
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible light-mediated intermolecular [2 + 2] photocycloaddition of 1-aryl-2-nitroethenes and olefins†
Lisa-Marie
Mohr
 ,
Andreas
Bauer
,
Christian
Jandl
and
Thorsten
Bach
*
,
Andreas
Bauer
,
Christian
Jandl
and
Thorsten
Bach
*
Department Chemie and Catalysis Research Center (CRC), Technische Universität München, 85747 Garching, Germany. E-mail: thorsten.bach@ch.tum.de; Fax: +49 89 28913315; Tel: +49 89 28913330
First published on 23rd July 2019
Abstract
Despite the importance of cyclobutanes there are not many direct [2 + 2] photocycloaddition reactions which can be performed with visible light in the absence of a catalyst. A notable exception is the reaction of 1-aryl-2-nitroethenes and olefins which can be performed at a wavelength of λ = 419 nm or λ = 424 nm in CH2Cl2 as the solvent. In the present study, a total of 15 1-aryl-2-nitroethenes were found to undergo a [2 + 2] photocycloaddition with 2,3-dimethyl-2-butene (28–86% yield) and a set of 12 olefins was studied in their photocycloaddition to 1-phenyl-2-nitroethene (37–88% yield). All mechanistic results are in agreement with a triplet reaction pathway and with the intermediacy of a 1,4-diradical.
Introduction
In 1887, when studying the properties of phenylnitroethylene (β-nitrostyrene, 1-phenyl-2-nitroethene), Priebs observed that the yellow coloured substrate was converted upon exposure to light to a colourless product which he suspected to be a polymer.1 Meisenheimer and Heim reinvestigated the reaction in 1907 proposing a dimeric product in analogy to the photodimer of cinnamic acid.2 They were not able to prove this hypothesis, however, and it took another 65 years until the cyclobutane structure of the photodimer was established by Shechter and co-workers.3 The [2 + 2] photodimerization was performed by exposure of solid trans-β-nitrostyrene to sunlight and the conversion was ca. 70% after 4–6 weeks of irradiation. The relative configuration of the two diastereomeric products was later established by Desiraju and Pedireddi in an X-ray crystallographic study.4Despite the fact that this precedence suggested that β-nitrostyrene can be involved in a [2 + 2] photocycloaddition reaction when exposed to visible light, the very few attempts to obtain [2 + 2] photocycloaddition products of β-nitrostyrene were performed with mercury lamps as UV irradiation sources. An initial report by Chapman and co-workers5 referred to work performed in the context of a Ph.D. thesis6 but did not provide any experimental details. In 1980, the Sakurai group described the [2 + 2] photocycloaddition of β-nitrostyrene with indene (Scheme 1) which was performed with a high-pressure mercury lamp in a pyrex vessel.7 Further photocycloaddition studies of 1-aryl-2-nitroethenes were reported by Ramkumar and Sankararaman (Michael type addition of silyl enol ethers to β-nitrostyrene),8 by Chapman and co-workers ([2 + 2] photocycloaddition of β-nitrostyrene and 2,3-dimethylbutadiene),9 and most recently by Ferreira and co-workers (Cr-catalysed [4 + 2] cycloaddition of trans-β-nitro-para-methoxystyrene and 1,3-dienes).10
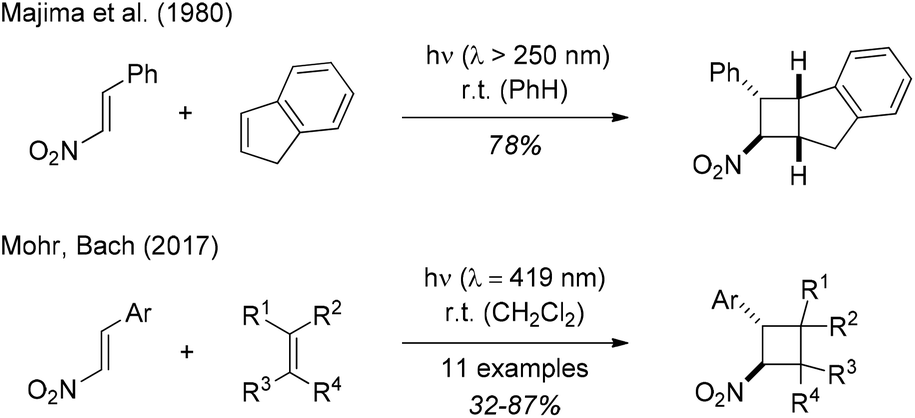 | ||
| Scheme 1 Previous studies on the title reaction. Visible light-mediated reactions were performed with fluorescent lamps (emission maximum: λ = 419 nm). | ||
We became interested in the intermolecular [2 + 2] photocycloaddition11 of 1-aryl-2-nitroethenes in the context of our work on visible light-mediated reactions.12 In preliminary studies (Scheme 1),13 we found that a smooth reaction occurred when the title compounds (c = 20 mM) were irradiated in a solution of the olefin (10 equiv.) in dichloromethane at λ = 419 nm. The reaction scope was limited, however, and the irradiation conditions were not fully optimized. We have now performed a more comprehensive array of experiments with a total number of 15 different 1-aryl-2-nitroethenes and with an additional set of 12 olefins. Moreover, further mechanistic studies were performed to shed light on the course of the [2 + 2] photocycloaddition. In this context, an unprecedented ring opening reaction of 1,1-dicyclopropylethylene was observed. Full details of our experimental work are presented in this account.
Reaction scope
The 1-aryl-2-nitroethenes 1 employed in our study (Fig. 1) were prepared from nitromethane and the respective aromatic aldehydes in a Henry reaction.14 The condensation was performed with ammonium acetate in nitromethane or in a mixture of nitromethane and acetic acid. A colour change of the refluxing solution indicated a successful elimination of the intermediate alcohol to the nitroethene which was isolated exclusively as the trans-isomer. Only the 2-thiophenyl product 1e was not accessible by this method and required the use of a stronger base (NaOH in MeOH)15 to induce the Henry reaction.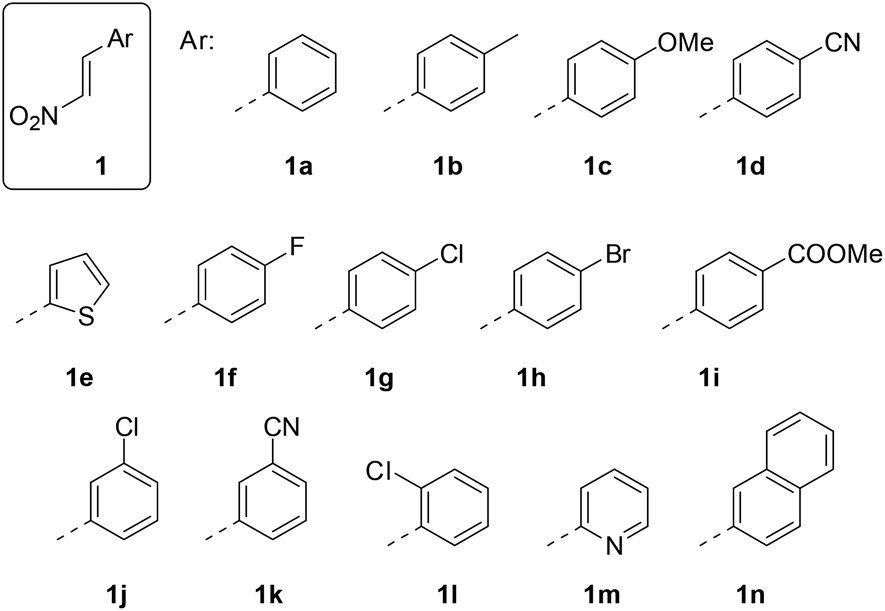 | ||
| Fig. 1 Structures of the 1-aryl-2-nitroethenes 1 employed in this study. | ||
UV/Vis-spectra16 of all 1-aryl-2-nitroethenes were recorded in dichloromethane solution and selected spectra are depicted in Fig. 2 (see the ESI† for all spectra). An electron withdrawing group at the phenyl group led to a hypsochromic shift relative to the parent compound 1a as shown for the 4-cyanophenyl derivative 1d. An electron donating group showed the opposite effect and the 4-methylphenyl (1b) and the 4-methoxyphenyl (1c) derivatives absorb at longer wavelength relative to 1a. The absorption coefficient is typically in the range of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–20000 M−1 cm−1 indicating that the absorption is due to an allowed transition (vide infra). All compounds are coloured which is in line with an – at least minimal – absorption in the visible range (λ > 380 nm).
000–20000 M−1 cm−1 indicating that the absorption is due to an allowed transition (vide infra). All compounds are coloured which is in line with an – at least minimal – absorption in the visible range (λ > 380 nm).
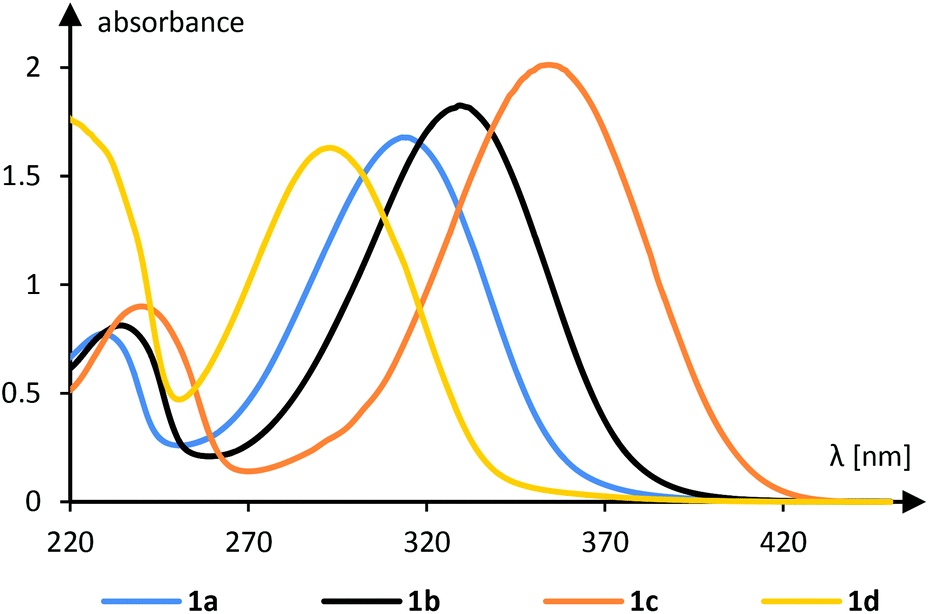 | ||
| Fig. 2 UV/Vis spectra of selected 1-aryl-2-nitroethenes (c = 1 mM, CH2Cl2). | ||
The previous preliminary irradiation experiments13 were conducted exclusively at room temperature with fluorescent lamps which display a relatively broad emission spectrum and an emission maximum at λ = 419 nm (Table 1, conditions A). In search for optimal conditions, we also performed the reaction with a light-emitting diode (LED) at λ = 424 nm at ambient temperature (conditions B) and at −78 °C (conditions C). Every substrate was tested under all conditions in its reaction with 2,3-dimethyl-2-butene and the best conditions for the individual substrate are recorded in Table 1 (for the complete set of results see the ESI†).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Cond.a | t [h] | Conv.b [%] | Product | Yieldc [%] |
| a The reactions were performed under conditions A, B, and C (see ESI† for further details). For each reaction the best conditions are listed in the table. Irradiation was discontinued after the indicated time period t. b The conversion is based on the amount of re-isolated starting material. c Yield of isolated product. d Olefinic by-product (11%), see narrative. | ||||||
| 1 | 1a | A | 12 | 100 | 2a | 59 |
| 2 | 1b | C | 3 | 100 | 2b | 67 |
| 3 | 1c | C | 4 | 100 | 2c | 77 |
| 4 | 1d | C | 6 | 100 | 2d | 44 |
| 5 | 1e | C | 2 | 100 | 2e | 86 |
| 6 | 1f | C | 4 | 100 | 2f | 50 |
| 7 | 1g | C | 4 | 100 | 2g | 54 |
| 8 | 1h | B | 6 | 100 | 2h | 58 |
| 9 | 1i | C | 5 | 100 | 2i | 33 |
| 10 | 1j | B | 7 | 73 | 2j | 38 |
| 11 | 1k | A | 7 | 100 | 2k | 35d |
| 12 | 1l | C | 7 | 57 | 2l | 28 |
| 13 | 1m | C | 24 | 100 | 2m | 66 |
| 14 | 1n | C | 3 | 100 | 2n | 79 |
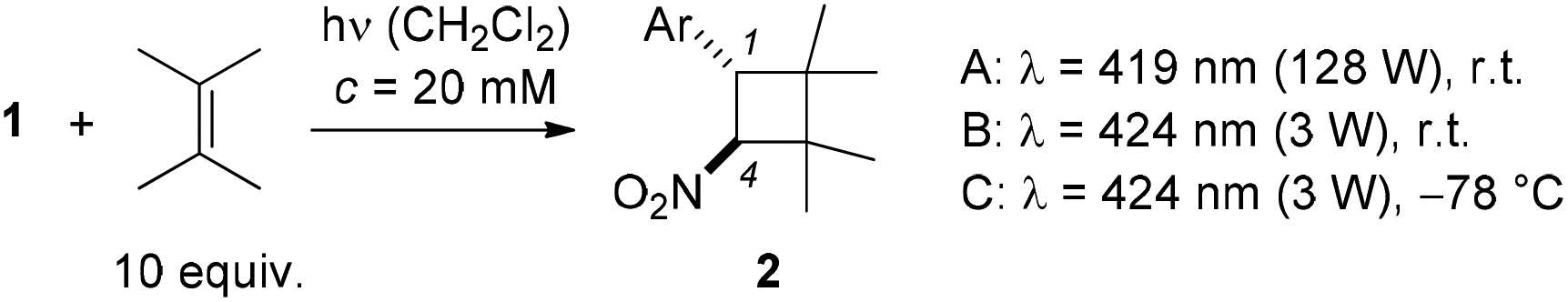
Unfunctionalized and heteroaromatic substrates (entries 1, 2, 5, 13 and 14) reacted consistently well and in good yields (59–86%). Methoxy and halogen substitution in para-position of the 1-phenyl-2-nitroethenes was compatible with the reaction (entries 3 and 6–8) and the respective products 2c, 2f–2h were obtained in moderate to good yields (50–77%). An electron withdrawing group (entries 4, 9 and 11) retarded the reaction slightly which reflects a smaller absorption cross section of the substrates in the visible range (cf. compound 1d in Fig. 2). In addition, side reactions were observed which were particularly significant for compound 1k (entry 11) and which will be discussed in the mechanistic section. The reactions of the meta- and ortho-chloro substituted 1-phenyl-2-nitroethenes (entries 10 and 12) proceeded sluggishly and were stopped after seven hours. Starting material was recovered as a mixture of the respective cis- and trans-compound. Likewise, whenever a reaction was stopped before completion, the recovered 1-aryl-2-nitroethenes were isolated as cis-/trans-mixtures. The composition in the photostationary state reflects the different absorption properties of the individual geometric isomers at the chosen irradiation wavelength.13,17 The only substrate which did not show any [2 + 2] photocycloaddition reaction was 1-(4′-N,N-dimethylamino)phenyl-2-nitroethene despite the fact that it displays a particularly extensive absorption in the visible region. There was no decomposition of starting material and it is likely that intramolecular relaxation pathways18 occur more rapidly than the intermolecular addition to the olefin. Products 2 were isolated as single diastereoisomers with the aryl group (Ar) and the nitro group in trans-position at the cyclobutane ring. This assignment was corroborated by NOE experiments which revealed a contact between the ortho protons at the C1 phenyl group and the proton at C4. It is also in line with the relative configuration found in previously reported [2 + 2] photocycloaddition products of trans-β-nitrostyrene (1a).7,9
In our preliminary communication, the reaction of trans-β-nitrostyrene (1a) with indene, vinyl ethyl ether, 2,3-dimethylbutadiene, and cyclopentene under visible light irradiation (conditions A) was reported.13Scheme 2 displays reactions of substrate 1a with olefins that had not been studied in previous work or that gave better yields under conditions B and C. Products 3a–3d were obtained as single isomers while cyclobutanes 3e–3h were formed as diastereomeric mixtures. It was possible in all cases to isolate the major isomer and to assign its relative configuration (see ESI† for further details). The given yield refers to the total yield of all diastereoisomers (dr = diastereomeric ratio).
