enzyme in a wholly desolvated state. The hydrophobicity of the active site (originating from the side chains of residues such as Trp12, Phe13, Leu124# and Met125#) does not favor the entrance of a water molecule and thereby prevents the hydrolysis of the intermediate, an event that takes place in glycosidases.18,19 Regarding the relative orientation between the first pair of reactive atoms, i.e. Glu98 OE1 and the anomeric C1′ atom of dIno (distance d1, Fig. 1A), in the Michaelis complex they are both well positioned and in close proximity (mean value of 3.94 Å, Fig. 1B). On the other hand, the two acid moieties that could provide the proton to either N3 or N7 of the leaving hypoxanthine in our simulations – the C-terminal carboxylic group of Tyr157# (d2) or the side chain of Asp72 (d3), respectively – were hydrogen-bonded to each of these nitrogen atoms of dIno along the simulation. In the case of d2 (N3), a small population of conformers was observed in which this interaction is lost (d2 > 2.5 Å) but replaced by a hydrogen bond to the carbonyl oxygen of dIno (d4 < 2.5 Å). In all cases, upon proton transfer the exiting hypoxanthine would tautomerize to the N9-protonated form, which is thermodynamically more stable.20 The structural analysis of similar enzymes from other families helped us to discard the option of Asp72 as the proton source since this residue is replaced by asparagine in similar purine NDT enzymes from Leishmania major (LmPDT)21 or Trypanosoma brucei (TbPDT),22 even though these enzymes share a highly conserved active site.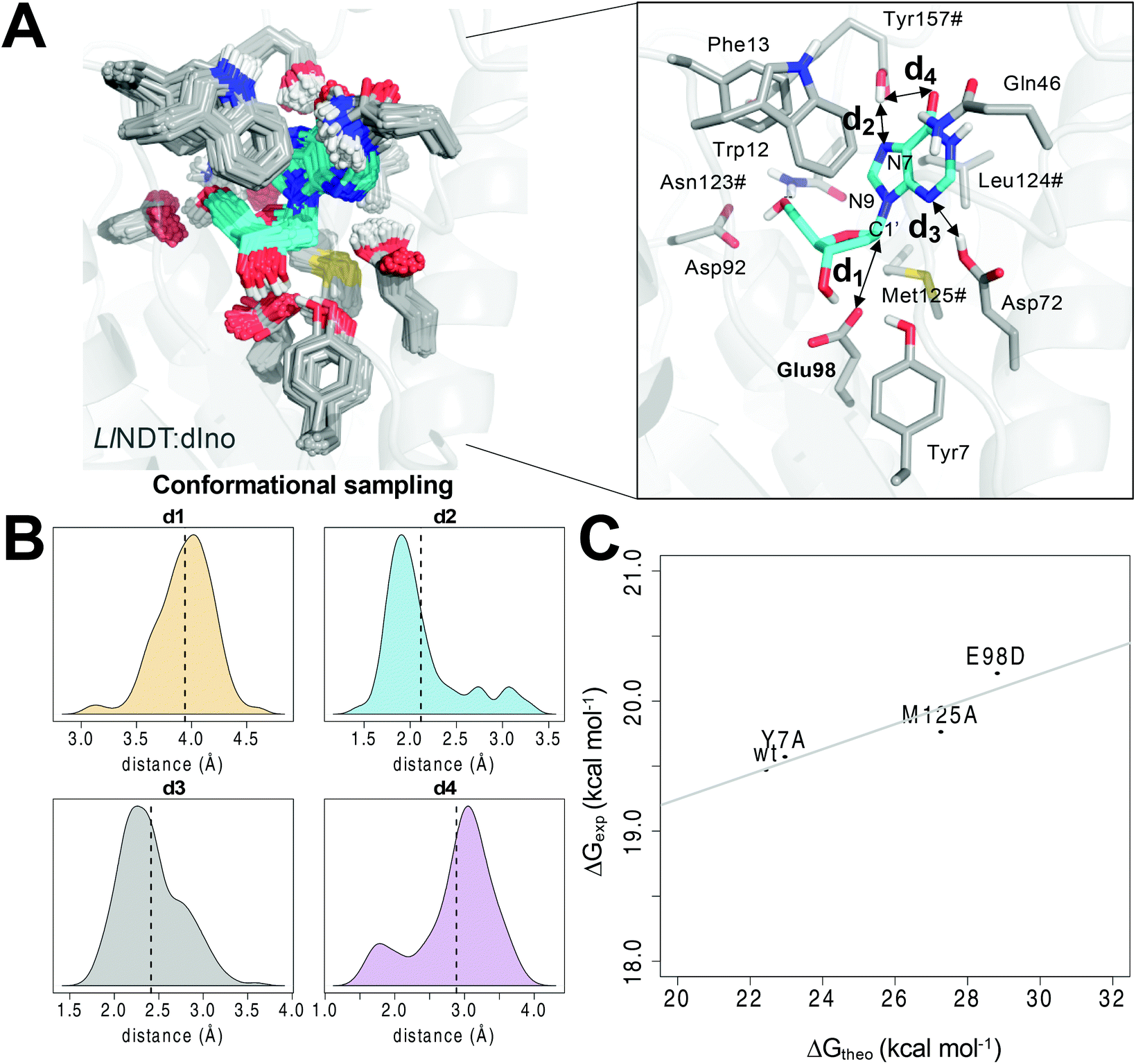 | ||
| Fig. 1 (A) Conformational sampling of the Michaelis complex I of dIno within the active site of LlNDT. (B) Distribution of the distances d1–d4 highlighted at the active site along the QM/MM MD simulation. The mean values are shown as vertical dotted lines. (C) Correlation between the computed (ΔG‡theo) and the experimental14 free energies of activation (ΔG‡exp) for the wild type and the Y7A, M125A and E98D variants of LlNDT. Asn123#, Leu124#, Met125# and Tyr157# are residues of the other subunit of the enzyme. | ||
From the former conformational sampling, we selected the 20 snapshots with the lowest values for the d1 and d2 distances (Fig. 1) in order to study the first half-reaction. Each of these frames was energy minimized using a QM/MM protocol (see Computational methods). After that, the first half reaction was simulated by means of steered QM/MM MD simulations using these 20 minimized snapshots as input files. After the analysis of the PMF profiles of the 20 simulated reactions (Fig. S2‡), one of these trajectories was selected to run an umbrella sampling QM/MM MD simulation.23 From the latter simulation, the free energy of activation value ΔG‡theo was obtained. We defined as two independent reaction coordinates the variation of the former distances, i.e. (i) the shortening of the distance separating OE1(Glu98) from C1′ of dIno leading to C–O bond formation (RC1 or d1) and (ii) the proton transfer from the carboxylic acid of the C-terminal Tyr157# to the nucleobase N7 atom (RC2 or d2). It must be stressed that the explicit enforcement of the second variable RC2 was needed to obtain a converged energy profile (Fig. S3‡). As a result, the calculated value of 22.43 ± 0.21 kcal mol−1 for the free energy barrier of LlNDT (Fig. S4‡) is only slightly above the experimental free energy of activation (19.48 ± 0.01 kcal mol−1)14 (wt in Fig. 1C and Table 1). Encouraged by this finding, we evaluated the impact of single residue changes within the active site on the free energy barrier of the first half-reaction (Fig. 1C). Thus, we simulated