
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Distortion of mannoimidazole supports a B2,5 boat transition state for the family GH125 α-1,6-mannosidase from Clostridium perfringens
Alexandra
Males
 a,
Gaetano
Speciale
b,
Spencer J.
Williams
*b and
Gideon J.
Davies
*a
a,
Gaetano
Speciale
b,
Spencer J.
Williams
*b and
Gideon J.
Davies
*a
aYork Structural Biology Laboratory, Department of Chemistry, The University of York, YO10 5DD York, UK. E-mail: gideon.davies@york.ac.uk
bSchool of Chemistry and Bio21 Molecular Science and Biotechnology Institute, University of Melbourne, Melbourne, Victoria 3010, Australia. E-mail: sjwill@unimelb.edu.au
First published on 13th August 2019
Abstract
Enzyme transition-state mimics can act as powerful inhibitors and allow structural studies that report on the conformation of the transition-state. Here, mannoimidazole, a mimic of the transition state of mannosidase catalyzed hydrolysis of mannosides, is shown to bind in a B2,5 conformation on the Clostridium perfringens GH125 α-1,6-mannosidase, providing additional evidence of a OS2–B2,5–1S5 conformational itinerary for enzymes of this family.
A conformational itinerary describes how a molecule changes shape along a reaction coordinate. For reactions catalysed by enzymes, the three-dimensional fold of the protein and its electrostatic microenvironment provides constraints such that well-defined conformational itineraries are observed. These conformational itineraries may differ from those observed for chemically-catalyzed reactions in the gas or condensed phase, but nonetheless must still obey stereoelectronic dictates of chemical reactivity. The study of conformational itineraries of enzymes has been assisted by the full spectrum of kinetic, computational and structural methods.1,2 In the case of glycoside hydrolases, conformational itineraries are often assigned through determination of the three-dimensional structures of ligands bound to enzymes, most commonly substrate (Michaelis), intermediate, and product complexes, and by using inhibitors that mimic the transition state.3
