
Stereoselective synthesis of unnatural (2S,3S)-6-hydroxy-4-sphingenine-containing sphingolipids†‡
Daniel
Leichnitz
 ,
Sebastian
Pflanze
and
Christine
Beemelmanns
*
,
Sebastian
Pflanze
and
Christine
Beemelmanns
*
Leibniz Institute for Natural Product Research and Infection Biology – Hans Knöll Institute, Beutenbergstraße 11a, D-07745 Jena, Germany. E-mail: Christine.beemelmanns@hki-jena.de
First published on 17th May 2019
Abstract
6-Hydroxy-(4E)-sphingenine-containing sphingolipids are found in mammalian and bacterial membranes and have multiple intra- and intercellular functions. Most sphingolipids contain a (2S,3R)-2-amino-1,3-diol core structure, but only limited examples of unnatural (2S,3S)-2-amino-1,3-diol derivates have so far been reported. Using an underexplored hydrozirconation–transmetalation reaction and an unusual three-step-one-pot deprotection sequence, we were able to synthesize several unnatural (2S,3S)-6-hydroxy-(4E)-sphingenine-containing sphingolipids in only three (protected) or four (deprotected) consecutive steps, respectively, including a fluoresence-labeled derivative suitable for future biological studies.
Introduction
Sphingolipids serve as essential structural components of eukaryotic and bacterial membranes and as important lipid mediators of many cellular functions, including proliferation, differentiation, and apoptosis.1–4 Sphingolipids (SLs) are of amphipathic character and are composed of an alkyl chain carrying a N-acylated 2-amino-1,3-diol containing head group (usually having 2S,3R configuration).5 Both alkyl chains may differ in chain length, saturation, and the presence of additional hydroxyl groups. While human SLs are usually built up from linear alkyl chains, bacteria-derived sphingolipids carry mostly branched alkyl chains.6Amongst the structural diverse group of SLs, 6-hydroxy-(4E)-sphingenine-containing derivatives (6-OH-SLs, Fig. 1) have caught the research interest in recent years.7 In particular, 6-OH-SL derivatives 1 and 2 were investigated intensively as they were identified as a major ceramide component of the human stratum corneum, and are proposed to regulate the water content of the epidermis.8,9 Decreasing levels of 6-OH-SLs within the skin layers were detected in many skin diseases. Hence, the dysbalance of sphingolipid content was correlated to the disease progression and intensity.7 These studies have also led to the development of synthetic “pseudoceramides” as ceramide replacements in the cosmetic industry.10 In addition to these research efforts, a better understanding of the physicochemical properties of 6-OH-SLs, as well as their biosynthetic regulation might help to design better treatments in skin diseases.
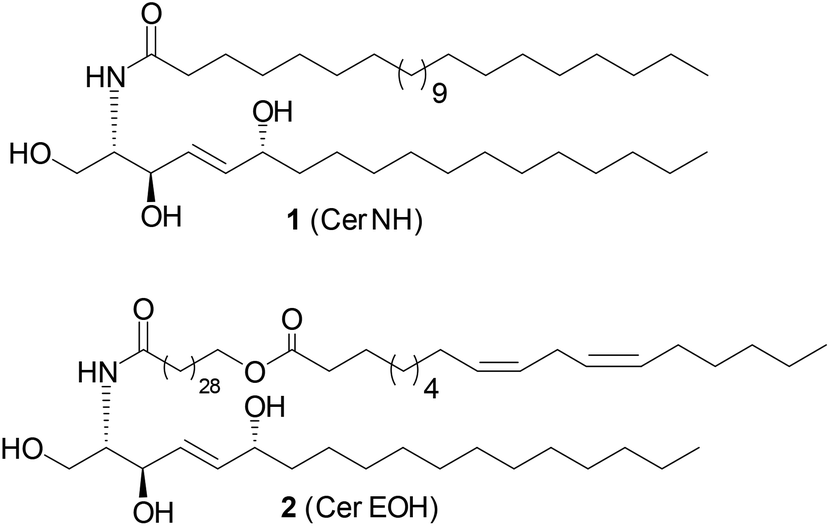 | ||
| Fig. 1 Naturally occurring 6-hydroxy-(4E)-sphingenine-containing SLs (6-OH-SLs) in humans.7 | ||
In a chemical ecology context, a bacterial-derived 6-hydroxy-(4E)-sphingenine derivative, a sulfonosphingolipid named RIF-2,11 was found to induce cell differentiation in a predatory choanoflagellate Salpingoeca rosetta, the closest living relative of animals. Subsequent ecological analysis of this peculiar group of sphingolipids hints to the importance of sphingolipids in cross-kingdom signalling pathways.4,12,13
Most synthetic approaches towards the (2S,3R)-6-hydroxysphingosine core structure rely on the stereoselective alkynylation of Garner's aldehyde 3, and reduction to the double bond in a second additional step (Scheme 1).14–16 More recent approaches included cross-metathesis reactions (Scheme 1, 6),17 or a [2,3]-Meisenheimer rearrangement followed by nucleophilic ring-opening reactions of N-activated aziridine derivatives 9.18
 | ||
| Scheme 1 (A) Retrosynthetic steps; (B) key steps of previously described 6-OH-sphingosine syntheses; (C) key reaction of this work. | ||
Despite these different synthetic methodologies, the preparation of 6-OH-SLs in sufficient amounts for bioactivity studies remains a challenge due to the acid-labile doubly hydroxylated allyl moiety. Furthermore, only limited examples of the so far unnatural (2S,3S)-stereoisomers have been described.
