
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Borylative cyclisation of diynes using BCl3 and borocations†
Andrew J.
Warner
a,
Kieron M.
Enright
a,
John M.
Cole
a,
Kang
Yuan
b,
John S.
McGough
a and
Michael J.
Ingleson
 *b
*b
aSchool of Chemistry, University of Manchester, Manchester, M13 9PL, UK
bSchool of Chemistry, University of Edinburgh, Edinburgh, EH9 3FJ, UK. E-mail: michael.ingleson@ed.ac.uk
First published on 20th May 2019
Abstract
The borylative cyclisation of 1,2-dialkynyl benzenes with BCl3 leads to dibenzopentalenes (via intramolecular SEAr) or benzofulvenes (via chloride addition) depending on substituents, with stabilised vinyl cation intermediates (e.g. with a p-MeO-C6H4-group) favouring the latter. The use of borocations leads to more selective dibenzopentalene formation, while other diyne frameworks undergo intramolecular SEAr selectively even with p-MeO groups.
The activation of a π nucleophile by a boron electrophile to induce cyclisation while forming C–Y (Y = C, N, S or O etc.) and C–B bonds concomitantly is an attractive method to make complex organoboranes. Termed borylative cyclisation, the catalyst-free version of this reaction1,2 requires sufficiently electrophilic boranes to proceed with pioneering work performed using B(C6F5)3.3 While notable, borylative cyclisation with B(C6F5)3 generally leads to [RB(C6F5)3]− containing products (though a carboboration step also occurs in some cases generating RB(C6F5)2 containing cyclised products).4 The utility of the [RB(C6F5)3]− moiety is much less developed relative to RB(OR)2 (for which there are a myriad of established functional group transformations)5 or RB(Aryl)2 (e.g. as an acceptor for generating low LUMO energy conjugated materials).6 Therefore the use of alternative boron electrophiles for borylative cyclisation that form, or can be transformed readily into, RB(OR)2 or RB(Aryl)2 has become an area of significant activity. While we and others have utilised BCl3,7 Blum and co-workers have exploited B-chloro-catecholborane (CatBCl) in borylative cyclisations,8 and a range of borylated systems now have been synthesised via borylative cyclisation methodologies.
In previous work of particular relevance to this study, Erker, Yamaguchi and co-workers used B(C6F5)3, and in one example BCl3, for the cyclisation of 1,2-dialkynyl benzenes to form dibenzopentalenes A and B (Scheme 1 inset top right).9 Due to our ongoing interest in low band gap boron containing polycyclic aromatic hydrocarbons,10 we were interested in extending BCl3 induced cyclization to related diynes substituted with donor (e.g. –OMe) groups. During this study certain diynes were found to form alternative borylative cyclisation products, specifically chlorinated benzofulvenes (Scheme 1, bottom). Herein we present our results into the borylative cyclisation of a range of diynes along with the development of methods to control, to some extent, the product distribution principally by varying the electrophile from BCl3 to a borocation.