GHs have been grouped into >150 families on the basis of primary sequence and ability to cleave specific glycoside substrates.4,5 As sequence defines the three-dimensional structure of proteins, family members typically adopt similar folds and possess highly conserved amino acids within their active site clefts. The chemical mechanisms through which family members perform catalysis tend to be conserved across a family and lead to substrate hydrolysis with either retention or inversion of anomeric stereochemistry.6 Inversion of stereochemistry occurs in a single step reaction when water acts as a nucleophile. Retention of stereochemistry is typically achieved through a two-step substitution mechanism involving participation by an enzymic nucleophile or by a pendant neighboring group on the substrate.6–8 Both pathways benefit from enzymatic amino acids that provide general acid catalysis to promote the departure of the anomeric groups, and general base catalysis to promote nucleophilic attack by water. In almost all cases the transition states possess oxocarbenium-ion-like character and the stability of this TS is greatest when partial double bond character is maximized between C1 and O5, which occurs when the system C5–O5–C1–C2 is planar.9 Such transition-state conformations include two boats: B2,5 and 2,5B; two half-chairs: 4H3 and 3H4 (Fig. 1a); and the related envelopes: 3E, E3, 4E and E4. As transition state mimicry is a powerful strategy for inhibitor design, efforts have been made to assign conformational itineraries, and in particular the TS conformation, for a large number of GH families.1,2
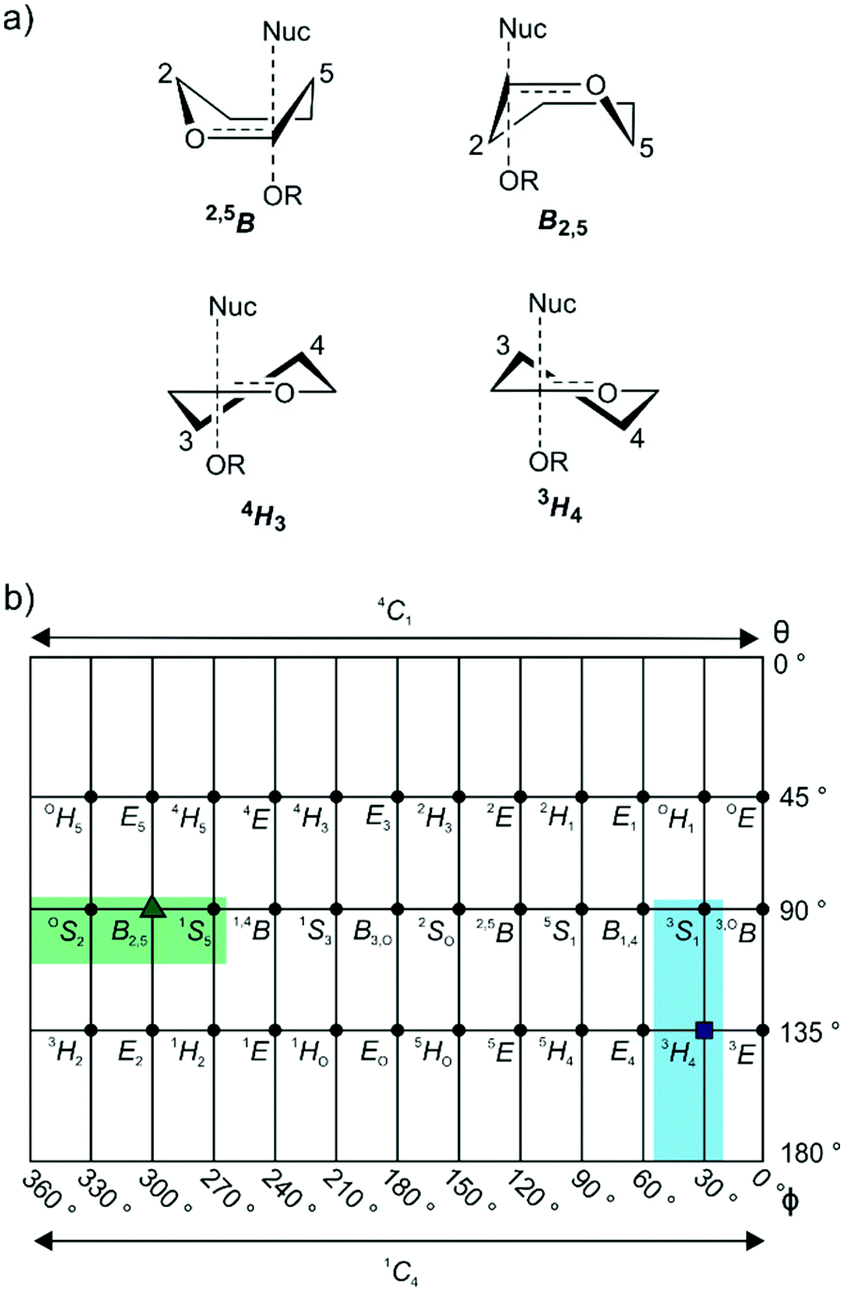 | ||
| Fig. 1 (a) Common half-chair and boat transition state conformations, illustrated for α-glycosidases. (b) Common mannosidase conformational itineraries shown on a Mercator plot with that for GH125 highlighted in green. Shown in blue is the 3S1–3H4–1C4 itinerary used by GH47 and 134 enzymes. The transition states are represented by a green triangle and a blue square. | ||
Mannosidases and mannanases catalyze the hydrolysis of mannoside linkages and these enzymes are found within 11 GH families. Enzymes within families GH2, 5, 26, 38, 76, 92, 99, 113, 125 and 134 perform catalysis through itineraries along the OS2–B2,5–1S5 region of conformational space whereas enzymes of families GH47 and 134 react through the 3S1–3H4–1C4 conformational axis (Fig. 1b). The use of mannoimidazole and analogues thereof have been instrumental in the assignment of conformational itineraries and transition state conformation (Table 1).