In light of our recent metabolomic studies characterizing novel bacterial sphingolipids, we aimed for a practical synthetic approach towards both stereoisomers, the more common (2S,3R) and less explored (2S,3S)-6-hydroxy-(4E)-sphingolipids, which would enable facile structure determination and bioactivity studies of unusual natural isolates.
Therefore, we set out to investigate the less explored hydrometalation–addition sequence of progaryl alcohols to aldehyde 3 in more detail as it combines the reduction of a triple bond and addition to aldehyde in one step, and thus reduces the number of synthetic steps (Scheme 1B).
Previous examples of hydrometalation–addition sequences in sphingolipid synthesis investigated almost exclusively non-substituted alkynes and reported partially contradictory yields and diastereoselectvity.19,20 Based on some preliminary tests however, we were confident that tuning of reaction parameters might allow selective access to both, the (2S,3R)- and (2S,3S)-6-hydroxysphingosine core structure.
Results and discussion
Our synthetic approach commenced with the synthesis of the protected propargylic alcohol 11a on gram scale (Scheme 2). The five-step sequence included a Friedel–Crafts type acylation of the previously synthesized fatty acid chloride of 13,21,22 and was followed by asymmetric transfer hydrogenation of the carbonyl group using RuCl[(R,R)-TsDPEN](mesitylene) (Noyori's catalyst),23 TMS-deprotection of the alkyne and then TBS-protection of the secondary alcohol in good overall yields. Similarly, propargylic alcohol 11b was available in five steps from tridecanoic acid in 82% yield.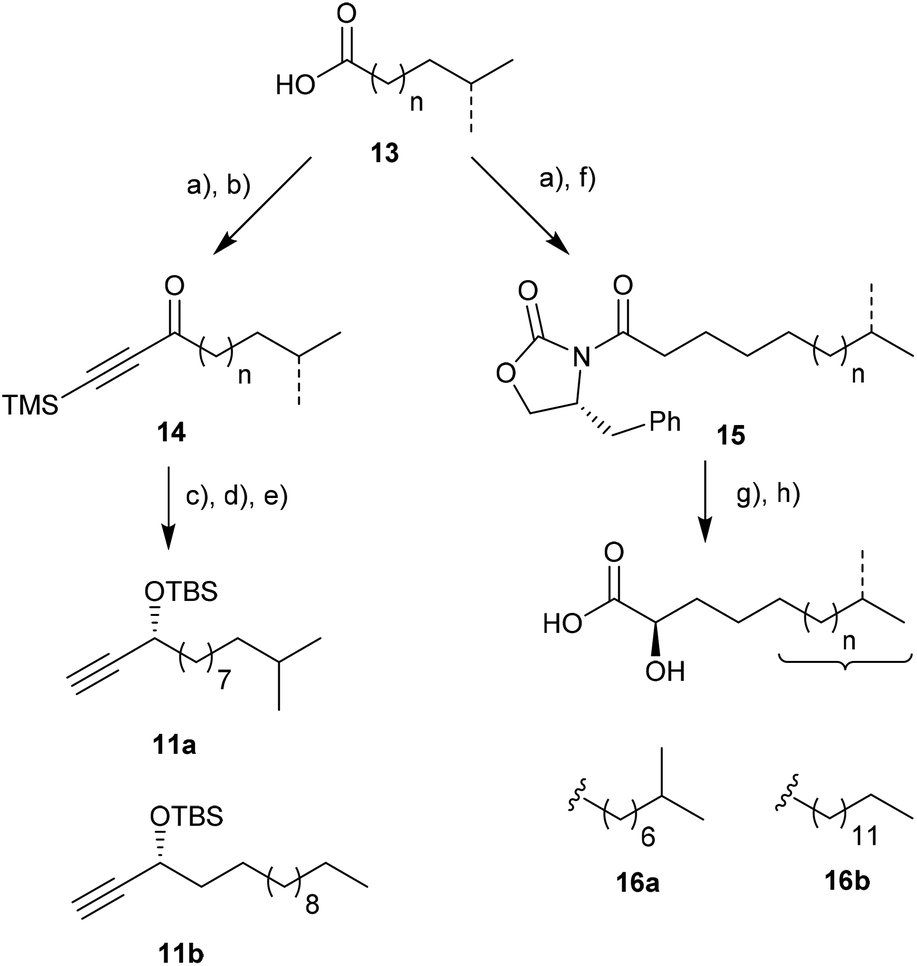 | ||
| Scheme 2 Synthesis of branched propargylic alcohol and α-hydroxylated fatty acids using the same starting material: (a) SOCl2, neat, r.t. 16 h; (b) AlCl3, bis(trimethylsilyl)acetylene, CH2Cl2, 0 °C → r.t., 3 h; for 14a 78% yield, for 14b 82% yield over two steps, respectively; (c) 2.0 mol% RuCl[(R,R)-TsDPEN](mesitylene), HCO2H, NEt3, i-PrOH, r.t., 6 h; (d) KOH, H2O/MeOH/CH2Cl2, r.t., 16 h; (e) TBSCl, DIPEA, 4-DMAP, DMF, r.t., 