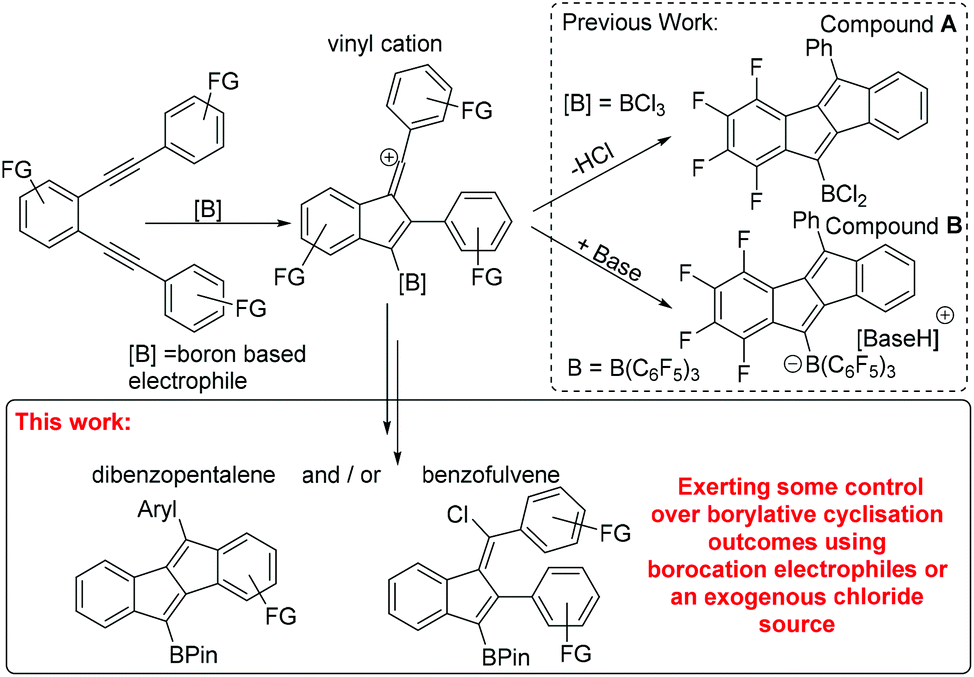 | ||
| Scheme 1 Previous work on borylative cyclisation and bottom inset this work. | ||
Results and discussion
The borylative cyclisation of diyne 1a, containing two para-methoxy groups (Fig. 1), was explored using BCl3 in dichloromethane (DCM). While C–B bond formation occurred (as indicated by a δ11B = 54 ppm consistent with an RBCl2 moiety) the major product was not the expected dibenzopentalene analogue of compound A. Addition of pinacol and Et3N followed by isolation of the pinacol boronate ester derivative enabled identification of the major product as the benzofulvene, 2-BPin, by single crystal X-ray diffraction (Fig. 1). This formulation was supported by mass and multinuclear NMR spectroscopy with 2-BPin isolated in 85% yield as a single isomer. The solid state structure of 2-BPin is unremarkable with both Ph groups and the Bpin moiety twisted out of the plane of the benzofulvene core to varying degrees to minimise steric interactions. Formation of related halogenated benzofulvenes, e.g.C (inset left Fig. 1), has been previously reported from the addition of HBr to ortho-bis(phenylethynyl)benzene, with alkyne protonation initiating the 5-endo-dig cyclisation.11 In contrast to the use of HBr (or I2),12 using BCl3 as the electrophile incorporates a nucleophilic C–B site in addition to an electrophilic C–X. The formation of 2-BCl2 presumably arises from a zwitterionic vinyl cation intermediate (Scheme 1, top middle) being trapped by chloride instead of undergoing intramolecular SEAr. Analysis of the NMR spectra of 2-BCl2 preparations prior to pinacol protection and for 2-BPin prior to purification revealed a second minor cyclisation product assigned as the dibenzopentalenes 3-BCl2 and 3-BPin (the latter supported by GC-MS), respectively. The ratio of 2-BPin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3-BPin in these mixtures from NMR spectroscopy and GC-MS is 17:1.
:3-BPin in these mixtures from NMR spectroscopy and GC-MS is 17:1.
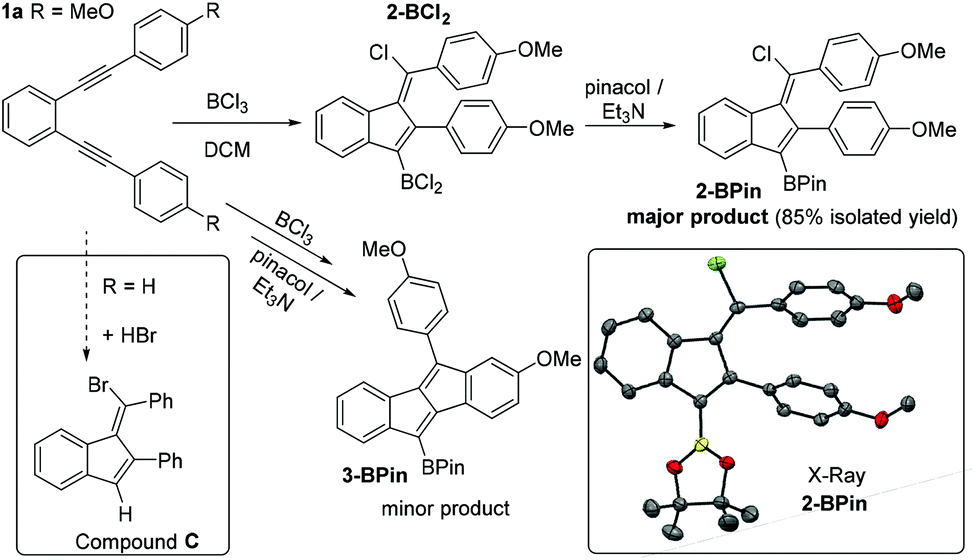 | ||
| Fig. 1 The functionalisation of 1a using BCl3 and subsequent pinacol protection. Inset right, solid state structure of 2-BPin, ellipsoids at the 50% probability level and hydrogens omitted for clarity. Inset left, previous work cyclising with HBr. | ||
The borylative cyclisation of diyne 1b using BCl3 to form compound A has been previously reported.9 This transformation was explored again herein to determine if a borylated benzofulvene analogous to 2-BCl2 was present at a low percentage using identical conditions to that for the formation of 2-BCl2. However, only compound A (and 4 post pinacol protection) was observed by multinuclear NMR spectroscopy under these conditions. Compound 4 was isolated in 92% yield and its formulation further confirmed by single crystal X-ray diffraction studies (Fig. 2, inset). The solid state metrics of 4 are unremarkable with core C–C distances comparable to that reported for compound B (Scheme 1).