the Tyr7Ala, Glu98Asp, and Met125Ala variants using the same dIno substrate and following the same procedure as for the wild type LlNDT enzyme (Fig. 1C and Fig. S5‡). The computed energy barriers correlate well with the experimental catalytic constants (R2 = 0.8489, Fig. 1C and Table 1).14 We observe that whereas the removal of one methylene in the side chain of the catalytic Glu98 (Glu98Asp variant) increases the energy barrier for the reaction (28.81 ± 0.33 kcal mol−1), the Tyr7Ala substitution barely affects the kinetics of the reaction (22.95 ± 0.10 kcal mol−1).14 This finding supports the view that the side chain of Tyr7 assists by promoting a correct orientation of the Glu98 carboxylate for the nucleophilic attack but does not directly participate in the catalytic cycle. Indeed, in our simulations of the Ty7Ala variant, the mobility of the attacking Glu98 side chain is increased and a water molecule enters the active site and occupies the space of the hydroxyl group of Tyr7 in the wild-type counterpart (Fig. S6‡). As far as Met125 is concerned, to the best of our knowledge, little or no attention has been paid to the role of this residue from the characteristic NLM motif in the reaction mechanism. We previously suggested6 that one of the lone pairs of the sulfur atom in the positionally equivalent methionine in TbPDT (Met128) was likely to stabilize the developing positive charge on the oxocarbenium II.24 Thus, we simulated the experimental Met125Ala and the virtual Met125Nle variants of LlNDT, in which the equivalent methionine had been replaced with either Ala or norleucine (Nle), respectively. This latter change retains the steric and hydrophobic character of the amino acid at position 125 but removes the putative stabilizing effect of the sulfur atom. In our model, the change to Ala leads to a much higher energy barrier (27.26 ± 0.19 kcal mol−1) – in the range of the Glu98Asp variant – whereas the replacement of Met with Nle slightly increases the energy barrier relative to that of the wild-type counterpart (23.07 ± 0.08 kcal mol−1). This suggests that Met125 not only contributes to the hydrophobic character of the active site but also provides electronic stabilization to the transition state by virtue of the lone orbitals of its sulfur atom.
| Variant | Experimental kcat (s−1)14 | ΔG‡exp![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (kcal mol−1) a (kcal mol−1) |
ΔG‡theo (kcal mol−1) |
|---|---|---|---|
a The experimental free energies of activation ΔG‡exp were computed using the Eyring equation 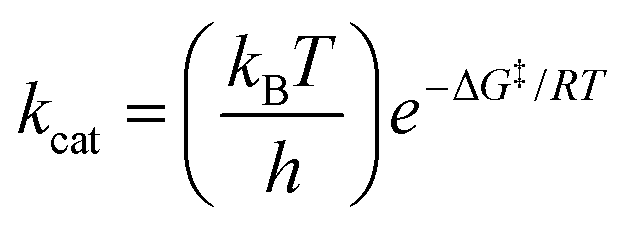 .
b Not reported. .
b Not reported.
|
|||
| wt | (3.20 ± 0.10) × 10−2 | 19.48 ± 0.01 | 22.43 ± 0.21 |
| Y7A | (2.69 ± 0.09) × 10−2 | 19.58 ± 0.01 | 22.95 ± 0.10 |
| E98D | (9.10 ± 0.50) × 10−3 | 20.22 ± 0.01 | 28.81 ± 0.33 |
| M125A | (1.95 ± 0.06) × 10−2 | 19.77 ± 0.01 | 27.26 ± 0.19 |
| M125Nle | n.r.b | — | 23.07 ± 0.08 |
Next, we explored the topicity of the reaction by identifying the reaction intermediate(s) (Fig. 2). Depending on the synchronization between the attack of OE1(Glu98) and the breaking of the glycosidic bond in dIno, the reaction can follow a single-displacement concerted mechanism (AN*DN or SN2-like) – like in most of the inverting glycosyltransferases24 – or a dissociative–associative stepwise mechanism (DN*AN or SN1-like).15,25 For NDTs this is still a matter of debate.12 In any case, the covalent glycosyl-enzyme intermediate III would be obtained as the product of the first half-reaction (Scheme 1).11 Nevertheless, depending on the reaction mechanism, the nature of the oxocarbenium species II would be a TS (SN2-like) or an intermediate (SN1-like).