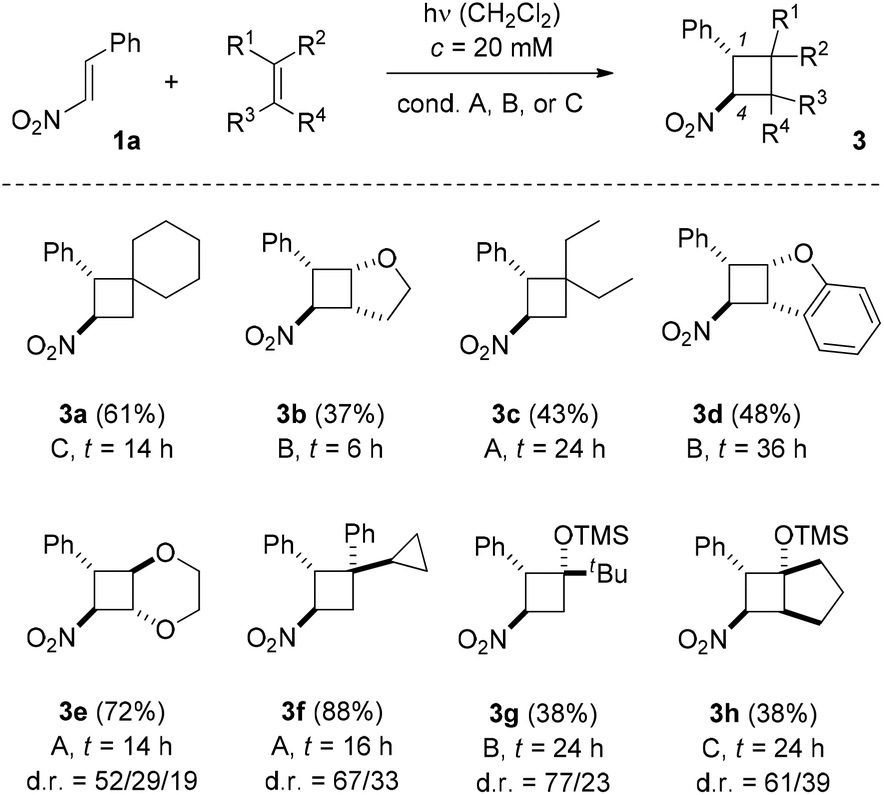 | ||
| Scheme 2 Intermolecular [2 + 2] photocycloaddition of 1-phenyl-2-nitroethene (1a) and various olefins: optimal reaction conditions for the individual olefins (t = reaction time, dr = diastereomeric ratio). | ||
Electron deficient olefins (e.g. 1,1-dichloroethene, methyl acrylate, allylic alcohol) showed no reaction in attempted intermolecular [2 + 2] photocycloaddition reactions with trans-β-nitrostyrene (1a). In the reaction to product 3f there was no indication for a ring opening of the cyclopropyl ring and seven-membered carbocyclic by-products were not detected. The fact that silyl enol ethers gave cyclobutanes 3g and 3h as the only isolable products was surprising. In previous photochemical studies,8 Michael addition products were observed suggesting an addition reaction of the silyl enol ether with opposite regioselectivity. For comparison, we prepared the Michael addition product of 1-(trimethylsilyloxy)cyclopentene and trans-β-nitrostyrene by a thermal reaction.19 However, this very same product was not detectable in the crude product mixture of the [2 + 2] photocycloaddition reaction neither by TLC nor by GLC analysis. It should be noted that different irradiation conditions (λ > 250 nm) and a different substrate stoichiometry (1a:silyl enol ether = 1:1) were used by Ramkumar and Sankararaman in their experiments.8 Still, it remains open why the regioselectivity should be completely reverted (vide infra). In all [2 + 2] photocycloaddition products 3 the better donor substituent of the former olefin is positioned at carbon atom C2 relative to carbon atom C4 which carries the nitro group.
Intramolecular [2 + 2] photocycloaddition reactions were attempted with 1-phenyl-2-nitroethenes that had an alkenyl group linked to the ortho position of the phenyl ring, such as substrate 1o20 (Scheme 3). Irrespective of the length of the tether there was no indication for an intramolecular reaction which could be due to the intrinsically low reactivity of a terminal olefin. An alternative explanation would be that initial C–C bond formation has to occur in the intramolecular reaction at the β-position of the nitrostyrene which might be electronically disfavored (vide infra). The chromophore of 1o is still reactive upon excitation as demonstrated by the intermolecular [2 + 2] photocycloaddition of 2,3-dimethyl-2-butene to product 2o.
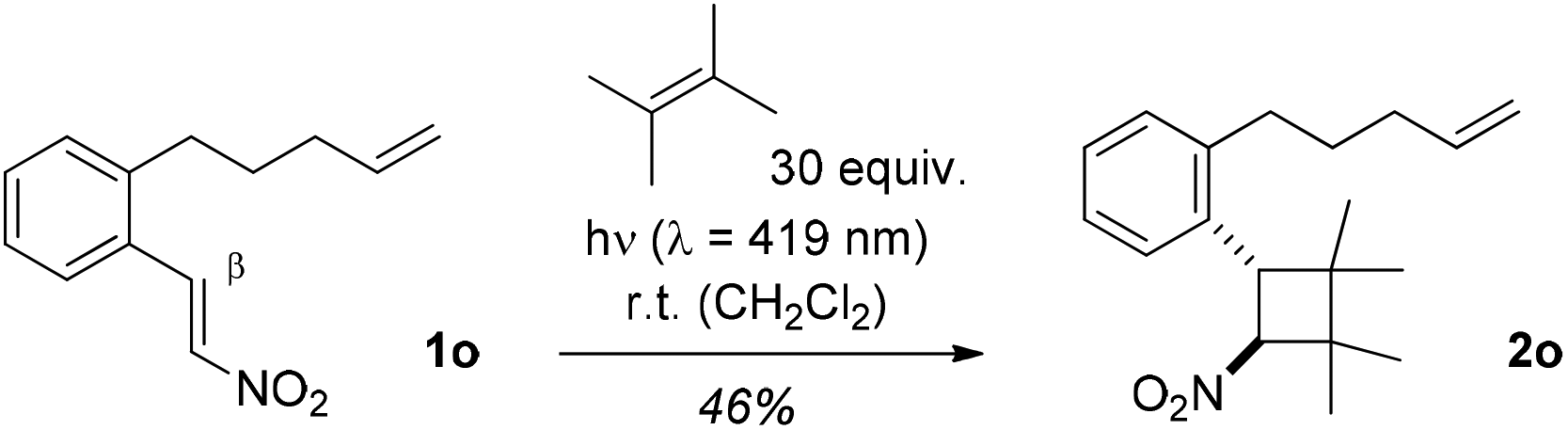 | ||
| Scheme 3 Inter- vs. intramolecular [2 + 2] photocycloaddition of 1-aryl-2-nitroethene 1o: exclusive formation of product 2o. | ||
Although synthetic applications of the nitrocyclobutanes were not in the focus of our current study, it was probed whether aminocyclobutanes would be accessible by a straightforward reduction.21 Gratifyingly, the reduction of nitrocyclobutane 1a, as a representative example, with zinc22 proceeded smoothly and without any loss of the stereochemical information. Product 4 was obtained in 77% yield (Scheme 4).
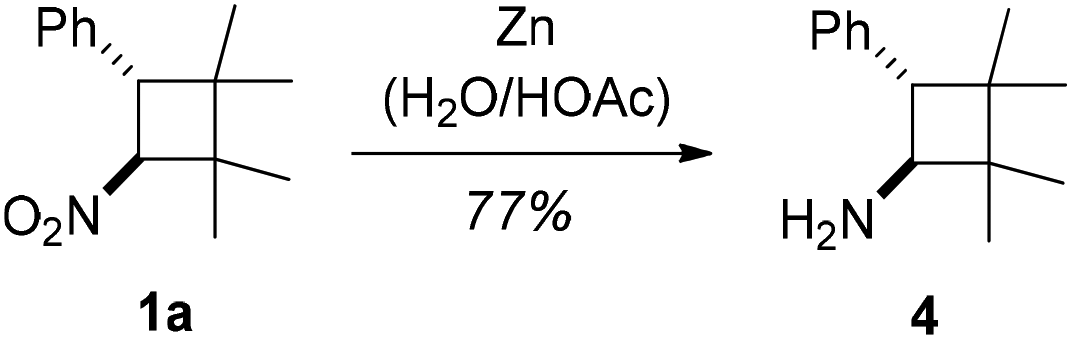 | ||
| Scheme 4 Reduction of nitrocyclobutane 1a to aminocyclobutane 4. | ||
Mechanistic studies
There is consensus in the literature that the longest wavelength absorption of 1-aryl-2-nitroethenes correlates to a ππ* transition into the respective first excited singlet state (S1).16,18 For trans-β-nitrostyrene (1a), calculations have been performed that allow to visualize the electron density in the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO).23 There is no detectable fluorescence for typical 1-aryl-2-nitroethenes if the energy of S1 is above 2.95 eV (285 kJ mol−1).16b Among other arguments, the fact, that fluorescence is not even observed in the solid state, suggests a rapid intersystem crossing (ISC) from the singlet to the triplet hypersurface leading to a population of the lowest lying triplet state (T1). Only 1-phenyl-2-nitroethenes with a strong electron donating group (e.g. NMe2) exhibit fluorescence with a fluorescence quantum yield of ca. 0.1 in benzene.18 The photophysical behaviour of 1-(4′-N,N-dimethylamino)phenyl-2-nitroethene has been studied by transient absorption spectroscopy. The ISC is extremely rapid [τ(S1) ≅ 6 ps] in a non-polar solvent (cyclohexane) and remains fast [τ(S1) ≅ 70 ps] in a solvent of moderate polarity.18The triplet energies of compounds 1a, 1c, and 1g have been determined from their phosphorescence emission at 77 K in an EtOH matrix to be E(T1) = 228 kJ mol−1, 226 kJ mol−1, and 219 kJ mol−1, respectively.16b We recorded the phosphorescence spectrum of compound 1a in an EtOH matrix at 77 K and obtained a value of E(T1) = 229 kJ mol−1 (see ESI†). The nature of the triplet state for compounds 1 has not been extensively explored. Cowley assigned to it an nπ* character which would be in accord with the high ISC rate and with the absence of fluorescence from S1.16b
Our mechanistic suggestion (Scheme 5) for the reaction course involves the nπ* triplet state 1 (T1) as the key intermediate which is accessed from 1 (S1) by ISC. Electron loss at the oxygen n orbitals and population of the π* orbital with an electron leads to electron deficiency at the α-carbon atom (photochemical Umpolung) which is the preferred position of olefin attack to generate triplet diradical 3D. ISC and subsequent ring closure lead to cyclobutane products but side reactions are possible from 3D prior or after ISC.
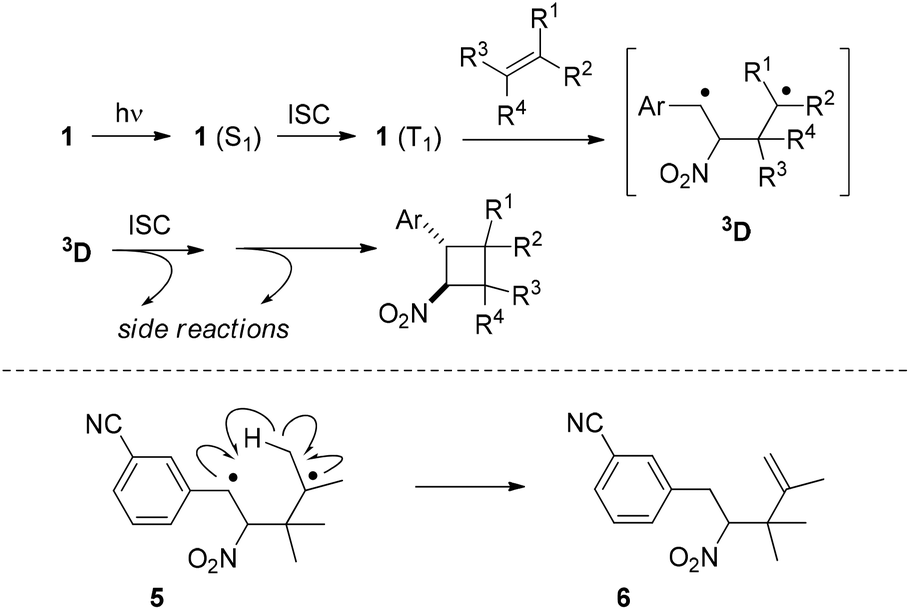 | ||
| Scheme 5 Suggested reaction course of the [2 + 2] photocycloaddition between 1-aryl-2-nitroethenes and olefins via triplet 1,4-diradical 3D (top); formation of by-product 6 (Table 1, entry 11) via 1,4-diradical 5 (bottom). | ||
Any detectable side reactions which occur from 3D support a pathway on the triplet hypersurface. As in our preliminary studies, there were again products of a photo-ene reaction24 isolated as by-products. In the present study, the reaction of substrate 1k (Table 1, entry 11) turned out to be particularly prone to undergo the reaction that likely involves an intramolecular hydrogen abstraction in 1,4-diradical 5 thus generating product 6.
Another way to substantiate the existence of a 1,4-diradical 3D is based on the ring opening of a cyclopropyl-substituted alkyl radical.25 In the reaction to product 3f, there was no indication for such a process, but when employing 1,1-dicyclopropylethylene26 as substrate a new product was isolated apart from the regular [2 + 2] photocycloaddition product 3i. Proof for its tricyclic structure 7 rests – apart from extensive NMR analysis – on the isolation of the related product 8 from the reaction between 1-(4′-methoxycarbonyl)phenyl-2-nitroethene (1i) and 1,1-dicyclopropylethylene (Scheme 6).
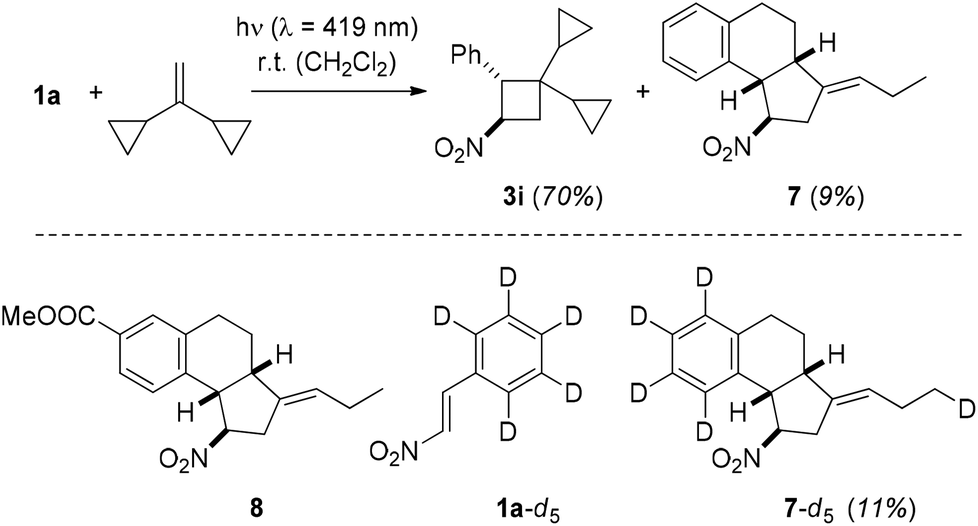 | ||
| Scheme 6 Intermolecular [2 + 2] photocycloaddition of 1-phenyl-2-nitroethene (1a) and 1,1-dicyclopropylethylene to product 3i and by-product 7 (top); by-products 8 obtained from 1-(4′-methoxycarbonyl)phenyl-2-nitroethene and 7-d5 obtained from deuterated substrate 1a-d5 (bottom). | ||
X-Ray crystallographic analysis of product 8 (Fig. 3) revealed the fact that both cyclopropyl rings had opened in the reaction sequence and that the exocyclic double bond was formally (E)-configured. In order to explore the fate of the hydrogen atom in the ortho position of the phenyl ring, which is involved in a C–C bond formation step, we submitted aldehyde 1a-d5 to the [2 + 2] photocycloaddition with 1,1-dicyclopropylethylene (Scheme 6). Product 7-d5 was isolated as by-product (11%) together with the major product 3i-d5 (61%). The deuterium atom was found at the terminal carbon atom of the ethyl group which is attached to the exocyclic (E)-double bond.