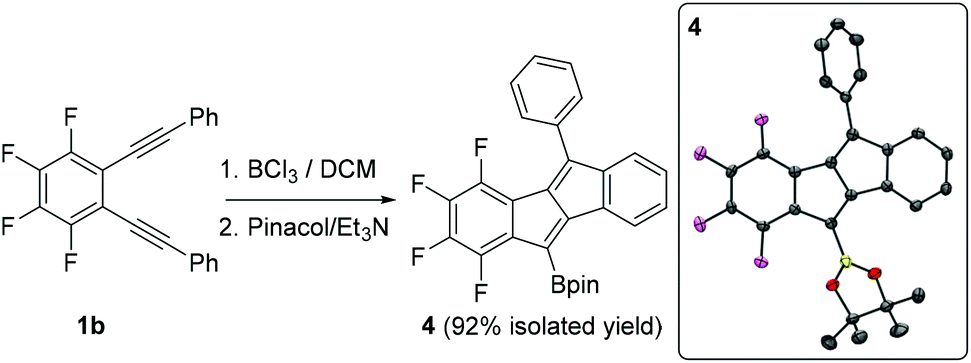 | ||
| Fig. 2 Borylative cyclisation of 1b. Inset right, the solid state structure of 4, ellipsoids at 50% probability level and hydrogens omitted for clarity. | ||
No intermediates were observed in the borylative cyclisation reactions starting from 1a or 1b and leaving 2-BCl2 in the presence of excess BCl3 for longer reaction times did not lead to any increase in the dibenzopentalene (disfavouring chloride abstraction from vinylC–Cl in 2-BCl2 by BCl3 generating the vinyl cation that subsequently undergoes SEAr). It should also be noted that the chloride present in the benzofulvene product is derived from BCl3 as closely comparable outcomes were observed on replacing DCM with o-dichlorobenzene (o-DCB, which is a solvent more resistant to C–Cl heterolysis).
The disparate outcomes observed commencing from diynes 1a and 1b are clearly due to the substituent electronic effects. This is attributed predominantly to the ability of the para MeO group to stabilise vinyl cation intermediates (the methoxy group will also have a small deactivating effect for SEAr due to the σ+meta value of +0.05). The reduced electrophilicity of the vinyl cation derived from 1a will increase its lifetime in solution thereby enabling a reaction with a chloride source to proceed. In contrast, the vinyl cation derived from 1b will be more electrophilic than that derived from 1a, thus the SEAr reaction will be more rapid (as found previously with related carbocations),13 outcompeting the chloride transfer reaction. DFT calculations at the M06-2X/6-311G(d,p) level with a polarisable continuum model (PCM) of DCM, revealed that the LUMO energies for the vinyl cations (which are dominated by pπ orbitals on the sp vinyl cation carbon and the adjacent aromatic ring in each case – see ESI†) vary significantly (Scheme 2). These energies will correlate with electrophilicity and the rates of reaction with π nucleophiles.13
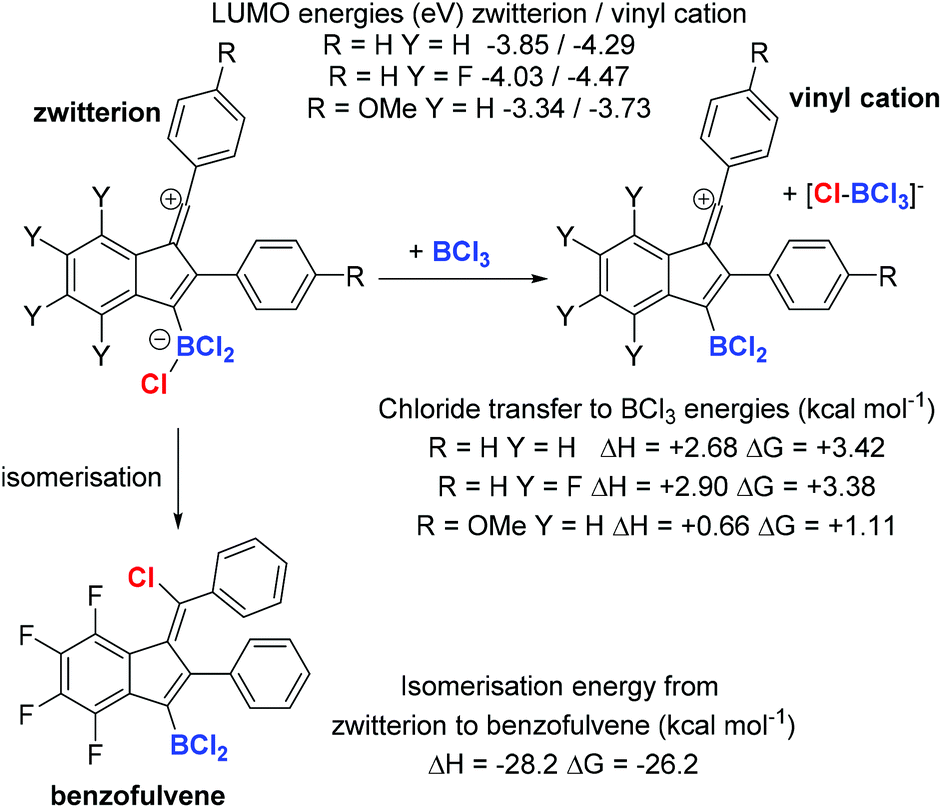 | ||
| Scheme 2 Calculations of the energy change in chloride transfer reactions (all at the M06-2x/6-311G(d,p)/PCM (DCM) level). In the vinyl cation the anion and cation were calculated at infinite distance. | ||
Another factor that was considered in effecting the product outcome was the identity (and nucleophilicity) of the chloride donor that forms the benzofulvene. Isodesmic calculations revealed that in each case the vinyl cation (Scheme 2 top right), is more Lewis acidic towards chloride than BCl3, thus the zwitterionic vinyl cation (Scheme 2 top left) will be favoured in solution over [vinyl-cation][BCl4]. Though it is notable that the vinyl cation derived from 1a and BCl3 have similar Lewis acidity towards chloride (ΔG = 1.11 kcal mol−1) thus both [vinyl cation][BCl4] and the zwitterion should be present in solution as intermediates during this reaction. Furthermore, the chloride ion affinity (CIA) of the vinyl cation derived from 1a is the lowest (of the three calculated), thus this zwitterion will be the better chloride donor and also have the highest concentration of [BCl4]− in solution (due to the lowest ΔG). This factor coupled with the relative LUMO energies presumably explains why the benzofulvene is formed from 1a but not 1b. Finally, the formation of the benzofulvene from the zwitterion was found to be highly exergonic. This is consistent with the formation of the benzofulvene being irreversible under these conditions even in the presence of BCl3 (hence there is no change in the benzofulvene/dibenzopentalene ratio from 1a in the presence of excess BCl3 over longer reaction times).