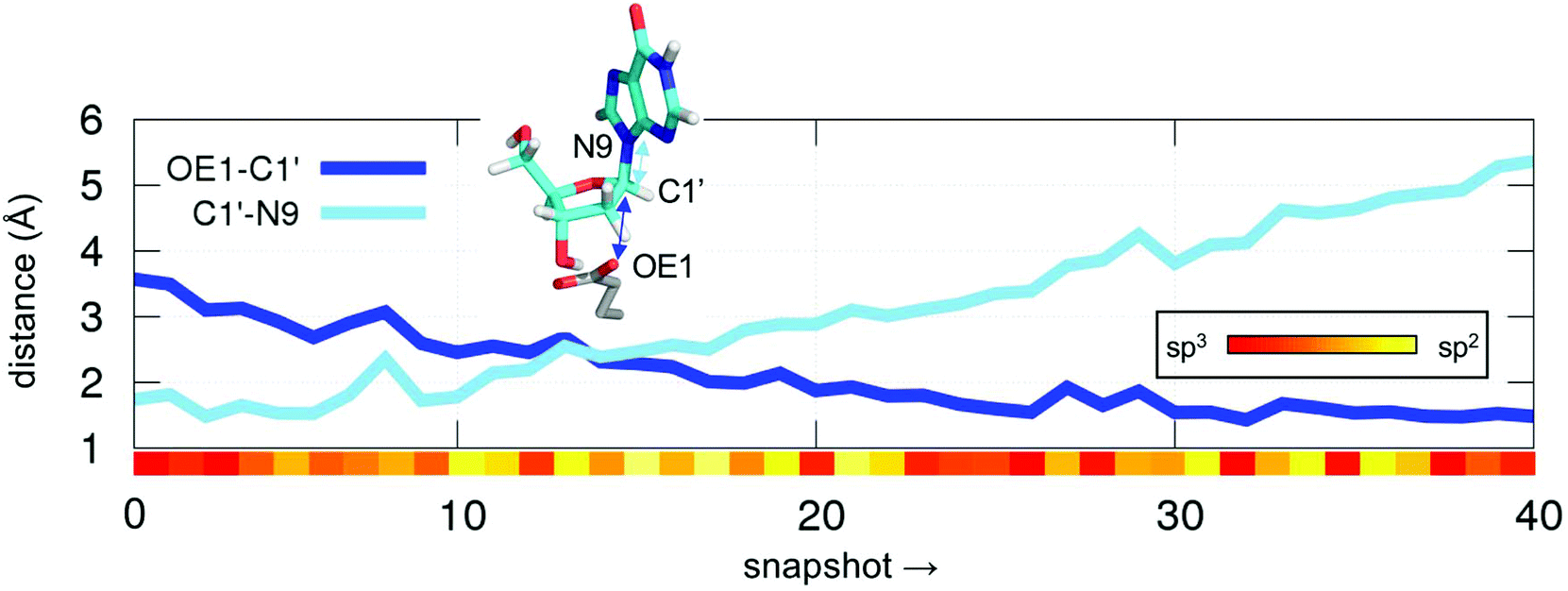 | ||
| Fig. 2 Evolution of distances between OE1–C1′ (dark blue line) and C1′–N9 (light blue line) atoms along a steered QM/MM MD simulation + umbrella sampling for the first half reaction. The variation of the two distances was defined as a linear combination of distances (LCOD). The degree of planarity on C1′ is shown as a heat map between sp3 (107.5°, red) and sp2 (120.0°, yellow) (coloured bar inserted in the body of the figure). | ||
First, we studied the synchronicity of the C1′–OE1 bond formation and C1′–N3 breakage (Fig. 2). At this point these two distances cross each other (2.4 Å), the leaving nucleobase is already gone but the attacking OE1 of Glu98 is still far from forming a bond with the anomeric carbon C1′, which has already an sp2 configuration (angle O2–C1′–H1 of ca. 117°, yellow bar in Fig. 2). The former angle is maintained above 115° for most of the simulation time until the new OE1–C1′ bond is formed, at which point it acquires an sp3 hybridization character (∼107°, red bar in Fig. 2). Therefore, the persistence of this planar intermediate together with a large C1′–N9 distance along the simulations were taken as indicators of an oxocarbenium intermediate rather than a TS for II (Scheme 1).
Second, we studied the synchronicity between the nucleophilic attack and the proton transfer events. To this end, we computed the two-dimensional minimum free energy pathway of the reaction by means of umbrella sampling simulations (Fig. 3). We obtained two possible pathways from I to III. The difference between them is at which point in time the proton transfer occurs. In the “north pathway”, the formation of the glycosidic bond precedes proton transfer. This pathway involves two local minima: IIA and IIB. Both of them are intimate ion pairs24,25 between the oxocarbenium ion and the anionic hypoxanthine. The Glu98 OE1 atom in IIA, which is oriented by Tyr7, does not form a bond with C1′ (distance of 2.28 Å) but the C1′–N3 glycosidic bond of dIno is already broken (2.19 Å). In IIB the attacking OE1 of Glu98 is closer to C1′ (1.68 Å), as is the nucleobase to the proton donor (2.16 Å). After the proton transfer, the reaction is completed, yielding III. In the “south pathway”, the proton transfer precedes the formation of the glycosidic bond. There is a local minimum I′ of similar energy to that of I but, in this conformation, the leaving nucleobase is closer to the proton donor, i.e. the Tyr157# carboxylic acid oxygen (2.39 Å vs. 3.50 Å in I). Thus, the proton is already transferred when the attacking Glu98 OE1 is still 1.97 Å apart (intermediate IIC). Nevertheless, in both pathways, the initial glycosidic bond (C1′–N7) is already broken when the attacking OE1 is still approaching at distances over 2.0 Å. Thus, the reaction mechanism may follow a stepwise SN1-like scheme as in some pyrimidine DNA hydrolases26 where the rate-limiting step is the cleavage/formation of the glycosidic bond. The optimized geometries of these intermediates obtained by means of the cluster method and their related energies are shown in Fig. 4.