3 h; for 11a 70% yield, for 11b 86% yield over three steps, respectively; (f) ((R)-4-benzyloxazolidin-2-one, n-BuLi, THF, −78 °C → r.t., 16 h; (g) KHMDS, THF, −78 °C, 30 min, then Davis’ oxaziridine, −78 °C, 5 min; (h) LiOH, THF/H2O2, r.t., 16 h; for 16a 22% yield, for 16b 2.5% over four steps, respectively. | ||
The stereoselective synthesis of the branched α-hydroxylated fatty acid 16a and 16b, necessary for N-acylation of the sphingosine moiety at a later stage, was achieved by Davis oxaziridine mediated α-oxidation of Evans-auxiliary modified fatty acids (Scheme 2).24,25 Subsequent liberation of the fatty acid was achieved by mild saponification.26
With alkynol 11a in hand we first applied the literature reported two-step alkynylation–reduction procedure using Garner's aldehyde 3, which resulted in the formation of 12a (dr 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 70% over two steps) in favour of the (2S,3R)-configuration (anti-12a).27 The anti-selectivity is generally explained by an attack of the nucleophile from the sterically least hindered side (re-side attack) via a non-chelation Felkin–Anh-type transition state.
:1, 70% over two steps) in favour of the (2S,3R)-configuration (anti-12a).27 The anti-selectivity is generally explained by an attack of the nucleophile from the sterically least hindered side (re-side attack) via a non-chelation Felkin–Anh-type transition state.
We then set out to test different hydrometallation reagents (e.g. Dibal-H, Red-Al etc.) and compared both, yields and diastereoselectivity of the reduction–addition sequence (see Table 1, and ESI‡). Amongst the tested reagents only Schwartz's reagent (Cp2ZrHCl) afforded a reasonable transformation of 11a to the desired vinyl-metal species I (Scheme 3) and up to 86% of alkene 17 were isolated (Table 1, entry 1) indicating a successful hydrozirconation reaction. It needs to be noted that in all further tested reaction conditions, alkene 17 was observed as side product in varying ratios.
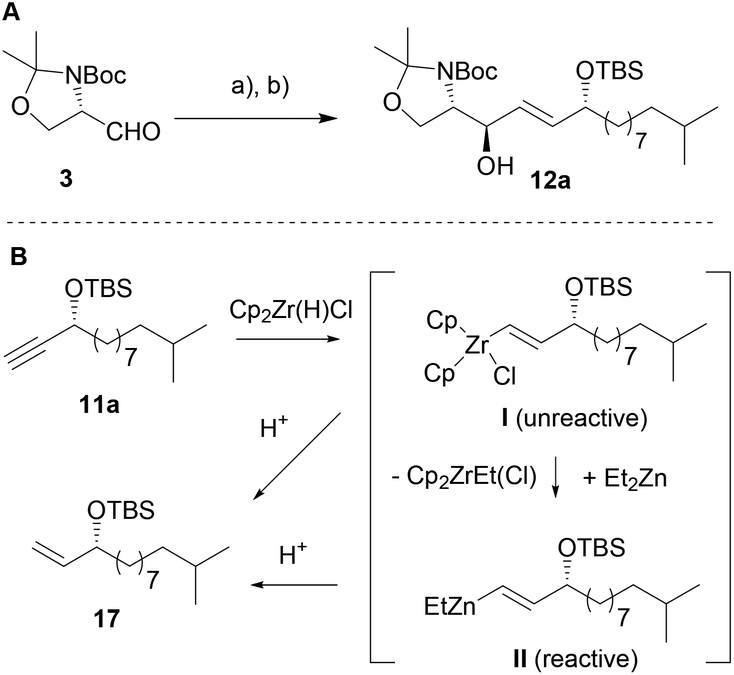 | ||