In the case of family GH125, the conformational itinerary remained elusive for many years. Initial X-ray structures of the foundation member CpGH125 exo-α-1,6-mannosidase (CpGH125) with the nonhydrolyzable substrate mimic methyl 1,6-α-thiomannobioside (1) (Protein Databank (PDB) ID: 3QT9) and deoxymannojirimycin (2) (PDB ID: 3QRY) revealed undistorted, ground-state 4C1 conformations (Fig. 2a).18 Instead, a computational approach involving ab initio QM/MM metadynamics was used to model the conformation of substrate, 1,6-α-mannobiose, in the enzyme active site, by in silico substitution of the sulfur in the experimentally determined X-ray structure.19 This calculation predicted that the O-glycoside favoured an OS2 conformation on-enzyme and hinted at an OS2 → B2,5‡ → 1S5 conformational itinerary, which was fully supported by QM/MM simulations of the reaction mechanism (Fig. 2b). Direct experimental support for this mechanism was obtained through determination of the Michaelis complex for the catalytically-disabled acid mutant (D220N) of CpGH125 in complex with 1,6-α-mannobiose, which revealed an OS2 conformation consistent with the computational predictions.19
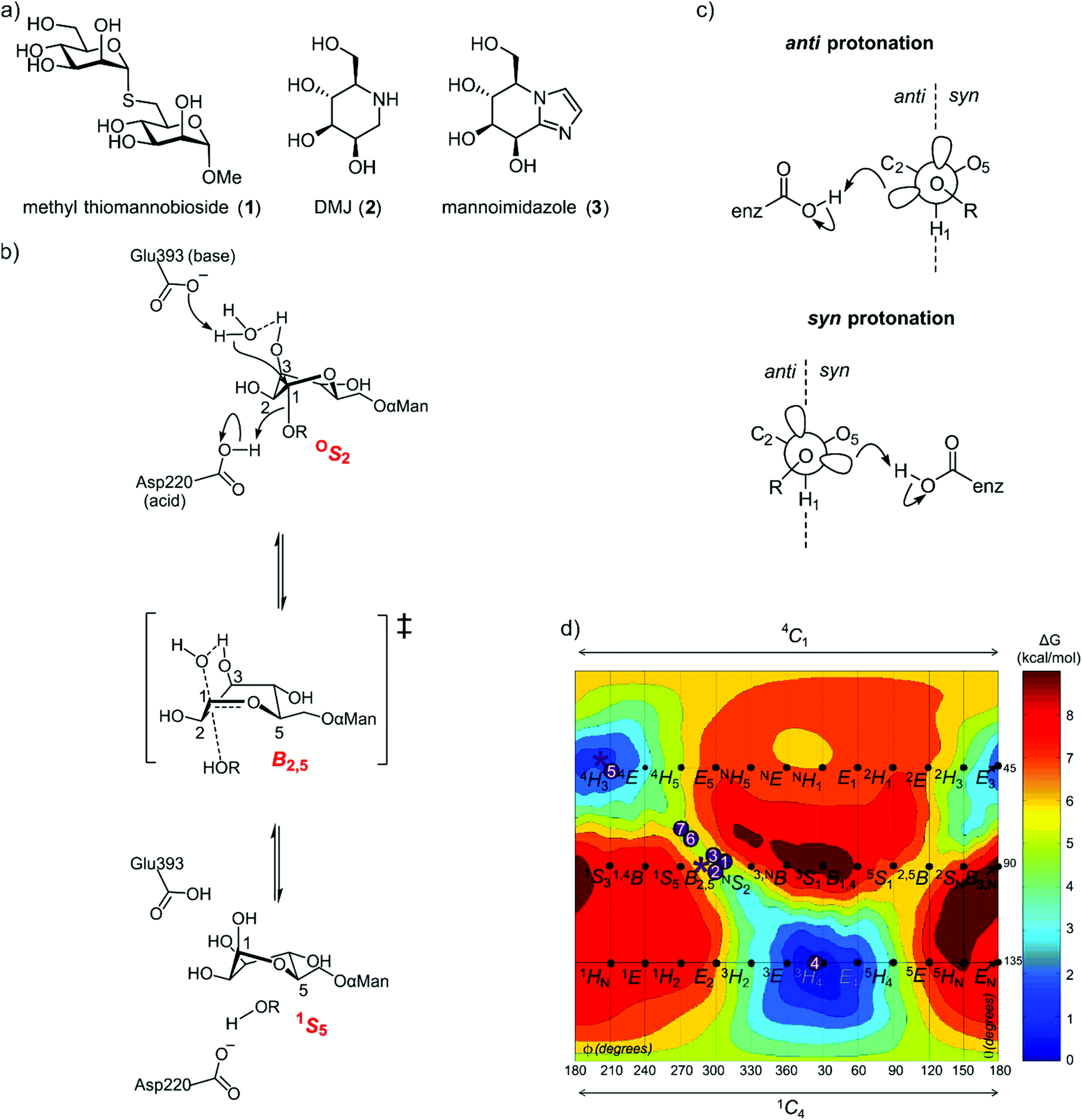 | ||
| Fig. 2 (a) Structures of methyl α-thiomannobioside, deoxymannojirimycin and mannoimidazole. (b) Mechanism showing distortion of mannose from an OS2 conformation to 1S5, via a B2,5 transition state, a credible conformational itinerary of GH125 α-mannosidases. (c) syn and anti trajectory of the protonator/general acid residue in respect to the carbohydrate ligand. (d) Conformational free energy landscape of mannoimidazole in complex with a variety of GH family mannosidases. 1 + 2: β-Man-1,4-MIm bound to GH26 CjMan26C and GH113 AaManA;33: MIm bound to GH2 BtMan2A;144: MIm bound to GH47 CkMan47;105 + 6: MIm bound to GH38 DmGManII;3,207: MIm bound to GH92 BtMan3990;12 * (asterisk) represent MIm bound to CpGH125 (this work). | ||
As the ultimate determinant of the activation barrier for catalysis, the TS is the cardinal feature of the conformational itinerary. Mannoimidazole (3) (MIm) has emerged as a powerful chemical probe for TS conformation of mannosidases owing to its flattened nature, with an sp2-hybridized anomeric centre and energetically feasible access to all of the possible stereoelectronically-compliant conformations expected for an oxocarbenium ion-like transition state (Fig. 2b).3 Indeed, for a single step reaction, determination of the conformation of the transition state allows prediction of the conformational itinerary (from substrate to product via the TS) by invoking the principle of least nuclear motion.21 This principle states that elementary reactions that involve the least change in atomic position and electronic configuration will be favoured. Given the importance of the TS conformation in a reaction coordinate, and the utility of MIm in assigning conformational itineraries, we chose to revisit family GH125 to gather additional evidence in support of the proposed conformational itinerary. Here we report an X-ray structure of mannoimidazole (3) bound to wild-type CpGH125 that provides compelling evidence in support of a ‘latitudinal’ OS2 → B2,5‡ → 1S5 conformational itinerary, complementing previous computational and structural data.