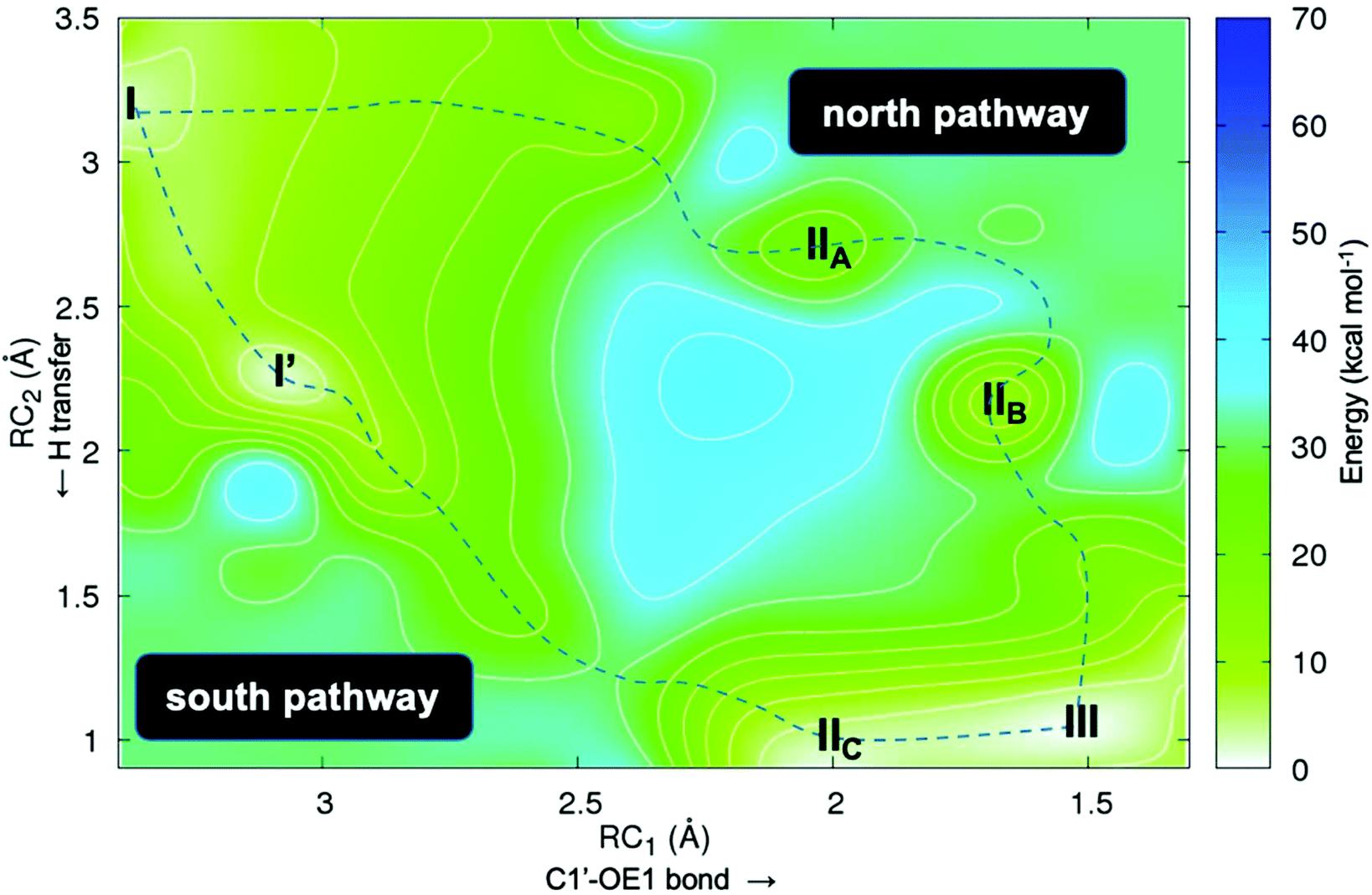 | ||
| Fig. 3 2D free energy plot (kcal mol−1) for the first half-reaction (Scheme 1) where RC1 is the attack of OE1 of Glu98 onto the anomeric C1′ carbon in dIno and RC2 is the proton transfer from the Tyr157# carboxylic acid group to N7 of dIno. | ||
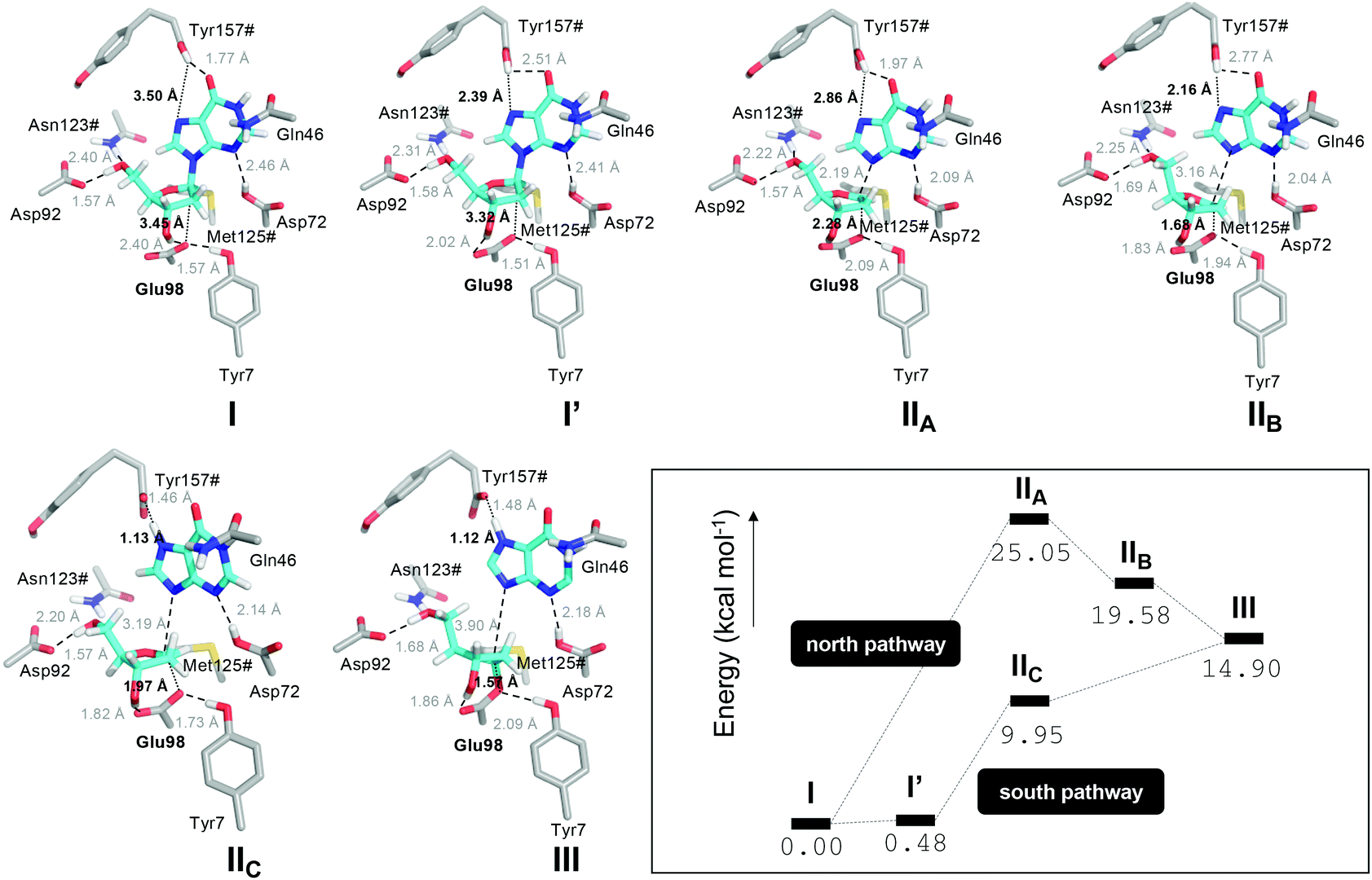 | ||
| Fig. 4 Optimized geometries (DLPNO-CCSD(T)/def2-TZVP//B3LYP-D3/def2-SVP) of states I, I′, IIA, IIB, IIC and III derived from the umbrella sampling QM/MM MD simulations and their corresponding energies (kcal mol−1). The energies were computed including the thermal correction to the total Gibbs free energy at 298 K. The transition states that connect intermediates I–III have not been characterized in the gas phase. | ||
The second half-reaction follows a similar mechanism but in the reverse direction (in Scheme S1‡ we summarize both first and second half-reactions). Nevertheless, the entering nucleobase in this step (i.e. adenine) will find a different shape and electrostatic distribution in complex III compared to I because the Y157# C-terminal carboxylic acid is deprotonated. As an example, we simulated the major tautomeric forms of adenine (protonated on either N7 or N9, and named AD7 and AD9, respectively) within the active site of the glycosylated LlNDT (Fig. S7‡). We corroborated this hypothesis since only tautomer AD7 (and not AD9) was able to adopt a pre-reactive near-attack orientation.