| Scheme 3 (A) Synthesis of protected (2S,3R)-6-hydroxy-sphingenine derivative 12a: (a) n-BuLi, 11a, THF, −78 °C → r.t., 3 h; (b) Red-Al, THF, −10 °C → 0 °C, 90 min, 70% yield over two steps, 18:1 dr (anti:syn). (B) Proposed organometallic intermediates. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Solvent | Temperature | Time [h] | Yield [%] (product) | Ratiob (a:b) |
|
| a Reactions performed on a 0.28 mmol scale (1 M solution of 3 and 11a). b Ratio determined by 1H-NMR (see ESI). c Reaction slowly warmed to r.t. overnight. d Reaction performed on a 0.87 mmol scale. | ||||||
| 1 | CH2Cl2 | No addition of Et2Zn | 0 °C to r.t. | 3 | 86 (17) | — |
| 2 | CH2Cl2 | — | −40 °C → r.t. | 4 | 33 (12) | 1.3:1.0 |
| 3 | CH2Cl2 | — | −40 °C → r.t. | 16 | 29 (12) | 1.3:1.0 |
| 4c | CH2Cl2 | — | −40 °C → r.t. | 16 | 50 (12) | 1.0:1.4 |
| 5c,d | CH2Cl2 | 11b used | −40 °C → r.t. | 16 | 61 (19) | 1.0:3.3 |
| 6 | Toluene | — | −40 °C → r.t. | 4 | 24 (12) | 2.4:1.0 |
| 7 | Toluene | 0.5 eq. Ti(Oi-Pr)4, 8 mol% (R,R)-18 | −78 °C → −20 °C | 16 | 39 (12) | 1.0:1.3 |
| 8 | Toluene | 0.5 eq. Ti(Oi-Pr)4, 8 mol% (S,S)-18 | −78 °C → −20 °C | 16 | 65 (12) | 1.0:2.4 |
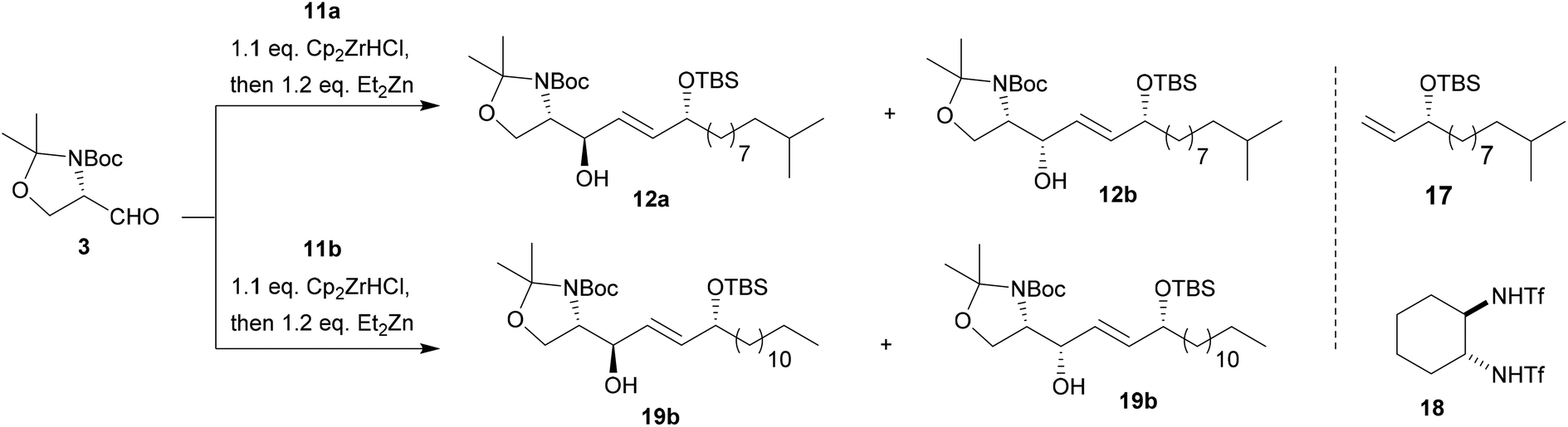
However, no addition to Garner's aldehyde 3 was observed presumably due to the weak nucleophilicity of intermediate I (Scheme 3). Testing of several literature known procedures revealed, that in situ transmetallation with ZnEt2 was required (II, Scheme 3) to achieve the addition reaction yielding 12 in moderate yields and with a slight preference for the anti-product 12a (entry 2, Table 1).28
To further improve the diastereoselectivity and yields of the addition reaction, we varied reaction time, temperature, and additives.29 The most pronounced increase in yield was observed, when the reaction mixture was slowly warmed to room temperature over a longer period of time after transmetallation (entry 4). These conditions also resulted in the slightly preferred formation of chelate-controlled syn-product 12b. Here, it can be speculated if chelation of the carboxy-group and the aldehyde occurs by the newly released Cp2Zr-species or the Zn-organyl itself.
