
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A hydroxamic-acid-containing nucleoside inhibits DNA repair nuclease SNM1A†
William
Doherty
 a,
Eva-Maria
Dürr
a,
Hannah T.
Baddock
b,
Sook Y.
Lee
bc,
Peter J.
McHugh
b,
Tom
Brown
c,
Mathias O.
Senge
d,
Eoin M.
Scanlan
a and
Joanna F.
McGouran
*a
a,
Eva-Maria
Dürr
a,
Hannah T.
Baddock
b,
Sook Y.
Lee
bc,
Peter J.
McHugh
b,
Tom
Brown
c,
Mathias O.
Senge
d,
Eoin M.
Scanlan
a and
Joanna F.
McGouran
*a
aSchool of Chemistry and Trinity Biomedical Sciences Institute, Trinity College Dublin, 152-160 Pearse St, Dublin 2, Ireland. E-mail: jmcgoura@tcd.ie
bDepartment of Oncology, MRC Weatherall Institute of Molecular Medicine, University of Oxford, John Radcliffe Hospital, Oxford OX3 9DS, UK
cDepartment of Chemistry, University of Oxford, 12 Mansfield Road, Oxford OX1 3TA, UK
dMolecular Medicine, Trinity Translational Medicine Institute, Trinity Centre for Health Sciences, Trinity College Dublin, The University of Dublin, St James's Hospital, Dublin 8, Ireland
First published on 5th August 2019
Abstract
Nine modified nucleosides, incorporating zinc-binding pharmacophores, have been synthesised and evaluated as inhibitors of the DNA repair nuclease SNM1A. The series included oxyamides, hydroxamic acids, hydroxamates, a hydrazide, a squarate ester and a squaramide. A hydroxamic acid-derived nucleoside inhibited the enzyme, offering a novel approach for potential therapeutic development through the use of rationally designed nucleoside derived inhibitors.
Introduction
SNM1A is a DNA damage repair enzyme implicated in interstrand crosslink (ICL) repair.1 It digests DNA with a 5′-to-3′ polarity past lesions by hydrolysing the phosphodiester backbone of DNA, producing predominantly mononucleotide products.2 ICL repair factors are associated with the ageing process,1 certain genetic diseases,3 and resistance to cancer therapy.4 As cells depleted in SNM1A show higher sensitivity to ICLs introduced by several anticancer crosslinking agents,5,6 SNM1A is a potential therapeutic target for treating cancers that have developed resistance to traditional DNA crosslinking agents. SNM1A interacts with long DNA strands via a positively charged patch on the enzyme's surface that binds the negatively charged DNA backbone. This leads to processive activity with higher molecular weight substrates, whereas no processivity is observed with small oligonucleotides.2 The 5′-phosphate group of the oligonucleotide substrate binds through H-bonding in the active site and is required for hydrolytic activity. The scissile phosphodiester is postulated to bind to the zinc metal centre prior to attack by an activated water molecule.2,7 There are a limited number of inhibitors of SNM1A. Among the known inhibitors are cephalosporins8 and the metal chelator o-phenanthroline.2 However, to date, there has not been a modified nucleoside inhibitor.We reasoned that the incorporation of compact zinc-binding pharmacophores9 at the 5′-position of a nucleoside would have the potential to block the zinc atom-containing active site of SNM1A and thus inhibit the digestion of single strands of DNA. The nucleobase and tetrahydrofuran ring would provide natural substrate recognition for the enzyme while the zinc-binding groups (ZBGs) would serve to both mimic the phosphate group and form a stable chelating interaction with the metal(s) in the active site. We focused on the installation of ZBGs and potential metal binding groups at the 5′-position of thymidine (Fig. 1). We aimed to incorporate the following bidentate chelating moieties: oxyamides, hydroxamic acids, hydroxamates, a hydrazide and a squarate ester/amide. These modifications will serve a dual purpose: both mimicking the 5′-phosphate group and binding the zinc in the active site of SNM1A. Hydroxamic acids are classic ZBGs with their incorporation featured in histone deacetylase inhibitors, for example.10 As a renowned metal-binding group11 we anticipated that hydroxamic acid derivatives could be good inhibitors of SNM1A. We also explored neutral oxyamide/hydroxamate moieties as potential inhibitors. Squaramides have been demonstrated to chelate metals with the added dimension of being phosphate mimics and thus display excellent precedent for incorporation into nucleoside-based inhibitors.12,13
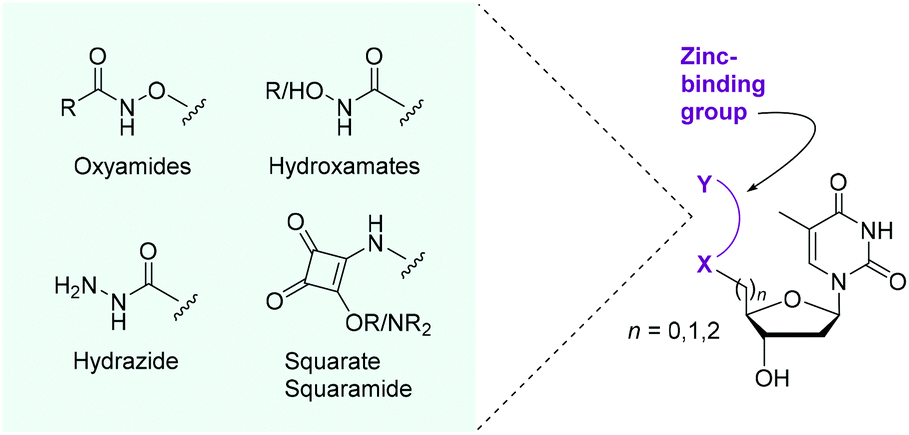 | ||
| Fig. 1 Proposed incorporation of ZBGs at the 5′-position of thymidine for the inhibition of the DNA repair nuclease SNM1A. | ||
In terms of inhibitor design, thymine was selected as the nucleobase as SNM1A has no reported sequence selectivity and the absence of a primary amine group in thymine allows for ease of synthesis and avoids protecting group manipulations on this moiety. The ZBG-modified nucleosides were tested as competitive inhibitors against oligonucleotide strands (20–21 nucleotides in length) which represents a natural substrate for SNM1A in assays with plate reader and gel electrophoresis readouts.
Results and discussion
As shown in Scheme 1, we focused our initial efforts on the synthesis of oxyamide-derived thymidine compounds. Using literature conditions,14 commercially available thymidine 1 was reacted with N-hydroxyphthalimide in a Mitsunobu reaction, followed by 3′-OH protection as a silyl ether and subsequent hydrazinolysis to give the oxyamine product 2 in 58% yield over 3 steps. This material was coupled with acetic acid and formic acid in 71% and 29% yield respectively using EDCI/HOAt-mediated coupling.15 Deprotection of the silyl ethers 3 and 4 using TBAF gave the hydroxy compounds 5 and 6 in excellent yields (92% and 93% respectively).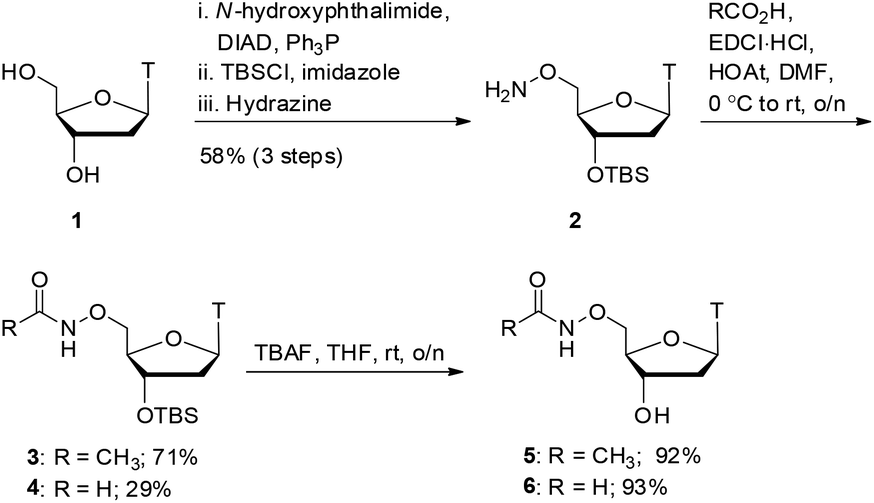 | ||
| Scheme 1 Acetyl and formyl oxyamide synthesis. T = thymine. | ||
With oxyamides 5 and 6 in hand, we then turned our attention to the synthesis of hydroxamic acid-derived nucleosides (Scheme 2). Using literature conditions, thymidine 1 was converted into the 3′-silyl ether 7 in 54% yield over 3 steps. This was achieved by 5′-OH protection of thymidine 1 with a dimethoxytrityl group,16 3′-OH protection as a silyl ether,17 and finally dimethoxytrityl deprotection.18 A TEMPO-BAIB-mediated oxidation in acetonitrile/water provided carboxylic acid198 which would later serve as a point of divergence in our syntheses. Coupling of carboxylic acid 8 with benzyloxyamine under EDCI/HOAt conditions provided the hydroxamate 9 in 76% yield. TBS deprotection was achieved using TBAF in tetrahydrofuran which gave the hydroxy compound 10 in 75% yield. The benzyl protecting group was removed via catalytic hydrogenation (Pd/C–H2) to give hydroxamic acid 11 in 96% yield.
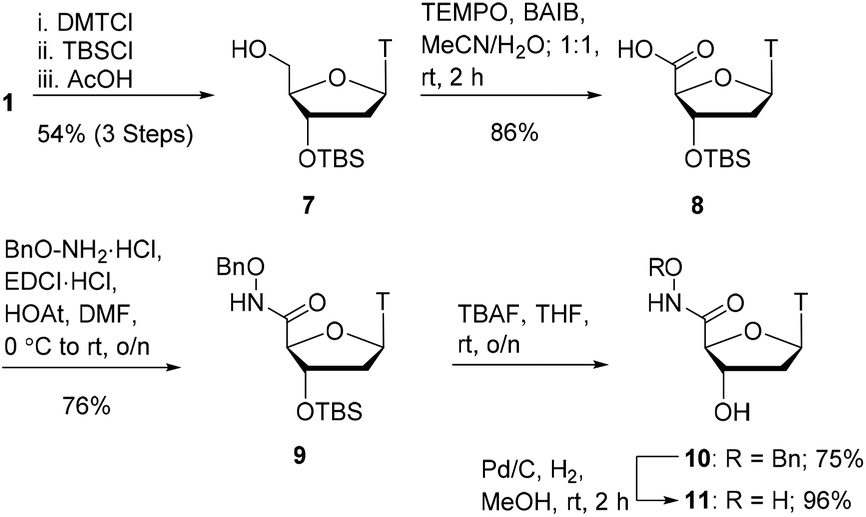 | ||
| Scheme 2 Hydroxamic acid synthesis. | ||
We postulated that modifying the length of the carbon chain attached to the hydroxamic acid would provide a valuable structure–activity-relationship. We therefore pursued 1- and 2-carbon homologues of compound 11 as shown in Scheme 3a. Initial attempts to oxidise alcohol 7 using TEMPO/BAIB in anhydrous dichloromethane failed to yield any aldehyde 12 but instead yielded multiple decomposition products. However, using Dess–Martin periodinane,20 we were able to access the aldehyde 12 in almost quantitative yield. Using Snowden's conditions,21 treatment of this freshly prepared aldehyde 12 with trichloroacetic acid and sodium trichloroacetate provided the trichloromethyl carbinol 13 as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.7 mixture of 5′-epimers in 34% yield. Treatment of this material with sodium borohydride and sodium hydroxide to access carboxylic acid 14 resulted in decomposition. After multiple unsuccessful attempts at optimising this reaction, we opted for a different approach.