Finally, and as a proof of concept of the applicability of our proposed reaction mechanism to other substrates, we simulated the glycosylation of the 4′-pyrimidone-substituted imidazole derivative 3s, which has been recently reported as a non-natural substrate for LlNDT (Fig. 5).27,28 Interestingly, the LlNDT-catalyzed glycosylation of 3s yields two products depending on the experimental conditions: the N1-isomer (5s) or the N3-isomer (4s), which are obtained under kinetic or thermodynamic control, respectively (Fig. 5).
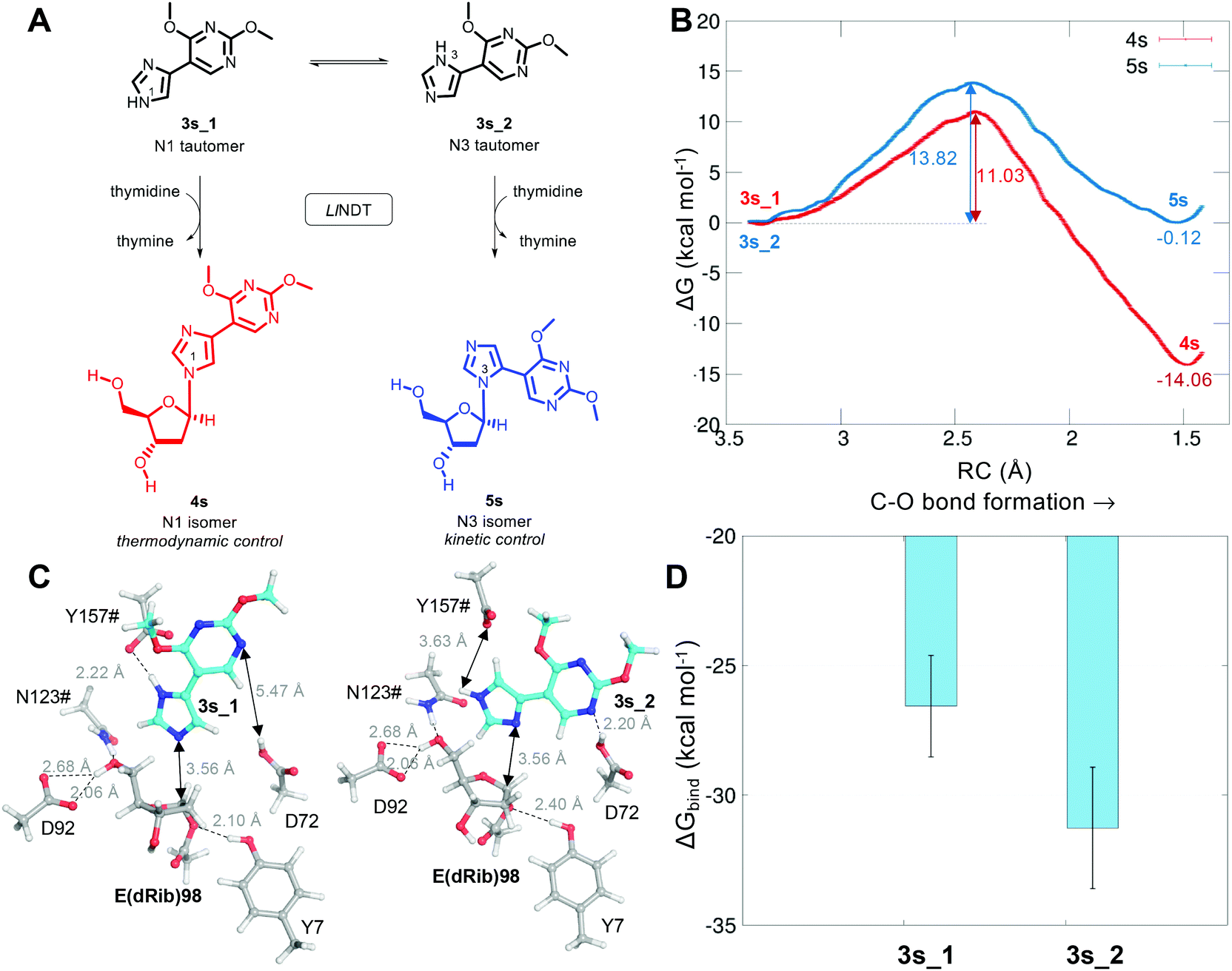 | ||
| Fig. 5 Glycosylation of the two tautomers 3s_1 and 3s_2 by LlNDT to yield 4s and 5s, respectively. (B) Computed free energy (kcal mol−1) profile for both reactions. (C) Michaelis complexes of 3s_1 and 3s_2 at the active site of LlNDT. (D) Mean value of the computed binding energy (kcal mol−1) along the MD simulations of the tautomer species 3s_1 and 3s_2 to LlNDT. | ||
As for the natural substrate deoxyadenosine (Ado) in the second half-reaction, we can now define both the nucleophilic attack of the nucleobase to C1′ and the proton transfer from Y157#. Based on our proposal of reaction mechanism, the umbrella sampling QM/MM MD computed free energies of activation for these LlNDT-catalyzed glycosylations are shown in Fig. 5A. The reaction towards 4s is thermodynamically favored (ΔG = −14.06 ± 0.11 kcal mol−1) compared to 3s (ΔG = −0.12 ± 0.03 kcal mol−1). Nevertheless, 4s also shows the lowest free energy of activation (11.03 ± 0.08 kcal mol−1) compared to 5s (13.82 ± 0.03 kcal mol−1). This means that the reaction towards 4s would be favored, in principle, in all cases. But why is 5s formed under kinetic conditions? The answer lies in substrate specificity. Of the two tautomers of 3s that can undergo the reaction, the isomer that would afford 5s (3s_2) binds more strongly (but in the same order) to LlNDT than does 3s_1 (Fig. 5B and C). Thus, the binding of 3s_2 would be favored over the binding of 3s_1 and 5s would be obtained in the short term. As soon as the system is allowed to equilibrate, the population of 3s_1 that binds to LlNDT would be transformed into 4s (product of thermodynamic control) in a process that is also kinetically favorable over 5s, pushing the global chemical equilibrium to the formation of 4s. Looking into the atomic detail, it can be seen that 3s_2 forms an extra hydrogen bond with Asp72 through its N10. This finding stresses the importance of considering the enzyme environment when studying reaction mechanisms.16
In summary, we have studied here the catalytic mechanism of 2′-deoxyribosyl transfer within the active site of LlNDT using an all-atom description of the reaction and hybrid QM/MM methods. The correlation with kinetic experimental values of the wild-type protein and the E98D, Y7A and M125A variants has allowed us to support the validity of the proposed mechanism for this family of enzymes. The reaction consists of two steps: a nucleophilic attack and a proton transfer with an oxocarbenium ion as the reaction intermediate. In addition, our model accounts for the substrate selectivity shown by LlNDT towards the reported non-natural substrates of the enzyme. Altogether, this full understanding of the all-atom reaction mechanism is expected to aid in guiding more efficient protein engineering campaigns.