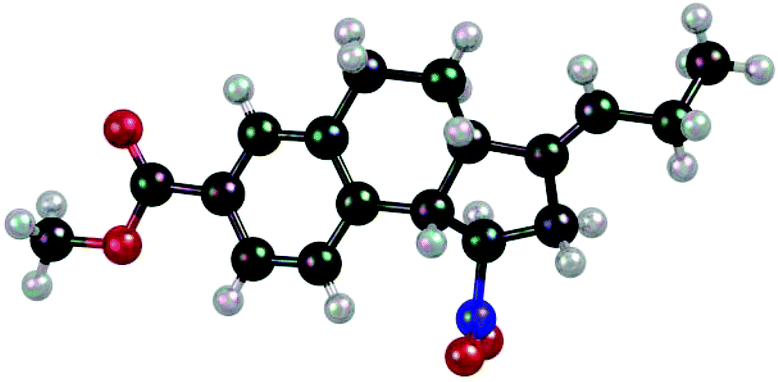 | ||
| Fig. 3 Structure of by-product 8 as obtained by single-crystal X-ray diffraction. | ||
Invoking a 1,4-diradical 9 for the reaction of 1a and 1,1-dicyclopropylethylene the formation of 7 can be tentatively explained by the reaction cascade depicted in Scheme 7. Ring opening of the cyclopropane leads to 1,7-diradical 10 which seems unsuited for seven-membered ring formation. Instead, the radical in α-position to the phenyl group attacks the double bond to produce 1,4-diradical 11 which opens to 1,7-diradical 12. The proximity of the primary radical center to the phenyl ring in this intermediate may initiate a stereoselective C–C bond formation with the former ortho hydrogen atom now being perfectly exposed for an intramolecular hydrogen abstraction. Indeed, molecular models suggest that this process is feasible in intermediate 13 leading to product 7.
 | ||
| Scheme 7 Suggested formation of by-product 7 from 1,4-diradical intermediate 9. | ||
The efficiency of the intermolecular [2 + 2] photocycloaddition is limited not only by the low absorption coefficient of the 1-aryl-2-nitroethenes but also by their rapid cis/trans isomerisation in the excited state.18 The quantum yield for the reaction 1a → 2a at λ = 382 nm was determined as 0.04 (see ESI† for further details). Due to the rapid isomerisation it is experimentally difficult to determine whether both isomers are involved in the [2 + 2] photocycloaddition but it is likely. Regarding the olefin, the stereochemical integrity during the [2 + 2] photocycloaddition is high. Reactions performed with either trans- or cis-β-methyl styrene (14, Scheme 8) delivered the cyclobutanes 3j and 3j′. The recovered olefin was still diastereomerically pure in either case which indicates that the olefin is not photochemically excited under the irradiation conditions.
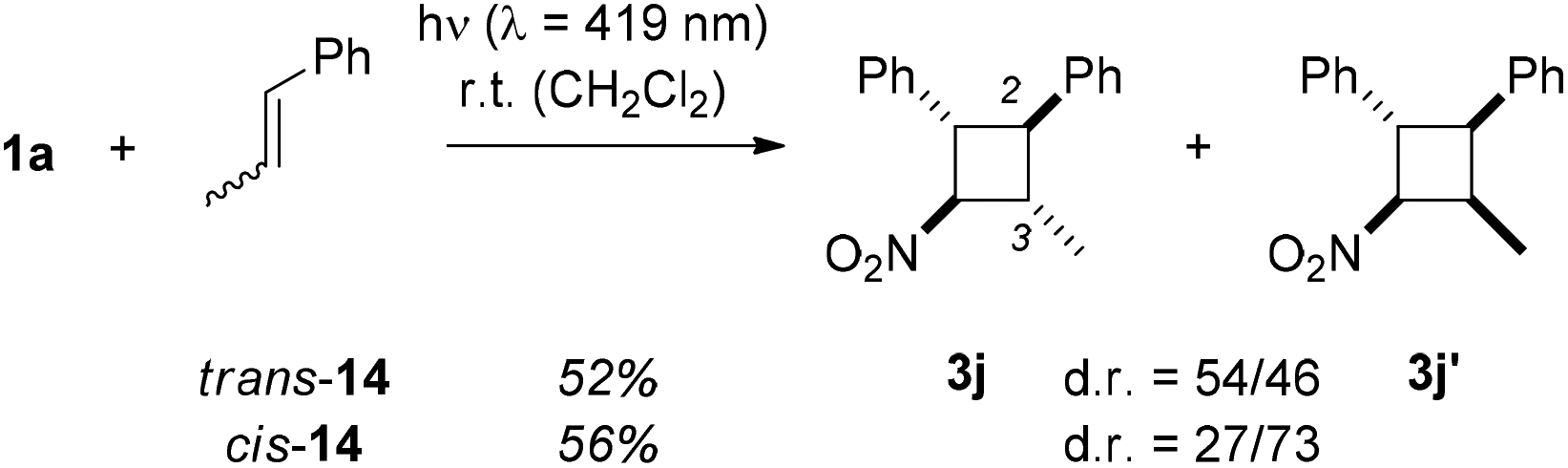 | ||
| Scheme 8 Intermolecular [2 + 2] photocycloaddition of 1-phenyl-2-nitroethene (1a) and β-methylstyrene (14): non-stereospecific reaction course, but incomplete stereoconvergence. | ||
The photocycloaddition of olefins 14 was not stereospecific regarding the olefinic double bond. Starting from trans-14 significant amounts of product 3j′ were obtained with the two substituents at C2 and C3 in the cis-position. Vice versa, cis-β-methyl styrene (cis-14) gave also notable quantities of the 2,3-trans-product 3j. In the absence of any detectable cis/trans isomerisation of β-methyl styrene during the reaction the non-stereospecifity is further evidence for the intermediacy of a triplet 1,4-diradical 3D in which rotation around single bonds in possible.27
A final comment is warranted on a possible involvement of single electron transfer (SET) processes. The redox potential of trans-β-nitrostyrene (1a) in its triplet state can be estimated by its triplet state energy E(T1) = 229 kJ mol−1 and by its ground state redox potential.28 Based on the known redox potential E1/2(1a/1a˙−) = −0.44 V (ref. 29) a calculated value E1/2(1a*/1a˙−) in the order of +1.90 V is obtained for the triplet state. Thermodynamically, the oxidation of several electron rich olefins with Eox(olefin˙+/olefin) < +1.90 V would thus seem feasible, e.g. of 2,3-dimethyl-2-butene (Eox = +1.50 V),30 2,3-dihydrofuran (Eox = +1.40 V),31 and 1-tert-butyl-1-(trimethylsilyloxy)ethene (Eox = +1.34 V).32 However, several other reactive olefins, e.g. methylenecyclohexane (Eox = +2.62 V),33 exhibit a redox potential far too high for an electron transfer to be possible. In addition, SET reactions34 are typically performed in polar solvents to assist charge separation which is more difficult in a nonpolar solvent. The fact that the observed photocycloaddition works also in benzene and that it is accelerated by a triplet sensitizer13 makes the involvement of SET processes unlikely. Further circumstantial evidence is based on the absence of by-products which would be expected in the reaction of dienes ([4 + 2] cycloaddition) and of 2,3-dimethyl-2-butene. The side products mentioned earlier (e.g. product 6, Scheme 5) should exhibit a different regioselectivity of addition35 had they been formed in an SET process.
Conclusions
To summarize, we have established a straightforward access to various 1-aryl-4-nitrocyclobutanes by a visible light-mediated intermolecular [2 + 2] photocycloaddition. The substituents are trans-positioned within the cyclobutane but it is known that the relative configuration can be inverted to cis by a deprotonation/protonation sequence.3 In addition, the nitro group can be interconverted to an amine if desired as has been shown in the present study by reduction to product 4. Accordingly, the method makes also trans- and cis-1-aryl-4-aminocyclobutanes accessible if desired.Although the nitro chromophore bears electronically some analogy to a carbonyl group, the photochemical behaviour of 1-phenyl-2-nitroethene is different from cinnamic aldehyde. While the latter compound does not form cyclobutanes upon direct irradiation36 the former compound and its analogues are suitable substrates for [2 + 2] photocycloaddition reactions, as shown in this study. For 1-aryl-2-nitroethenes, an intrinsic feature of their excited state seems to be the propensity to react only with electron rich olefins.
Experimental
General information
All preparations and manipulations of air and moisture sensitive compounds were carried out in flame dried glassware under an argon atmosphere using standard Schlenk techniques. Dry solvents were either obtained water and oxygen free by a Braun MB SPS purification system or from commercial sources (see ESI†). Irradiation experiments were either performed in a Rayonet RPR-100 photochemical reactor (Southern New England Ultra Violet Company, Branford, CT, USA) at λmax = 419 nm (16 lamps, cool white, 8 W, Osram)37 or with a high power light emitting diode (LED) at λmax = 424 nm (Roithner Lasertechnik, 350 mA, UF ∼ 3.4 V). Flash column chromatography was performed with silica 60 (Merck, 230–400 mesh) as the stationary phase. Infrared (IR) spectra were recorded on a PerkinElmer Frontier IR FTR spectrometer by ATR technique. Nuclear magnetic resonance (NMR) spectra were recorded at room temperature either on a Bruker AVHD 300, AVHD 400, AVHD 500 or an AV 500 cryo. 1H NMR spectra were referenced to the residual proton signal of the respective solvent. 13C NMR spectra were referenced to the 13C-D multiplet of the solvent employed. Assignment and multiplicity of the 13C NMR signals were determined by two-dimensional NMR experiments (COSY, HSQC, HMBC). The relative configuration of diastereoisomers was corroborated by NOESY. Melting points were determined using a Kofler melting point apparatus (“Thermopan”, Reichert), with a range quoted to the nearest whole number. Mass spectrometry (MS) was performed on a GC-coupled Agilent system (EI, 70 eV). High resolution mass spectrometry (HRMS) was performed on a Thermo Scientific LTQ FT Ultra (ESI) or a Thermo Scientific DFS HRMS spectrometer (EI). UV/Vis spectra were measured on a PerkinElmer Lambda 35 UV/Vis spectrometer.Experimental Procedures
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (cm−1) = 3079, 2967, 2871, 1538, 1510, 1460, 1371, 1226, 1151, 1135, 844, 761; 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.09–7.00 (m, 4H, Har), 4.85 (d, 3J = 10.1 Hz, 1H, H-4′), 3.92 (d, 3J = 10.1 Hz, 1H, H-1′), 1.24 (s, 3H, CH3-2′), 1.16 (s, 3H, CH3-3′), 1.14 (s, 3H, CH3-2′), 0.70 (s, 3H, CH3-3′); 13C NMR (101 MHz, CDCl3): δ (ppm) = 162.1 (d, 1JCF = 254.4 Hz, C-1), 132.2 (s, C-4), 128.5 (d, 3JCF = 7.9 Hz, C-3, C-5), 115.6 (d, 2JCF = 21.4 Hz, C-2, C–6), 85.2 (d, C-4′), 48.9 (d, C-1′), 45.0 (s, C-3′), 39.3 (s, C-2′), 24.2 [q, (C-3′)CH3], 22.8 [q, (C-2′)CH3], 21.4 [q, (C-3′)CH3], 19.4 [q, (C-2′)CH3]; MS (EI): m/z (%) = 205 (45) [M − NO2]+, 163 (100) [M − NO2 − C3H6]+, 106 (54) [C7H6F]+; HRMS (EI, 70 eV): calcd for C14H18FNO2+ [M]+: 251.1316; found: 251.1316.
(cm−1) = 3073, 2972, 2958, 1539, 1494, 1370, 1153, 1135, 1089, 877, 839, 767; 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.33–7.29 (m, 2H, H-2, H-6), 7.06–7.02 (m, 2H, H-3, H-5), 4.85 (d, 3J = 10.0 Hz, 1H, H-4′), 3.92 (d, 3J = 10.0 Hz, 1H, H-1′), 1.24 (s, 3H, CH3-2′), 1.17 (s, 3H, CH3-3′), 1.15 (s, 3H, CH3-2′), 0.70 (s, 3H, CH3-3′); 13C NMR (101 MHz, CDCl3): δ (ppm) = 134.9 (s, C-4), 133.0 (s, C-1), 128.9 (d, 2C, C-2, C-6), 128.4 (d, 2C, C-3, C-5), 84.9 (d, C-4′), 49.0 (d, C-1′), 45.1 (s, C-3′), 39.4 (s, C-2′), 24.3 [q, (C-3′)CH3], 22.8 [q, (C-2′)CH3], 21.5 [q, (C-3′)CH3], 19.5 [q, (C-2′)CH3]; MS (EI): m/z (%) = 221 (29) [M − NO2]+, 179 (100) [M − NO2 − C3H6]+, 125 (64) [C7H6Cl]+; HRMS (EI, 70 eV): calcd for C14H18ClNO2+ [M]+: 267.1021; found: 267.1021.
(cm−1) = 2962, 2925, 1552, 1464, 1376, 1259, 1072, 1010, 861, 797; 1H NMR (500 MHz, CDCl3): δ [ppm] = 7.48–7.44 (m, 2H, H-2, H-6), 7.00–6.96 (m, 2H, H-3, H-5), 4.85 (d, 3J = 10.1 Hz, 1H, H-4′), 3.90 (d, 3J = 10.1 Hz, 1H, H-1′), 1.23 (s, 3H, CH3-3′), 1.17 (s, 3H, CH3-2′), 1.14 (s, 3H, CH3-3′), 0.70 (s, 3H, CH3-2′); 13C NMR (101 MHz, CDCl3): δ (ppm) = 135.5 (s, C-1), 131.8 (d, 2C, C-2, C-6), 128.7 (d, 2C, C-3, C-5), 121.1 (s, C-4), 84.8 (d, C-4′), 49.1 (d, C-1′), 45.1 (s, C-3′), 39.4 (s, C-2′), 24.3 [q, (C-3′)CH3], 22.8 [q, (C-2′)CH3], 21.5 [q, (C-3′)CH3], 19.5 [q, (C-2′)CH3]; MS (EI): m/z (%) = 265 (20) [M − NO2]+, 168 (100) [M − NO2 − Br]+, 143 (56) [M − NO2 − Br − C3H6]+; HRMS (ESI): calcd for C14H1979BrNO2+ [M + H]+: 312.0594; found: 312.0593, calcd for C14H1981BrNO2+ [M + H]+: 314.0573; found: 314.0573.
(cm−1) = 2954, 1717, 1541, 1433, 1371, 1277, 1181, 1151, 1138, 1110, 1017, 860, 756; 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.00 (d, 3J = 7.3 Hz, 2H, H-2, H-6), 7.18 (d, 3J = 7.3 Hz, 2H, H-3, H-5), 4.92 (d, 3J = 9.9 Hz, 1H, H-4′), 4.01 (d, 3J = 9.9 Hz, 1H, H-1′), 1.24 (s, 3H, CH3-3′), 1.20 (s, 3H, CH3-2′), 1.16 (s, 3H, CH3-3′), 0.69 (s, 3H, CH3-2′); 13C NMR (101 MHz, CDCl3): δ (ppm) = 167.0 (s, CH3CO2Ar), 141.8 (s, C-1), 130.0 (d, 2C, C-2, C-6), 129.1 (s, C-4), 127.0 (d, 2C, C-3, C-5), 84.6 (d, C-4′), 53.2 (q, CH3CO2Ar) 49.5 (d, C-1′), 45.1 (s, C-2′), 39.7 (s, C-3′), 24.3 [q, (C-3′)CH3], 22.7 [q, (C-2′)CH3], 21.5 [q, (C-2′)CH3], 19.5 [q, (C-3′)CH3]; MS (EI): m/z (%) = 245 (92) [M − NO2]+, 203 (39) [M − NO2 − C3H6]+, 171 (51), 159 (34), 84 (100) [C6H12]+, 69 (35); HRMS (ESI): calcd for C16H22NO4+ [M + H]+ 292.1543; found: 292.1544.