:1.7 mixture of 5′-epimers in 34% yield. Treatment of this material with sodium borohydride and sodium hydroxide to access carboxylic acid 14 resulted in decomposition. After multiple unsuccessful attempts at optimising this reaction, we opted for a different approach.
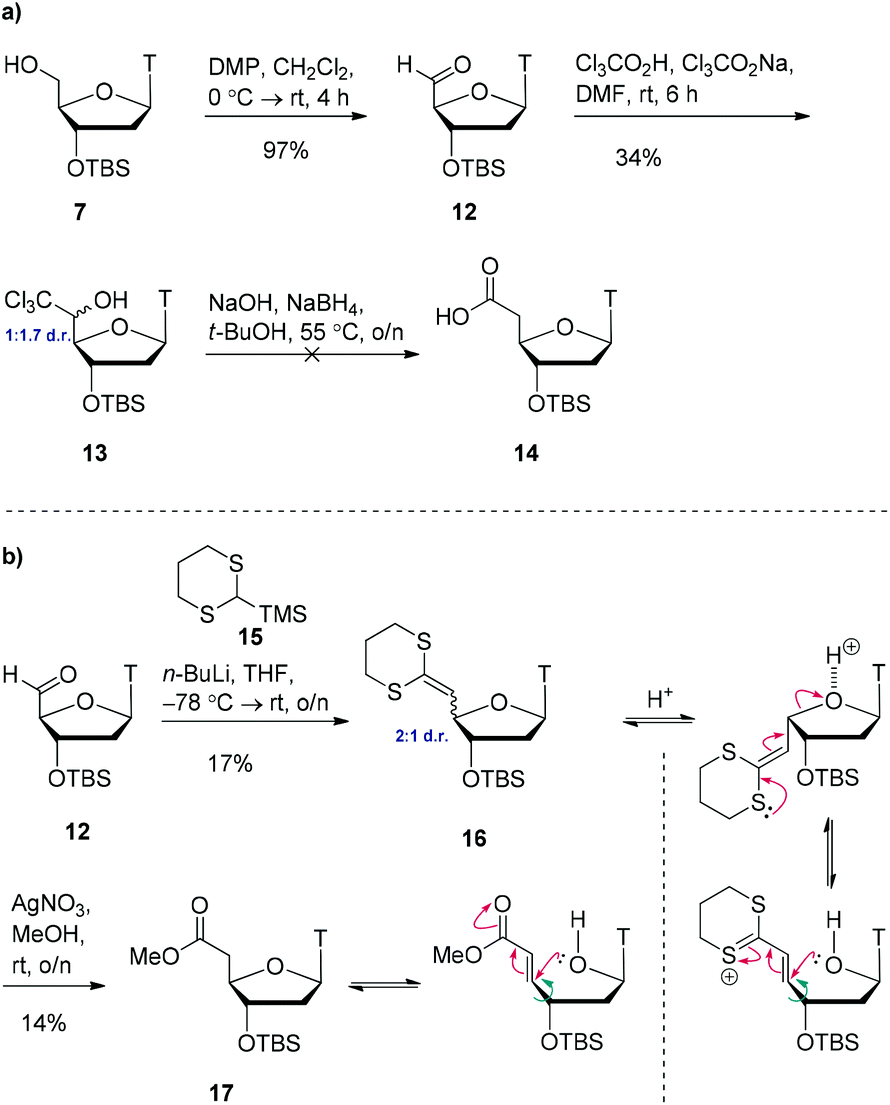 | ||
| Scheme 3 (a) Attempted one-carbon homologation via trichloromethylcarbinol 13. (b) One-carbon homologation via Peterson olefination and postulated epimerisation mechanisms. | ||
Starting from aldehyde 12, a Peterson olefination with commercially available silane 15 gave the ketene-dithioacetal 16 in low yield (Scheme 3b). This compound was found to be highly acid sensitive and as such it was isolated as a 2:1 mixture of 4′-epimers. This epimerisation event occurred during column chromatography as analysis of the crude reaction mixture by 1H NMR spectroscopy indicated the presence of one diastereoisomer. Attempts at purification on alumina resulted in decomposition. Due to the acid sensitivity of this molecule, it was used immediately in the next step (carried forward as a mixture of 4′-epimers). We initially attempted to convert the ketene-dithioacetal 16 into the corresponding methyl ester 17 using CuSO4·5H2O in refluxing methanol,22 however this resulted in decomposition of the starting material. Switching to AgNO3 in refluxing methanol23 for one hour (or rt overnight) did provide product 17, but in very low yield (14%). Interestingly, analysis of the 1H NMR spectrum of the complex crude reaction mixture showed a virtually 1:1 mixture of 4′-epimers. Remarkably, after column chromatography on silica gel, only one epimer was isolated. We speculate that upon exposure of this molecule to silica gel, a β-elimination-type reaction could be occurring, delivering only one epimer (determined by NOE). However, given the poor yields for this sequence, we do not view this route as viable to access the 1-carbon homologue of compound 11.
At this point, we turned our focus to the 2-carbon homologation (Scheme 4). Starting from aldehyde 12, using adapted literature conditions,24 a Wittig reaction with stablised ylide 18 gave the α,β-unsaturated benzyl ester 19 in 81% yield. Catalytic hydrogenation of this material with Pd/C delivered the global hydrogenation product 20 (saturated carboxylic acid) in virtually quantitative yield. EDCI-mediated coupling of carboxylic acid 20 with benzyloxyamine provided the hydroxamate product 21 in 54% yield. The silyl ether in compound 21 was removed with TBAF to provide alcohol 22 in 80% yield. Finally, hydrogenolysis of the benzyl group with Pd/C–H2 furnished the final hydroxamic acid product 23 in 88% yield.
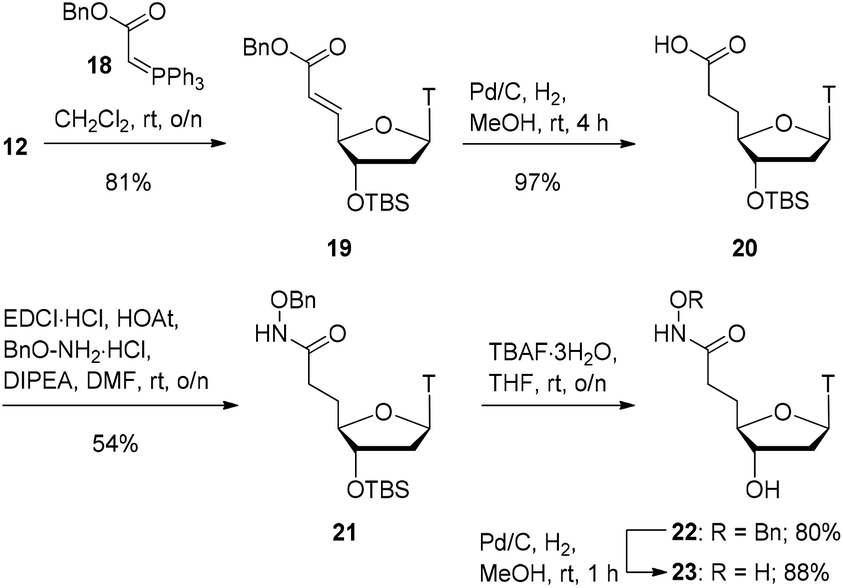 | ||
| Scheme 4 Synthesis of two-carbon homologue 23. | ||
Alongside hydroxamic acids 11 and 23, we explored the idea of capping the hydroxyl portion of the hydroxamic acid with labile and non-labile groups. It was postulated that the labile acetate group on the hydroxamic could be cleaved in buffer during the assay and could have pro-drug potential. In order to discern the importance the free hydroxy group has in the inhibition of SNM1A, we also pursued an O-methylated hydroxamic acid. As shown in Scheme 5a, hydroxamate 9 was reduced by catalytic hydrogenation which gave hydroxamic acid 24 in 88% yield. Acetylation of compound 24 with acetic anhydride capped the hydroxamic acid moiety as an acetate giving compound 25 in 89% yield. Achieving high yields for the silyl deprotection of this compound proved to be a considerable challenge.
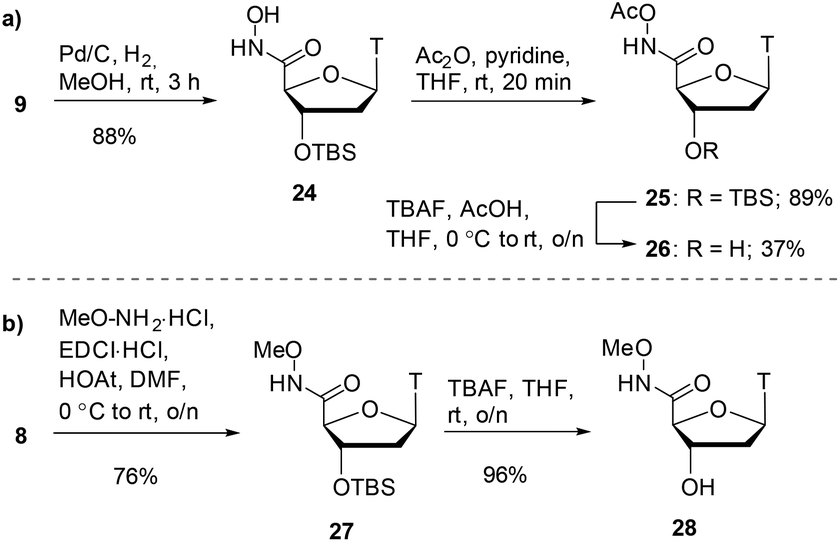 | ||
| Scheme 5 a) Acetate-capped hydroxamic acid synthesis. (b) Methyl-capped hydroxamic acid synthesis. | ||
Optimisation was required for this transformation as use of TBAF alone did not sufficiently deprotect the silyl ether. The addition of acetic acid improved the yields of alcohol 26 to 37% yield. As shown in Scheme 5b, the final hydroxamate in the series was a methyl-capped hydroxamic acid. We performed a coupling reaction of carboxylic acid 8 with methoxyamine which gave the methoxyamide product 27 in 76% yield. Deprotection of the silyl ether with TBAF proceeded smoothly to give the alcohol compound 28 in 96% yield.