Computational methods
System setup and MD simulations
The Cartesian coordinates of the purine 2′-deoxyribosyl transferase of Lactobacillus leichmannii (LlNDT) in complex with its substrate 5-methyl-2′-deoxypseudouridine (PDB id. 1F8Y)10 were used as templates for the LlNDT:dIno complex. dIno was docked manually into the active site of the enzyme by means of structural best-fit superposition onto the former nucleoside. Care was taken to protonate properly the titratable residues at the active site using the server H++ 3.0.29 The ground state geometries of the ligand dIno, adenine, the ribosylated glutamic acid, the norleucine residue and 3s_1 and 3s_2 were first optimized in the gas phase and their electrostatic potentials were computed at the standard level of theory (HF/6-31G**//HF/3-21G) and fitted to the atoms as RESP charges using the program antechamber (AmberTools18, URL:ambermd.org). The atoms of the ligands and of the former amino acids were described as AMBER atom types. The leaprcff14SB force field was used in all the MD simulations. The MD simulations were run on GPUs using the pmemd.cuda module of Amber17 in the Single-Precision-Fixed-Precision (SPFP) mode. The dIno:LlNDT complex was simulated as a protein dimer. dIno (first half reaction) and dAdo (second half reaction) were placed in each of the active sites and the total complex was embedded in a truncated octahedral box of ca. 13000 TIP3P water molecules30 that extended 12 Å away from any solute atom and 10 Na+ ions were added to ensure charge neutrality. The system was relaxed by energy minimization in three consecutive steps (3 × 5000 cycles), in which after the first 1000 cycles the minimization method was switched from steepest descendent to conjugate gradient. The resulting system was heated from 100 to 300 K during 200 ps with a time step of 0.2 fs and with the position of all the solute atoms restrained with a harmonic potential of 20 kcal mol−1 Å−2. The Berendsen thermostat31 was employed for temperature regulation and the simulation was run with a fixed volume (NVT ensemble). The harmonic restraints were gradually reduced in four steps from 40 to 10 kcal mol−1 Å−2. Then, the density of the system was equilibrated for 20 ps using a time step of 0.2 fs by fixing the pressure, using the Berendsen thermostat with isotropic pressure scaling (NPT ensemble), and allowing the volume of the box to change. The system was simulated for 10 ns at 300 K with a time step of 2 fs. A harmonic restraint of 5 kcal mol−1 Å−2 was imposed on the alpha carbon atoms of the protein in order to maintain the secondary structure. The cutoff distance for the nonbonded interactions was 10 Å and the periodic boundary conditions were used. Electrostatic interactions were treated by using the smooth particle mesh Ewald (PME) method32 with a grid spacing of 1 Å. The SHAKE algorithm was applied to all bonds involving hydrogen atoms. The replacement of residues 7, 98 and 125 by Asp, Tyr and Ala/Nle, respectively, was carried out in PyMOL, minimizing the number of steric clashes. Then, the protocol followed with the wild-type enzyme being applied to the three variants.