Results and discussion
Glycoimidazoles have been argued to act as glycosidase inhibitors through ‘lateral’ protonation of the imidazole nitrogen lone pair by the enzymatic general acid approaching in the plane of the imidazole ring.22 In fact, this interaction served as the basis for an early classification of glycosidases into syn and anti protonators, based on whether their general acid residues are situated syn or anti to the substrate C1–O5 bond (Fig. 2c).23 However, analysis of published complexes with GH125 enzymes shows that the general acid residue is located below the mean plane of the ring,18,19 suggesting that there are poor prospects for binding of MIm to CpGH125. Yet, there are now several examples of glycoimidazoles binding to glycosidases with good affinity even without achieving a prototypical interaction between the enzymatic general acid and the imidazole nitrogen, such as β-glucosidases of family GH116,24 α-mannanases of family GH9925 and α-mannosidases of family GH4710 (Fig. 2d). The success of these cases inspired us to investigate binding of MIm to CpGH125.We studied the thermodynamics for binding of MIm to CpGH125 using isothermal titration calorimetry (Fig. 3a). Analysis of the calorimogram using a one site binding model allowed prediction of binding at 3.2 sites with an overall KD value of 1.6 mM. Using a sequential binding site model, individual dissociation constants can be calculated at each site yielding values of 40 μM, 105 μM, and 3.1 mM. However, whilst this is indicative of multiple binding sites, it is not clear that the individual KD values are accurately determined by the data. In order to gain evidence that binding by MIm, as shown by ITC, results in enzyme inhibition, we determined its IC50 value. At a concentration of 0.5 mM DNPMan, we determined an IC50 of 1.7 mM (Fig. 3b), consistent with the overall ITC figure of 1.6 mM; this IC50 value cannot be converted to a KI value since the substrate does not give saturation kinetics and so KM cannot be determined.
 | ||
| Fig. 3 (a) Raw thermodynamic data obtained by isothermal titration calorimetry. Top: injection profile with the baseline in red; bottom: titration curve with the fitting curve in blue. (b) IC50 curve of MIm against GH125 using the monosaccharide substrate DNPMan. | ||
Binding stoichiometry of >1 for basic sugar-shaped heterocycles is unusual and suggests that the ligand may not solely be binding at the −1 subsite. Previous work has identified three subsites, −1, +1 and +2, in CpGH125 through a complex with 1,6-α-mannotriose (PDB ID: 5M7Y),19 and studies of a disaccharide substrate that binds in the −1/+1 subsites revealed saturation kinetics, whereas a monosaccharide substrate, 2,4-dinitrophenyl α-mannoside (DNPMan), that bound only in the −1 subsite did not.18,26 Previous poor electron density in the +2 subsite suggests that this may represent the low affinity binding site, a conclusion supported by subsequent X-ray crystallographic studies (vide infra). The two tighter affinities therefore should likely reflect binding at the −1 and +1 subsites; however, we cannot draw conclusions as to which value is assigned to each of these subsites. Nevertheless, the interesting ITC data encouraged efforts to determine the 3D structure of a complex of CpGH125 with mannoimidazole.