Further to the hydroxamic acid and hydroxamate series, the hydrazide moiety is also known to be a potent zinc binder.9 As shown in Scheme 6, coupling of carboxylic acid 8 with Fmoc-protected hydrazine (see ESI† for synthesis) gave the protected hydrazide product 29 in 48% yield. Deprotection of the 3′-silyl ether proved to be a difficult transformation and a range of conditions were explored including various acids, TBAF and a combination of TBAF and acid. Ultimately, the use of TBAF with acetic acid gave the alcohol product 30 in a modest 26% yield. The Fmoc group was removed with piperidine to afford the free hydrazide product 31 in 76% yield.
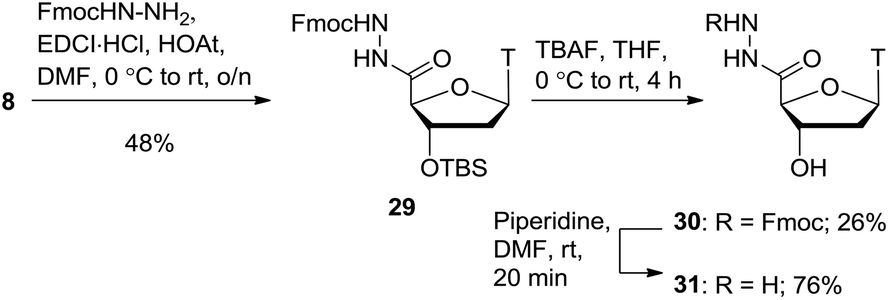 | ||
| Scheme 6 Hydrazide synthesis. | ||
The squaramide functionality is a known phosphate bioisostere in nucleoside chemistry12,13 and has also found other varied uses in medicinal chemistry.25 Furthermore, hydroxylamine derived squaramides have been demonstrated to possess metal and zinc-binding capabilities.26–29
As shown in Scheme 7, starting from thymidine 1, using literature conditions, we converted the 5′-OH group into an amino group through an iodination/displacement/reduction sequence in 71% yield over 3 steps.30–32 Using literature conditions,13 coupling of amine 32 with diethyl squarate under basic conditions provided the squarate ester 33 in 61% yield. This material was subsequently converted into the squaramide 34 in 74% yield.
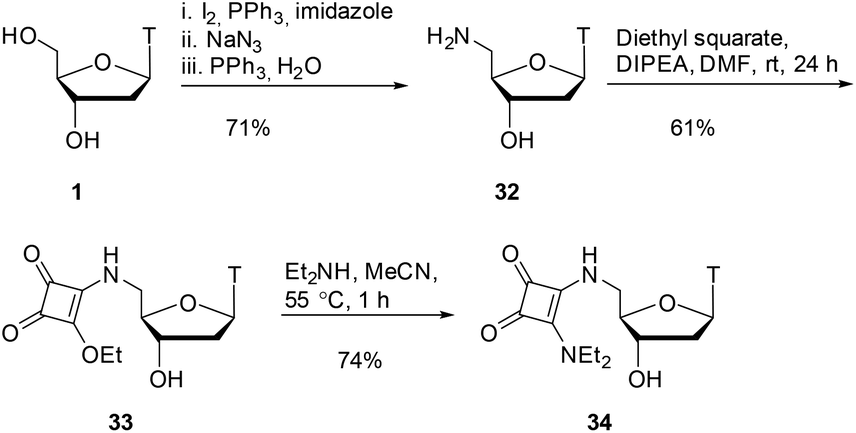 | ||
| Scheme 7 Squarate ester and squaramide synthesis. | ||
With a diverse range of 5′-modified nucleosides in hand, their ability to inhibit the human exonuclease SNM1A was evaluated (Fig. 2). As SNM1A has been documented to efficiently digest single-stranded DNA (ssDNA) substrates, we synthesised a ssDNA oligonucleotide containing the Cy3 fluorophore at the 3′-end of the oligonucleotide (compound 35). This compound served as a substrate for the enzyme and could be used to monitor the activity of our inhibitor series by gel electrophoresis. SNM1A was incubated with inhibitors 5, 6, 11, 23, 26, 28, 31, 33 and 34, respectively at 1 mM concentrations for 5 minutes at 37 °C. After inhibitor pre-incubation, oligonucleotide strand 35 was introduced to the reactions to compete with the inhibitors. After 1 hour, the assay was stopped and the extent of digestion of the oligonucleotide 35 was analysed by denaturing gel electrophoresis. As a control, thymidine 1 was included in the assay to ensure any inhibitory effect could be attributed to the modifications on the nucleoside rather than simply non-covalent interaction between the enzyme and the thymine nucleobase and the tetrahydrofuran ring.
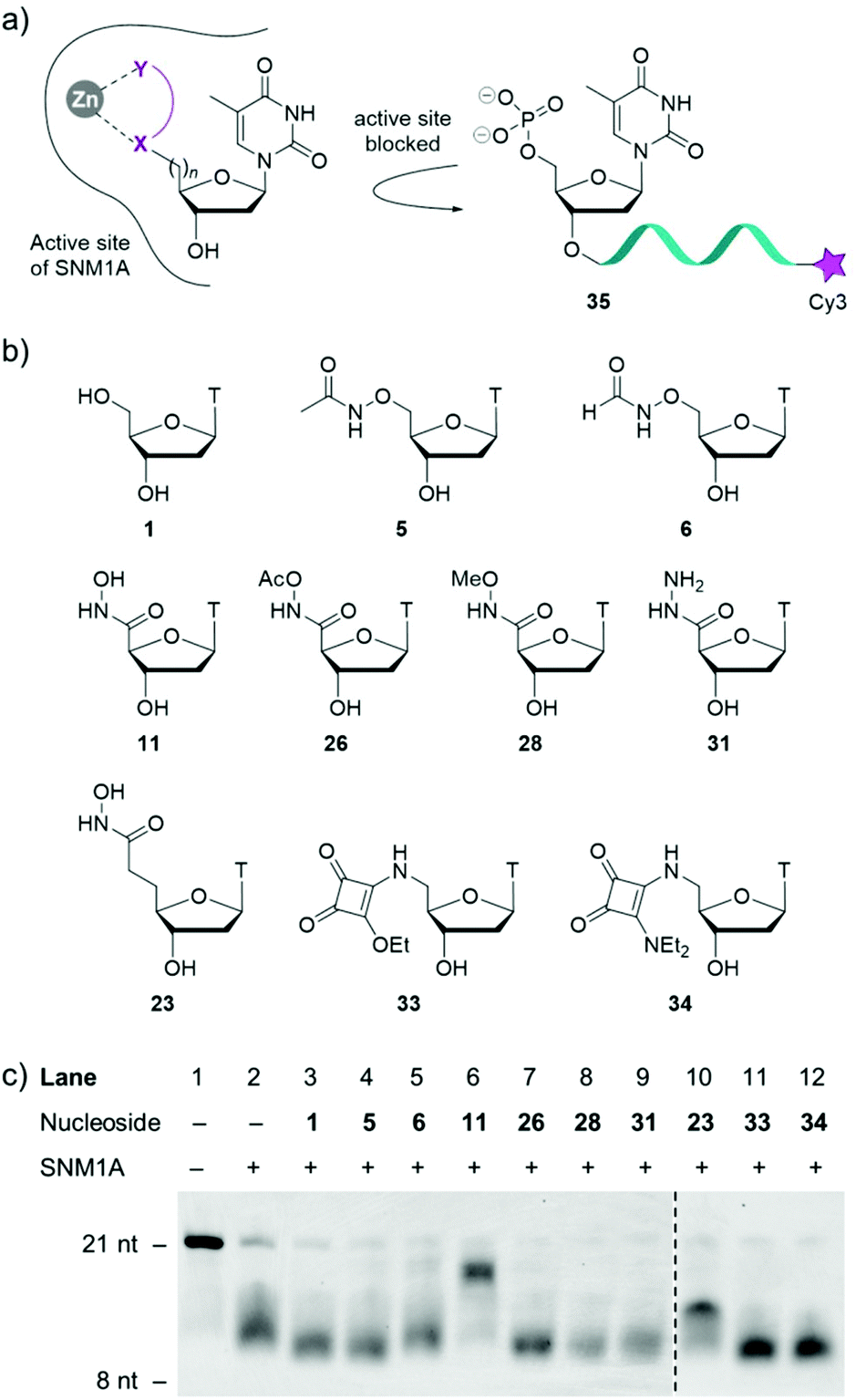 | ||
| Fig. 2 Biological evaluation of modified nucleosides 5, 6, 11, 23, 26, 28, 31, 33 and 34 as competitive inhibitors of SNM1A. (a) Postulated inhibition mechanism. (b) Modified nucleosides evaluated as inhibitors of SNM1A. (c) Denaturing PAGE gel of the extent of digestion of oligonucleotide 35. SNM1A was pre-incubated with inhibitors for 5 minutes at 1 mM concentrations. Oligonucleotide 35 was then added and the assay was halted after one hour. nt = nucleotides. | ||
As shown in Fig. 2c, the results (acquired in duplicate) demonstrate that hydroxamic acids 11 and 23 act as inhibitors of SNM1A as the oligonucleotide is not fully digested in this assay. The difference in digestion of oligonucleotide 35 in the presence of the two hydroxamic acids 11 and 23 highlights the importance of the position of the hydroxamic acid group relative to the nucleoside. In the presence of compound 11 the oligonucleotide products obtained are only slightly shorter than the substrate 35, indicating that hydroxamic acid 11 competes with the 21-nucleotide substrate. The reaction yields shorter fragments in the presence of nucleoside 23, which implies that compound 23 is unable to compete with the 21 mer 35 or the longer oligonucleotides that result from the initial hydrolysis events. However, compound 23 inhibits the hydrolysis of already shortened oligonucleotides, which benefit less from distal binding sites of SNM1A.