To probe substituent effects further and guided by these calculations three other diynes, 1c–1e (Scheme 3), were synthesised and investigated. These were chosen to either stabilise vinyl cations to some extent or to be less Lewis acidic at boron relative to vinyl cations derived from 1b. Compounds 1c and 1d have para F and Me substituents, respectively, while 1e is the unsubstituted congener that was previously calculated and shown to form benzofulvene C on reaction with HBr (Fig. 1). Notably, F and Me have distinct σ+ values (Me σ+para = −0.31 σ+meta = −0.10, F σ+para = −0.07 F σ+meta = +0.35), with both groups able to stabilise the vinyl cation to some extent (albeit less than MeO, σ+para = −0.78), but Me is activating while F is deactivating, with respect to SEAr in the meta position. In each case the diyne was combined with BCl3 in DCM and after all starting diyne was consumed (by in situ NMR spectroscopy) the products were converted to the pinacol boronate esters which were analysed as crude mixtures to determine the ratio of dibenzopentalene:benzofulvene by NMR spectroscopy and GC-MS. This revealed that for fluoro substituted 1c the benzofulvene 5 was the major product (ca. 90%). However, for 1d and 1e the dibenzopentalene products were the major products but were only formed in a slightly larger amount than the benzofulvene products. Combined these results indicate that deactivating the aromatic system towards SEAr (e.g. using a meta fluorine relative to the site of SEAr) is also an effective way to achieve preferential benzofulvene formation, more effective than vinyl cation stabilisation using a p-Me-C6H4 group in this case.
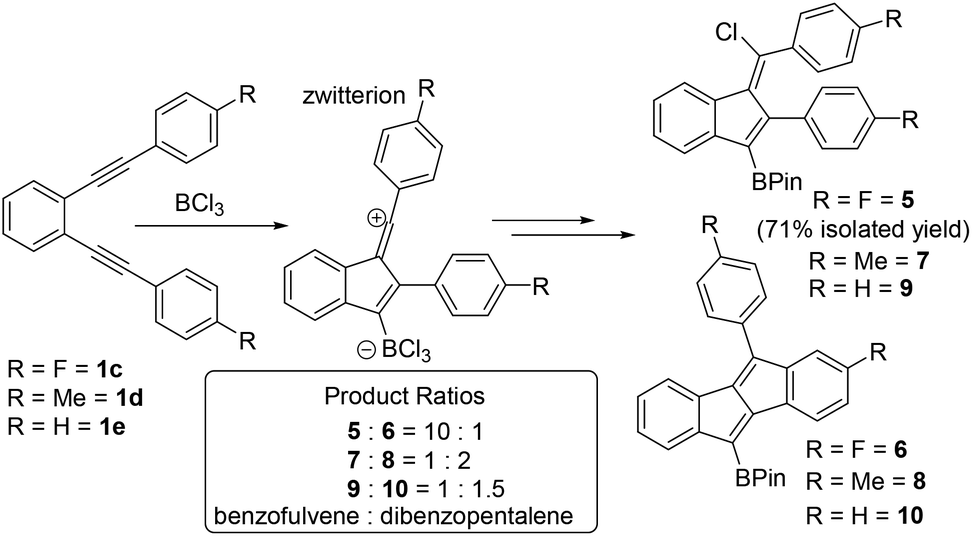 | ||
| Scheme 3 Product ratios from the borylative cyclisation of 1c–1e. | ||
In previous work the frustrated Lewis pair (FLP) combination of B(C6F5)3/P(o-tolyl)3 transformed 1e selectively (97% isolated yield) to the dibenzopentalene analogue of compound B.9 The effect of using the FLP combination of BCl3/2,4,6-tritert-butylpyridine (TBP) on product distribution was investigated as in weakly basic media the deprotonation of an arenium cation (a key step in dibenzopentalene formation) can become the rate determining step in SEAr.14 Therefore the inclusion of TBP as a Brønsted base was hoped to favour dibenzopentalene formation. However, using BCl3/TBP combinations led to comparable ratios of benzofulvene:dibenzopentalene starting from 1d and 1e as just using BCl3 (>two equivalents of BCl3 were used in these reactions as the by-product formed from SEAr is [TBPH][BCl4], thus use of only one equiv. BCl3 leads to incomplete consumption of the starting diyne).