(cm−1) = 3066, 2967, 1598, 1538, 1371, 1151, 1138, 1081, 877, 814, 772; 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.29–7.22 (m, 2H, Har), 7.09–7.08 (m, 1H, Har), 6.99 (dtd, 3J = 7.1 Hz, 4J = 1.8 Hz, 0.8 Hz, 1H, Har), 4.86 (d, 3J = 10.0 Hz, 1H, H-4′), 3.93 (d, 3J = 10.0 Hz, 1H, H-1′), 1.24 (s, 3H, CH3-3′), 1.18 (s, 3H, CH3-2′), 1.14 (s, 3H, CH3-3′), 0.72 (s, 3H, CH3-2′); 13C NMR (101 MHz, CDCl3): δ (ppm) = 138.6 (s, C-3), 134.7 (s, C-1), 129.9 (d, CarH), 127.3 (d, CarH), 127.2 (d, CarH), 125.2 (d, CarH), 84.7 (d, C-4′), 49.2 (d, C-1′), 45.0 (s, C-3′), 39.5 (s, C-2′), 24.3 [q, (C-2′)CH3], 22.7 [q, (C-3′)CH3], 21.5 [q, (C-2′)CH3], 19.4 [q, (C-3′)CH3]; MS (EI): m/z (%) = 221 (32) [M − NO2]+, 179 (100) [M − NO2 − C3H6]+, 125 (32) [C7H6Cl]+; HRMS (ESI): calcd for C14H19ClNO2+ [M + H]+: 268.1099; found: 268.1098.
(cm−1) = 3079, 2960, 2231, 1533, 1372, 1151, 1136, 793; 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.59–7.55 (m, 1H, Har), 7.46 (td, 3J = 7.7 Hz, 4J = 0.6 Hz, 1H, Har), 7.41–7.39 (m, 1H, Har), 7.36–7.33 (m, 1H, Har), 4.87 (d, 3J = 10.0 Hz, 1H, H-4′), 3.97 (d, 3J = 10.0 Hz, 1H, H-1′), 1.25 (s, 3H, CH3-3′), 1.20 (s, 3H, CH3-2′), 1.16 (s, 3H, CH3-3′), 0.71 (s, 3H, CH3-2′); 13C NMR (101 MHz, CDCl3): δ (ppm) = 138.2 (s, C-3), 131.5 (d, CarH), 130.9 (d, CarH), 130.6 (d, CarH), 129.6 (d, CarH), 118.7 (s, CarCN), 113.0 (s, C-1), 84.4 (d, C-4′), 49.1 (d, C-1′), 45.2 (s, C-3′), 39.6 (s, C-2′), 24.3 [q, (C-2′)CH3], 22.7 [q, (C-3′)CH3], 21.5 [q, (C-2′)CH3], 19.4 [q, (C-3′)CH3]; MS (EI): m/z (%) = 212 (32) [M − NO2]+, 170 (100) [M − NO2 − C3H6]+, 116 (20) [C8H6N]+; HRMS (ESI): calcd for C15H19N2O2+ [M + H]+: 259.1440; found: 259.1442. 6: Rf = 0.53 (P/Et2O = 9/1); IR: (cm−1) = 2923, 2231, 1549, 1365, 1148, 905, 798, 691; 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.54 (dd, 3J = 7.6 Hz, 4J = 1.6 Hz, 1H, H-4)*, 7.41–7.38 (m, 2H, H-2, H-5), 7.35 (d, 3J = 8.0 Hz, 1H, H-6)*, 5.04 (s, 1H, CHH-5′), 5.00 (s, 1H, CHH-5′), 4.76 (dd, 3J = 12.0 Hz, 2.2 Hz, 1H, H-2′), 3.32 (dd, 2J = 15.0, 3J = 11.9 Hz, 1H, CHH-1′), 2.94 (dd, 2J = 15.0, 3J = 2.2 Hz, 1H, CHH-1′), 1.87 (d, 4J = 1.4 Hz, 3H, CH3-4′), 1.29 (s, 3H, CH3-3′), 1.23 (s, 3H, CH3-3′) (* the assignments are interconvertible); 13C NMR (126 MHz, CDCl3): δ (ppm) = 147.3 (s, C-4′), 138.3 (s, C-3), 133.3 (d, C-6)*, 132.4 (d, C-2), 131.2 (d, C-4)*, 129.9 (d, C-5), 118.6 (s, CN), 114.5 (t, C-5′), 113.2 (s, C-1), 96.0 (d, C-2′), 43.0 (s, C-3′), 34.4 (t, C-1′), 24.7 [q, (C-3′)CH3], 21.6 [q, (C-3′)CH3], 19.6 [q, (C-4′)CH3] (* the assignments are interconvertible); MS (EI): m/z (%) = 196 (15) [C14H14N]+, 170 (58) [C12H12N]+, 116 (100) [C8H6N]+; HRMS (ESI): calcd for C15H19N2O2+ [M + H]+: 259.1441; found: 259.1439.
(cm−1) = 3061, 2973, 2960, 1541, 1372, 1153, 1135, 1033, 876, 807, 754; 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.41 (dd, 3J = 7.6 Hz, 4J = 1.6 Hz, 1H, H-6), 7.28–7.18 (m, 2H, H-5, H-4), 7.16 (dd, 3J = 7.5 Hz, 4J = 1.9 Hz, 1H, H-3), 4.99 (d, 3J = 10.2 Hz, 1H, H-4′), 4.43 (d, 3J = 10.2 Hz, 1H, H-1′), 1.26 (s, 3H, CH3-2′), 1.24 (s, 3H, CH3-3′), 1.19 (s, 3H, CH3-3′), 0.73 (s, 3H, CH3-2′); 13C NMR (101 MHz, CDCl3): δ (ppm) = 134.7 (s, C-2), 134.1 (s, C-1), 130.3 (d, C-6), 128.4 (d, C-5/C-4)*, 128.1 (d, C-3), 126.8 (d, C-5/C-4)*, 84.4 (d, C-4′), 46.8 (d, C-1′), 44.5 (s, C-2′), 40.5 (s, C-3′), 24.9 [q, (C-3′)CH3], 22.7 [q, (C-2′)CH3], 21.8 [q, (C-2′)CH3], 19.4 [q, (C-3′)CH3] (* the assignments are interconvertible); MS (EI): m/z (%) = 221 (32) [M − NO2]+, 179 (100) [M − NO2 − C3H6]+, 125 (28) [C7H6Cl]+; HRMS (ESI): calcd for C14H19ClNO2+ [M + H]+: 268.1099; found: 268.1094.
(cm−1) = 3445, 3008, 2930, 1597, 1448, 1386, 1225, 1052, 1025, 992, 760; 1H NMR (500 MHz, CDCl3): δ (ppm) 8.59–8.37 (m, 2H, H-5, H-6), 7.43 (dt, 3J = 7.9 Hz, 4J = 2.0 Hz, 1H, H-3), 7.31–7.27 (m, 1H, H-4), 4.90 (d, 3J = 10.0 Hz, 1H, H-4′), 3.96 (d, 3J = 10.0 Hz, 1H, H-1′), 1.25 (s, 3H, CH3-2′), 1.19 (s, 3H, CH3-3′), 1.16 (s, 3H, CH3-2′), 0.73 (s, 3H, CH3-3′); 13C NMR (126 MHz, CDCl3): δ (ppm) = 148.7 (d, C-5/C-6)*, 148.6 (d, C-5/C-6)*, 134.5 (d, C-3), 131.9 (s, C-2), 123.4 (d, C-4), 84.2 (d, C-4′), 47.4 (d, C-1′), 45.3 (s, C-3′), 39.4 (s, C-2′), 24.2 [q, (C-2′)CH3], 22.7 [q, (C-3′)CH3], 21.6 [q, (C-2′)CH3], 19.3 [q, (C-3′)CH3] (* the assignments are interconvertible); MS (EI): m/z (%) = 188 (100) [M − NO2]+, 146 (72) [M − NO2 − C3H6]+, 132 (40) [C9H10N]+; HRMS (ESI): calcd for C13H19N2O2+ [M + H]+: 235.1441; found: 235.1441.
(cm−1) = 2954, 1537, 1459, 1367, 1139, 858, 810, 755; 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.85–7.79 (m, 3H, Har), 7.54–7.52 (br. s., 1H, Har), 7.51–7.43 (m, 2H, Har), 7.26–7.23 (m, 1H, Har), 5.06 (d, 3J = 10.1 Hz, 1H, H-4′), 4.14 (d, 3J = 10.1 Hz, 1H, H-1′), 1.28 (s, 3H, CH3-2′), 1.26 (s, 3H, CH3-3′), 1.20 (s, 3H, CH3-2′), 0.73 (s, 3H, CH3-3′); 13C NMR (101 MHz, CDCl3): δ (ppm) = 134.2 (s, C-2), 133.5 (s, C-5/C-10)*, 132.6 (s, C-5/C-10)*, 128.4 (d, CarH), 127.8 (d, CarH), 126.5 (d, CarH), 125.9 (d, CarH), 125.7 (d, CarH), 125.1 (d, CarH), 85.0 (d, C-4′), 49.6 (d, C-1′), 45.1 (s, C-3′), 39.5 (s, C-2′), 24.4 [q, (C-2′)CH3], 22.8 [q, (C-3′)CH3], 21.6 [q, (C-2′)CH3], 19.5 [q, (C-3′)CH3] (* the assignments are interconvertible); MS (EI): m/z (%) = 237 (28) [M − NO2]+, 181 (100) [M − NO2 − C4H8]+, 127 (10) [C10H7]+; HRMS (ESI): calcd for C18H22NO2+ [M + H]+: 284.1645; found: 284.1646.
:88). The analytical data obtained matched those reported in the literature.13
:56). Rf = 0.45 (P/Et2O = 19/1); IR: (cm−1) = 3063, 3030, 2965, 1542, 1455, 1366, 784; 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.34 (t, 3J = 7.5 Hz, 2H, meta-Har), 7.29–7.24 (m, 1H, para-Har), 7.23–7.20 (m, 2H, ortho-Har), 5.27 (virt. q, 3J ≅ 3J = 8.7 Hz, 1H, H-4′), 3.98 (d, 3J = 9.1 Hz, 1H, H-1′), 2.40 (dd, 2J = 11.9 Hz, 3J = 8.5 Hz, 1H, CHH-3), 2.30 (dd, 2J = 11.9 Hz, 3J = 8.6 Hz, 1H, CHH-3), 1.76 (dq, 2J = 14.8 Hz, 3J = 7.5 Hz, 1H, CHHCH3), 1.64 (dq, 2J = 14.8 Hz, 3J = 7.5 Hz, 1H, CHHCH3), 1.30–1.19 (m, 2H, CH2CH3), 0.96 (t, 3J = 7.5 Hz, 3H, CH2CH3), 0.60 (t, 3J = 7.4 Hz, 3H, CH2CH3); 13C NMR (126 MHz, CDCl3): δ (ppm) = 136.6 (s, Car), 128.6 (d, 2C, meta-CarH), 127.5 (d, 2C, ortho-CarH), 127.2 (d, para-CarH), 76.6 (d, C-4′), 53.8 (d, C-1′), 41.8 (s, C-2′), 33.7 (t, C-3′), 31.7 (t, CH2CH3), 26.4 (t, CH2CH3), 8.62 (q, CH2CH3), 7.98 (q, CH2CH3); MS (EI): m/z (%) = 187 (4) [M − NO2]+, 157 (12) [M − NO2 − C2H6]+, 117 (100) [C9H9]+; HRMS (ESI): calcd for C14H20NO2+ [M + H]+: 234.1488; found: 234.1489.
(cm−1) = 3062, 3032, 2923, 1543, 1474, 1368, 1218, 1095, 1051, 1019, 814; 1H NMR (500 MHz, C6D6): δ (ppm) = 6.98–6.96 (m 3H, meta-Har, para-Har), 6.84 (t, 3J = 7.5 Hz, 1H, H-5), 6.79 (d, 3J = 7.5 Hz, 1H, H-4), 6.61–6.56 (m, 2H, ortho-Har), 6.45 (t, 3J = 7.5 Hz, 1H, H-6), 6.29 (d, 3J = 7.5 Hz, 1H, H-7), 5.13 (dd, 3J = 7.4 Hz, 4.2 Hz, 1H, H-2a), 4.97 (ddd, 3J = 9.4 Hz, 4.2 Hz, 4J = 1.5 Hz, 1H, H-1), 3.72 (virt. t, 3J ≅ 3J = 9.3 Hz, 1H, H-2), 3.50 (virt. t, 3J ≅ 3J = 8.4 Hz, 1H, H-7b); 13C NMR (101 MHz, C6D6): δ (ppm) = 160.9 (s, C-3a), 135.4 (s, C-2′), 129.6 (d, C-5), 128.6 (d, meta-CarH/para-CarH)*, 128.3 (d, C-7), 127.9 (d, meta-CarH/para-CarH)*, 127.6 (d, 2C, ortho-CarH)*, 124.9 (s, C-7a), 121.6 (d, C-6), 111.5 (d, C-4), 85.6 (d, C-1), 80.6 (d, C-2a), 45.6 (d, C-2), 44.6 (d, C-7b) (* the exact assignment was not possible due to significant overlap with the solvent C6D6); MS (EI): m/z (%) = 221 (12) [M − NO2]+, 118 (100) [C8H6O]+, 90 (8); HRMS (ESI): calcd for C16H14NO3+ [M + H]+: 268.0968; found: 268.0970.
:19:29) as an orange coloured oil. Starting material 1a was recovered as a mixture of isomers (4.50 mg, 30.2 μmol, 15%, cis/trans = 55:45). NMR data are given for the major diastereoisomer depicted in Scheme 2. Rf = 0.06 (P/Et2O = 4/1); IR: (cm−1) = 3031, 2923, 1545, 1375, 1132, 1043, 874, 751; 1H NMR (400 MHz, C6D6): δ (ppm) = 7.02–6.93 (m, 5H, Har), 4.26 (virt. t, 3J ≅ 3J = 7.8 Hz, 1H, H-7), 3.81 (virt. t, 3J ≅ 3J = 8.6 Hz, 1H, H-6), 3.56 (dd, 3J = 9.8 Hz, 7.3 Hz, 1H, H-8), 3.40–3.28 (m, 2H, CHH-3, CHH-4), 3.19–3.15 (m, 2H, CHH-3, CHH-4), 2.99 (d, 3J = 9.8 Hz, 1H, H-1); 13C NMR (101 MHz, C6D6): δ (ppm) = 136.5 (s, Car), 129.0 (d, 2C, ortho-CarH)*, 127.9 (d, 2C, meta-CarH)*, 126.9 (d, para-CarH), 82.3 (d, C-7), 77.9 (d, C-6), 75.2 (d, C-1), 68.3 (t, C-3), 68.1 (t, C-4), 51.2 (d, C-8) (* the assignments are interconvertible); MS (EI): m/z (%) = 235 (16) [M]+, 189 (52) [M − NO2]+, 117 (72) [C9H9]+, 91 (100) [C7H7]+; HRMS (ESI): calcd for C12H14NO4+ [M + H]+: 236.0917; found: 236.0918.