However, higher yields in favour of the syn-product trend were observed when alkynol 11b was used instead of 11a and the reaction was performed at larger scale (entry 5). Addition of stronger Lewis acids such as ZnBr2 known to increase yields and selectivity resulted in only low yields (<10%) presumably due to side reactions such deprotection/isomerization of the propargyl alcohol 11 (ESI‡).16,20
Using toluene as solvent resulted in moderate amounts of 12 in favour of the anti-product 12a. However, addition of Ti(OiPr)4 as a chelating Lewis acid and the chiral ligand (R,R)-1730 caused a change in selectivity towards syn-isomer 12b and an increase in total yield (entry 7). Interestingly, enantiomeric ligand (S,S)-17 caused the preferred formation of syn-isomer 12b as well with a significant rise in total yield (entry 8), indicating a better matching ligand–substrate pair. Overall, our results indicate that the investigated hydrozirconation sequence can be tuned towards the preferred formation of the unnatural syn-isomers in one step with good overall yields. Thus, further reactions towards the desired 6-hydroxy-sphingenine were carried out on a 0.87 mmol scale using CH2Cl2 as solvent. Slow increase of reaction temperature over a period of 16 h resulted in overall good yields of 60–65% with an average dr of 1:3 in favour of the syn-product.
We then continued the synthesis of unnatural syn-6-OH-sphingolipids by double deprotection of the acetonide and Boc-group of 12b to generate the free amine suitable for acylation reaction. Standard deprotection conditions, such as acidic conditions, resulted in the loss of the TBS and acetonide-protecting groups prior to carbamate cleavage. Additionally, we observed isomerization and migration of the 6-hydroxy group leading to the undesired amine 20 as undefined isomeric mixture (Scheme 4). Based on literature reports that described the selective transesterification of t-butyl-esters and -carbamates to their respective silyl analogues by strong silylating agents,31 we applied the reaction conditions to 12b. We speculated that 12b undergoes the selective removal of the Boc-group and acetonide,32 and at the same time TBS-protection of the second allylic alcohol might occur.