QM/MM MD simulations
The MD simulation protocol was run iteratively to refine the Michaelis complex between dIno and the protein for the first half reaction. To do this, the distances between the two-reactant atoms of the first half reaction (OE1 of Glu98 and the C1′ anomeric carbon of dIno) were monitored as well as all the distances between the residues at the active site and the ligand. Then, a snapshot was selected and further simulated for 10 ps in a QM/MM MD simulation without any restraint except for that on the alpha carbon atoms of the protein (5 kcal mol−1 Å−2). For the first half reaction, the ligand (dIno), the side chains of the residues Glu98, Asp72, Asn123#, Asp92 and Tyr7 and the carboxylate of Tyr157# were included in the QM region (78 atoms). 7 linking atoms were introduced after truncation between Csp3 atoms. For the second half reaction, the same side chains were included in the QM region together with the ligand (dAdo, 3s_1 or 3s_2) but with a glycosylated Glu98. In all cases, the amino acid side chains included into the QM region were cut from the β-carbon atoms and care was taken to avoid cutting bonds where any of the atoms was a heteroatom. The QM region was treated using the self-consistent-charge density-functional tight-binding method (SCC-DFTB)33 using the DFTB3 formulation34 and the rest of the system was treated classically as described above. In particular, the set of parameters for O, C, N and H atoms of the third-order parametrization for organic and biological systems (3OB) were used for the QM region.35 The effect of the environment on the QM region was included using the electrostatic embedding scheme. A time step of 0.5 fs and the Berendsen thermostat with isotropic pressure scaling (NPT ensemble) were used. A snapshot was saved every 50 fs. All the calculations were run using the AMBER:DFTB3 scheme implemented in AMBER17.33,36Multiconformational sampling and steered QM/MM MD simulation of the enzymatic reaction
In order to understand the dynamics of the enzyme, we selected the 20 snapshots of the former 10ps-QM/MM MD simulation of the Michaelis complex where the distances between (i) the attacking carboxylic oxygen of Glu98 and the anomeric carbon C1′ of dIno and (ii) the hydrogen of the carboxylic acid of Tyr157# and the N7 of hypoxanthine were shorter. Then, each of these 20 snapshots were used as an input for a QM/MM MD simulation of 500 cycles of which the first were run using the steepest descent algorithm and the last 300 with the conjugate gradient method. The first half reaction was simulated by steered QM/MM MD simulations where the reaction coordinate (RC) was defined as a linear combination of two distance variables: (i) the shortening of the distance between OE1(Glu98) and C1′ of dIno so as to form the C–O bond (RC1) and (ii) the proton transfer from the carboxylic acid of the C-terminal Tyr157# to the nucleobase N7 atom (RC2) (Fig. 1). All reactions were simulated for 2 ps and a harmonic force constant of 500 and 300 kcal mol−1 Å−2 was defined for the first and the second variable, respectively. No restraints were imposed on the enzyme during the steered QM/MM MD simulation except for those defining the reaction coordinate. The inclusion of the proton transfer into the global definition of RC was needed to complete the reaction. Fig. S3‡ shows the PMF profiles computed for the same 20 initial snapshots using either only 1 variable (RC1) or 2 variables (RC1 + RC2). As can be observed, without RC2 (Fig. S3,‡ left), the energy profiles do not decrease in energy after the first local maximum.Umbrella QM/MM MD sampling
For each of the simulated variants, we selected a representative trajectory with a PMF value close to the average PMF of the 20 trajectories. Then, 40 snapshots of the steered QM/MM MD simulation were printed and subjected to an umbrella sampling QM/MM MD protocol using the same definition for the reaction coordinates RC1 and RC2. Each of the windows was allowed to oscillate for 5 ps around the imposed restraints. In order to relax the system, the harmonic constraints were reduced to 200 kcal mol−1 Å−2 and 100 kcal mol−1 Å−2 for RC1 and RC2, respectively. A time step of 0.2 fs was employed. All the simulations were performed at 300 K, using the Berendsen thermostat. The PME and SHAKE algorithms were deactivated in the QM region. A significant overlap between the consecutive window histograms was ensured to obtain a consistent free-energy profile. The computed ΔGtheo values shown in Table 1 were obtained after computing the free energy profile by the Weighted Histogram Analysis Method (WHAM).11,37 A Monte Carlo bootstrap error analysis was carried out (20 trials). The 2D-free energy map shown in Fig. 3 for the wild-type LlNDT was computed including a total of 250 windows obtained from 7 steered QM/MM MD simulations. The 2D free energy profile was calculated using the variational free energy profile (vFEP) method.38Free energy calculations
The binding energies of substrates 3s_1 and 3s_2 with LlNDT were calculated with the MM-ISMSA tool39 with explicit water molecules as the solvent. 200 snapshots extracted from 2 ns windows of the MD simulations of each of the ligand:enzyme complexes were used for the binding energy calculations.Cluster method (quantum mechanics geometry optimization)
The geometries of the intermediates I, I′, IIA, IIB, IIC and III found in the 2D free-energy map shown in Fig. 3 were optimized in the gas phase using density functional theory (DFT) with the hybrid functional B3LYP40 including the Grimme dispersion correction D3.41 A total of 120 atoms were selected for these optimizations, as shown in Fig. 4. The def2-SVP basis set was used on the C, H, N, O and S atoms.42 In order to preserve the fingerprint of the protein on the conformation of the active site, all the carbon atoms in these complexes (except for C1′) were frozen during optimization under vacuum. The remaining atoms (N, O, S, H) were allowed to relax. The energies were computed including the thermal correction to the total Gibbs free energy at 298 K by means of a calculation of the frequencies in the ground state. The optimized geometries of the intermediates did not show any negative frequency. These calculations were performed with the software Gaussian16, revision A.03.43 Besides, the optimized geometries were used as the input for a single point calculation using coupled cluster (CC) theory (coupled cluster singles, doubles, and perturbative triples (CCSD(T))) together with the domain-based local pair natural orbital (DLPNO) approach implemented in ORCA 4.0.144 and the large Ahlrichs’ basis set def2-TZVP42 and the def2-TZVP/C auxiliary basis.45 The final energies were corrected with the thermal correction to the total Gibbs free energy at 298 K.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the FWF (Lise Meitner Program M 2260) to PAS-M, the Spanish Ministerio de Economía y Competitividad (SAF2015-64629-C2-2-R) to F. G. and Grants XSAN001906 from the Santander Foundation to J. F. L. is gratefully acknowledged. We thank the University of Vienna for computational resources at the Institute of Theoretical Chemistry.References
- J. Schmid, D. Heider, N. J. Wendel, N. Sperl and V. Sieber, Front. Microbiol., 2016, 7, 182 Search PubMed.
- J. B. McArthur and X. Chen, Biochem. Soc. Trans., 2016, 44, 129–142 CrossRef CAS PubMed.
- L. Raich, A. Nin-Hill, A. Ardèvol and C. Rovira, in Methods in Enzymology, ed. G. A. Voth, Academic Press, 2016, vol. 577, pp. 159–183 Search PubMed.
- N. Crespo, P. A. Sánchez-Murcia, F. Gago, J. Cejudo-Sanches, M. A. Galmes, J. Fernández-Lucas and J. M. Mancheño, Appl. Microbiol. Biotechnol., 2017, 101, 7187–7200 CrossRef CAS PubMed.