All the other inhibitors (5, 6, 26, 28, 31, 33 and 34) in this assay show virtually no inhibition of SNM1A. Formyloxyamide 6 does show marginal levels of enzyme inhibition but given the relatively weak inhibitory potential of this compound, we opted not to pursue this for further analysis.
Acetamide derivative 5 displays no inhibition and both labile and non-labile capped hydroxamate 26 and 28, respectively show no inhibition, indicating the crucial role the free hydroxy group has in enzyme inhibition. Further corroborating evidence for this is that replacing the OH with NH2 (i.e. hydrazide 31) results in no inhibition of SNM1A. Squarate ester 33 and squaramide 34 display no activity in this assay.
With hydroxamic acids 11 & 23 showing the most initial promise, we opted to further investigate the extent to which it could inhibit SNM1A through determination of IC50 values (Fig. 3). Thymidine 1 was included as a control for this experiment, and for inhibitors 11 & 23 IC50 determination was carried out using a real time fluorescence assay in a manner analogous to Lee et al. with a determined IC50 of 139 μM for compound 11 with no discernible inhibition of SNM1A by either control compound 1 or hydroxamic acid 23 (Fig. 3).8 To evaluate the membrane permeability of compound 11, a parallel artificial membrane permeability assay (PAMPA) was performed.33 Nucleoside 11 was unable to cross the artificial membrane (see ESI†), presumably due to the hydrophilic nature of the hydroxamic acid moiety. A pro-drug approach where the active moiety is masked by a labile protecting group, as envisaged for compound 26, could be used to overcome this limitation. As evident by the lack of activity of compound 26 (Fig. 2c, lane 7), the nature of the protecting group is crucial to the successful release of the active compound. The results of the permeability assay indicate that a significant increase in lipophilicity is required for the successful diffusion of derivatives of compound 11 across a membrane.
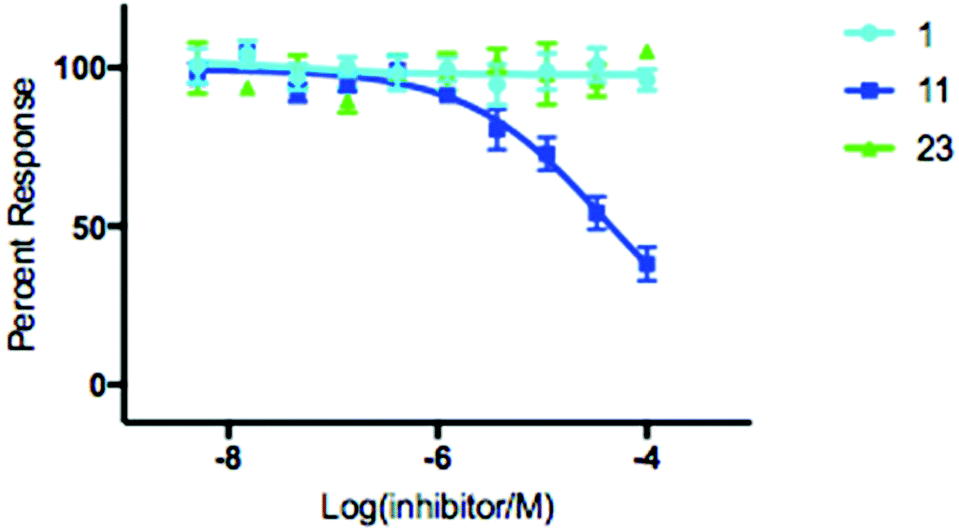 | ||
| Fig. 3 IC50 determination of modified nucleoside 11 with SNM1A. Error bars generated from 4 independent repeats. | ||
Finally, inhibitor 11 was examined in a gel-based assay across concentrations from 1 mM to 3 μM, with thymidine 1 (1 mM) included as a control. Hydroxamic acid 11 was incubated with SNM1A for 5 minutes at 37 °C, in decreasing concentrations, after which time oligonucleotide 35 was added to each reaction. After one hour, the reaction mixture was halted and the extent of digestion of the oligonucleotide was analysed by gel electrophoresis. This experiment was run in duplicate. As can be seen from Fig. 4, hydroxamic acid 11 is an effective inhibitor of SNM1A at higher concentrations (1 mM–0.1 mM) and has a marginal effect as low as 33 μM.
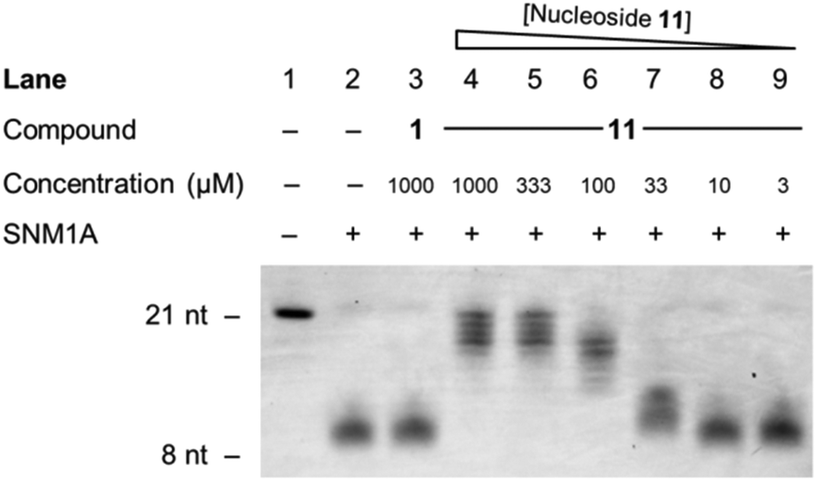 | ||
| Fig. 4 Denaturing PAGE electrophoresis gel of the extent of digestion of oligonucleotide 35 with decreasing concentrations of hydroxamic acid 11 (1 mM to 3 μM). SNM1A was pre-incubated with inhibitors for 5 minutes. Oligonucleotide 35 was then added and the assay was halted after one hour. nt = nucleotides. | ||
Conclusions
In summary, we have synthesised a range of zinc-binding pharmacophores incorporated at the 5′-position of thymidine and these were tested against the human exonuclease SNM1A for inhibition. The hydroxamic acid-derived nucleoside 11 proved to be a novel competitive inhibitor of SNM1A with an IC50 of 139 μM and is the first nucleoside-based inhibitor of the exonuclease. The free hydroxy portion of the hydroxamic acid group is essential to activity, as capped or protected hydroxamates 26 and 28 are not active in this assay. Furthermore, the proximity of the hydroxamic acid group to the nucleoside ring proved to be crucial as hydroxamic acid 23 showed significantly less inhibition of SNM1A. Surprisingly, the hydrazide containing nucleoside 31 showed no activity in this assay, highlighting the crucial role the oxygen of the hydroxamic acid plays in SNM1A inhibition. It is proposed that the mechanism of inhibition of this enzyme is via the occupation of the active site and complexation of the zinc ion(s). Hydroxamic acid 11 serves as a lead compound and further optimisation such as utilising other nucleobases or labile protecting groups could improve the activity and membrane permeability of this molecule. Compound 11 could find application in a combination therapy for cancers which are resistant to interstrand cross-linking agents and is currently under investigation in our laboratory.Experimental
Biological evaluation
:1, 1.4 mL 5× TBE (0.45 M Tris, 0.45 M boric acid, 0.01 M EDTA pH 8.0), 0.6 mL H2O] in 1× TBE at 150 V for 100 minutes alongside bromophenol blue and xylene cyanol as markers for 8 nucleotides and 28 nucleotides respectively and imaged using Typhoon FLA 9500.
Synthetic procedures
:1) showed the consumption of starting material (Rf = 0.5) and the formation of product (Rf = 0.1). The reaction mixture was diluted with EtOAc (100 mL) and washed with sat. aq. NaHCO3 solution (100 mL). The aqueous layer was extracted with EtOAc (2 × 100 mL) and the combined organic extracts were washed with brine (100 mL), dried over Na2SO4, filtered and solvent was removed in vacuo to give the crude product. Purification by column chromatography (CH2Cl2/MeOH; 98:2) gave formyloxyamide 4 as a white foam (300 mg, 29%). IR νmax 3198, 2955, 2928, 2856, 1690, 1470, 1370, 1276, 1254, 1199, 1135, 1086, 1032, 952, 836, 780 cm−1. 1H NMR (400 MHz, CDCl3): δ = 0.09 (s, 6H, 2 × CH3TBS), 0.89 (s, 9H, t-BuTBS), 1.94 (s, 3H, CH3T), 2.23–2.33 (m, 2H, H-2′a + H-2′b), 4.01–4.02 (m, 1H, H-4′), 4.13 (dd, J = 10.6, 3.3 Hz, 1H, H-5′a), 4.21 (dd, J = 10.6, 2.1 Hz, 1H, H-5′b), 4.56–4.63 (m, 1H, H-3′), 6.13 (app. t, J = 7.1 Hz, 1H, H-1′), 7.40 (s, 1H, H-6), 7.99 (s, 1H, CHO), 8.32 (s, 1H, O–NH), 8.76 (s, 1H, NH-3) ppm. 13C NMR (100 MHz, CDCl3): δ = −4.7 (CH3TBS), −4.5 (CH3TBS), 12.6 (CH3T), 18.1 (qC, t-BuTBS), 25.8 (t-BuTBS), 40.5 (C-2′), 71.6 (C-3′), 75.8 (C-5′), 85.6 (C-4′), 86.9 (C-1′), 111.6 (C-5), 137.0 (C-6), 150.4 (C-2), 157.5 (CHO), 163.6 (C-4) ppm. HRMS (ESI+): m/z calc. 422.1718 [M + Na]+, found: 422.1719.
:1) showed the complete consumption of the starting material (Rf = 0.4) and the formation of the product (Rf = 0.2). Silica (∼1 g) was added to the flask and solvent was removed in vacuo to give the crude product. Purification by column chromatography (CH2Cl2/MeOH; 85:15) gave alcohol 6 as a white hygroscopic foam (189 mg, 93%). IR νmax 3389, 3181, 3054, 2922, 2875, 1718, 1655, 1476, 1272, 1093, 1078 cm−1. 1H NMR (600 MHz, DMSO-d6): δ = 1.79 (d, J = 0.9 Hz, 3H, CH3T), 2.06–2.09 (m, 2H, H-2′a + H-2′b), 3.92–3.96 (m, 2H, H-4′ + H-5′a), 4.00–4.03 (m, 1H, H-5′b), 4.26–4.28 (m, 1H, H-3′), 5.40 (s (br), 1H, OH), 6.19 (app. t, J = 7.0 Hz, 1H, H-1′), 7.45 (s (br), 0.15H, H-6), 7.62 (d, J = 0.9 Hz, 0.85H, H-6), 7.88 (s, 0.8H, CHO), 8.42 (s, 0.2H, CHO), 11.13–11.39 (m, 2H, NH-3 + O–NH) ppm. Note: compound 6 exhibits rotamers in NMR spectroscopy.