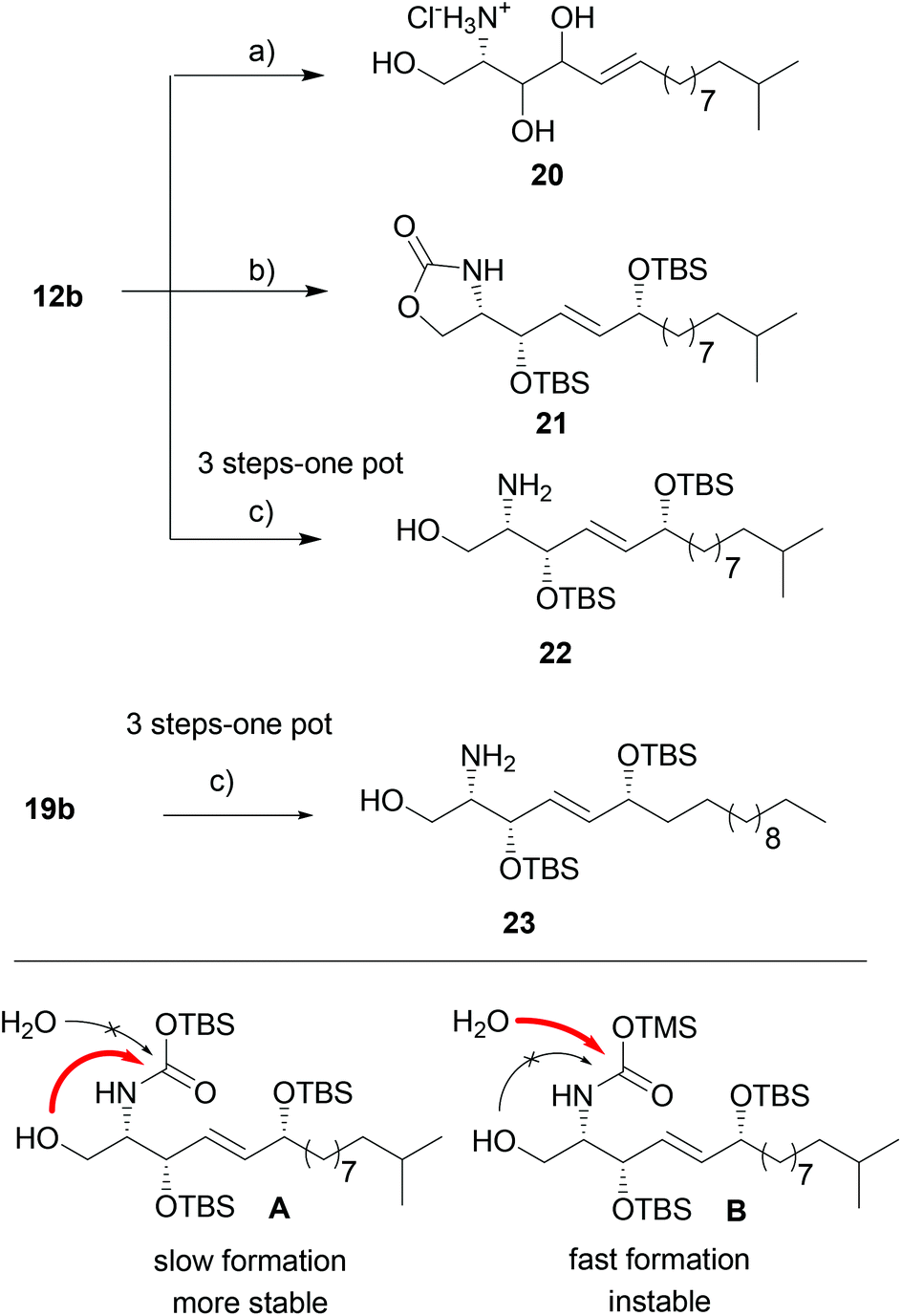 | ||
| Scheme 4 Optimization of deprotection sequence: (a) AcCl, MeOH, 0 °C → r.t., 1 h 32%; (b) TBSOTf excess, 2,6-lutidine, CH2Cl2, 0 °C, 2 h, 92%; (c) TBSOTf, 2,6-lutidine, 4-DMAP (10 mol%), then TMSOTf, CH2Cl2, 0 °C, 1 h; for 22 46% yield, for 23 78% yield. | ||
And indeed, treatment of 12b with an excess of TBSOTf resulted in a deprotection–protection sequence. However carbamate 21 was isolated as only product. Carbamate formation can be explained by a slow formation of the silylcarbamate followed by a relatively fast nucleophilic attack of the presumably unprotected alcohol in 1-position (intermediate A, Scheme 4). To prevent the side reaction, we sought to first apply equimolar amounts of TBSOTf to protect the secondary alcohol, and then add an excess of the more reactive TMSOTf to generate the less stable TMS-carbamate. Indeed, using this three-step one-pot sequence we were able to isolate free amines 22 and 23 in satisfying 46% and 78% overall yield, respectively.
To finalize the synthesis of 6-OH-SLs, we coupled the capnine base to the respective fatty acids using standard peptide coupling procedure with HBTU in DMF or DCC in CH2Cl2. Global deprotection was achieved with TBAF/THF for compound 24 and with HF/pyridine for compounds 28 and 30 (Scheme 5). As we are currently envisaging investigation into the biological activity of our unusual syn-sphingolipid derivatives, we also pursued the introduction of a fluorescent label. Previous studies showed that 6-NBD-hexanoic acid-modified spingosine allowed for the visualization of the Golgi apparatus in living cells.33 Thus, we integrated the same fluorescent probe into one of our sphingosine analogues (Scheme 5). Furthermore, we prepared derivative 32 carrying an alkyne moiety that could allow the introduction of tags within living organisms using modified click-reaction conditions.34
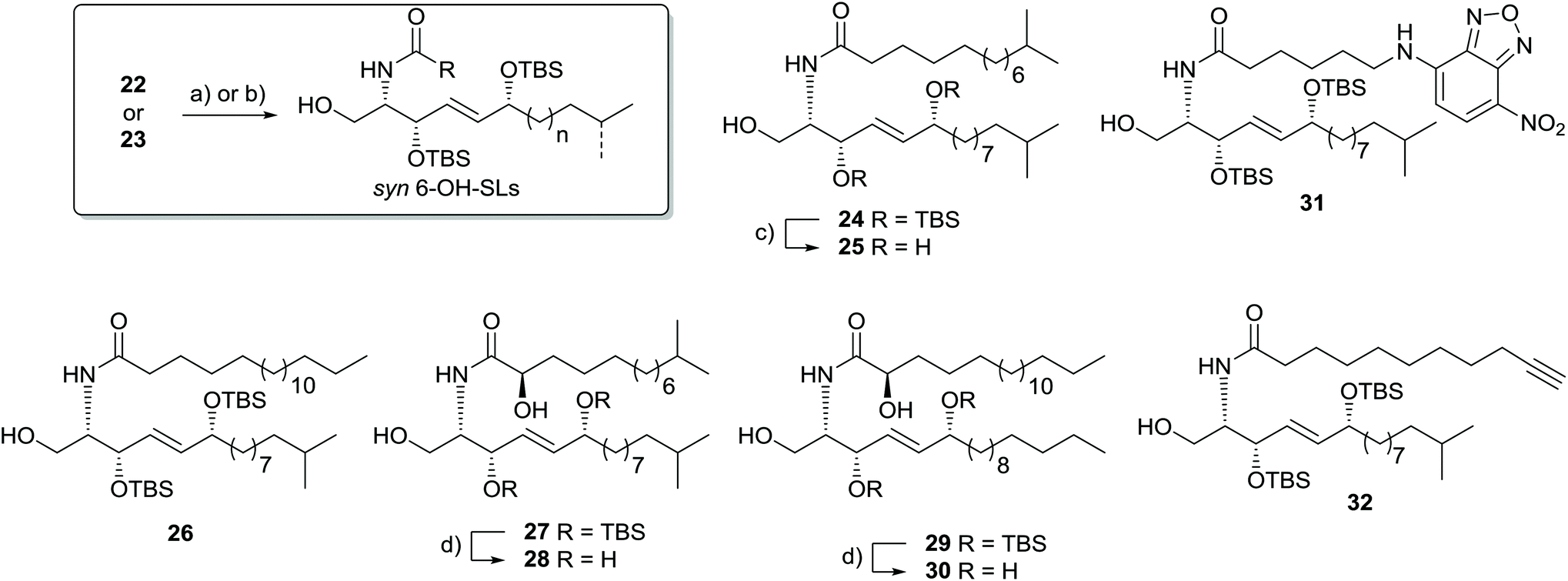 | ||
| Scheme 5 Synthesis of unnatural (3S)-6-OH-SLs: (a) carboxylic acids, DCC, DIPEA, CH2Cl2, 0 °C → r.t., 20 h; for 24 (13-methyltetradecanoic acid) 35% yield, for 26 (stearic acid), 30% yield, for 31 (6-((7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)amino)hexanoic acid) 50% yield; (b) carboxylic acids, HBTU, DMF, 0 °C → r.t., 2 h; for 27 ((R)-2-hydroxy-13-methyltetradecanoic acid) 47% yield, for 29 ((R)-2-hydroxyoctadecanoic acid) 25% yield, for 32 (undec-10-ynoic acid) 46% yield; (c) TBAF, THF, r.t., 16 h, 22% yield; (d) HF/pyridine, THF, 0 °C → r.t., 16 h; for 28 11% yield, for 30 8% yield. | ||