:33) as a pale-yellow coloured oil. NMR data are given for the major diastereoisomer depicted in Scheme 2. Rf = 0.69 (P/Et2O = 9/1); IR: (cm−1) = 3028, 1542, 1496, 1368, 1028, 824, 770; 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.17–7.13 (m, 6H, Har), 7.00–6.98 (m, 2H, Har), 6.83–6.77 (m, 2H, Har), 5.13 (virt. q, 3J ≅ 3J = 9.0 Hz, 1H, H-3′), 4.07 (d, 3J = 9.5 Hz, 1H, H-4′), 3.01 (dd, 2J = 12.3 Hz, 3J = 8.1 Hz, 1H, CHH-2′), 2.60 (dd, 2J = 12.4 Hz, 3J = 9.2 Hz, 1H, CHH-2′), 1.43 [tt, 3J = 8.3 Hz, 5.6 Hz, 1H, CH(CH2)2], 0.77–0.66 [m, 2H, CH(CH2)2], 0.57 [virt. tt, 2J ≅ 3J ≅ 3J = 8.6 Hz, 3J = 5.5 Hz, 1H, CH(CH2)2], 0.44 [virt. dq, 2J = 9.0 Hz, 3J ≅ 3J = 5.5 Hz, 1H, CH(CH2)2]; 13C NMR (101 MHz, CDCl3): δ (ppm) = 141.1 (s, C-1a), 136.2 (s, C-4a), 128.4 (d, 2C, CarH), 128.3 (d, 2C, CarH), 128.0 (d, 2C, CarH), 127.8 (d, CarH), 127.5 (d, CarH), 126.8 (d, 2C, CarH), 76.9 (d, C–3′), 55.1 (d, C-4′), 47.3 (s, C-1′), 32.1 (t, C-2′), 22.6 [d, CH(CH2)2], 3.21 [t, CH(CH2)2], 2.11 [t, CH(CH2)2]; MS (EI): m/z (%) = 247 (4) [M − NO2]+, 205 (32) [M − NO2 − C3H6]+, 117 (100) [C9H9]+; HRMS (ESI): calcd for C19H20NO2+ [M + H]+: 294.1488; found: 294.1488.
:23) as a yellow coloured oil. NMR data are given for the major diastereoisomer depicted in Scheme 2. Rf = 0.70 (P/Et2O = 19/1); IR: (cm−1) = 3063, 3031, 2958, 1546, 1480, 1395, 1368, 1252, 1146, 1029, 870, 833; 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.33–7.31 (m, 5H, Har), 5.13 (virt. q, 3J ≅ 3J = 8.5 Hz, 1H, H-3), 4.24 (d, 3J = 8.6 Hz, 1H, H-2), 2.93 (dd, 2J = 13.2 Hz, 3J = 8.2 Hz, 1H, CHH-4), 2.50 (dd, 2J = 13.2 Hz, 3J = 8.6 Hz, 1H, CHH-4), 0.97 [s, 9H, C(CH3)3], 0.12 [s, 9H, OSi(CH3)3]; 13C NMR (126 MHz, CDCl3): δ (ppm) = 136.4 (s, Car), 129.0 (d, 2C, ortho-CarH), 128.1 (d, 2C, meta-CarH), 127.3 (d, para-CarH), 83.3 (s, C-1), 78.2 (d, C-3), 52.2 (d, C-2), 37.9 [s, C(CH3)3], 34.3 (t, C-4), 25.8 [q, 3C, C(CH3)3], 2.6 [q, 3C, OSi(CH3)3]; MS (EI): m/z (%) = 275 (17) [M − NO2]+, 219 (18) [M − NO2 − C(CH3)3]+, 117 (100) [C9H9]+, 73 (36) [Si(CH3)3]+; HRMS (ESI): calcd for C17H28NO3Si+ [M + H]+: 322.1833; found: 322.1833.
:39) as a colourless oil. NMR data are given for the major diastereoisomer depicted in Scheme 2. Rf = 0.70 (P/Et2O = 9/1); IR: (cm−1) = 3004, 2926, 1542, 1497, 1364, 1264, 1045, 1018, 891, 822, 758; 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.38–7.32 (m, 2H, meta-Har), 7.29–7.25 (m, 3H, ortho-Har, para-Har), 5.35 (dd, 3J = 10.1 Hz, 8.8 Hz, 1H, H-6), 4.06 (d, 3J = 8.8 Hz, 1H, H-7), 3.09 (virt. t, 3J ≅ 3J = 9.2 Hz, 1H, H-5), 2.00–1.91 (m, 4H, CH2-3, CHH-2, CHH-4), 1.89–1.77 (m, 1H, CHH-2), 1.62–1.44 (m, 1H, CHH-4), −0.14 [s, 9H, OSi(CH3)3]; 13C NMR (126 MHz, CDCl3): δ (ppm) = 136.5 (s, Car), 129.5 (d, 2C, ortho-CarH), 128.8/128.5 (d, 2C meta-CarH), 127.5/127.3 (d, para-CarH), 83.5 (s, C-1), 81.0 (d, C-6), 51.9 (d, C-7), 49.9 (d, C-5), 40.1 (t, CH2-2), 26.1 (t, CH2-4), 25.9 (t, CH2-3), 1.64 [q, 3C, OSi(CH3)3]; MS (EI): m/z (%) = 259 (88) [M − NO2]+, 169 (60) [M − NO2 − OSi(CH3)3]+, 91 (28) [C7H7]+, 73 (100) [Si(CH3)3]+; HRMS (ESI): calcd for C16H24NO3Si+ [M + H]+: 306.1522; found: 306.1522.
(cm−1) = 3066, 2931, 2870, 1542, 1460, 1371, 993, 912, 751; 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.15–7.09 (m, 3H, H-3, H-4, H-5), 7.07–7.03 (m, 1H, H-6), 5.83 (ddt, 3J = 17.0 Hz, 10.2 Hz, 6.7 Hz, 1H, H-4′), 5.02 (virt. dq, 3J = 17.2 Hz, 2J ≅ 4J = 1.7 Hz, 1H, CHH-5′), 4.98–4.92 (m, 2H, CHH-5′, H-4′′), 4.15 (d, 3J = 10.2 Hz, 1H, H-1′′), 2.76 (ddd, 2J = 14.0 Hz, 3J = 10.5 Hz, 5.3 Hz, 1H, CHH-1′), 2.46 (ddd, 2J = 14.0 Hz, 3J = 10.6 Hz, 6.0 Hz, 1H, CHH-1′), 2.19–2.03 (m, 2H, H-3′), 1.82–1.72 (m, 1H, CHH-2′), 1.70–1.58 (m, 1H, CHH-2′), 1.18 (s, 3H, CH3-3′′), 1.10 (s, 3H, CH3-3′′), 1.09 (s, 3H, CH3-2′′), 0.67 (s, 3H, CH3-2′′); 13C NMR (126 MHz, CDCl3): δ (ppm) = 141.7 (s, C-1), 138.6 (d, C-4′), 133.2 (s, C-2), 129.8 (d, C-6), 127.1 (d, C-5)*, 127.0 (d, C-3), 125.9 (d, C-4)*, 115.3 (t, C-5′), 85.3 (d, C-4′′), 45.6 (d, C-1′′), 44.4 (s, C-2′′), 40.1 (s, C-3′′), 33.8 (t, C-3′), 32.4 (t, C-1′), 30.6 (t, C-2′), 24.6 [q, (C-2′′)CH3], 22.8 [q, (C-3′′)CH3], 21.8 [q, (C-2′′)CH3], 19.4 [q, (C-3′′)CH3] (* the assignments are interconvertible); MS (EI): m/z (%) = 301 (2) [M]+, 255 (20) [M − NO2]+, 199 (49) [M − NO2 − C3H6]+, 143 (100) [C11H11]+; HRMS (ESI): calcd for C19H27NO2+ [M + H]+: 302.2115; found: 302.2115.
(cm−1) = 3060, 2959, 2866, 2604, 1566, 1458, 1449, 1358, 1337, 1270, 1132, 885, 810; 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.29–7.21 (m, 2H, meta-HPh), 7.19–7.08 (m, 3H, ortho-HPh, para-HPh), 3.37 (d, 3J = 9.9 Hz, 1H, H-1), 2.86 (d, 3J = 9.9 Hz, 1H, H-4), 1.51 (br. s, 2H, NH2), 1.03 (s, 3H, CH3-2), 1.01 (s, 3H, CH3-3), 0.93 (s, 3H, CH3-2), 0.59 (s, 3H, CH3-3); 13C NMR (101 MHz, CDCl3): δ (ppm) = 139.6 (s, CPh), 128.3 (d, 2C, meta-CPhH), 127.7 (d, 2C, ortho-CPhH), 126.1 (d, para-CPhH), 56.8 (d, C-4), 55.5 (d, C-1), 41.7 (s, C-2), 39.6 (s, C-3), 24.2 [q, (C-3)CH3], 22.6 [q, (C-2)CH3], 21.1 [q, (C-3)CH3], 18.7 [q, (C-2)CH3]; MS (EI, 70 eV): m/z (%) = 132 (5) [M − C4H9N]+, 119 (100) [M − C4H9N − CH3]+, 91 (13) [C7H7]+, 71 (31) [C4H9]+, 56 (11); HRMS (ESI): calcd for C14H22N+ [M + H]+: 204.1741; found: 204.1748.
(cm−1) = 3079, 3004, 1542, 1449, 1369, 1017, 822, 758; 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.44–7.20 (m, 5H, Har), 5.20 (virt. q, 3J ≅ 3J = 8.7 Hz, 1H, H-4′), 3.97 (d, 3J = 9.2 Hz, 1H, H-1′), 2.06 (dd, 2J = 12.2 Hz, 3J = 8.7 Hz, 1H, CHH-3′), 1.71 (dd, 3J = 12.2 Hz, 3J = 8.3 Hz, 1H, CHH-3′), 1.09 [tt, 3J = 8.4 Hz, 5.5 Hz, 1H, CH(CH2)2], 0.60–0.25 [m, 6H, CH(CH2)2, CH(CH2)2], 0.20–0.13 [m, 1H, CH(CH2)2]; 13C NMR (101 MHz, CDCl3): δ (ppm) = 136.4 (s, Car), 128.5 (d, 2C, meta-CarH), 127.7 (d, 2C, ortho-CarH), 127.2 (d, para-CarH), 76.6 (d, C-4′), 55.5 (d, C-1′), 41.7 (s, C-2′), 27.8 (t, C-3′), 20.2 [d, CH(CH2)2], 14.6 [d, CH(CH2)2], 1.93 [t, CH(CH2)2], 1.74 [t, CH(CH2)2], 0.90 [t, CH(CH2)2], 0.72 [t, CH(CH2)2]; MS (EI): m/z (%) = 211 (4) [M − NO2]+, 169 (16) [M − NO2 − C3H6]+, 117 (100) [C9H9]+; HRMS (ESI): calcd for C16H20NO2+ [M + H]+: 258.1448; found: 258.1449. 7: Rf = 0.69 (P/Et2O = 19/1); IR: (cm−1) = 3419, 2928, 1722, 1547, 1367, 1023, 856, 791; 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.17–7.05 (m, 4H, Har), 5.45 (ttd, 3J = 7.1 Hz, 4J = 2.5 Hz, 1.6 Hz, 1H, C
(cm−1) = 3079, 2967, 2871, 1538, 1510, 1460, 1371, 1226, 1151, 1135, 844, 761; 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.09–7.00 (m, 4H, Har), 4.85 (d, 3J = 10.1 Hz, 1H, H-4′), 3.92 (d, 3J = 10.1 Hz, 1H, H-1′), 1.24 (s, 3H, CH3-2′), 1.16 (s, 3H, CH3-3′), 1.14 (s, 3H, CH3-2′), 0.70 (s, 3H, CH3-3′); 13C NMR (101 MHz, CDCl3): δ (ppm) = 162.1 (d, 1JCF = 254.4 Hz, C-1), 132.2 (s, C-4), 128.5 (d, 3JCF = 7.9 Hz, C-3, C-5), 115.6 (d, 2JCF = 21.4 Hz, C-2, C–6), 85.2 (d, C-4′), 48.9 (d, C-1′), 45.0 (s, C-3′), 39.3 (s, C-2′), 24.2 [q, (C-3′)CH3], 22.8 [q, (C-2′)CH3], 21.4 [q, (C-3′)CH3], 19.4 [q, (C-2′)CH3]; MS (EI): m/z (%) = 205 (45) [M − NO2]+, 163 (100) [M − NO2 − C3H6]+, 106 (54) [C7H6F]+; HRMS (EI, 70 eV): calcd for C14H18FNO2+ [M]+: 251.1316; found: 251.1316.
(cm−1) = 3073, 2972, 2958, 1539, 1494, 1370, 1153, 1135, 1089, 877, 839, 767; 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.33–7.29 (m, 2H, H-2, H-6), 7.06–7.02 (m, 2H, H-3, H-5), 4.85 (d, 3J = 10.0 Hz, 1H, H-4′), 3.92 (d, 3J = 10.0 Hz, 1H, H-1′), 1.24 (s, 3H, CH3-2′), 1.17 (s, 3H, CH3-3′), 1.15 (s, 3H, CH3-2′), 0.70 (s, 3H, CH3-3′); 13C NMR (101 MHz, CDCl3): δ (ppm) = 134.9 (s, C-4), 133.0 (s, C-1), 128.9 (d, 2C, C-2, C-6), 128.4 (d, 2C, C-3, C-5), 84.9 (d, C-4′), 49.0 (d, C-1′), 45.1 (s, C-3′), 39.4 (s, C-2′), 24.3 [q, (C-3′)CH3], 22.8 [q, (C-2′)CH3], 21.5 [q, (C-3′)CH3], 19.5 [q, (C-2′)CH3]; MS (EI): m/z (%) = 221 (29) [M − NO2]+, 179 (100) [M − NO2 − C3H6]+, 125 (64) [C7H6Cl]+; HRMS (EI, 70 eV): calcd for C14H18ClNO2+ [M]+: 267.1021; found: 267.1021.
(cm−1) = 2962, 2925, 1552, 1464, 1376, 1259, 1072, 1010, 861, 797; 1H NMR (500 MHz, CDCl3): δ [ppm] = 7.48–7.44 (m, 2H, H-2, H-6), 7.00–6.96 (m, 2H, H-3, H-5), 4.85 (d, 3J = 10.1 Hz, 1H, H-4′), 3.90 (d, 3J = 10.1 Hz, 1H, H-1′), 1.23 (s, 3H, CH3-3′), 1.17 (s, 3H, CH3-2′), 1.14 (s, 3H, CH3-3′), 0.70 (s, 3H, CH3-2′); 13C NMR (101 MHz, CDCl3): δ (ppm) = 135.5 (s, C-1), 131.8 (d, 2C, C-2, C-6), 128.7 (d, 2C, C-3, C-5), 121.1 (s, C-4), 84.8 (d, C-4′), 49.1 (d, C-1′), 45.1 (s, C-3′), 39.4 (s, C-2′), 24.3 [q, (C-3′)CH3], 22.8 [q, (C-2′)CH3], 21.5 [q, (C-3′)CH3], 19.5 [q, (C-2′)CH3]; MS (EI): m/z (%) = 265 (20) [M − NO2]+, 168 (100) [M − NO2 − Br]+, 143 (56) [M − NO2 − Br − C3H6]+; HRMS (ESI): calcd for C14H1979BrNO2+ [M + H]+: 312.0594; found: 312.0593, calcd for C14H1981BrNO2+ [M + H]+: 314.0573; found: 314.0573.
(cm−1) = 2954, 1717, 1541, 1433, 1371, 1277, 1181, 1151, 1138, 1110, 1017, 860, 756; 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.00 (d, 3J = 7.3 Hz, 2H, H-2, H-6), 7.18 (d, 3J = 7.3 Hz, 2H, H-3, H-5), 4.92 (d, 3J = 9.9 Hz, 1H, H-4′), 4.01 (d, 3J = 9.9 Hz, 1H, H-1′), 1.24 (s, 3H, CH3-3′), 1.20 (s, 3H, CH3-2′), 1.16 (s, 3H, CH3-3′), 0.69 (s, 3H, CH3-2′); 13C NMR (101 MHz, CDCl3): δ (ppm) = 167.0 (s, CH3CO2Ar), 141.8 (s, C-1), 130.0 (d, 2C, C-2, C-6), 129.1 (s, C-4), 127.0 (d, 2C, C-3, C-5), 84.6 (d, C-4′), 53.2 (q, CH3CO2Ar) 49.5 (d, C-1′), 45.1 (s, C-2′), 39.7 (s, C-3′), 24.3 [q, (C-3′)CH3], 22.7 [q, (C-2′)CH3], 21.5 [q, (C-2′)CH3], 19.5 [q, (C-3′)CH3]; MS (EI): m/z (%) = 245 (92) [M − NO2]+, 203 (39) [M − NO2 − C3H6]+, 171 (51), 159 (34), 84 (100) [C6H12]+, 69 (35); HRMS (ESI): calcd for C16H22NO4+ [M + H]+ 292.1543; found: 292.1544.