13C NMR (150 MHz, DMSO-d6): δ = 12.1 (CH3T), 39.0 (C-2′), 70.6 (C-3′), 76.1 (C-5′), 83.9 (C-4′), 84.1 (C-1′), 109.9 (C-5), 136.1 (C-6), 150.5 (C-2), 157.4 (CHO), 163.8 (C-4), 165.0 (CHO) ppm. HRMS (ESI+): m/z calc. 308.0853 [M + Na]+, found: 308.0856.
:1) showed the complete consumption of starting material (Rf = 0.2) and the formation of product (Rf = 0.7). The reaction mixture was diluted with EtOAc (20 mL) and washed with H2O (20 mL). The aqueous layer was extracted with EtOAc (2 × 20 mL) and the combined organic layers were washed with brine (20 mL), dried over MgSO4, filtered and solvent was removed in vacuo to give the crude product. Purification by column chromatography (PE/EtOAc; 1:2) gave benzyloxyamide 9 as a white foam (286 mg, 76%). IR νmax 3187, 2929, 2856, 1664, 1470, 1363, 1277, 1252, 1228, 1134, 1066, 993, 920, 832, 778, 742 cm−1. 1H NMR (400 MHz, CDCl3): δ = 0.11 (s, 3H, CH3TBS), 0.12 (s, 3H, CH3TBS), 0.89 (s, 9H, t-BuTBS), 1.92 (s, 3H, CH3T), 2.01 (dd, J = 12.9, 4.8 Hz, 1H, H-2′a), 2.54 (ddd, J = 12.5, 9.9, 5.2 Hz, 1H, H-2′b), 4.23–4.28 (m, 1H, H-4′), 4.53 (app. d, J = 4.9 Hz, 1H, H-3′), 4.90 (d, J = 11.1 Hz, 1H, Habenzyl), 4.99 (d, J = 11.1 Hz, 1H, Hbbenzyl), 5.94 (dd, J = 9.5, 5.1 Hz, 1H, H-1′), 7.24 (s, 1H, H-6), 7.31–7.39 (m, 3H, HAr), 7.40–7.45 (m, 2H, HAr), 8.50 (s, 1H, NH-3), 9.76 (s, 1H, O–NH) ppm. 13C NMR (100 MHz, CDCl3): δ = −4.8 (CH3TBS), −4.7 (CH3TBS), 12.5 (CH3T), 18.1 (qC, t-BuTBS), 25.8 (t-BuTBS), 37.7 (C-2′), 75.6 (C-3′), 78.4 (CH2benzyl), 86.4 (C-4′), 90.9 (C-1′), 111.8 (C-5), 128.7 (CHAr), 128.9 (CHAr), 129.4 (CHAr), 135.2 (CAr), 138.5 (C-6), 150.5 (C-2), 163.4 (C-4), 167.3 (C-5′) ppm. HRMS (APCI−): m/z calc. 474.2066 [M − H]−, found: 474.2073.
:1) indicated complete consumption of the starting material (Rf = 0.7) and the formation of product (Rf = 0.4). Solvent was removed in vacuo and the residue was directly purified by column chromatography (CH2Cl2/MeOH; 9:1) to give free alcohol 10 as a white solid (21 mg, 75%). M.p. 215–222 °C (decomp.). IR νmax 3431, 3227, 3058, 2923, 1709, 1655, 1639, 1513, 1474, 1407, 1385, 1293, 1270, 1218, 1132, 1052, 1015 cm−1. 1H NMR (400 MHz, CD3OD): δ = 1.91 (d, J = 0.9 Hz, 3H, CH3T), 2.23 (ddd, J = 13.7, 5.8, 2.2 Hz, 1H, H-2′a), 2.33 (ddd, J = 13.7, 8.6, 5.4 Hz, 1H, H-2′b), 4.17 (d, J = 1.5 Hz, 1H, H-4′), 4.40–4.45 (m, 1H, H-3′), 4.90 (s, 2H, Hbenzyl), 6.37 (dd, J = 8.6, 5.8 Hz, 1H, H-1′), 7.32–7.41 (m, 3H, HAr), 7.42–7.48 (HAr), 8.00 (d, J = 0.9 Hz, 1H, H-6) ppm. 13C NMR (100 MHz, CD3OD): δ = 12.6 (CH3T), 39.8 (C-2′), 75.4 (C-3′), 79.1 (CH2benzyl), 85.4 (C-4′), 88.1 (C-1′), 111.8 (C-5), 129.5 (CHAr), 129.8 (CHAr), 130.5 (CHAr), 136.7 (CAr), 138.9 (C-6), 152.5 (C-2), 166.3 (C-4), 169.5 (C-5′) ppm. HRMS (ESI−): m/z calc. 360.1201 [M − H]−, found: 360.1199.
:1) indicated complete consumption of the starting material (Rf = 0.4) and the formation of product (Rf = 0.1). The reaction mixture was filtered through Celite® and the filter cake was washed with MeOH (2 × 5 mL). The filtrate was concentrated in vacuo to give hydroxamic acid 11 as a white solid (13 mg, 96%). M.p. 168–172 °C (decomp.). IR νmax 3490, 3279, 3203, 3061, 2930, 1678, 1652, 1637, 1568, 1478, 1413, 1277, 1238, 1086, 1046, 961, 796 cm−1. 1H NMR (600 MHz, DMSO-d6): δ = 1.77 (s, 3H, CH3T), 2.13 (ddd, J = 13.3, 5.6, 1.8 Hz, 1H, H-2′a), 2.22 (ddd, J = 13.3, 8.6, 5.1 Hz, 1H, H-2′b), 4.07–4.11 (m, 1H, H-4′), 4.27–4.31 (m, 1H, H-3′), 5.68 (s, 1H, OH-3′), 6.32 (dd, J = 8.6, 5.6 Hz, 1H, H-1′), 8.16 (s, 1H, H-6), 9.15 (s (br), 1H, O–NH), 11.08 (s (br), 1H, N–OH), 11.29 (s, 1H, NH-3) ppm. 13C NMR (150 MHz, DMSO-d6): δ = 12.4 (CH3T), 39.1 (C-2′), 73.4 (C-3′), 83.5 (C-4′), 84.9 (C-1′), 109.5 (C-5), 136.6 (C-6), 150.6 (C-2), 163.7 (C-4), 166.8 (C-5′) ppm. HRMS (APCI−): m/z calc. 270.0732 [M − H]−, found: 270.0723.
:1) indicated the formation of two diastereomeric products (Rf = 0.4, 0.3). The reaction mixture was diluted with Et2O (10 mL) and washed with sat. aq. NaHCO3 solution (3 × 10 mL), brine (10 mL), dried over Na2SO4, filtered and solvent was removed in vacuo to give the crude product. Purification by column chromatography (PE/EtOAc; 1:1) gave trichloromethyl carbinol 13 (1:1.7 mixture of diastereomers) as a white foam (157 mg, 34%). A small amount of the less polar diastereoisomer [PE/EtOAc; 1:1 (Rf = 0.4)] was isolated by column chromatography which was used for characterisation. IR νmax 3352, 2955, 2930, 2886, 2858, 1655, 1472, 1278, 1254, 1201, 1125, 1054, 1004, 834, 816, 777 cm−1. 1H NMR (400 MHz, CDCl3): δ = 0.11 (s, 3H, CH3TBS), 0.12 (s, 3H, CH3TBS), 0.90 (s, 9H, t-BuTBS), 1.92 (s, 3H, CH3T), 2.11–2.18 (m, 1H, H-2′a), 2.59–2.68 (m, 1H, H-2′b), 4.16–4.23 (m, 1H, H-5′), 4.49–4.56 (m, 2H, H-4′ + H-3′), 4.87 (d, J = 7.4 Hz, 1H, OH-5′), 5.93 (app. t, J = 7.1 Hz, 1H, H-1′), 7.24 (s, 1H, H-6), 8.86 (s, 1H, NH-3) ppm. 13C NMR (100 MHz, CDCl3): δ = –4.6 (CH3TBS), −4.5 (CH3TBS), 12.5 (5-CH3), 18.1 (qC, t-BuTBS), 25.8 (t-BuTBS), 38.5 (C-2′), 75.3 (C-3′), 82.0 (C-5′), 84.8 (C-4′), 91.1 (C-1′), 101.5 (Cl3C), 111.5 (C-5), 138.6 (C-6), 150.5 (C-2), 163.7 (C-4) ppm. HRMS (ESI+): m/z calc. 495.0647 [M + Na]+, found: 495.0657.
:1) indicated the formation of product (Rf = 0.3). The reaction mixture was quenched with half-saturated NH4Cl solution (10 mL) and extracted with Et2O (2 × 10 mL). The combined ethereal layers were washed with brine (10 mL), dried over MgSO4, filtered and solvent was removed in vacuo to give the crude product as an orange oil. Purification by column chromatography (PE/EtOAc; 1:1) gave ketene dithioacetal 16 (mixture of 4′-epimers; dr 2:1) as an unstable yellow foam (135 mg, 17%). This material was used immediately for the next step. IR νmax 3178, 2955, 2928, 2855, 1685, 1583, 1470, 1421, 1362, 1271, 1251, 1190, 1108, 1046, 915, 833, 776 cm−1. 1H NMR (400 MHz, CDCl3): δ = 0.06 (s, 2H, CH3TBS), 0.07 (s, 2H, CH3TBS), 0.09 (s, 2H, CH3TBS), 0.89 (s, 6H, t-BuTBS), 0.89 (s, 3H, t-BuTBS), 1.94 (d, J = 1.2 Hz, 3H, CH3T), 2.05 (dd, J = 13.5, 6.6 Hz, 0.67H, H-2′a), 2.09–2.23 (m, 2.33H, H-2′a + CH2dithiane), 2.37 (ddd, J = 13.5, 6.3, 3.9 Hz, 0.67H, H-2′b), 2.51 (ddd, J = 13.7, 6.0, 1.1 Hz, 0.33H, H-2′b), 2.81–3.06 (m, 4H, CH2Sdithiane), 4.15 (app. dt, J = 6.3, 3.9 Hz, 0.67H, H-3′), 4.36–4.40 (m, 0.33H, H-3′), 4.83 (dd, J = 8.9, 3.6 Hz, 0.67H, H-4′), 5.14 (dd, J = 8.0, 3.1 Hz, 0.33H, H-4′), 5.77 (d, J = 8.9 Hz, 0.67H, H-5′), 5.97 (d, J = 8.0 Hz, 0.33H, H-5′), 6.17 (app. t, J = 6.6 Hz, 0.67H, H-1′), 6.19–6.23 (m, 0.33H, H-1′), 7.18 (d, J = 1.2 Hz, 0.67H, H-6), 7.23 (d, J = 1.2 Hz, 0.33H, H-6), 8.56 (s, 1H, NH-3) ppm. Note: 1H NMR spectrum complicated due to mixture of 4′-epimers. HRMS (ESI+): m/z calc. 479.1465 [M + Na]+, found: 479.1457.