Conclusions
In summary, we investigated a hydrozirconation–addition reaction using Garner's aldehyde 3 and TBS-protected propargylic alcohols to prepare preferentially unnatural protected (3S)-6-OH-sphingenine derivatives. Using subsequently a 3-step-one-pot deprotection sequence, we were able to synthesize several novel (3S)-6-OH-SLs in only eight steps (TBS-protected, longest linear sequence) or nine steps (fully deprotected, longest linear sequence). Two of the synthesized derivatives carried an α-hydroxylated fatty acid, one a fatty acid having a terminal propargyl-group and one already containing a fluorophore.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the Leibniz Association and the Deutsche Forschungsgemeinschaft (DFG, Grant BE4799/2-1, EXSPHINGO). DL is generously supported by the International Leibniz Research School for Microbial and Biomolecular Interactions (ILRS). We thank L. Raguž (HKI) and A. Perner (HKI) for HRMS measurements and H. Heinecke (HKI) for NMR measurements.Notes and references
- T. Kolter and K. Sandhoff, Angew. Chem., Int. Ed., 1999, 38, 1532 CrossRef CAS PubMed.
- S. T. Pruett, A. Bushnev, K. Hagedorn, M. Adiga, C. A. Haynes, M. C. Sullards, D. C. Liotta and A. H. Merrill, J. Lipid Res., 2008, 49, 1621 CrossRef CAS PubMed.
- S. A. Morad and M. C. Cabot, Nat. Rev. Cancer, 2013, 13, 51 CrossRef CAS PubMed.
- J. B. Parsons and C. O. Rock, Prog. Lipid Res., 2013, 52, 249 CrossRef CAS PubMed.
- D. Leichnitz, L. Raguž and C. Beemelmanns, Chem. Soc. Rev., 2017, 46, 6330 RSC.
- Y. A. Hannun and L. M. Obeid, Nat. Rev. Mol. Cell Biol., 2008, 9, 139 CrossRef CAS PubMed.
- A. Kováčik, J. Roh and K. Vávrová, ChemBioChem, 2014, 15, 1555 CrossRef PubMed.
- D. T. Downing, M. E. Stewart, P. W. Wertz, S. W. Colton, W. Abraham and J. S. Strauss, J. Invest. Dermatol., 1987, 88, 2s CrossRef CAS PubMed.
- E. N. Tessema, T. Gebre-Mariam, R. H. H. Neubert and J. Wohlrab, Skin Pharmacol. Physiol., 2017, 30, 115 CrossRef CAS PubMed.