(cm−1) = 3066, 2967, 1598, 1538, 1371, 1151, 1138, 1081, 877, 814, 772; 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.29–7.22 (m, 2H, Har), 7.09–7.08 (m, 1H, Har), 6.99 (dtd, 3J = 7.1 Hz, 4J = 1.8 Hz, 0.8 Hz, 1H, Har), 4.86 (d, 3J = 10.0 Hz, 1H, H-4′), 3.93 (d, 3J = 10.0 Hz, 1H, H-1′), 1.24 (s, 3H, CH3-3′), 1.18 (s, 3H, CH3-2′), 1.14 (s, 3H, CH3-3′), 0.72 (s, 3H, CH3-2′); 13C NMR (101 MHz, CDCl3): δ (ppm) = 138.6 (s, C-3), 134.7 (s, C-1), 129.9 (d, CarH), 127.3 (d, CarH), 127.2 (d, CarH), 125.2 (d, CarH), 84.7 (d, C-4′), 49.2 (d, C-1′), 45.0 (s, C-3′), 39.5 (s, C-2′), 24.3 [q, (C-2′)CH3], 22.7 [q, (C-3′)CH3], 21.5 [q, (C-2′)CH3], 19.4 [q, (C-3′)CH3]; MS (EI): m/z (%) = 221 (32) [M − NO2]+, 179 (100) [M − NO2 − C3H6]+, 125 (32) [C7H6Cl]+; HRMS (ESI): calcd for C14H19ClNO2+ [M + H]+: 268.1099; found: 268.1098.
(cm−1) = 3079, 2960, 2231, 1533, 1372, 1151, 1136, 793; 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.59–7.55 (m, 1H, Har), 7.46 (td, 3J = 7.7 Hz, 4J = 0.6 Hz, 1H, Har), 7.41–7.39 (m, 1H, Har), 7.36–7.33 (m, 1H, Har), 4.87 (d, 3J = 10.0 Hz, 1H, H-4′), 3.97 (d, 3J = 10.0 Hz, 1H, H-1′), 1.25 (s, 3H, CH3-3′), 1.20 (s, 3H, CH3-2′), 1.16 (s, 3H, CH3-3′), 0.71 (s, 3H, CH3-2′); 13C NMR (101 MHz, CDCl3): δ (ppm) = 138.2 (s, C-3), 131.5 (d, CarH), 130.9 (d, CarH), 130.6 (d, CarH), 129.6 (d, CarH), 118.7 (s, CarCN), 113.0 (s, C-1), 84.4 (d, C-4′), 49.1 (d, C-1′), 45.2 (s, C-3′), 39.6 (s, C-2′), 24.3 [q, (C-2′)CH3], 22.7 [q, (C-3′)CH3], 21.5 [q, (C-2′)CH3], 19.4 [q, (C-3′)CH3]; MS (EI): m/z (%) = 212 (32) [M − NO2]+, 170 (100) [M − NO2 − C3H6]+, 116 (20) [C8H6N]+; HRMS (ESI): calcd for C15H19N2O2+ [M + H]+: 259.1440; found: 259.1442. 6: Rf = 0.53 (P/Et2O = 9/1); IR: (cm−1) = 2923, 2231, 1549, 1365, 1148, 905, 798, 691; 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.54 (dd, 3J = 7.6 Hz, 4J = 1.6 Hz, 1H, H-4)*, 7.41–7.38 (m, 2H, H-2, H-5), 7.35 (d, 3J = 8.0 Hz, 1H, H-6)*, 5.04 (s, 1H, CHH-5′), 5.00 (s, 1H, CHH-5′), 4.76 (dd, 3J = 12.0 Hz, 2.2 Hz, 1H, H-2′), 3.32 (dd, 2J = 15.0, 3J = 11.9 Hz, 1H, CHH-1′), 2.94 (dd, 2J = 15.0, 3J = 2.2 Hz, 1H, CHH-1′), 1.87 (d, 4J = 1.4 Hz, 3H, CH3-4′), 1.29 (s, 3H, CH3-3′), 1.23 (s, 3H, CH3-3′) (* the assignments are interconvertible); 13C NMR (126 MHz, CDCl3): δ (ppm) = 147.3 (s, C-4′), 138.3 (s, C-3), 133.3 (d, C-6)*, 132.4 (d, C-2), 131.2 (d, C-4)*, 129.9 (d, C-5), 118.6 (s, CN), 114.5 (t, C-5′), 113.2 (s, C-1), 96.0 (d, C-2′), 43.0 (s, C-3′), 34.4 (t, C-1′), 24.7 [q, (C-3′)CH3], 21.6 [q, (C-3′)CH3], 19.6 [q, (C-4′)CH3] (* the assignments are interconvertible); MS (EI): m/z (%) = 196 (15) [C14H14N]+, 170 (58) [C12H12N]+, 116 (100) [C8H6N]+; HRMS (ESI): calcd for C15H19N2O2+ [M + H]+: 259.1441; found: 259.1439.
(cm−1) = 3061, 2973, 2960, 1541, 1372, 1153, 1135, 1033, 876, 807, 754; 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.41 (dd, 3J = 7.6 Hz, 4J = 1.6 Hz, 1H, H-6), 7.28–7.18 (m, 2H, H-5, H-4), 7.16 (dd, 3J = 7.5 Hz, 4J = 1.9 Hz, 1H, H-3), 4.99 (d, 3J = 10.2 Hz, 1H, H-4′), 4.43 (d, 3J = 10.2 Hz, 1H, H-1′), 1.26 (s, 3H, CH3-2′), 1.24 (s, 3H, CH3-3′), 1.19 (s, 3H, CH3-3′), 0.73 (s, 3H, CH3-2′); 13C NMR (101 MHz, CDCl3): δ (ppm) = 134.7 (s, C-2), 134.1 (s, C-1), 130.3 (d, C-6), 128.4 (d, C-5/C-4)*, 128.1 (d, C-3), 126.8 (d, C-5/C-4)*, 84.4 (d, C-4′), 46.8 (d, C-1′), 44.5 (s, C-2′), 40.5 (s, C-3′), 24.9 [q, (C-3′)CH3], 22.7 [q, (C-2′)CH3], 21.8 [q, (C-2′)CH3], 19.4 [q, (C-3′)CH3] (* the assignments are interconvertible); MS (EI): m/z (%) = 221 (32) [M − NO2]+, 179 (100) [M − NO2 − C3H6]+, 125 (28) [C7H6Cl]+; HRMS (ESI): calcd for C14H19ClNO2+ [M + H]+: 268.1099; found: 268.1094.
(cm−1) = 3445, 3008, 2930, 1597, 1448, 1386, 1225, 1052, 1025, 992, 760; 1H NMR (500 MHz, CDCl3): δ (ppm) 8.59–8.37 (m, 2H, H-5, H-6), 7.43 (dt, 3J = 7.9 Hz, 4J = 2.0 Hz, 1H, H-3), 7.31–7.27 (m, 1H, H-4), 4.90 (d, 3J = 10.0 Hz, 1H, H-4′), 3.96 (d, 3J = 10.0 Hz, 1H, H-1′), 1.25 (s, 3H, CH3-2′), 1.19 (s, 3H, CH3-3′), 1.16 (s, 3H, CH3-2′), 0.73 (s, 3H, CH3-3′); 13C NMR (126 MHz, CDCl3): δ (ppm) = 148.7 (d, C-5/C-6)*, 148.6 (d, C-5/C-6)*, 134.5 (d, C-3), 131.9 (s, C-2), 123.4 (d, C-4), 84.2 (d, C-4′), 47.4 (d, C-1′), 45.3 (s, C-3′), 39.4 (s, C-2′), 24.2 [q, (C-2′)CH3], 22.7 [q, (C-3′)CH3], 21.6 [q, (C-2′)CH3], 19.3 [q, (C-3′)CH3] (* the assignments are interconvertible); MS (EI): m/z (%) = 188 (100) [M − NO2]+, 146 (72) [M − NO2 − C3H6]+, 132 (40) [C9H10N]+; HRMS (ESI): calcd for C13H19N2O2+ [M + H]+: 235.1441; found: 235.1441.
(cm−1) = 2954, 1537, 1459, 1367, 1139, 858, 810, 755; 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.85–7.79 (m, 3H, Har), 7.54–7.52 (br. s., 1H, Har), 7.51–7.43 (m, 2H, Har), 7.26–7.23 (m, 1H, Har), 5.06 (d, 3J = 10.1 Hz, 1H, H-4′), 4.14 (d, 3J = 10.1 Hz, 1H, H-1′), 1.28 (s, 3H, CH3-2′), 1.26 (s, 3H, CH3-3′), 1.20 (s, 3H, CH3-2′), 0.73 (s, 3H, CH3-3′); 13C NMR (101 MHz, CDCl3): δ (ppm) = 134.2 (s, C-2), 133.5 (s, C-5/C-10)*, 132.6 (s, C-5/C-10)*, 128.4 (d, CarH), 127.8 (d, CarH), 126.5 (d, CarH), 125.9 (d, CarH), 125.7 (d, CarH), 125.1 (d, CarH), 85.0 (d, C-4′), 49.6 (d, C-1′), 45.1 (s, C-3′), 39.5 (s, C-2′), 24.4 [q, (C-2′)CH3], 22.8 [q, (C-3′)CH3], 21.6 [q, (C-2′)CH3], 19.5 [q, (C-3′)CH3] (* the assignments are interconvertible); MS (EI): m/z (%) = 237 (28) [M − NO2]+, 181 (100) [M − NO2 − C4H8]+, 127 (10) [C10H7]+; HRMS (ESI): calcd for C18H22NO2+ [M + H]+: 284.1645; found: 284.1646.
:88). The analytical data obtained matched those reported in the literature.13
:56). Rf = 0.45 (P/Et2O = 19/1); IR: (cm−1) = 3063, 3030, 2965, 1542, 1455, 1366, 784; 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.34 (t, 3J = 7.5 Hz, 2H, meta-Har), 7.29–7.24 (m, 1H, para-Har), 7.23–7.20 (m, 2H, ortho-Har), 5.27 (virt. q, 3J ≅ 3J = 8.7 Hz, 1H, H-4′), 3.98 (d, 3J = 9.1 Hz, 1H, H-1′), 2.40 (dd, 2J = 11.9 Hz, 3J = 8.5 Hz, 1H, CHH-3), 2.30 (dd, 2J = 11.9 Hz, 3J = 8.6 Hz, 1H, CHH-3), 1.76 (dq, 2J = 14.8 Hz, 3J = 7.5 Hz, 1H, CHHCH3), 1.64 (dq, 2J = 14.8 Hz, 3J = 7.5 Hz, 1H, CHHCH3), 1.30–1.19 (m, 2H, CH2CH3), 0.96 (t, 3J = 7.5 Hz, 3H, CH2CH3), 0.60 (t, 3J = 7.4 Hz, 3H, CH2CH3); 13C NMR (126 MHz, CDCl3): δ (ppm) = 136.6 (s, Car), 128.6 (d, 2C, meta-CarH), 127.5 (d, 2C, ortho-CarH), 127.2 (d, para-CarH), 76.6 (d, C-4′), 53.8 (d, C-1′), 41.8 (s, C-2′), 33.7 (t, C-3′), 31.7 (t, CH2CH3), 26.4 (t, CH2CH3), 8.62 (q, CH2CH3), 7.98 (q, CH2CH3); MS (EI): m/z (%) = 187 (4) [M − NO2]+, 157 (12) [M − NO2 − C2H6]+, 117 (100) [C9H9]+; HRMS (ESI): calcd for C14H20NO2+ [M + H]+: 234.1488; found: 234.1489.
(cm−1) = 3062, 3032, 2923, 1543, 1474, 1368, 1218, 1095, 1051, 1019, 814; 1H NMR (500 MHz, C6D6): δ (ppm) = 6.98–6.96 (m 3H, meta-Har, para-Har), 6.84 (t, 3J = 7.5 Hz, 1H, H-5), 6.79 (d, 3J = 7.5 Hz, 1H, H-4), 6.61–6.56 (m, 2H, ortho-Har), 6.45 (t, 3J = 7.5 Hz, 1H, H-6), 6.29 (d, 3J = 7.5 Hz, 1H, H-7), 5.13 (dd, 3J = 7.4 Hz, 4.2 Hz, 1H, H-2a), 4.97 (ddd, 3J = 9.4 Hz, 4.2 Hz, 4J = 1.5 Hz, 1H, H-1), 3.72 (virt. t, 3J ≅ 3J = 9.3 Hz, 1H, H-2), 3.50 (virt. t, 3J ≅ 3J = 8.4 Hz, 1H, H-7b); 13C NMR (101 MHz, C6D6): δ (ppm) = 160.9 (s, C-3a), 135.4 (s, C-2′), 129.6 (d, C-5), 128.6 (d, meta-CarH/para-CarH)*, 128.3 (d, C-7), 127.9 (d, meta-CarH/para-CarH)*, 127.6 (d, 2C, ortho-CarH)*, 124.9 (s, C-7a), 121.6 (d, C-6), 111.5 (d, C-4), 85.6 (d, C-1), 80.6 (d, C-2a), 45.6 (d, C-2), 44.6 (d, C-7b) (* the exact assignment was not possible due to significant overlap with the solvent C6D6); MS (EI): m/z (%) = 221 (12) [M − NO2]+, 118 (100) [C8H6O]+, 90 (8); HRMS (ESI): calcd for C16H14NO3+ [M + H]+: 268.0968; found: 268.0970.
:19:29) as an orange coloured oil. Starting material 1a was recovered as a mixture of isomers (4.50 mg, 30.2 μmol, 15%, cis/trans = 55:45). NMR data are given for the major diastereoisomer depicted in Scheme 2. Rf = 0.06 (P/Et2O = 4/1); IR: (cm−1) = 3031, 2923, 1545, 1375, 1132, 1043, 874, 751; 1H NMR (400 MHz, C6D6): δ (ppm) = 7.02–6.93 (m, 5H, Har), 4.26 (virt. t, 3J ≅ 3J = 7.8 Hz, 1H, H-7), 3.81 (virt. t, 3J ≅ 3J = 8.6 Hz, 1H, H-6), 3.56 (dd, 3J = 9.8 Hz, 7.3 Hz, 1H, H-8), 3.40–3.28 (m, 2H, CHH-3, CHH-4), 3.19–3.15 (m, 2H, CHH-3, CHH-4), 2.99 (d, 3J = 9.8 Hz, 1H, H-1); 13C NMR (101 MHz, C6D6): δ (ppm) = 136.5 (s, Car), 129.0 (d, 2C, ortho-CarH)*, 127.9 (d, 2C, meta-CarH)*, 126.9 (d, para-CarH), 82.3 (d, C-7), 77.9 (d, C-6), 75.2 (d, C-1), 68.3 (t, C-3), 68.1 (t, C-4), 51.2 (d, C-8) (* the assignments are interconvertible); MS (EI): m/z (%) = 235 (16) [M]+, 189 (52) [M − NO2]+, 117 (72) [C9H9]+, 91 (100) [C7H7]+; HRMS (ESI): calcd for C12H14NO4+ [M + H]+: 236.0917; found: 236.0918.