Preformed crystals of CpGH125 recombinantly expressed in E. coli were soaked with MIm and the structure of the complex was solved at 1.85 Å resolution (Table 2). The crystal form contains 2 monomers in the asymmetric unit. Density for MIm was observed both in the −1 and +1 subsites, but not in the +2 subsite (Fig. 4a). The MIm molecule in the −1 subsite bound in a B2,5 conformation and the MIm molecule in the +1 subsite bound in a 4H3 conformation. The occupancy of MIm in the −1 subsite was estimated to be 80% and 70% for monomer A and B, respectively; the occupancy of MIm in the +1 subsite was 100% for both monomers. These observations are consistent with tight binding of MIm at the −1/+1 subsites, and weaker binding at the +2 subsite.
 | ||
| Fig. 4 (a) Complex of CpGH125 with mannoimidazole in the −1 and +1 subsites. The residues of the enzyme that form hydrogen bonding interactions and close contacts with mannoimidazole (<3 Å distance) are shown in blue and the nucleophilic water molecule is depicted by a red sphere. The electron density map is a REFMAC maximum-likelihood/σA-weighted 2 Fo − Fc synthesis contoured at 0.75 electrons Å−3 and 1.20 electrons Å−3 clipped to MIm in the −1 and +1 subsite, respectively. (b) Superposition of CpGH125 in complex with MIm (monomer A) and 1,6-α-mannobiose (PDB ID: 5M7I), shown in purple. Hydrogen bonds shown in black between the catalytic residues of CpGH125 and nucleophilic water. | ||
MIm molecules in each site engage with a range of active site residues (Fig. 4a). Key interactions with the −1 subsite MIm include the carbonyl oxygen of Ser219 with O2, Oδ1 of Asn302 with O3, Oδ1 and Oδ2 of Asp65 with O4 and O6, respectively, and NH2 of Arg405 with O6. The general base Glu393 is 2.3 Å away from a water poised for inline attack at the ‘anomeric carbon’ of MIm. Major interactions with MIm in subsite +1 include Oδ1 of Asp195 with O2, Glu129 Oε1 with O3 and Oε2 with O4, and the terminal amine nitrogen, NZ of Lys131 and NH2 of Arg188 with O3. The catalytic amino acid, Asp220, interacts with O6 of MIm in the +1 subsite. Trp116 is stacked parallel to the imidazole of MIm, with CZ2 of Trp119 and N2 of MIm spaced 3.79 Å apart.
Overlay of the complex of MIm with wildtype CpGH125 with that of the D220N mutant of the same enzyme with 1,6-α-mannobiose (PDB ID: 5M7I) is shown in Fig. 4b. This shows that the two MIm molecules bind such that their carbohydrate rings are well-matched with the equivalent mannose rings of 1,6-α-mannobiose. Moreover, the general acid (Asp220; or variant Asn220), and general base (Glu393) residues occupy similar positions in space. This overlay highlights that the interaction of Asp220 with the +1 subsite MIm molecule, in the complex determined here, can be considered equivalent to the interaction of Asn220 with the inter-glycosidic oxygen of 1,6-α-mannobiose seen in the CpGH125-D220N complex. Both complexes include a well-defined water molecule hydrogen-bonded to Glu393 and O3 of the −1 subsite MIm that is poised to undertake a nucleophilic attack at the anomeric centre. The interaction of this water molecule with O3 assists in orientating the water molecule to perform an in-line attack on C1 (Fig. 2b).