:1) indicated complete consumption of starting material (Rf = 0.35) and the formation of product (Rf = 0.3). The reaction mixture was filtered through Celite® and the cake was washed with EtOAc (5 mL). The filtrate was azeotropically evaporated with toluene (10 mL). The residue was re-dissolved in EtOAc (10 mL) and the organic layer was washed with brine (5 mL), dried over MgSO4, filtered and solvent was removed in vacuo to give the crude product as a complex mixture. Purification by column chromatography (PE/EtOAc; 1:1) gave methyl ester 17 as a colourless wax (11 mg, 14%). IR νmax 2954, 2929, 2856, 1686, 1464, 1438, 1362, 1306, 1271, 1192, 1174, 1049, 984, 834, 777 cm−1. 1H NMR (400 MHz, CDCl3): δ = 0.05 (s, 3H, CH3TBS), 0.10 (s, 3H, CH3TBS), 0.90 (s, 9H, t-BuTBS), 1.94 (d, J = 1.0 Hz, 3H, CH3T), 2.15 (ddd, J = 14.0, 7.4, 4.7 Hz, 1H, H-2′a), 2.49 (ddd, J = 14.0, 6.1, 1.3 Hz, 1H, H-2′b), 2.70 (dd, J = 16.0, 5.9 Hz, 1H, H-5′a), 2.76 (dd, J = 16.0, 6.4 Hz, 1H, H-5′b), 3.71 (s, 3H, OCH3), 4.48–4.52 (m, 1H, H-3′), 4.60 (app. td, J = 6.4, 3.2 Hz, 1H, H-4′), 6.15 (dd, J = 7.4, 6.1 Hz, 1H, H-1′), 7.18 (d, J = 1.0 Hz, 1H, H-6), 8.33 (s, 1H, NH-3) ppm. 13C NMR (100 MHz, CDCl3): δ = −5.1 (CH3TBS), −4.5 (CH3TBS), 12.8 (CH3T), 18.1 (qC, t-BuTBS), 25.8 (t-BuTBS), 34.2 (C-5′), 42.7 (C-2′), 51.9 (OCH3), 72.5 (C-3′), 81.0 (C-4′), 86.5 (C-1′), 111.0 (C-5), 135.3 (C-6), 150.1 (C-2), 163.7 (C-4), 171.6 (COester) ppm. HRMS (ESI+): m/z calc. 421.1765 [M + Na]+, found: 421.1770.
:1) showed the formation of product (Rf = 0.4). Solvent was removed in vacuo to give the crude product. Purification by column chromatography (PE/EtOAc; 1:1) gave the α,β-unsaturated ester 19 as a white foam (761 mg, 81%). IR νmax 3348, 3204, 2954, 2929, 2887, 2857, 1719, 1688, 1652, 1473, 1361, 1267, 1241, 1176, 1028, 833 cm−1. 1H NMR (400 MHz, CDCl3) δ = 0.08 (s, 6H, 2 × CH3TBS), 0.89 (s, 9H, t-BuTBS), 1.92 (d, J = 1.2 Hz, 3H, CH3T), 2.12 (app. dt, J = 13.6, 6.7 Hz, 1H, H-2′a), 2.31 (ddd, J = 13.6, 6.5, 4.3 Hz, 1H, H-2′b), 4.24 (app. dt, J = 6.7, 4.3 Hz, 1H, H-3′), 4.39 (app. td, J = 5.2, 1.7 Hz, 1H, H-4′), 5.22 (s, 2H, Hbenzyl), 6.16 (dd, J = 15.7, 1.7 Hz, 1H, H-α), 6.29 (t, J = 6.7 Hz, 1H, H-1′), 7.00 (dd, J = 15.7, 5.2 Hz, 1H, H-β), 7.09 (d, J = 1.2 Hz, 1H, H-6), 7.31–7.40 (m, 5H, HAr), 8.74 (s, 1H, NH-3) ppm. 13C NMR (100 MHz, CDCl3) δ = −4.7 (CH3TBS), −4.5 (CH3TBS), 12.8 (CH3T), 18.1 (qC, t-BuTBS), 25.8 (t-BuTBS), 40.3 (C-2′), 66.7 (CH2benzyl), 75.1 (C-3′), 85.27 (C-1′), 85.33 (C-4′), 111.6 (C-5), 122.4 (CH-α), 128.4 (CHAr), 128.5 (CHAr), 128.7 (CHAr), 135.2 (C-6), 135.8 (CAr), 144.1 (CH-β), 150.2 (C-2), 163.6 (C-4), 165.7 (COester). HRMS (ESI+): m/z calc. 509.2078 [M + Na]+, found: 509.2074.
:1) showed complete consumption of the starting material (Rf = 0.8) and the formation of product (Rf = 0.2). The reaction mixture was filtered through Celite® and the filter cake was washed with methanol (3 × 10 mL). The filtrate was concentrated in vacuo to give carboxylic acid 20 as a white foam (300 mg, 97%). IR νmax 3179, 2954, 2930, 1686, 1472, 1273, 1259, 1095, 1062, 834, 777 cm−1. 1H NMR (400 MHz, DMSO-d6): δ = 0.08 (s, 3H, CH3TBS), 0.08 (s, 3H, CH3TBS), 0.87 (s, 9H, t-BuTBS), 1.71–1.94 (m, 5H, H-5′ + CH3T), 2.02 (ddd, J = 13.0, 6.5, 4.1 Hz, 1H, H-2′a), 2.21–2.36 (m, 3H, H-2′b + CH2-α), 3.61–3.69 (m, 1H, H-4′), 4.20–4.27 (m, 1H, H-3′), 6.09 (app. t, J = 6.5 Hz, 1H, H-1′), 7.42 (s, 1H, H-6), 11.29 (s, 1H, NH-3) ppm. 13C NMR (100 MHz, DMSO-d6): δ = −4.9 (CH3TBS), −4.7 (CH3TBS), 12.1 (CH3T), 17.6 (qC, t-BuTBS), 25.7 (t-BuTBS), 28.1 (C-5′), 30.2 (CH2-α), 38.7 (C-2′), 74.4 (C-3′), 83.4 (C-1′), 84.9 (C-4′), 109.8 (C-5), 136.2 (C-6), 150.4 (C-2), 163.7 (C-4), 174.1 (CO2H) ppm. HRMS (ESI+): m/z calc. 421.1765 [M + Na]+, found: 421.1767.
:1) indicated consumption of starting material (Rf = 0.2) and the formation of product (Rf = 0.7). The reaction mixture was diluted with EtOAc (20 mL) and washed successively with NaHCO3 sat. aq. solution (10 mL) and brine (10 mL). The layers were separated and the organic layer was dried over MgSO4, filtered and solvent was removed in vacuo to give the crude product. Purification by column chromatography (PE/EtOAc; 1:2 → EtOAc) gave hydroxamate 21 as a white foam (175 mg, 54%). IR νmax 3188, 2953, 2929, 2856, 1655, 1471, 1362, 1273, 1252, 1195, 833, 777 cm−1. 1H NMR (600 MHz, DMSO-d6): δ = 0.08 (s, 3H, CH3TBS), 0.09 (s, 3H, CH3TBS), 0.87 (s, 9H, t-BuTBS), 1.71–1.78 (m, 1H, H-5′a), 1.80 (s, 3H, CH3T), 1.86–1.93 (m, 1H, H-5′b), 1.99–2.14 (m, 3H, H-2′a + CH2-α), 2.25 (app. dt, J = 13.6, 6.9 Hz, 1H, H-2′b), 3.62 (app. dt, J = 8.4, 3.8 Hz, 1H, H-4′), 4.21 (app. dt, J = 6.9, 3.8 Hz, 1H, H-3′), 4.75 (d, J = 11.1 Hz, 1H, Habenzyl), 4.77 (d, J = 11.1 Hz, 1H, Hbbenzyl), 6.10 (app. t, J = 6.9 Hz, 1H, H-1′), 7.31–7.39 (m, 5H, HAr), 7.42 (s, 1H, H-6), 10.99 (s, 1H, NH-3), 11.28 (s, 1H, O–NH) ppm. 13C NMR (150 MHz, DMSO-d6): δ = −4.8 (Si–CH3), −4.7 (Si–CH3), 12.1 (CH3T), 17.6 (qC, t-BuTBS), 25.7 (t-BuTBS), 28.4 (C-5′), 28.9 (CH2-α), 38.7 (C-2′), 74.5 (C-3′), 76.7 (CH2benzyl), 83.4 (C-1′), 85.1 (C-4′), 109.8 (C-5), 128.2 (CHAr), 128.3 (CHAr), 128.7 (CHAr), 136.0 (CAr), 136.2 (C-6), 150.4 (C-2), 163.7 (C-4), 168.9 (CO–NH–O) ppm. HRMS (ESI−): m/z calc. 502.2379 [M − H]−, found: 502.2379.
:1) indicated consumption of starting material (Rf = 0.6) and the formation of product (Rf = 0.2). Solvent was removed in vacuo to give the crude product. Purification by column chromatography (EtOAc/MeOH; 9:1) gave alcohol 22 as a white solid (105 mg, 80%). M.p. 176–180 °C (decomp.). IR νmax 3443, 3212, 2932, 1711, 1647, 1473, 1262, 1048 cm−1. 1H NMR (600 MHz, DMSO-d6): δ = 1.71–1.78 (m, 1H, H-5′a), 1.80 (s, 3H, CH3T), 1.86–1.94 (m, 1H, H-5′b), 2.00–2.12 (m, 3H, H-2′a + CH2-α), 2.16 (app. dt, J = 13.7, 6.9 Hz, 1H, H-2′b), 3.61 (app. dt, J = 8.4, 3.8 Hz, 1H, H-4′), 4.04 (app. dt, J = 7.0, 3.8 Hz, 1H, H-3′), 4.76 (s, 2H, CH2benzyl), 5.27 (s (br), 1H, OH-3′), 6.12 (app. t, J = 6.9 Hz, 1H, H-1′), 7.32–7.39 (m, 5H, HAr), 7.41 (s, 1H, H-6), 11.00 (s (br), 1H, NH-3), 11.26 (s (br), 1H, O–NH) ppm. 13C NMR (150 MHz, DMSO-d6): δ = 12.1 (CH3T), 28.7 (C-5′), 28.9 (CH2-α), 38.4 (C-2′), 73.0 (C-3′), 76.8 (CH2benzyl), 83.3 (C-1′), 85.2 (C-4′), 109.8 (C-5), 128.2 (CHAr), 128.3 (CHAr), 128.8 (CHAr), 136.08 (CAr), 136.1 (C-6), 150.5 (C-2), 163.7 (C-4), 169.0 (CO–NH–O) ppm. HRMS (APCI−): m/z calc. 388.1514 [M − H]−, found: 388.1502.