:33) as a pale-yellow coloured oil. NMR data are given for the major diastereoisomer depicted in Scheme 2. Rf = 0.69 (P/Et2O = 9/1); IR: (cm−1) = 3028, 1542, 1496, 1368, 1028, 824, 770; 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.17–7.13 (m, 6H, Har), 7.00–6.98 (m, 2H, Har), 6.83–6.77 (m, 2H, Har), 5.13 (virt. q, 3J ≅ 3J = 9.0 Hz, 1H, H-3′), 4.07 (d, 3J = 9.5 Hz, 1H, H-4′), 3.01 (dd, 2J = 12.3 Hz, 3J = 8.1 Hz, 1H, CHH-2′), 2.60 (dd, 2J = 12.4 Hz, 3J = 9.2 Hz, 1H, CHH-2′), 1.43 [tt, 3J = 8.3 Hz, 5.6 Hz, 1H, CH(CH2)2], 0.77–0.66 [m, 2H, CH(CH2)2], 0.57 [virt. tt, 2J ≅ 3J ≅ 3J = 8.6 Hz, 3J = 5.5 Hz, 1H, CH(CH2)2], 0.44 [virt. dq, 2J = 9.0 Hz, 3J ≅ 3J = 5.5 Hz, 1H, CH(CH2)2]; 13C NMR (101 MHz, CDCl3): δ (ppm) = 141.1 (s, C-1a), 136.2 (s, C-4a), 128.4 (d, 2C, CarH), 128.3 (d, 2C, CarH), 128.0 (d, 2C, CarH), 127.8 (d, CarH), 127.5 (d, CarH), 126.8 (d, 2C, CarH), 76.9 (d, C–3′), 55.1 (d, C-4′), 47.3 (s, C-1′), 32.1 (t, C-2′), 22.6 [d, CH(CH2)2], 3.21 [t, CH(CH2)2], 2.11 [t, CH(CH2)2]; MS (EI): m/z (%) = 247 (4) [M − NO2]+, 205 (32) [M − NO2 − C3H6]+, 117 (100) [C9H9]+; HRMS (ESI): calcd for C19H20NO2+ [M + H]+: 294.1488; found: 294.1488.
:23) as a yellow coloured oil. NMR data are given for the major diastereoisomer depicted in Scheme 2. Rf = 0.70 (P/Et2O = 19/1); IR: (cm−1) = 3063, 3031, 2958, 1546, 1480, 1395, 1368, 1252, 1146, 1029, 870, 833; 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.33–7.31 (m, 5H, Har), 5.13 (virt. q, 3J ≅ 3J = 8.5 Hz, 1H, H-3), 4.24 (d, 3J = 8.6 Hz, 1H, H-2), 2.93 (dd, 2J = 13.2 Hz, 3J = 8.2 Hz, 1H, CHH-4), 2.50 (dd, 2J = 13.2 Hz, 3J = 8.6 Hz, 1H, CHH-4), 0.97 [s, 9H, C(CH3)3], 0.12 [s, 9H, OSi(CH3)3]; 13C NMR (126 MHz, CDCl3): δ (ppm) = 136.4 (s, Car), 129.0 (d, 2C, ortho-CarH), 128.1 (d, 2C, meta-CarH), 127.3 (d, para-CarH), 83.3 (s, C-1), 78.2 (d, C-3), 52.2 (d, C-2), 37.9 [s, C(CH3)3], 34.3 (t, C-4), 25.8 [q, 3C, C(CH3)3], 2.6 [q, 3C, OSi(CH3)3]; MS (EI): m/z (%) = 275 (17) [M − NO2]+, 219 (18) [M − NO2 − C(CH3)3]+, 117 (100) [C9H9]+, 73 (36) [Si(CH3)3]+; HRMS (ESI): calcd for C17H28NO3Si+ [M + H]+: 322.1833; found: 322.1833.
:39) as a colourless oil. NMR data are given for the major diastereoisomer depicted in Scheme 2. Rf = 0.70 (P/Et2O = 9/1); IR: (cm−1) = 3004, 2926, 1542, 1497, 1364, 1264, 1045, 1018, 891, 822, 758; 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.38–7.32 (m, 2H, meta-Har), 7.29–7.25 (m, 3H, ortho-Har, para-Har), 5.35 (dd, 3J = 10.1 Hz, 8.8 Hz, 1H, H-6), 4.06 (d, 3J = 8.8 Hz, 1H, H-7), 3.09 (virt. t, 3J ≅ 3J = 9.2 Hz, 1H, H-5), 2.00–1.91 (m, 4H, CH2-3, CHH-2, CHH-4), 1.89–1.77 (m, 1H, CHH-2), 1.62–1.44 (m, 1H, CHH-4), −0.14 [s, 9H, OSi(CH3)3]; 13C NMR (126 MHz, CDCl3): δ (ppm) = 136.5 (s, Car), 129.5 (d, 2C, ortho-CarH), 128.8/128.5 (d, 2C meta-CarH), 127.5/127.3 (d, para-CarH), 83.5 (s, C-1), 81.0 (d, C-6), 51.9 (d, C-7), 49.9 (d, C-5), 40.1 (t, CH2-2), 26.1 (t, CH2-4), 25.9 (t, CH2-3), 1.64 [q, 3C, OSi(CH3)3]; MS (EI): m/z (%) = 259 (88) [M − NO2]+, 169 (60) [M − NO2 − OSi(CH3)3]+, 91 (28) [C7H7]+, 73 (100) [Si(CH3)3]+; HRMS (ESI): calcd for C16H24NO3Si+ [M + H]+: 306.1522; found: 306.1522.
(cm−1) = 3066, 2931, 2870, 1542, 1460, 1371, 993, 912, 751; 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.15–7.09 (m, 3H, H-3, H-4, H-5), 7.07–7.03 (m, 1H, H-6), 5.83 (ddt, 3J = 17.0 Hz, 10.2 Hz, 6.7 Hz, 1H, H-4′), 5.02 (virt. dq, 3J = 17.2 Hz, 2J ≅ 4J = 1.7 Hz, 1H, CHH-5′), 4.98–4.92 (m, 2H, CHH-5′, H-4′′), 4.15 (d, 3J = 10.2 Hz, 1H, H-1′′), 2.76 (ddd, 2J = 14.0 Hz, 3J = 10.5 Hz, 5.3 Hz, 1H, CHH-1′), 2.46 (ddd, 2J = 14.0 Hz, 3J = 10.6 Hz, 6.0 Hz, 1H, CHH-1′), 2.19–2.03 (m, 2H, H-3′), 1.82–1.72 (m, 1H, CHH-2′), 1.70–1.58 (m, 1H, CHH-2′), 1.18 (s, 3H, CH3-3′′), 1.10 (s, 3H, CH3-3′′), 1.09 (s, 3H, CH3-2′′), 0.67 (s, 3H, CH3-2′′); 13C NMR (126 MHz, CDCl3): δ (ppm) = 141.7 (s, C-1), 138.6 (d, C-4′), 133.2 (s, C-2), 129.8 (d, C-6), 127.1 (d, C-5)*, 127.0 (d, C-3), 125.9 (d, C-4)*, 115.3 (t, C-5′), 85.3 (d, C-4′′), 45.6 (d, C-1′′), 44.4 (s, C-2′′), 40.1 (s, C-3′′), 33.8 (t, C-3′), 32.4 (t, C-1′), 30.6 (t, C-2′), 24.6 [q, (C-2′′)CH3], 22.8 [q, (C-3′′)CH3], 21.8 [q, (C-2′′)CH3], 19.4 [q, (C-3′′)CH3] (* the assignments are interconvertible); MS (EI): m/z (%) = 301 (2) [M]+, 255 (20) [M − NO2]+, 199 (49) [M − NO2 − C3H6]+, 143 (100) [C11H11]+; HRMS (ESI): calcd for C19H27NO2+ [M + H]+: 302.2115; found: 302.2115.
(cm−1) = 3060, 2959, 2866, 2604, 1566, 1458, 1449, 1358, 1337, 1270, 1132, 885, 810; 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.29–7.21 (m, 2H, meta-HPh), 7.19–7.08 (m, 3H, ortho-HPh, para-HPh), 3.37 (d, 3J = 9.9 Hz, 1H, H-1), 2.86 (d, 3J = 9.9 Hz, 1H, H-4), 1.51 (br. s, 2H, NH2), 1.03 (s, 3H, CH3-2), 1.01 (s, 3H, CH3-3), 0.93 (s, 3H, CH3-2), 0.59 (s, 3H, CH3-3); 13C NMR (101 MHz, CDCl3): δ (ppm) = 139.6 (s, CPh), 128.3 (d, 2C, meta-CPhH), 127.7 (d, 2C, ortho-CPhH), 126.1 (d, para-CPhH), 56.8 (d, C-4), 55.5 (d, C-1), 41.7 (s, C-2), 39.6 (s, C-3), 24.2 [q, (C-3)CH3], 22.6 [q, (C-2)CH3], 21.1 [q, (C-3)CH3], 18.7 [q, (C-2)CH3]; MS (EI, 70 eV): m/z (%) = 132 (5) [M − C4H9N]+, 119 (100) [M − C4H9N − CH3]+, 91 (13) [C7H7]+, 71 (31) [C4H9]+, 56 (11); HRMS (ESI): calcd for C14H22N+ [M + H]+: 204.1741; found: 204.1748.
(cm−1) = 3079, 3004, 1542, 1449, 1369, 1017, 822, 758; 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.44–7.20 (m, 5H, Har), 5.20 (virt. q, 3J ≅ 3J = 8.7 Hz, 1H, H-4′), 3.97 (d, 3J = 9.2 Hz, 1H, H-1′), 2.06 (dd, 2J = 12.2 Hz, 3J = 8.7 Hz, 1H, CHH-3′), 1.71 (dd, 3J = 12.2 Hz, 3J = 8.3 Hz, 1H, CHH-3′), 1.09 [tt, 3J = 8.4 Hz, 5.5 Hz, 1H, CH(CH2)2], 0.60–0.25 [m, 6H, CH(CH2)2, CH(CH2)2], 0.20–0.13 [m, 1H, CH(CH2)2]; 13C NMR (101 MHz, CDCl3): δ (ppm) = 136.4 (s, Car), 128.5 (d, 2C, meta-CarH), 127.7 (d, 2C, ortho-CarH), 127.2 (d, para-CarH), 76.6 (d, C-4′), 55.5 (d, C-1′), 41.7 (s, C-2′), 27.8 (t, C-3′), 20.2 [d, CH(CH2)2], 14.6 [d, CH(CH2)2], 1.93 [t, CH(CH2)2], 1.74 [t, CH(CH2)2], 0.90 [t, CH(CH2)2], 0.72 [t, CH(CH2)2]; MS (EI): m/z (%) = 211 (4) [M − NO2]+, 169 (16) [M − NO2 − C3H6]+, 117 (100) [C9H9]+; HRMS (ESI): calcd for C16H20NO2+ [M + H]+: 258.1448; found: 258.1449. 7: Rf = 0.69 (P/Et2O = 19/1); IR: (cm−1) = 3419, 2928, 1722, 1547, 1367, 1023, 856, 791; 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.17–7.05 (m, 4H, Har), 5.45 (ttd, 3J = 7.1 Hz, 4J = 2.5 Hz, 1.6 Hz, 1H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2CH3), 4.90 (virt. q, 3J ≅ 3J = 7.3 Hz, 1H, H-1), 3.90 (virt. t, 3J ≅ 3J = 7.5 Hz, 1H, H-9b), 3.07–2.94 (m, 2H, H-3a, CHH-2), 2.90 (dddd, 2J = 17.3 Hz, 3J = 7.9 Hz, 4J = 2.7 Hz, 1.4 Hz, 1H, CHH-2), 2.80–2.75 (m, 1H, CHH-5), 2.73–2.68 (m, 1H, CHH-5), 2.07–1.98 (m, 2H, CCHCH2CH3), 1.84 (ddt, 2J = 13.8 Hz, 3J = 6.2 Hz, 4.7 Hz, 1H, CHH-4), 1.67 (dtd, 2J = 13.8 Hz, 3J = 9.5 Hz, 4.8 Hz, 1H, CHH-4), 1.00 (t, 3J = 7.5 Hz, 3H, CH3); 13C NMR (126 MHz, CDCl3): δ (ppm) = 138.7 (s, C-3), 137.1 (s, C-5a), 134.4 (s, C-9a), 129.3 (d, C-6), 128.6 (d, C-9), 127.2 (d, C-8), 126.6 (d, C-7), 125.9 (d, CCHCH2CH3), 92.2 (d, C-1), 47.7 (d, C-9b), 42.0 (d, C-3a), 34.6 (t, C-2), 27.8 (t. C-5), 27.3 (t, C-4), 22.8 (t, CCHCH2CH3), 14.1 (q, CH3); MS (EI): m/z (%) = 181 (100) [M − NO2 − C2H4]+, 167 (40) [C13H11]+, 128 (16) [C10H8]+; HRMS (ESI): calcd for C16H20NO2+ [M + H]+: 258.1448; found: 258.1449.
(cm−1) = 3423, 2955, 1720, 1550, 1437, 1368, 1284, 1105, 762; 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.80–7.77 (m, 2H, H-6, H-8), 7.14 (d, 3J = 8.6 Hz, 1H, H-9), 5.46 (virt. tq, 3J = 6.9 Hz, 4J ≅ 4J = 2.4 Hz, 1H, CCHCH2CH3), 4.88 (virt. q, 3J ≅ 3J = 7.3 Hz, 1H, H-1), 3.96–3.91 (m, 1H, H-9b), 3.90 (s, 3H, CO2CH3), 3.11–2.95 (m, 2H, H-3a, CHH-2), 2.95–2.80 (m, 2H, CHH-2, CHH-5), 2.78–2.66 (m, 1H, CHH-5), 2.08–1.96 (m, 2H, CCHCH2CH3), 1.91–1.81 (m, 1H, CHH-4), 1.68 (dtd, 2J = 13.9 Hz, 3J = 9.3 Hz, 4.8 Hz, 1H, CHH-4), 1.00 (t, 3J = 7.5 Hz, 3H, CCHCH2CH3); 13C NMR (126 MHz, CDCl3): δ (ppm) = 138.7 (s, C-3), 137.1 (s, C-5a), 134.4 (s, C-9a), 129.3 (d, C-6), 128.6 (d, C–9), 127.2 (d, C-8), 126.6 (d, C-7), 125.9 (d, CCHCH2CH3), 92.2 (d, C-1), 52.3 (q, CO2CH3) 47.7 (d, C-9b), 42.0 (d, C-3a), 34.6 (t, C-2), 27.8 (t. C-5), 27.3 (t, C-4), 22.8 (t, CCHCH2CH3), 14.1 (q, CCHCH2CH3); MS (EI): m/z (%) = 284 (19) [M − OCH3]+, 253 (54), 149 (100) [C9H9O2]+, 115 (49), 91 (63) [C7H7]+; HRMS (ESI): calcd for C18H22NO2+ [M + H]+: 316.1543; found: 316.1545.
(cm−1) = 3418, 2928, 1711, 1547, 1368, 1261, 1024, 858, 803, 752; 1H NMR (400 MHz, CDCl3): δ (ppm) = 5.45 (tq, 3J = 7.1 Hz, 4J = 2.4 Hz, 1H, CCHCH2CH3), 4.90 (virt. q, 3J ≅ 3J = 7.4 Hz, 1H, H-1), 3.90 (virt. t, 3J ≅ 3J = 7.6 Hz, 1H, H-9b), 3.08–2.94 (m, 2H, H-3a, CHH-2), 2.90 (dd, 2J = 17.3 Hz, 3J = 7.8 Hz, 1H, CHH-2), 2.81–2.75 (m, 1H, CHH-5), 2.69 (ddd, 2J = 16.4 Hz, 3J = 9.1 Hz, 4.8 Hz, 1H, CHH-5), 2.05–1.98 (m, 2H, CCHCH2CH3), 1.89–1.79 (m, 1H, CHH-4), 1.67 (dtd, 2J = 14.0 Hz, 3J = 9.4 Hz, 4.8 Hz, 1H, CHH-4), 0.98 (tt, 3J = 7.6 Hz, 2J = 2.1 Hz, 2H, CH2D); 13C NMR (101 MHz, CDCl3): δ (ppm) = 138.8 (s, C-3), 137.0 (s, C-5a), 134.4 (s, C-9a), 126.0 (d, CCHCH2CH2D), 92.2 (d, C-1), 47.6 (d, C-9b), 42.1 (d, C-3a), 34.6 (t, C-2), 27.7 (t. C-5), 27.3 (t, C-4), 22.8 (t, CCHCH2CH2D), 14.1 (t, 1JCD = 19.4 Hz, CCHCH2CH2D) (the aromatic signals of carbon atoms linked to deuterium atoms were not visible in the 13C-NMR spectrum); MS (EI): m/z (%) = 215 (65) [M − NO2]+, 185 (100) [M − NO2 − C2H4]+, 171 (36), 132 (20) [C10H4D4]+, 95 (6) [C7H3D4]+; HRMS (ESI): calcd for C16H15D5NO2+ [M + H]+: 263.1802; found: 263.1804.