Nerinckx generalized the concept of syn/anti protonation to consider divergence from canonical orientation in the mean plane of the ring to include orientations in a syn or anti hemisphere (Fig. 2c).27 According to this extended concept, CpGH125 can be considered a syn protonator and with the present work CpGH125 joins a growing list examples of glycosidases that can bind glycoimidazoles even without achieving lateral protonation. While binding is seen in this and related cases, presumably the potency of binding is less than what might be expected for an ideally located general acid that can achieve such an interaction, an expectation that is borne-out by much tighter binding to canonical anti-protonators such as Bt3130 and Bt3965 from Bacteroides thetaiotaomicron GH92 (Ki = 1.0 and 0.4 μM, respectively) whereas other α-mannosidases from B. thetaiotaomicron have a Ki values ranging from 40–6000 μM.12 Also, imidazole-substituted mannoimidazoles can exhibit even tighter binding, for example, the β-mannosidase BtMan2A (from family GH2) binds phenethylmannoimidazole with Ki = 57 nM compared to binding to mannoimidazole with Ki = 1400 nM.14 In this case since the inhibitor lies across both the −1 and +1 subsite, the number of interactions the inhibitor can make with the protein increases. We highlight one important anomalous case, in which a complex of a disaccharide MIm analogue with BxGH99 1,2-α-mannanase revealed an unusual 2H3/E3 conformation possibly due to steric clashes with an active site residue, Tyr252.25 In this case no KD value could be measured by ITC indicating poor affinity for the enzyme.
Conclusions
The present work builds upon prior computational and structural studies of CpGH125 focussed on the Michaelis complex that provided evidence for an OS2 → B2,5‡ → 1S5 conformational itinerary (Fig. 4). The present TS mimicking complex with MIm in a B2,5 conformation provides compelling evidence for this conformation in the transition state of this enzyme. Since the free energy landscape3 for MIm reveals that the most stable conformations for this inhibitor in the gas phase are the 4H3 and 3H4 conformations, with the B2,5 conformation some 6 kcal mol−1 less stable, the observation of a B2,5 conformation provides persuasive evidence that this conformation is induced upon binding because of the enzyme's strong preference for stabilizing a transition state with this conformation. Combined with the previously published Michaelis complex in an OS2 conformation, we now have crystallographic ‘snapshots’ of states that together define two of the three critical points along the reaction coordinate of family GH125 enzymes.Experimental
Expression, purification and crystallisation of CpGH125WT
Wild-type CpGH125 was expressed and purified under the same conditions as the mutant, CpGH125-D220N.19CpGH125, at 12 mg ml−1, was crystallised under 0.2 M MgCl2, 0.1 M HEPES pH 7.0 and 24% PEG 3350, protein![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :reservoir ratio 0.3:0.5 μL conditions and subsequently soaked with mannoimidazole. Crystals were soaked in 5 mM inhibitor for 2 h at 20 °C and then cryoprotected stepwise in 25% ethylene glycol before snap freezing in liquid nitrogen.
:reservoir ratio 0.3:0.5 μL conditions and subsequently soaked with mannoimidazole. Crystals were soaked in 5 mM inhibitor for 2 h at 20 °C and then cryoprotected stepwise in 25% ethylene glycol before snap freezing in liquid nitrogen.
Data were collected on Diamond MX beamline I24 and processed using xia2 3dii.28 Integrated data were processed using the CCP4 software suite;29 the programs used were aimless,30,31 MOLREP32 with 5M7I as the reference model and cycles of REFMAC533–38 and COOT39 for refinement of the structure. CpGH125 structures figures were produced using CCP4 mg.40 Coordinate and observed structure-factor amplitudes have been deposited on the PDB with accession code 6RQK.