:1) indicated complete consumption of starting material (Rf = 0.4) and the formation of product (Rf = 0.2). The reaction mixture was filtered and the filtrate was concentrated in vacuo to give hydroxamic acid 23 as white solid (19 mg, 88%). M.p. 169–174 °C (decomp.). IR νmax 3394, 3210, 3049, 1654, 1476, 1420, 1262, 1078 cm−1. 1H NMR (600 MHz, DMSO-d6): δ = 1.68–1.77 (m, 1H, H-5′a), 1.80 (s, 3H, CH3T), 1.85–1.93 (m, 1H, H-5′b), 1.98–2.12 (m, 3H, CH2-α + H-2′a), 2.17 (app. dt, J = 13.7, 7.0 Hz, 1H, H-2′b), 3.60 (app. dt, J = 8.8, 4.4 Hz, 1H, H-4′), 4.01–4.07 (m, 1H, H-3′), 5.27 (d, J = 4.4 Hz, 1H, OH-3′), 6.12 (app. t, J = 7.0 Hz, 1H, H-1′), 7.40 (s, 1H, H-6), 8.69 (s, 0.9H, N–OH), 9.04 (s, 0.1H, N–OH), 9.81 (s, 0.1H, O–NH), 10.38 (s, 0.9H, O–NH), 11.28 (s, 1H, NH-3) ppm. Note: compound 3 exhibits rotamers in NMR spectroscopy. 13C NMR (150 MHz, DMSO-d6): δ = 12.1 (CH3T), 28.9 (CH2-α), 29.0 (C-5′), 38.4 (C-2′), 73.0 (C-3′), 83.3 (C-1′), 85.1 (C-4′), 109.9 (C-5), 136.1 (C-6), 150.5 (C-2), 163.7 (C-4), 168.6 (CO–NH–O) ppm. HRMS (ESI+): m/z calc. 322.1010 [M + Na]+, found: 322.0996.
:1 → 4:1) gave alcohol 26 as a white powder (94 mg, 37%). M.p. 122–125 °C (decomp.). IR νmax 3456, 3230, 3060, 1799, 1706, 1654, 1636, 1476, 1408, 1366, 1334, 1293, 1274, 1216, 1177, 1132, 1110, 955, 778 cm−1. 1H NMR (400 MHz, DMSO-d6): δ = 1.76 (s, 3H, CH3T), 2.14 (dd, J = 7.2, 3.2 Hz, 2H, H-2′a + H-2′b), 2.19 (s, 3H, CH3acetate), 4.27–4.30 (m, 1H, H-4′), 4.39–4.44 (m, 1H, H-3′), 5.76 (d, J = 4.4 Hz, 1H, OH-3′), 6.37 (t, J = 7.3 Hz, 1H, H-1′), 7.91 (s, 1H, H-6), 11.34 (s, 1H, NH-3), 12.21 (s (br), 1H, O–NH) ppm. 13C NMR (100 MHz, DMSO-d6): δ = 12.3 (CH3T), 18.1 (CH3acetate), 38.3 (C-2′), 73.6 (C-3′), 83.3 (C-4′), 85.2 (C-1′), 109.7 (C-5), 136.4 (C-6), 150.7 (C-2), 163.7 (C-4), 167.4 (C-5′), 168.3 (COacetate) ppm. HRMS (APCI−): m/z calc. 312.0837 [M − H]−, found: 312.0829.
:1) indicated complete consumption of the starting material (Rf = 0.2) and the formation of product (Rf = 0.6). The reaction mixture was diluted with EtOAc (30 mL) and washed with H2O (30 mL). The aqueous layer was extracted with EtOAc (2 × 30 mL) and the combined organic layers were washed with brine (30 mL), dried over MgSO4, filtered and solvent was removed in vacuo to give the crude product. Purification by column chromatography (CH2Cl2/MeOH; 199:1 → 19:1) gave methoxyamide 27 as a white foam (331 mg, 76%). IR νmax 3184, 2930, 2857, 1664, 1471, 1277, 1252, 1228, 1191, 1134, 1066, 994, 919, 832, 777 cm−1. 1H NMR (400 MHz, CDCl3): δ = 0.13 (s, 3H, CH3TBS), 0.14 (s, 3H, CH3TBS), 0.90 (s, 9H, t-BuTBS), 1.95 (s, 3H, CH3T), 2.05 (dd, J = 12.9, 4.8 Hz, 1H, H-2′a), 2.67 (ddd, J = 12.9, 9.8, 5.1 Hz, 1H, H-2′b), 3.79 (s, 3H, OCH3), 4.28–4.33 (m, 1H, H-4′), 4.64 (d, J = 4.8 Hz, 1H, H-3′), 5.95 (dd, J = 9.8, 5.1 Hz, 1H, H-1′), 7.26 (s, 1H, H-6), 8.69 (s, 1H, O–NH), 10.03 (s, 1H, NH-3) ppm. Note: H-6 coincident with residual CHCl3 in the 1H NMR spectrum. 13C NMR (100 MHz, CDCl3): δ = −4.8 (CH3TBS), −4.7 (CH3TBS), 12.5 (CH3T), 18.1 (qC, t-BuTBS), 25.8 (t-BuTBS), 37.6 (C-2′), 64.5 (OCH3), 75.6 (C-3′), 86.5 (C-4′), 91.4 (C-1′), 111.9 (C-5), 138.8 (C-6), 150.7 (C-2), 163.5 (C-4), 167.3 (C-5′) ppm. HRMS (APCI−): m/z calc. 398.1753 [M − H]−, found: 398.1756.
:1) indicated complete consumption of the starting material (Rf = 0.5) and the formation of product (Rf = 0.1). Methanol (5 mL), followed by silica (∼3 g) were added to the flask. Solvent was removed in vacuo to give the crude product which was purified by column chromatography (CH2Cl2/MeOH; 19:1 → 9:1) to give alcohol 28 as a white foam (211 mg, 96%). IR νmax 3469, 3424, 3234, 3061, 2937, 1655, 1637, 1512, 1475, 1405, 1335, 1293, 1270, 1222, 1134, 1051, 967, 920, 795, 774 cm−1. 1H NMR (400 MHz, DMSO-d6): δ = 1.78 (s, 3H, CH3T), 2.12 (ddd, J = 13.3, 5.7, 1.7 Hz, 1H, H-2′a), 2.22 (ddd, J = 13.3, 8.6, 5.2 Hz, 1H, H-2′b), 3.62 (s, 3H, OCH3), 4.05 (d, J = 1.1 Hz, 1H, H-4′), 4.33–4.38 (m, 1H, H-3′), 5.67 (d, J = 4.1 Hz, 1H, OH-3′), 6.32 (dd, J = 8.6, 5.7 Hz, 1H, H-1′), 8.00 (s, 1H, H-6), 11.33 (s (br), 1H, NH-3), 11.54 (s (br), 1H, O–NH) ppm. 13C NMR (100 MHz, DMSO-d6): δ = 12.4 (CH3T), 38.6 (C-2′), 63.3 (O–CH3), 73.3 (C-3′), 83.4 (C-4′), 85.1 (C-1′), 109.6 (C-5), 136.6 (C-6), 150.6 (C-2), 163.7 (C-4), 166.8 (C-5′) ppm. HRMS (APCI−): m/z calc. 284.0888 [M − H]−, found: 284.0892.
:1) indicated complete consumption of the starting material (Rf = 0.2) and the formation of product (Rf = 0.7). The reaction mixture was diluted with EtOAc (20 mL) and washed with H2O (20 mL). The aqueous layer was extracted with EtOAc (2 × 20 mL) and the combined organic layers were washed with brine (20 mL), dried over MgSO4, filtered and solvent was removed in vacuo to give the crude product. Purification by column chromatography (PE/EtOAc; 1:2) gave hydrazide 29 as a white foam (414 mg, 70%). IR νmax 3244, 2952, 2929, 2857, 1677, 1470, 1450, 1247, 1101, 1070, 995, 834, 778, 758, 739 cm−1. 1H NMR (600 MHz, CDCl3): δ = 0.13 (s, 3H, CH3TBS), 0.15 (s, 3H, CH3TBS), 0.91 (s, 9H, t-BuTBS), 1.89 (s, 3H, CH3T), 2.07 (d, J = 9.6 Hz, 1H, H-2′a), 2.43–2.58 (m, 1H, H-2′b), 4.24 (t, J = 7.2 Hz, 1H, CHFmoc), 4.40–4.48 (m, 3H, CH2Fmoc + H-4′), 4.75–4.86 (m, 1H, H-3′), 6.22–6.39 (m, 1H, H-1′), 7.05 (s, 1H, NHFmoc), 7.29 (app. t, J = 7.5 Hz, 2H, HAr), 7.39 (app. t, J = 7.5 Hz, 2H, HAr), 7.45 (s, 1H, H-6), 7.57 (d, J = 7.5 Hz, 1H, HAr), 7.59 (d, J = 7.5 Hz, 1H, HAr), 7.75 (d, J = 7.5 Hz, 2H, HAr), 8.82 (s (br), 1H, CO–NH), 8.97 (s (br), 1H, NH-3) ppm. 13C NMR (150 MHz, CDCl3): δ = −4.8 (CH3TBS), −4.7 (CH3TBS), 12.3 (CH3T), 18.1 (qC, t-BuTBS), 25.9 (t-BuTBS), 37.9 (C-2′), 47.0 (CHFmoc), 68.3 (CH2Fmoc), 75.6 (C-3′), 86.6 (C-4′), 89.4 (C-1′), 112.4 (C-5), 120.2 (CHAr), 125.2 (CHAr), 125.3 (CHAr), 127.3 (CHAr), 128.0 (CHAr), 137.5 (C-6), 141.39 (CAr), 141.43 (CAr), 143.5 (CAr), 143.6 (CAr), 151.0 (C-2), 156.4 (COFmoc), 163.6 (C-4), 169.9 (C-5′) ppm. HRMS (ESI+): m/z calc. 629.2402 [M + Na]+, found: 629.2410.