:54) as a colourless oil. Starting material was recovered as cis-isomer cis-1a (6.60 mg, 42.3 μmol, 22%). General procedure for the [2 + 2] photocycloaddition of 1a to cis-β-methylstyrene: a solution of nitroethene (29.8 mg, 200 μmol, 1.00 equiv.) and cis-β-methylstyrene (260 μL, 236 mg, 2.00 mmol, 10.0 equiv.) in dichloromethane (5 mL, c = 20 mM) was irradiated at λmax = 419 nm for twelve hours at room temperature. Purification by column chromatography (P/Et2O = 50/1) yielded 3j/3j′ (45.8 mg, 1.12 mmol, 56%, dr = 77:23) as a colourless oil. Starting material was recovered as cis-isomer cis-1a (6.80 mg, 45.6 μmol, 23%). The analytical data obtained matched those reported in the literature.13
CHCH2CH3), 4.90 (virt. q, 3J ≅ 3J = 7.3 Hz, 1H, H-1), 3.90 (virt. t, 3J ≅ 3J = 7.5 Hz, 1H, H-9b), 3.07–2.94 (m, 2H, H-3a, CHH-2), 2.90 (dddd, 2J = 17.3 Hz, 3J = 7.9 Hz, 4J = 2.7 Hz, 1.4 Hz, 1H, CHH-2), 2.80–2.75 (m, 1H, CHH-5), 2.73–2.68 (m, 1H, CHH-5), 2.07–1.98 (m, 2H, CCHCH2CH3), 1.84 (ddt, 2J = 13.8 Hz, 3J = 6.2 Hz, 4.7 Hz, 1H, CHH-4), 1.67 (dtd, 2J = 13.8 Hz, 3J = 9.5 Hz, 4.8 Hz, 1H, CHH-4), 1.00 (t, 3J = 7.5 Hz, 3H, CH3); 13C NMR (126 MHz, CDCl3): δ (ppm) = 138.7 (s, C-3), 137.1 (s, C-5a), 134.4 (s, C-9a), 129.3 (d, C-6), 128.6 (d, C-9), 127.2 (d, C-8), 126.6 (d, C-7), 125.9 (d, CCHCH2CH3), 92.2 (d, C-1), 47.7 (d, C-9b), 42.0 (d, C-3a), 34.6 (t, C-2), 27.8 (t. C-5), 27.3 (t, C-4), 22.8 (t, CCHCH2CH3), 14.1 (q, CH3); MS (EI): m/z (%) = 181 (100) [M − NO2 − C2H4]+, 167 (40) [C13H11]+, 128 (16) [C10H8]+; HRMS (ESI): calcd for C16H20NO2+ [M + H]+: 258.1448; found: 258.1449.
(cm−1) = 3423, 2955, 1720, 1550, 1437, 1368, 1284, 1105, 762; 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.80–7.77 (m, 2H, H-6, H-8), 7.14 (d, 3J = 8.6 Hz, 1H, H-9), 5.46 (virt. tq, 3J = 6.9 Hz, 4J ≅ 4J = 2.4 Hz, 1H, CCHCH2CH3), 4.88 (virt. q, 3J ≅ 3J = 7.3 Hz, 1H, H-1), 3.96–3.91 (m, 1H, H-9b), 3.90 (s, 3H, CO2CH3), 3.11–2.95 (m, 2H, H-3a, CHH-2), 2.95–2.80 (m, 2H, CHH-2, CHH-5), 2.78–2.66 (m, 1H, CHH-5), 2.08–1.96 (m, 2H, CCHCH2CH3), 1.91–1.81 (m, 1H, CHH-4), 1.68 (dtd, 2J = 13.9 Hz, 3J = 9.3 Hz, 4.8 Hz, 1H, CHH-4), 1.00 (t, 3J = 7.5 Hz, 3H, CCHCH2CH3); 13C NMR (126 MHz, CDCl3): δ (ppm) = 138.7 (s, C-3), 137.1 (s, C-5a), 134.4 (s, C-9a), 129.3 (d, C-6), 128.6 (d, C–9), 127.2 (d, C-8), 126.6 (d, C-7), 125.9 (d, CCHCH2CH3), 92.2 (d, C-1), 52.3 (q, CO2CH3) 47.7 (d, C-9b), 42.0 (d, C-3a), 34.6 (t, C-2), 27.8 (t. C-5), 27.3 (t, C-4), 22.8 (t, CCHCH2CH3), 14.1 (q, CCHCH2CH3); MS (EI): m/z (%) = 284 (19) [M − OCH3]+, 253 (54), 149 (100) [C9H9O2]+, 115 (49), 91 (63) [C7H7]+; HRMS (ESI): calcd for C18H22NO2+ [M + H]+: 316.1543; found: 316.1545.
(cm−1) = 3418, 2928, 1711, 1547, 1368, 1261, 1024, 858, 803, 752; 1H NMR (400 MHz, CDCl3): δ (ppm) = 5.45 (tq, 3J = 7.1 Hz, 4J = 2.4 Hz, 1H, CCHCH2CH3), 4.90 (virt. q, 3J ≅ 3J = 7.4 Hz, 1H, H-1), 3.90 (virt. t, 3J ≅ 3J = 7.6 Hz, 1H, H-9b), 3.08–2.94 (m, 2H, H-3a, CHH-2), 2.90 (dd, 2J = 17.3 Hz, 3J = 7.8 Hz, 1H, CHH-2), 2.81–2.75 (m, 1H, CHH-5), 2.69 (ddd, 2J = 16.4 Hz, 3J = 9.1 Hz, 4.8 Hz, 1H, CHH-5), 2.05–1.98 (m, 2H, CCHCH2CH3), 1.89–1.79 (m, 1H, CHH-4), 1.67 (dtd, 2J = 14.0 Hz, 3J = 9.4 Hz, 4.8 Hz, 1H, CHH-4), 0.98 (tt, 3J = 7.6 Hz, 2J = 2.1 Hz, 2H, CH2D); 13C NMR (101 MHz, CDCl3): δ (ppm) = 138.8 (s, C-3), 137.0 (s, C-5a), 134.4 (s, C-9a), 126.0 (d, CCHCH2CH2D), 92.2 (d, C-1), 47.6 (d, C-9b), 42.1 (d, C-3a), 34.6 (t, C-2), 27.7 (t. C-5), 27.3 (t, C-4), 22.8 (t, CCHCH2CH2D), 14.1 (t, 1JCD = 19.4 Hz, CCHCH2CH2D) (the aromatic signals of carbon atoms linked to deuterium atoms were not visible in the 13C-NMR spectrum); MS (EI): m/z (%) = 215 (65) [M − NO2]+, 185 (100) [M − NO2 − C2H4]+, 171 (36), 132 (20) [C10H4D4]+, 95 (6) [C7H3D4]+; HRMS (ESI): calcd for C16H15D5NO2+ [M + H]+: 263.1802; found: 263.1804.
:54) as a colourless oil. Starting material was recovered as cis-isomer cis-1a (6.60 mg, 42.3 μmol, 22%). General procedure for the [2 + 2] photocycloaddition of 1a to cis-β-methylstyrene: a solution of nitroethene (29.8 mg, 200 μmol, 1.00 equiv.) and cis-β-methylstyrene (260 μL, 236 mg, 2.00 mmol, 10.0 equiv.) in dichloromethane (5 mL, c = 20 mM) was irradiated at λmax = 419 nm for twelve hours at room temperature. Purification by column chromatography (P/Et2O = 50/1) yielded 3j/3j′ (45.8 mg, 1.12 mmol, 56%, dr = 77:23) as a colourless oil. Starting material was recovered as cis-isomer cis-1a (6.80 mg, 45.6 μmol, 23%). The analytical data obtained matched those reported in the literature.13
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the European Research Council under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 665951 – ELICOS) is gratefully acknowledged.Notes and references
- B. Priebs, Justus Liebigs Ann. Chem., 1884, 225, 319–364 CrossRef.
- J. Meisenheimer and F. Heim, Justus Liebigs Ann. Chem., 1907, 355, 260–267 CrossRef.
- D. B. Miller, P. W. Flanagan and H. Shechter, J. Am. Chem. Soc., 1972, 94, 3912–3918 CrossRef CAS.
- G. R. Desiraju and V. R. Pedireddi, J. Chem. Soc., Chem. Commun., 1989, 1112–1113 RSC.
- O. L. Chapman, A. A. Griswold, E. Hoganson, G. Lenz and J. Reasoner, Pure Appl. Chem., 1964, 9, 585–590 CAS.
- E. D. Hoganson, Ph.D. thesis, Iowa State University, 1965 Search PubMed.
- T. Majima, C. Pac and H. Sakurai, J. Am. Chem. Soc., 1980, 102, 5265–5273 CrossRef CAS.
- D. Ramkumar and S. Sankararaman, J. Chem. Soc., Perkin Trans. 2, 1996, 939–941 RSC.
- A. A. Russell, O. L. Chapman, J. T. Magner and M. Selke, J. Chem. Educ., 1996, 73, 854–856 CrossRef.
- S. M. Stevenson, R. F. Higgins, M. P. Shores and E. M. Ferreira, Chem. Sci., 2017, 8, 654–660 RSC.
- Review: S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS PubMed.
- Recent examples: (a) S. Stegbauer, C. Jandl and T. Bach, Angew. Chem., Int. Ed., 2018, 57, 14593–14596 CrossRef CAS PubMed; (b) A. Hölzl-Hobmeier, A. Bauer, A. V. Silva, S. M. Huber, C. Bannwarth and T. Bach, Nature, 2018, 564, 240–243 CrossRef PubMed.
- L. Mohr and T. Bach, Synlett, 2017, 28, 2946–2950 CrossRef CAS.
- L. Henry, Comptes Rendus, 1895, 120, 1265–1268 CAS.
- M. Sathish, J. Chetna, N. H. Krishna, N. Shankaraiah, A. Alarifi and A. Kamal, J. Org. Chem., 2016, 81, 2159–2165 CrossRef CAS PubMed.
- (a) E. A. Braude, E. R. H. Jones and G. G. Rose, J. Chem. Soc., 1947, 1104–1105 RSC; (b) D. J. Cowley, J. Chem. Soc., Perkin Trans. 2, 1975, 1576–1580 RSC.
- (a) A. L. Bluhm and J. Weinstein, J. Am. Chem. Soc., 1965, 87, 5511–5512 CrossRef CAS; (b) J. A. Sousa, J. Weinstein and A. L. Bluhm, J. Org. Chem., 1969, 34, 3320–3323 CrossRef CAS; (c) D. B. Miller, P. W. Flanagan and H. Shechter, J. Org. Chem., 1976, 41, 2112–2120 CrossRef CAS; (d) M. Z. Kassaee and E. Vessally, J. Photochem. Photobiol., A, 2005, 172, 331–336 CrossRef CAS.
- A. Nandi, R. Ghosh and D. K. Palit, J. Photochem. Photobiol., A, 2016, 321, 171–179 CrossRef CAS and references cited therein.
- S. Chandrasekhar, B. Tiwari, B. B. Parida and C. R. Reddy, Tetrahedron: Asymmetry, 2008, 19, 495–499 CrossRef CAS.
- Nitroethene 1o was prepared from the corresponding aldehyde ( M. M. Coulter, P. K. Dornan and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 6932–6933 CrossRef CAS PubMed ) in a Henry reaction.
- (a) G. W. Kabalka and R. S. Varma, Org. Prep. Proced. Int., 1987, 19, 283–328 CrossRef CAS; (b) R. Ballini, E. Marcantoni and M. Petrini, in Amino Group Chemistry: From Synthesis to the Life Sciences, ed. A. Ricci, Wiley-VCH, Weinheim, 2008, pp. 93–148 Search PubMed.
- G. Talavera, E. Reyes, J. L. Vicario and L. Carrillo, Angew. Chem., Int. Ed., 2012, 51, 4104–4107 CrossRef CAS PubMed.
- (a) A. Barański and E. Cholewka, Chem. Pap., 1991, 45, 449–455 Search PubMed; (b) S.-Q. Zhang, H.-G. Wang, K.-M. Pei, X. Zheng and D. L. Phillips, J. Chem. Phys., 2007, 126, 194505 CrossRef PubMed.
- S. Ahuja, R. Raghunathan, E. Kumarasamy, S. Jockusch and J. Sivaguru, J. Am. Chem. Soc., 2018, 140, 13185–13189 CrossRef CAS PubMed and references cited therein.
- (a) A. Rudolph and A. C. Weedon, Can. J. Chem., 1990, 68, 1590–1597 CrossRef CAS; (b) S. Hu and D. C. Neckers, J. Org. Chem., 1997, 62, 755–757 CrossRef CAS PubMed; (c) C. Y. Gan and J. N. Lambert, J. Chem. Soc., Perkin Trans. 1, 1998, 2363–2372 RSC; (d) S. C. Coote, A. Pöthig and T. Bach, Chem. – Eur. J., 2015, 21, 6906–6912 CrossRef CAS PubMed.
- G. Hu, J. Xu and P. Li, Org. Lett., 2014, 16, 6036–6039 CrossRef CAS PubMed.
- (a) W. L. Dilling, T. E. Tabor, F. P. Boer and P. P. North, J. Am. Chem. Soc., 1970, 92, 1399–1400 CrossRef CAS; (b) N. P. Peet, R. L. Cargill and D. F. Bushey, J. Org. Chem., 1973, 38, 1218–1221 CrossRef CAS; (c) N. C. Yang, M. Kimura and W. Eisenhardt, J. Am. Chem. Soc., 1973, 95, 5058–5060 CrossRef CAS; (d) J. F. D. Kelly, J. M. Kelly and T. B. H. McMurry, J. Chem. Soc., Perkin Trans. 2, 1999, 1933–1941 RSC.
- (a) D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259–271 CrossRef CAS; (b) D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC; (c) L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed.
- All redox potentials are given as potentials against a saturated calomel electrode (SCE). For the redox potential of 1a, see: J. A. Squella, J. C. Sturm, B. Weiss-Lopez, M. Bontá and L. J. Núñez-Vergara, J. Electroanal. Chem., 1999, 466, 90–98 CrossRef CAS.
- M. Patz, H. Mayr, J. Maruta and S. Fukuzumi, Angew. Chem., Int. Ed., 1995, 34, 1225–1227 CrossRef CAS.
- C. Huo, X. Jia, W. Zhang, L. Yang, J. Lü and Z.-L. Liu, Synlett, 2004, 251–254 CAS.
- S. Fukuzumi, M. Fujita, J. Otera and Y. Fujita, J. Am. Chem. Soc., 1992, 114, 10271–10278 CrossRef CAS.
- N. P. Schepp and L. J. Johnston, J. Am. Chem. Soc., 1996, 118, 2872–2881 CrossRef CAS.
- (a) M. A. Fox, Photochem. Photobiol., 1990, 52, 617–627 CrossRef CAS; (b) F. Müller and J. Mattay, Chem. Rev., 1993, 93, 99–117 CrossRef; (c) M. Julliard and M. Chanon, Chem. Rev., 1983, 83, 425–506 CrossRef CAS; (d) N. Hoffmann, J. Photochem. Photobiol., C, 2008, 9, 43–60 CrossRef CAS; (e) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- F. D. Lewis and R. J. DeVoe, Tetrahedron, 1982, 38, 1069–1077 CrossRef CAS.
- For an example of a sensitized intermolecular [2 + 2] photocycloaddition, see: T. Lei, C. Zhou, M.-Y. Huang, L.-M. Zhao, B. Yang, C. Ye, H. Xiao, Q.-Y. Meng, V. Ramamurthy, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2017, 56, 15407–15410 CrossRef CAS PubMed.
- For the emission spectrum, see: R. Alonso and T. Bach, Angew. Chem., Int. Ed., 2014, 53, 4368–4371 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and full characterization for all starting materials (1) and products (2, 3, 4, 6, 7), emission spectrum of 1a, quantum yield for 2a. CCDC 1915359. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/C9OB01146C |
| This journal is © The Royal Society of Chemistry 2019 |