Measurement of dissociation constant (KD) by isothermal titration calorimetry
CpGH125 and MIm were buffer matched by dialysis into 20 mM HEPES pH 7.0 and 200 mM NaCl. MIm was added by syringe to a solution of CpGH125 in the calorimeter cell using a MicroCal ITC200 calorimeter at 25 °C with 20 injections. The concentration of CpGH125 was 200 μM and the ligand concentration was varied between 4 mM and 36 mM to obtain a consistent KD value. The binding affinity was obtained using one site fitting and a sequential binding fit (3 sites) within the Origin7 software. The association constant (KA) was converted to a dissociation constant using the equation KD = 1/KA. An average over 5 experiments was taken to obtain accurate KD values for the three subsites.Measurement of inhibition constant (IC50) of MIm
Enzyme assays were performed in 40 μL quartz cuvettes with path lengths of 10 mm. Change in absorbance values at 405 nm were monitored using a Cary UV-Vis spectrophotometer. Reactions were initiated by addition of CpGH125 at 9.05 μM to a solution of 0.5 mM DNPMan in PBS buffer (pH 7.4) at 37 °C. Rates were measured by observing change in absorption for 3 min. Each reaction was performed in triplicate and averaged, and the data fit to a Hill plot.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by Biotechnology and Biological Sciences Research Council (grant BB/M011151/1) for support AM. SJW is funded by the Australian Research Council (DP180101957, DP160100597). GJD is the Royal Society Ken Murray Research Professor. We thank Diamond Light Source for access to beamline I24 (proposal number mx-9948-71) that contributed to the results presented here and Johan Turkenburg and Sam Hart for coordinating data collection. PDB accession code: 6RQK. We would also like to thank Zach Armstrong for support with the IC50 enzyme assay.Notes and references
- G. J. Davies, A. Planas and C. Rovira, Acc. Chem. Res., 2012, 45, 308–316 CrossRef CAS PubMed.
- G. Speciale, A. J. Thompson, G. J. Davies and S. J. Williams, Curr. Opin. Struct. Biol., 2014, 28, 1–13 CrossRef CAS PubMed.
- R. J. Williams, J. Iglesias-Fernández, J. Stepper, A. Jackson, A. J. Thompson, E. C. Lowe, J. M. White, H. J. Gilbert, C. Rovira, G. J. Davies and S. J. Williams, Angew. Chem., Int. Ed., 2014, 53, 1087–1091 CrossRef CAS PubMed.
- V. Lombard, H. Golaconda Ramulu, E. Drula, P. M. Coutinho and B. Henrissat, Nucleic Acids Res., 2014, 42, D490–D495 CrossRef CAS PubMed.
- B. Henrissat and A. Bairoch, Biochem. J., 1996, 316(Pt 2), 695–696 CrossRef PubMed.
- D. E. Koshland, Biol. Rev., 1953, 28, 416–436 CrossRef CAS.
- M. L. Sinnott, Chem. Rev., 1990, 90, 1171–1202 CrossRef CAS.
- J. D. McCarter and G. Stephen Withers, Curr. Opin. Struct. Biol., 1994, 4, 885–892 CrossRef CAS PubMed.
- R. J. Woods, C. W. Andrews and J. P. Bowen, J. Am. Chem. Soc., 1992, 114, 859–864 CrossRef CAS.
- A. J. Thompson, J. Dabin, J. Iglesias-Fernández, A. Ardèvol, Z. Dinev, S. J. Williams, O. Bande, A. Siriwardena, C. Moreland, T.-C. Hu, D. K. Smith, H. J. Gilbert, C. Rovira and G. J. Davies, Angew. Chem., Int. Ed., 2012, 51, 10997–11001 CrossRef CAS PubMed.
- A. J. Thompson, G. Speciale, J. Iglesias-Fernández, Z. Hakki, T. Belz, A. Cartmell, R. J. Spears, E. Chandler, M. J. Temple, J. Stepper, H. J. Gilbert, C. Rovira, S. J. Williams and G. J. Davies, Angew. Chem., Int. Ed., 2015, 54, 5378–5382 CrossRef CAS PubMed.
- Y. Zhu, M. D. L. Suits, A. J. Thompson, S. Chavan, Z. Dinev, C. Dumon, N. Smith, K. W. Moremen, Y. Xiang, A. Siriwardena, S. J. Williams, H. J. Gilbert and G. J. Davies, Nat. Chem. Biol., 2010, 6, 125–132 CrossRef CAS PubMed.
- Y. Jin, M. Petricevic, A. John, L. Raich, H. Jenkins, L. Portela De Souza, F. Cuskin, H. J. Gilbert, C. Rovira, E. D. Goddard-Borger, S. J. Williams and G. J. Davies, ACS Cent. Sci., 2016, 2, 896–903 CrossRef CAS PubMed.
- L. E. Tailford, W. A. Offen, N. L. Smith, C. Dumon, C. Morland, J. Gratien, M.-P. Heck, R. V. Stick, Y. Blériot, A. Vasella, H. J. Gilbert and G. J. Davies, Nat. Chem. Biol., 2008, 4, 306–312 CrossRef CAS PubMed.