- J. Del Arco, P. A. Sánchez-Murcia, J. M. Mancheño, F. Gago and J. Fernández-Lucas, Appl. Microbiol. Biotechnol., 2018, 102, 6947–6957 CrossRef CAS PubMed.
- E. Pérez, P. A. Sánchez-Murcia, J. Jordaan, M. D. Blanco, J. M. Mancheño, F. Gago and J. Fernández-Lucas, ChemCatChem, 2018, 10, 4406–4416 CrossRef.
- R. Tadeusz, L.-M. Ewa, K. Anna and R. Ewa, Curr. Med. Chem., 2006, 13, 3165–3189 CrossRef.
- P. A. Kaminski, P. Dacher, L. Dugué and S. Pochet, J. Biol. Chem., 2008, 283, 20053–20059 CrossRef CAS PubMed.
- P. A. Kaminski and G. Labesse, J. Biol. Chem., 2013, 288, 6534–6541 CrossRef CAS PubMed.
- S. Armstrong, W. J. Cook, S. A. Short and S. E. Ealick, Structure, 1996, 4, 97–107 CrossRef CAS PubMed.
- R. Anand, P. A. Kaminski and S. E. Ealick, Biochemistry, 2004, 43, 2384–2393 CrossRef CAS PubMed.
- A. Fresco-Taboada, I. de la Mata, M. Arroyo and J. Fernández-Lucas, Appl. Microbiol. Biotechnol., 2013, 97, 3773–3785 CrossRef CAS PubMed.
- D. J. T. Porter, B. M. Merrill and S. A. Short, J. Biol. Chem., 1995, 270, 15551–15556 CrossRef CAS PubMed.
- S. A. Short, S. R. Armstrong, S. E. Ealick and D. J. T. Porter, J. Biol. Chem., 1996, 271, 4978–4987 CrossRef CAS PubMed.
- T. Hansen, L. Lebedel, W. A. Remmerswaal, S. van der Vorm, D. P. A. Wander, M. Somers, H. S. Overkleeft, D. V. Filippov, J. Désiré, A. Mingot, Y. Bleriot, G. A. van der Marel, S. Thibaudeau and J. D. C. Codée, ACS Cent. Sci., 2019, 5, 781–788 CAS.
- S. J. Benkovic, G. G. Hammes and S. Hammes-Schiffer, Biochemistry, 2008, 47, 3317–3321 CrossRef CAS PubMed.
- R. Lonsdale, J. N. Harvey and A. J. Mulholland, Chem. Soc. Rev., 2012, 41, 3025–3038 RSC.
- V. L. Y. Yip and S. G. Withers, Org. Biomol. Chem., 2004, 2, 2707–2713 RSC.
- D. L. Zechel and S. G. Withers, Acc. Chem. Res., 2000, 33, 11–18 CrossRef CAS.
- J. Holguin and R. Cardinaud, Eur. J. Biochem., 1975, 54, 515–520 CrossRef CAS PubMed.
- W. Shi, V. L. Schramm and S. C. Almo, J. Biol. Chem., 1999, 274, 21114–21120 CrossRef CAS PubMed.
- J. Bosch, M. A. Robien, C. Mehlin, E. Boni, A. Riechers, F. S. Buckner, W. C. Van Voorhis, P. J. Myler, E. A. Worthey, G. DeTitta, J. R. Luft, A. Lauricella, S. Gulde, L. A. Anderson, O. Kalyuzhniy, H. M. Neely, J. Ross, T. N. Earnest, M. Soltis, L. Schoenfeld, F. Zucker, E. A. Merritt, E. Fan, C. L. Verlinde and W. G. Hol, J. Med. Chem., 2006, 49, 5939–5946 CrossRef CAS PubMed.
- J. Gao, S. Ma, D. T. Major, K. Nam, J. Pu and D. G. Truhlar, Chem. Rev., 2006, 106, 3188–3209 CrossRef CAS PubMed.
- C. Breton, S. Fournel-Gigleux and M. M. Palcic, Curr. Opin. Struct. Biol., 2012, 22, 540–549 CrossRef CAS.
- A. Ardèvol, J. Iglesias-Fernández, V. Rojas-Cervellera and C. Rovira, Biochem. Soc. Trans., 2016, 44, 51–60 CrossRef.
- R. M. Werner and J. T. Stivers, Biochemistry, 2000, 39, 14054–14064 CrossRef CAS PubMed.
- S. Vichier-Guerre, L. Dugué, F. Bonhomme and S. Pochet, Org. Biomol. Chem., 2016, 14, 3638–3653 RSC.
- S. Vichier-Guerre, L. Dugué, F. Bonhomme and S. Pochet, Org. Biomol. Chem., 2017, 15, 8193–8203 RSC.
- R. Anandakrishnan, B. Aguilar and A. V. Onufriev, Nucleic Acids Res., 2012, 40, W537–W541 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. v. Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- G. d. M. Seabra, R. C. Walker, M. Elstner, D. A. Case and A. E. Roitberg, J. Phys. Chem. A, 2007, 111, 5655–5664 CrossRef PubMed.
- M. Gaus, Q. Cui and M. Elstner, J. Chem. Theory Comput., 2011, 7, 931–948 CrossRef CAS PubMed.
- M. Gaus, A. Goez and M. Elstner, J. Chem. Theor. Comput., 2013, 9, 338–354 CrossRef CAS PubMed.
- R. C. Walker, M. F. Crowley and D. A. Case, J. Comput. Chem., 2008, 29, 1019–1031 CrossRef CAS PubMed.
- A. Grossfield, WHAM: the weighted histogram analysis method, version 2.0.9 Search PubMed.
- T.-S. Lee, B. K. Radak, M. Huang, K.-Y. Wong and D. M. York, J. Chem. Theor. Comput., 2014, 10, 24–34 CrossRef CAS PubMed.
- J. Klett, A. Núñez-Salgado, H. G. Dos Santos, Á. Cortés-Cabrera, A. Perona, R. Gil-Redondo, D. Abia, F. Gago and A. Morreale, J. Chem. Theory Comput., 2012, 8, 3395–3408 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 (Revision A.03), Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- A. Hellweg, C. Hättig, S. Höfener and W. Klopper, Theor. Chem. Acc., 2007, 117, 587–597 Search PubMed.
Footnotes |
| † Dedicated to the memory of Prof. Dr Ángel Vidal. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ob01315f |
| This journal is © The Royal Society of Chemistry 2019 |