With diynes 1d and 1e giving mixtures containing significant amounts of benzofulvene and dibenzopentalene products (which proved inseparable in our hands partly due to column chromatography leading to formation of protodeborylated products) alternative borylative cyclisation conditions were sought that would strongly favour formation of only one cyclised product. Considering a mechanism involving a vinyl cation intermediate two options were considered: (i) using boron electrophiles that form poorer chloride (or other anion) donors during the cyclisation process to favour dibenzopentalene formation. (ii) Increasing the concentration of chloride donor to favour benzofulvene formation, e.g. by adding [BCl4]− salts. Borocations of the general formula [Y2B(amine)]+ were selected for the first approach. Initial attempts focused on using borocations containing catechol (Cat) substituents, with the chelating diol disfavouring anion transfer reactions. However, [CatB(2-DMAP)][AlCl4] (2-DMAP = 2-(N,N-dimethylamino)-pyridine) led to no reaction with 1d at room temperature or at 60 °C for 24 h. Other more electrophilic catechol based borocations,15e.g. [CatB(NEt3)][AlCl4], led to very slow reactions at room temperature, with heating furnishing multiple intractable products in our hands.
From previous work, dihalo-borocations are more electrophilic than catechol derivatives thus the boronium salt [(2-DMAP)BCl2][AlCl4],7b and the borenium salt [Cl2B(2,6-lutidine)][AlCl4]16 were investigated. In comparison to [BCl4]− and [RBCl3]− these borocations will form weaker chloride donors during cyclisation, as loss of chloride, e.g. from compounds with the general structure D (Fig. 3, right), would lead to dications, which will have a high chloride ion affinity (this is supported by the chloride ion affinity of the dication derived from D by loss of chloride (and where the amine is pyridine) being 23.8 kcal mol−1 greater than that of BCl3). Furthermore, the vinyl cation D (where the amine is pyridine) will also be more electrophilic than the comparable zwitterion (calculations support this with a LUMO energy of −4.13 eV significantly lower in energy than that for the comparable zwitterion, −3.85 eV). Finally, AlCl3 is also a strong Lewis acid towards chloride, notably stronger than BCl3 (by 25 kcal mol−1),17 so [AlCl4]− will also be a poor chloride donor.
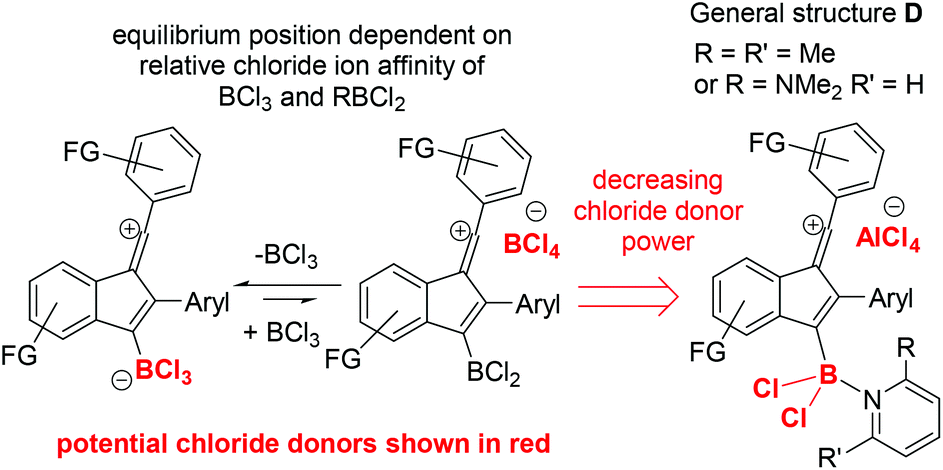 | ||
| Fig. 3 Potential sources of chloride during the borylative cyclisation of diynes 1x using different boron electrophiles. | ||
Using the boronium salt [Cl2B(2-DMAP)][AlCl4] in the cyclisation of 1d led to more selective formation of dibenzopentalene 8, with a 1:10 ratio of 7:8 observed (by NMR spectroscopy and GC-MS). In contrast, the use of [Cl2B(2-DMAP)][AlCl4] in the cyclisation of 1e led to a smaller change in the ratio of benzofulvene to dibenzopentalene (Scheme 4, inset for ratios). Therefore the borenium salt [Cl2B(2,6-lutidine)][AlCl4] was utilised as this will form an even poorer chloride donor (as 2,6-lutidine is a poorer nucleophile than 2-DMAP). Indeed using [Cl2B(2,6-lutidine)][AlCl4] the selectivity for the dibenzopentalene product 10 increased, with the ratio of 9:10 now 1:7. Thus it is possible to exert significant control over the product distribution in borylative cyclisation using borocations to increase the amount of dibenzopentalene products from 1d and 1e. However, attempts to use either of the [Cl2B(amine)]+ borocations to initiate cyclisation of 1a led to complex mixtures containing minimal dibenzopentalene, in part due to competitive ether cleavage, consistent with the reduced functional group tolerance of borocations relative to BCl3.7b,15
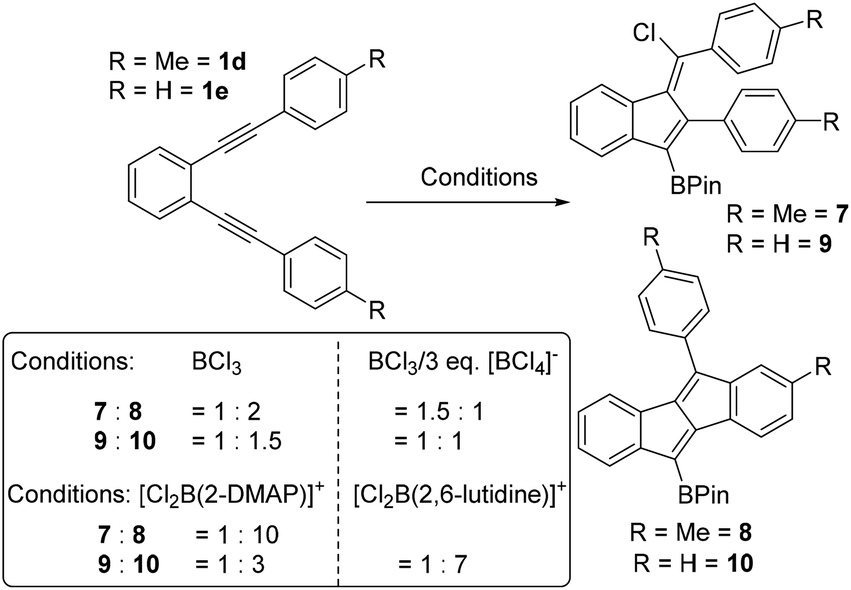 | ||
| Scheme 4 Change in product ratio on altering the boron electrophile or by the addition of an exogenous chloride source. | ||
Attempts to increase the amount of benzofulvene product formed by addition of an exogenous chloride donor were partially successful. For example, while the borylative cyclisation of 1d using BCl3 in the presence of three equivalents of [NBu4][BCl4] did lead to a change in the product ratio, the chlorinated benzofulvene 7:dibenzopentalene 8 ratio was only 3:2. With diyne 1e a small change also was observed with the ratio of 9:10 using BCl3/three equiv. of [NBu4][BCl4] being 1:1. Compound 5 was formed more selectively (with no 6 observed by NMR spectroscopy) when cyclisation using BCl3 was repeated in the presence of 3 equiv. [NBu4][BCl4]. However, attempts to form the benzofulvene derived from 1b were unsuccessful using BCl3/3 equiv. [NBu4][BCl4] with the dibenzopentalene product still formed in excellent conversion, presumably due to the intermediacy of a highly electrophilic vinyl cation that leads to rapid intramolecular SEAr. Thus increasing the chloride donor concentration using [BCl4]− has only a modest effect on selectivity. The use of “better” halide donors (i.e. free halide as in previously reported systems using HBr or I2)11,12 is not possible in this system as these would react with BCl3 (to form [X-BCl3]−, X = halide). Furthermore, the use of weaker Lewis acids e.g. PhBCl2 led to complex mixtures and significant amounts of unreacted diyne 1d after 18 h. Therefore the ability to access other benzofulvenes is limited via this methodology to requiring substituents that strongly stabilise the vinyl cation or significantly disfavour SEAr.
With an understanding of substituent effects in hand for aryl substituted diynes, the heteroaromatic bis-thienyl derivative 1f, was investigated next (Scheme 5). The 3-thienyl derivative was selected to preclude any S → BCl3 interactions directing reactivity and altering the reaction outcome.91f contains an electron rich thiophene heteroaromatic able to stabilise a vinyl cation to a greater extent (relative to benzene), however, the higher nucleophilicity of thiophene and its lower aromaticity (relative to benzene) will favour the SEAr reaction. Indeed on addition of BCl3 to 1f the SEAr product, 11, was the major product with no benzofulvene product observed by NMR spectroscopy. Using other conditions discussed above led to no observation of a benzofulvene product by NMR spectroscopy and GC-MS.
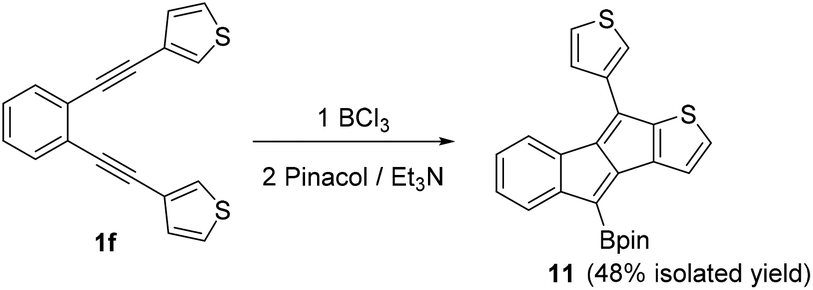 | ||
| Scheme 5 Borylative cyclisation of diyne 1f. | ||
Finally, other diynes were explored to determine if p-MeO-phenyl substituents would again lead to a chloro incorporated product (e.g. inset Scheme 6) by stabilising a vinyl cation intermediate during borylative cyclisation. Compound E had been previously shown to undergo cyclisation with BCl3 readily and was isolated as compound F post protection.7b Thus the p-MeO analogue, 12 was synthesised, however, this underwent cyclisation to give the product from intramolecular SEAr exclusively, with 13 isolated in 79% yield. No difference in product distribution was observed on performing the cyclisation with BCl3 in the presence of 3 equiv. of [NBu4][BCl4].
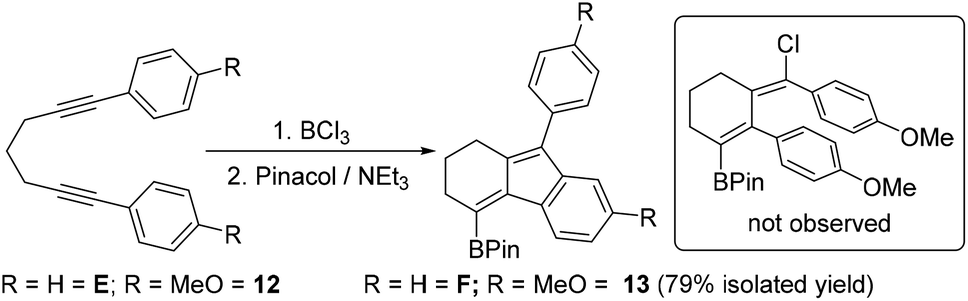 | ||
| Scheme 6 Borylative cyclisation of E (previous work) and 12 (this work). | ||
Hypothesising that the absence of any vinyl chloride product derived from 12 maybe due to the flexibility in the partially saturated 12 leading to a low barrier for the intramolecular SEAr step we investigated another fully Csp2 based diyne substituted with p-MeO-C6H4 groups, 14. However, addition of BCl3 to this diyne also led exclusively to the SEAr product, with no chloro-vinyl product observed, even when the reaction with BCl3 was performed in the presence of 3 equiv. of [NBu4][BCl4]. Compound 15 can be isolated post pinacol protection in 82% yield and is analogous to the cyclisation product derived from 14 using I2 as the electrophile.18 Again the BCl3 route is complementary to I2 induced cyclisation as the former installs a C-BPin nucleophilic moiety instead of an electrophilic C–I unit. We attribute the absence of any chlorine incorporated product in the borylative cyclisation of 14 with BCl3 to a lower barrier to SEAr due to the vinyl cation intermediate containing a six membered ring (ring A, inset Fig. 4) in contrast to a five membered ring in the vinyl cation derived from 1a (ring B). The latter may result in a higher barrier for the intramolecular SEAr step due to a less favourable geometry derived from the relative bond angles around rings A and B.
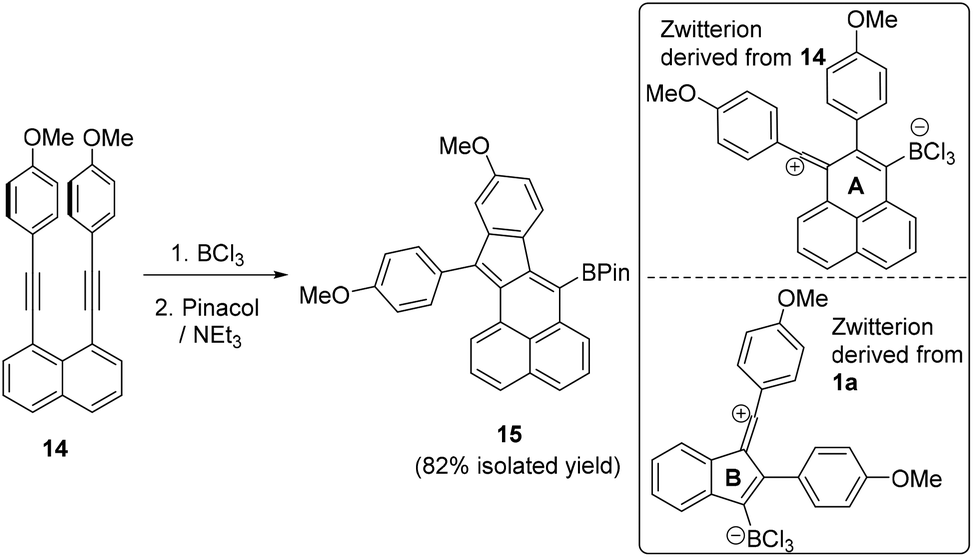 | ||
| Fig. 4 Borylative cyclisation of 14. Inset right, the zwitterionic vinyl cations derived from 1a and 14 on combination with BCl3. | ||
Conclusions
In conclusion the product outcome from the borylative cyclisation of 1,2-dialkynylbenzenes with BCl3 is highly dependent on substituents, with more strongly electrophilic vinyl cation intermediates favouring dibenzopentalene formation. In contrast, vinyl cations that are stabilised by donor groups (e.g. p-MeO-C6H4), or contain groups deactivating towards SEAr lead to benzofulvene products. The product distribution can be controlled to some extent by variation in the boron electrophile and by addition of an exogenous chloride donor ([BCl4]−) that does not shut down borylative cyclisation by reaction with the electrophile. However, the reaction outcome divergence could not be extended to two other diynes substituted with p-MeO-C6H4 groups presumably due to lower barriers to the SEAr reaction in these systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research leading to these results received funding from the ERC under framework 7 (Grant no. 305868) and the Horizon 2020 Research and Innovation Program (Grant no. 769599).References
- For reviews on metal catalysed borylative cyclisation: (a) E. Buñuel and D. J. Cárdenas, Chem. – Eur. J., 2018, 24, 11239 CrossRef; (b) E. Buñuel and D. J. Cárdenas, Eur. J. Org. Chem., 2016, 5446 CrossRef; (c) For a recent example of a radical borylative cyclization process see: M. Shimoi, K. Maeda, S. J. Geib, D. P. Curran and T. Taniguchi, Angew. Chem., Int. Ed., 2019, 58, 6357 CrossRef CAS PubMed.
- For a review covering metal catalysed and metal free routes see: A. Issaian, K. N. Tu and S. A. Blum, Acc. Chem. Res., 2017, 50, 2598 CrossRef CAS PubMed.
- For reviews on borylative cyclisation: (a) R. L. Melen, Chem. Commun., 2014, 50, 1161 RSC; (b) J. R. Lawson and R. L. Melen, Inorg. Chem., 2017, 56, 8627 CrossRef CAS PubMed; (c) For one early example see: T. Voss, C. Chen, G. Kehr, E. Nauha, G. Erker and D. W. Stephan, Chem. – Eur. J., 2010, 16, 3005 CrossRef CAS PubMed. B(C6F5)3 can effect cyclo-isomerisation via borylative cyclisation: (d) S. Tamke, Z.-W. Qu, N. A. Sitte, U. Flörke, S. Grimme and J. Paradies, Angew. Chem., Int. Ed., 2016, 55, 4336 CrossRef CAS PubMed; (e) Y. Soltani, L. C. Wilkins and R. L. Melen, Angew. Chem., Int. Ed., 2017, 56, 11995 CrossRef CAS.
- For a review on carboboration (including with cyclisation) see: G. Kehr and G. Erker, Chem. Sci., 2016, 7, 56 RSC.
- Boronic Acids: Preparation and Applications, ed. D. G. Hall, Wiley-VCH, Weinheim, Germany, 2nd edn, 2011 Search PubMed.
- Main Group Strategies towards Functional Hybrid Materials, ed. F. Jäkle and T. Baumgartner, Wiley and Sons, 2018 Search PubMed.
- For borylative cyclisation using BCl3 see: (a) C.-H. Yang, Y.-S. Zhang, W.-W. Fan, G.-Q. Liu and Y.-M. Li, Angew. Chem., Int. Ed., 2015, 54, 12636 CrossRef CAS PubMed; (b) A. J. Warner, J. R. Lawson, V. Fasano and M. J. Ingleson, Angew. Chem., Int. Ed., 2015, 54, 11245 CrossRef CAS; (c) A. J. Warner, A. Churn, J. S. McGough and M. J. Ingleson, Angew. Chem., Int. Ed., 2017, 56, 354 CrossRef CAS PubMed; (d) J. Lv, B. Zhao, L. Liu, Y. Han, Y. Yuan and Z. Shi, Adv. Synth. Catal., 2018, 360, 4054 CrossRef CAS.
- For a recent example of borylative cyclisation using CatBCl see: H. Bel Abed and S. A. Blum, Org. Lett., 2018, 20, 6673 CrossRef CAS . For earlier examples see ref. 2.
- (a) C. Chen, M. Harhausen, R. Liedtke, K. Bussmann, A. Fukazawa, S. Yamaguchi, J. L. Petersen, C. G. Daniliuc, R. Fröhlich, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2013, 52, 5992 CrossRef CAS PubMed; (b) C. Chen, M. Harhausen, A. Fukazawa, S. Yamaguchi, R. Fröhlich, C. G. Daniliuc, J. L. Petersen, G. Kehr and G. Erker, Chem. – Asian J., 2014, 9, 1671 CrossRef CAS PubMed.
- R. J. Kahan, W. Hirunpinyopas, J. Cid, M. J. Ingleson and R. A. W. Dryfe, Chem. Mater., 2019, 31, 1891 CrossRef CAS.
- H. W. Whitlock, P. E. Sandvick, L. E. Overman and P. B. Reichardt, J. Org. Chem., 1969, 34, 879 CrossRef CAS.
- R. K. Sauthwal, A. B. Danodia, M. Patel, S. Kumar and A. K. Verma, Chem. – Asian J., 2016, 11, 3001 CrossRef PubMed.
- L.-G. Zhou, W. Liao and Z.-X. Yu, Asian J. Org. Chem., 2012, 1, 336 CrossRef.
- For a discussion on how low basicity media effect SEAr see: T. S. De Vries, A. Prokofjevs, J. N. Harvey and E. Vedejs, J. Am. Chem. Soc., 2009, 131, 14679 CrossRef PubMed.
- V. Bagutski, A. Del Grosso, J. Ayuso Carrillo, I. A. Cade, M. D. Helm, J. R. Lawson, P. J. Singleton, S. A. Solomon, T. Marcelli and M. J. Ingleson, J. Am. Chem. Soc., 2013, 135, 474 CrossRef CAS PubMed.
- J. R. Lawson, E. R. Clark, I. A. Cade, S. A. Solomon and M. J. Ingleson, Angew. Chem., Int. Ed., 2013, 52, 7518 CrossRef CAS PubMed.
- E. R. Clark, A. Del Grosso and M. J. Ingleson, Chem. – Eur. J., 2013, 19, 2462 CrossRef CAS PubMed.
- X. Chen, P. Lu and Y. Wang, Chem. – Eur. J., 2011, 17, 8105 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, NMR spectra, crystallographic data and Cartesian coordinates for all calculations. CCDC 1529716 and 1529715. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ob00991d |
| This journal is © The Royal Society of Chemistry 2019 |