- T. Förster, The Chemistry of Natural and Synthetic Skin Barrier Lipids, in Cosmetic Lipids and the Skin Barrier, ed. H. Möller, CRC Press, 2001, Cosmet. Sci. Technol. Ser., vol. 24, p. 1 Search PubMed.
- A. Woznica, A. M. Cantley, C. Beemelmanns, E. Freinkman, J. Clardy and N. King, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7894 CrossRef CAS PubMed.
- A. M. Cantley, A. Woznica, C. Beemelmanns, N. King and J. Clardy, J. Am. Chem. Soc., 2016, 138, 4326 CrossRef CAS PubMed.
- M. McFall-Ngai, M. G. Hadfield, T. C. G. Bosch, H. V. Carey, T. Domazet-Lošo, A. E. Douglas, N. Dubilier, G. Eberl, T. Fukami, S. F. Gilbert, U. Hentschel, N. King, S. Kjelleberg, A. H. Knoll, N. Kremer, S. K. Mazmanian, J. L. Metcalf, K. Nealson, N. E. Pierce, J. F. Rawls, A. Reid, E. G. Ruby, M. Rumpho, J. G. Sanders, D. Tautz and J. J. Wernegreen, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3229 CrossRef CAS PubMed.
- J. S. Yadav, V. Geetha, A. Krishnam Raju, D. Gnaneshwar and S. Chandrasekhar, Tetrahedron Lett., 2003, 44, 2983 CrossRef CAS.
- J. Chun, H.-S. Byun and R. Bittman, J. Org. Chem., 2003, 68, 348 CrossRef CAS PubMed.
- A. Kovácik, L. Opálka, M. Silarová, J. Roh and K. Vávrová, RSC Adv., 2016, 6, 73343 RSC.
- P. Wisse, M. A. R. de Geus, G. Cross, A. M. C. H. van den Nieuwendijk, E. J. van Rooden, R. J. B. H. N. van den Berg, J. M. F. G. Aerts, G. A. van der Marel, J. D. C. Codée and H. S. Overkleeft, J. Org. Chem., 2015, 80, 7258 CrossRef CAS PubMed.
- S. K. Liew, S. J. Kaldas and A. K. Yudin, Org. Lett., 2016, 18, 6268 CrossRef CAS PubMed.
- Y. Iwayama, H. Ando, H. Ishida and M. Kiso, Chem. – Eur. J., 2009, 15, 4637 CrossRef CAS PubMed.
- T. Murakami, R. Hirono and K. Furusawa, Tetrahedron, 2005, 61, 9233 CrossRef CAS.
- C. Beemelmanns, A. Woznica, R. A. Alegado, A. M. Cantley, N. King and J. Clardy, J. Am. Chem. Soc., 2014, 136, 10210 CrossRef CAS PubMed.
- D. R. M. Walton and F. Waugh, J. Organomet. Chem., 1972, 37, 45 CrossRef CAS.
- N. Arai, H. Satoh, N. Utsumi, K. Murata, K. Tsutsumi and T. Ohkuma, Org. Lett., 2013, 15, 3030 CrossRef CAS PubMed.
- D. A. Evans, J. Am. Chem. Soc., 1985, 107, 4346 CrossRef CAS.
- F. A. Davis, U. K. Nadir and E. W. Kluger, J. Chem. Soc., Chem. Commun., 1977, 25 RSC.
- D. A. Evans, T. C. Britton and J. A. Ellman, Tetrahedron Lett., 1987, 28, 6141 CrossRef CAS.
- K. Mori and Y. Masuda, Tetrahedron Lett., 2003, 44, 9197 CrossRef CAS.
- R. S. Coleman and A. J. Carpenter, Tetrahedron Lett., 1992, 33, 1697 CrossRef CAS.
- M. Passiniemi and A. M. Koskinen, Beilstein J. Org. Chem., 2013, 9, 2641 CrossRef PubMed.
- H. Takahashi, T. Kawakita, M. Ohno, M. Yoshioka and S. Kobayashi, Tetrahedron, 1992, 48, 5691 CrossRef CAS.
- M. Sakaitani and Y. Ohfune, J. Org. Chem., 1990, 55, 870 CrossRef CAS.
- K. W. C. Poon, K. M. Lovell, K. N. Dresner and A. Datta, J. Org. Chem., 2008, 73, 752 CrossRef CAS PubMed.
- N. Lipsky and R. Pagano, Science, 1985, 228, 745 CrossRef CAS PubMed.
- J. Fink and J. Seibel, Biol. Chem., 2018, 399, 1157 CAS.
Footnotes |
| † Dedicated to Prof. Hans-Ulrich Reissig on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ob00990f |
| This journal is © The Royal Society of Chemistry 2019 |