:1) indicated complete consumption of the starting material (Rf = 0.7) and the formation of product (Rf = 0.3). Toluene (5 mL) was added to the flask, followed by silica (∼0.5 g) and solvent was removed in vacuo. Purification by column chromatography (CH2Cl2/MeOH; 9:1) gave hydrazide 31 as a white powder (56 mg, 76%). M.p. 188–192 °C (decomp.). IR νmax 3476, 3320, 3205, 3053, 2927, 1650, 1635, 1535, 1475, 1411, 1341, 1275, 1197, 1132, 1077, 1006, 960, 797 cm−1. 1H NMR (400 MHz, DMSO-d6): δ = 1.77 (d, J = 0.9 Hz, 3H, CH3T), 2.11 (ddd, J = 13.4, 5.6, 2.1 Hz, 1H, H-2′a), 2.21 (ddd, J = 13.4, 8.6, 5.0 Hz, 1H, H-2′b), 4.16 (d, J = 1.2 Hz, 1H, H-4′), 4.25–4.29 (m, 1H, H-3′), 4.39 (s (br), 2H, NH2), 5.65 (d, J = 4.3 Hz, 1H, OH-3′), 6.31 (dd, J = 8.6, 5.6 Hz, 1H, H-1′), 8.21 (d, J = 0.9 Hz, 1H, H-6), 9.54 (s, 1H, CO–NH), 11.28 (s, 1H, NH-3) ppm. 13C NMR (100 MHz, DMSO-d6): δ = 12.5 (CH3T), 38.9 (C-2′), 73.4 (C-3′), 84.3 (C-4′), 85.1 (C-1′), 109.5 (C-5), 136.9 (C-6), 150.6 (C-2), 163.8 (C-4), 169.3 (C-5′) ppm. HRMS (ESI+): m/z calc. 271.1037 [M + H]+, found: 271.1034.
:1) showed complete consumption of the starting material (Rf = 0.3) and formation of the product (Rf = 0.1). The orange solution was concentrated to give the desired product 34 as a red solid (79 mg, 74%). M.p. 207–209 °C. IR νmax 3438, 3213, 2978, 1790, 1712, 1651, 1559, 1517, 1442, 1275, 1227, 1083, 1063, 567 cm−1. 1H NMR (600 MHz, DMSO-d6): δ = 1.13 (t, J = 7.2 Hz, 6H, CH3Et), 1.78 (d, J = 1.0 Hz, 3H, CH3T), 2.04–2.11 (m, 2H, H-2′a + H-2′b), 3.45–3.60 (m, 4H, CH2Et), 3.73 (app. dt, J = 13.6, 5.0 Hz, 1H, H-5′a), 3.87 (ddd, J = 7.1, 5.0, 3.3 Hz, 1H, H-4′), 3.93 (app. dt, J = 13.6, 7.1 Hz, 1H, H-5′b), 4.19–4.22 (m, 1H, H-3′), 5.35 (d, J = 3.3 Hz, 1H, OH-3′), 6.14 (app. t, J = 7.1 Hz, 1H, H-1′), 7.42 (d, J = 1.0 Hz, 1H, H-6), 7.68–7.72 (m, 1H, NHSq), 11.30 (s, 1H, NH-3) ppm. 13C NMR (150 MHz, DMSO-d6): δ = 12.0 (CH3T), 15.0 (CH3Et), 38.5 (C-2′), 43.5 (CH2Et), 45.5 (C-5′), 70.6 (C-3′), 83.9 (C-1′), 85.3 (C-4′), 109.7 (C-5), 135.8 (C-6), 150.4 (C-2), 163.7 (C-4), 166.9 (CSq1), 167.0 (CSq2), 181.8 (CSq4), 182.5 (CSq3) ppm. HRMS (ESI+): m/z calc. 393.1769 [M + H]+, found: 393.1756.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by The Wellcome Trust ISSF (JFM & WD), Trinity College Dublin with an Ussher Postgraduate fellowship (EMD), Science Foundation Ireland (IvP 13/IA/1894, MOS), (15/CDA/3310 (EMS) and CRUK (C9047/A24759, HTB & PJM). We thank Opher Gileadi (SGC) for providing us with recombinant SNM1A enzyme and Marcin Bielinski for their assistance with the IC50 determination assays.Notes and references
- U. B. Abdullah, J. F. McGouran, S. Brolih, D. Ptchelkine, A. H. El-Sagheer, T. Brown and P. J. McHugh, EMBO J., 2017, 36, 2047 CrossRef CAS PubMed.
- B. Sengerová, C. K. Allerston, M. Abu, S. Y. Lee, J. Hartley, K. Kiakos, C. J. Schofield, J. A. Hartley, O. Gileadi and P. J. McHugh, J. Biol. Chem., 2012, 287, 26254 CrossRef PubMed.
- T. Iyama, S. Y. Lee, B. R. Berquist, O. Gileadi, V. A. Bohr, M. M. Seidman, P. J. McHugh and D. M. Wilson III, Nucleic Acids Res., 2015, 43, 247 CrossRef CAS PubMed.
- T. Helleday, E. Petermann, C. Lundin, B. Hodgson and R. A. Sharma, Nat. Rev. Cancer, 2008, 8, 193 CrossRef CAS PubMed.
- A. T. Wang, B. Sengerová, E. Cattell, T. Inagawa, J. M. Hartley, K. Kiakos, N. A. Burgess-Brown, L. P. Swift, J. H. Enzlin, C. J. Schofield, O. Gileadi, J. A. Hartley and P. J. McHugh, Genes Dev., 2011, 25, 1859 CrossRef CAS PubMed.
- J. Hejna, S. Philip, J. Ott, C. Faulkner and R. Moses, Nucleic Acids Res., 2007, 35, 6115 CrossRef CAS PubMed.
- C. K. Allerston, S. Y. Lee, J. A. Newman, C. J. Schofield, P. J. McHugh and O. Gileadi, Nucleic Acids Res., 2015, 43, 11047 CrossRef CAS PubMed.
- S. Y. Lee, J. Brem, I. Pettinati, T. D. W. Claridge, O. Gileadi, C. J. Schofield and P. J. McHugh, Chem. Commun., 2016, 52, 6727 RSC.
- For a review of zinc-binding metalloproteinase inhibitors see: J. A. Jacobsen, J. L. Major Jourden, M. T. Miller and S. M. Cohen, Biochim. Biophys. Acta, Mol. Cell Res., 2010, 1803, 72 CrossRef CAS PubMed.
- C. Monneret, Eur. J. Med. Chem., 2005, 40, 1 CrossRef CAS PubMed.
- C. J. Marmion, D. Griffith and K. B. Nolan, Eur. J. Inorg. Chem., 2004, 3003 CrossRef CAS.
- K. Sato, K. Seio and M. Sekine, J. Am. Chem. Soc., 2002, 124, 12715 CrossRef CAS PubMed.
- K. Seio, T. Miyashita, K. Sato and M. Sekine, Eur. J. Org. Chem., 2005, 5163 CrossRef CAS.
- S. Peyrat and J. Xie, Synthesis, 2012, 44, 1718 CrossRef CAS.
- E. M. Dürr, W. Doherty, S. Y. Lee, A. H. El-Sagheer, A. Shivalingam, P. J. McHugh, T. Brown and J. F. McGouran, ChemistrySelect, 2018, 3, 12824 CrossRef.
- B. S. Ross, M. Han and V. T. Ravikumar, Nucleosides, Nucleotides Nucleic Acids, 2006, 25, 765 CrossRef CAS PubMed.
- J. D'Onofrio, L. De Napoli, G. Di Fabio and D. Montesarchio, Synlett, 2006, 845 Search PubMed.
- K. Satou, Y. Komatsu, T. Torizawa, K. Kato, I. Shimada, O. Nikaido and E. Ohtsuka, Tetrahedron Lett., 2000, 41, 2175 CrossRef CAS.
- A. Varizhuk, S. Kochetkova, N. Kolganova, E. Timofeev and V. Florentiev, Nucleosides, Nucleotides Nucleic Acids, 2011, 30, 31 CrossRef CAS PubMed.
- V. Samano and M. J. Robins, J. Org. Chem., 1990, 55, 5186 CrossRef CAS.
- L. R. Cafiero and T. S. Snowden, Org. Lett., 2008, 10, 3853 CrossRef CAS PubMed.
- S. Hanessian, D. K. Maji, S. Govindan, R. Matera and M. Tintelnot-Blomley, J. Org. Chem., 2010, 75, 2861 CrossRef CAS PubMed.
- T. Saito, T. Suzuki, M. Morimoto, C. Akiyama, T. Ochiai, K. Takeuchi, T. Matsumoto and K. Suzuki, J. Am. Chem. Soc., 1998, 120, 11633 CrossRef CAS.
- V. Kotikam and E. Rozners, Org. Lett., 2017, 19, 4122 CrossRef CAS PubMed.
- R. I. Storer, C. Aciro and L. H. Jones, Chem. Soc. Rev., 2011, 40, 2330 RSC.
- N. C. Lim, M. D. Morton, H. A. Jenkins and C. Brückner, J. Org. Chem., 2003, 68, 9233 CrossRef CAS PubMed.
- M. B. Onaran, A. B. Comeau and C. T. Seto, J. Org. Chem., 2005, 70, 10792 CrossRef CAS PubMed.
- S. Hanessian, V. Vinci, L. Auzzas, M. Marzi and G. Giannini, Bioorg. Med. Chem. Lett., 2006, 16, 4784 CrossRef CAS PubMed.
- J. Charton, B. P. Deprez and R. F. Deprez-Poulain, Bioorg. Med. Chem. Lett., 2008, 18, 4968 CrossRef CAS PubMed.
- S. K. V. Vernekar, L. Qiu, J. Zacharias, R. J. Geraghty and Z. Wang, MedChemComm, 2014, 5, 603 RSC.
- K. Suthagar and A. J. Fairbanks, Chem. Commun., 2017, 53, 713 RSC.
- K. Suthagar, W. Jiao, H. Munier-Lehmann and A. J. Fairbanks, Carbohydr. Res., 2018, 457, 32 CrossRef CAS PubMed.
- D. Schmidt and J. Lynch, Millipore Corporation Application Note, 2003, Lit. No. AN1728EN00.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for known compounds, NMR spectra of novel compounds, PAMPA experimental details. See DOI: 10.1039/c9ob01133a |
| This journal is © The Royal Society of Chemistry 2019 |