- F. Vincent, T. M. Gloster, J. Macdonald, C. Morland, R. V. Stick, F. M. V. Dias, J. A. M. Prates, C. M. G. A. Fontes, H. J. Gilbert and G. J. Davies, ChemBioChem, 2004, 5, 1596–1599 CrossRef CAS PubMed.
- A. Tankrathok, J. Iglesias-Fernandez, R. J. Williams, S. Pengthaisong, S. Baiya, Z. Hakki, R. C. Robinson, M. Hrmova, C. Rovira, S. J. Williams and J. R. Ketudat Cairns, ACS Catal., 2015, 5, 6041–6051 CrossRef CAS.
- F. Cuskin, A. Baslé, S. Ladevèze, A. M. Day, H. J. Gilbert, G. J. Davies, G. Potocki-Véronèse and E. C. Lowe, J. Biol. Chem., 2015, 290, 25023–25033 CrossRef CAS PubMed.
- K. J. Gregg, W. F. Zandberg, J.-H. Hehemann, G. E. Whitworth, L. Deng, D. J. Vocadlo and A. B. Boraston, J. Biol. Chem., 2011, 286, 15586–15596 CrossRef CAS PubMed.
- S. Alonso-Gil, A. Males, P. Z. Fernandes, S. J. Williams, G. J. Davies and C. Rovira, J. Am. Chem. Soc., 2017, 139, 1085–1088 CrossRef CAS PubMed.
- D. A. Kuntz, C. A. Tarling, S. G. Withers and D. R. Rose, Biochemistry, 2008, 47, 10058–10068 CrossRef CAS PubMed.
- M. L. Sinnott, Adv. Phys. Org. Chem., 1988, 24, 113–204 CrossRef CAS.
- A. Varrot, M. Schülein, M. Pipelier, A. Vasella and G. J. Davies, J. Am. Chem. Soc., 1999, 121, 2621–2622 CrossRef CAS.
- T. D. Heightman and A. T. Vasella, Angew. Chem., Int. Ed., 1999, 38, 750–770 CrossRef CAS PubMed.
- R. Charoenwattanasatien, S. Pengthaisong, I. Breen, R. Mutoh, S. Sansenya, Y. Hua, A. Tankrathok, L. Wu, C. Songsiriritthigul, H. Tanaka, S. J. Williams, G. J. Davies, G. Kurisu and J. R. K. Cairns, ACS Chem. Biol., 2016, 11, 1891–1900 CrossRef CAS PubMed.
- P. Z. Fernandes, M. Petricevic, L. Sobala, G. J. Davies and S. J. Williams, Chem. – Eur. J., 2018, 24, 7464–7473 CrossRef CAS PubMed.
- L. Deng, P. Tsybina, K. J. Gregg, R. Mosi, W. F. Zandberg, A. B. Boraston and D. J. Vocadlo, Bioorg. Med. Chem., 2013, 21, 4839–4845 CrossRef CAS PubMed.
- W. Nerinckx, T. Desmet, K. Piens and M. Claeyssens, FEBS Lett., 2005, 579, 302–312 CrossRef CAS PubMed.
- G. Winter, J. Appl. Crystallogr., 2010, 43, 186–190 CrossRef CAS.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- P. Evans, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, 62, 72–82 CrossRef PubMed.
- P. R. Evans and G. N. Murshudov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1204–1214 CrossRef CAS PubMed.
- A. Vagin and A. Teplyakov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 22–25 CrossRef CAS PubMed.
- G. N. Murshudov, P. Skubák, A. A. Lebedev, N. S. Pannu, R. A. Steiner, R. A. Nicholls, M. D. Winn, F. Long and A. A. Vagin, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 355–367 CrossRef CAS PubMed.
- G. N. Murshudov, A. A. Vagin and E. J. Dodson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53, 240–255 CrossRef CAS PubMed.
- N. S. Pannu, G. N. Murshudov, E. J. Dodson and R. J. Read, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1998, 54, 1285–1294 CrossRef CAS PubMed.
- M. D. Winn, G. N. Murshudov and M. Z. Papiz, Methods Enzymol., 2003, 374, 300–321 CAS.
- A. A. Vagin, R. A. Steiner, A. A. Lebedev, L. Potterton, S. McNicholas, F. Long and G. N. Murshudov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2184–2195 CrossRef PubMed.
- R. A. Nicholls, F. Long and G. N. Murshudov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 404–417 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- S. McNicholas, E. Potterton, K. S. Wilson and M. E. M. Noble, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 386–394 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |