
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA detailed mechanism of the oxidative half-reaction of D-amino acid oxidase: another route for flavin oxidation†
Dóra Judit
Kiss
 ab and
György G.
Ferenczy
*b
ab and
György G.
Ferenczy
*b
aDoctoral School of Chemistry, Eötvös Loránd University, Pázmány s 1/A, H-1117, Budapest, Hungary. E-mail: kiss.dora.judit@ttk.mta.hu
bMedicinal Chemistry Research Group, Research Centre for Natural Sciences, Hungarian Academy of Sciences, Magyar tudósok krt 2, H-1117, Budapest, Hungary. E-mail: ferenczy.gyorgy@ttk.mta.hu
First published on 5th August 2019
Abstract
D-Amino acid oxidase (DAAO) is a flavoenzyme whose inhibition is expected to have therapeutic potential in schizophrenia. DAAO catalyses hydride transfer from the substrate to the flavin in the reductive half-reaction, and the flavin is reoxidized by O2 in the oxidative half-reaction. Quantum mechanical/molecular mechanical calculations were performed and their results together with available experimental information were used to elucidate the detailed mechanism of the oxidative half-reaction. The reaction starts with a single electron transfer from FAD to O2, followed by triplet–singlet transition. FAD oxidation is completed by a proton coupled electron transfer to the oxygen species and the reaction terminates with H2O2 formation by proton transfer from the oxidized substrate to the oxygen species via a chain of water molecules. The substrate plays a double role by facilitating the first electron transfer and by providing a proton in the last step. The mechanism differs from the oxidative half-reaction of other oxidases.
Introduction
D-Amino acid oxidase (DAAO) regulates the D-amino acid levels in the brain, kidney and liver by degrading them through oxidative deamination.1D-Serine, the main substrate of DAAO in the brain, is the co-agonist of the N-methyl-D-aspartic acid (NMDA) receptor,2 whose malfunction is linked to the negative, positive and cognitive symptoms of schizophrenia.3 Increased DAAO expression manifested in post-mortem brain samples of schizophrenic patients.4 Moreover, genetic studies have identified association between schizophrenia and single nucleotide polymorphisms in DAAO.5 These findings suggest that raising the D-serine level through DAAO inhibition offers a potential therapeutic route to alleviate the symptoms of schizophrenia. Although the validation of the efficacy of DAAO inhibition therapy requires further studies,6 significant effort has been devoted to the development of novel, small molecule inhibitors of DAAO.7 However, until now only a single molecule (benzoate) has reached the clinical trial phase of development.8 Owing to the intense research on DAAO inhibition significant knowledge on the structure and function of DAAO enzymes has accumulated.DAAO belongs to the amino oxidase family of flavoproteins and it catalyses the degradation of D-amino acids into imino acids, which are further converted into α-keto acids in solution non-enzymatically. In humans, the enzyme functions as a homodimer,9 and in addition to the active site pocket, it has a pocket with unknown functionality at the dimer interface.10 Human DAAO binds FAD, the natural cofactor, with fairly low affinity (8 μM Kd), though substrate binding decreases the Kd to 0.3 μM.9,11 The active site residues are highly conserved across species suggesting a common reaction mechanism. DAAO works optimally around pH 8, where the amino acids bind negatively charged to the enzyme.12 The bound substrate is coplanar with the isoalloxazine moiety of the FAD and interacts by hydrogen bonds with the conserved arginine (Arg283 in human DAAO (hDAAO) and Arg285 in Rhodotorula gracilis DAAO (RgDAAO)), glycine backbone and water (Fig. 1a).
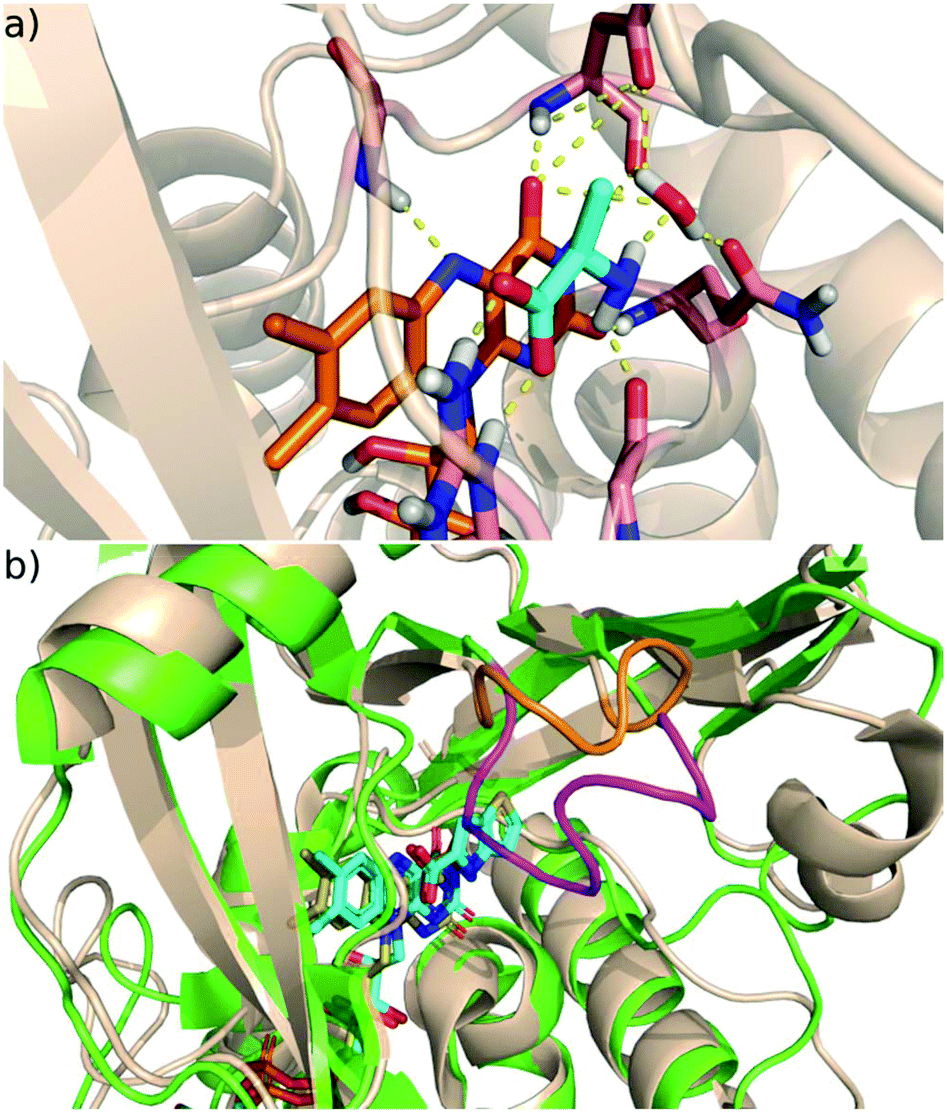 | ||
| Fig. 1 (a) The active site of DAAO with bound D-alanine (PDB code 1C0P; FAD orange, D-alanine cyan, protein wheat cartoon, H-bonds dashed yellow lines. (b) The active site and its environment of the human (green) and Rhodotorula gracilis (tan) DAAO; the active site loop in the human enzyme is coloured purple and the corresponding shorter segment in Rg is orange. The FAD cofactor and the bound ligand are coloured cyan (human) and yellow (Rg). | ||
A notable feature of mammalian DAAOs is the presence of a flexible active site loop13 missing in yeast DAAOs (Fig. 1b). This structural variation is proposed to be responsible for the differences in the rate limiting step of the DAAO catalysed reaction14–16 that is the product release for mammalian DAAO,17 and the reductive half-reaction (see below) for yeast DAAO.18
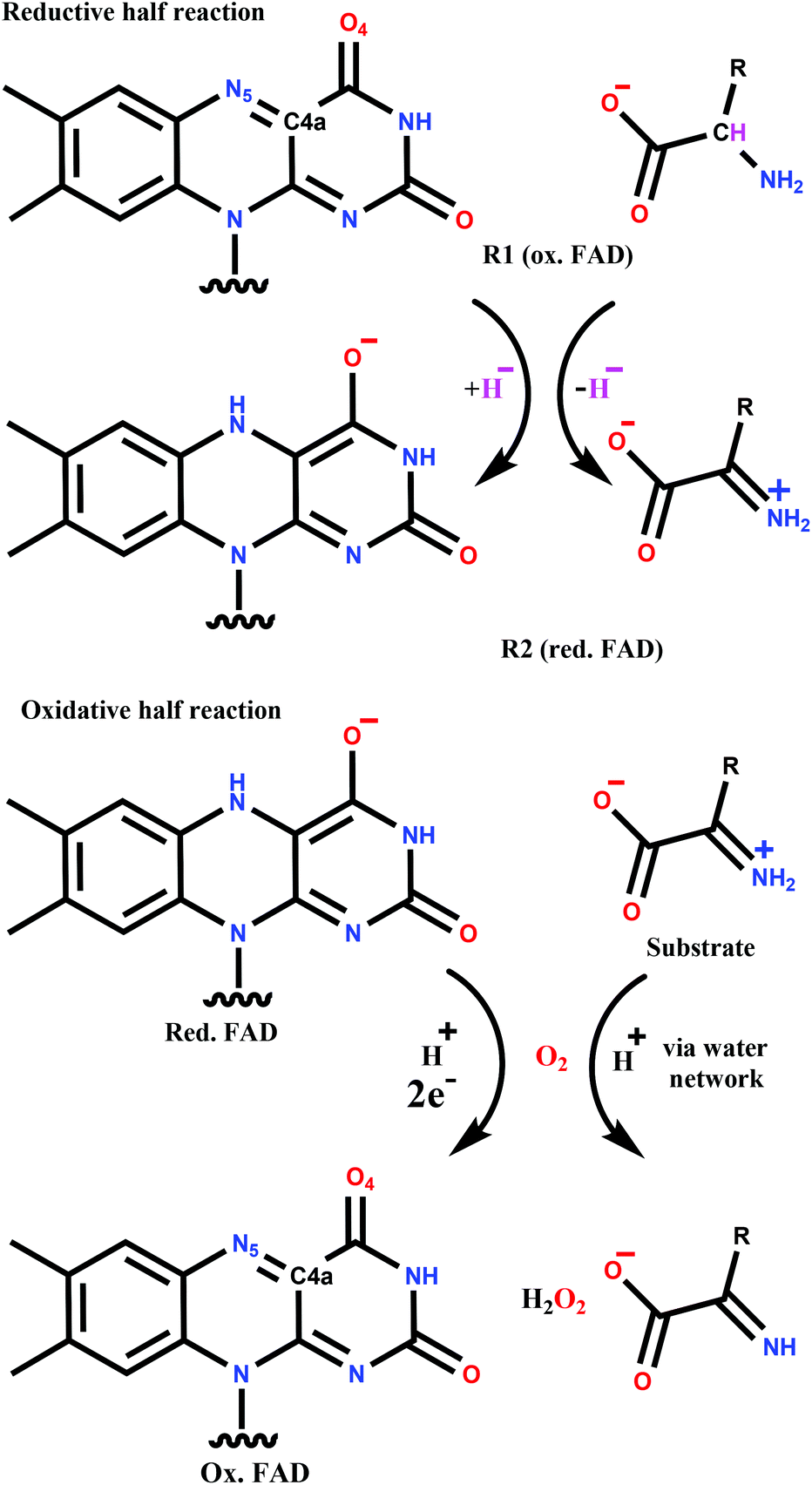
The catalytic cycle can be divided into two parts (Scheme 1). The reductive half-reaction includes the reduction of FAD by the substrate and leads to the anionic hydroquinone state of FAD. Both experimental (kinetic studies, X-ray structures) and quantum mechanics (QM) based calculations support a hydride transfer mechanism from the substrate,15,19,20 which is a general feature of flavoprotein oxidases.21,22
 | ||
| Scheme 1 The general mechanism of the DAAO catalysed reaction. | ||
The mechanism of reoxidation of reduced FAD with molecular oxygen (oxidative half-reaction) is still the subject of intense research.22–25 The two groups of flavoproteins that readily react with O2, namely monooxygenases and oxidases, follow different mechanisms leading to different products. Both are proposed to start with a single electron transfer from the flavin to the oxygen resulting in a caged diradical pair.22,23,26–28 The reaction with monooxygenases proceeds through a covalent intermediate (C4a-(hydro)peroxy adduct; see Scheme 1 for atom numbering) and completes with water formation. In contrast, the covalent adduct is typically not detected with oxidases. The end-product in the oxidative half-reaction of oxidases is hydrogen-peroxide that is formed together with the reconstituted FAD. This general picture of the mechanism has been challenged by recent combined experimental and computational studies suggesting that the first step is a proton coupled electron transfer to O2 in p-hydroxyphenylacetate 3-hydroxylase29 and pyranose 2-oxidase.30 Moreover, in contrast to many other oxidases, the formation of the C4a-(hydro)peroxy adduct was observed for pyranose 2-oxidase.31
The O2 molecule accesses the FAD buried in the interior of the protein by diffusion through tunnels. Molecular dynamics studies identified oxygen tunnels and two high affinity oxygen sites in DAAO. Moreover, mutations near the highest affinity oxygen locus at the FAD Si side were shown to influence O2 reactivity.32 The oxygen probably approaches “edge-on” the flavin, which was proposed to be characteristic of oxidases and might facilitate the non-covalent pathway.24 Stopped-flow kinetic studies suggest that the reoxidation process progresses differently in the absence and in the presence of the oxidized substrate; the reoxidation of the substrate-enzyme complex is faster than that of the free reduced DAAO9,18,33 showing that the presence of the ligand is beneficial in the oxidative half-reaction.
To the best of our knowledge, the mechanism of the oxidative half-reaction was investigated in detail in three oxidases, namely pyranose 2-oxidase,30,31 choline oxidase34 and aryl-alcohol oxidase.35 It was found that their mechanisms concerning the sequence and relative energetics of the elementary steps are different. In the present study QM/MM calculations were performed for the oxidative half-reaction of DAAO. Mechanistic details of this reaction are not readily attainable for experiments, primarily because the first step is rate limiting and no intermediates of the subsequent steps are observed.18,36 The combination of computational and experimental results was used to identify the reaction steps and to explore the role of the ligand, as well as the electronic states and configurations of the intermediates. The details of the reaction are compared to the mechanism of flavin oxidation found in other oxidases.
Computational methods
Model building
The models applied in the calculations were based on the high-resolution X-ray structure of the Rhodotorula gracilis DAAO (RgDAAO) (PDB code 1C0P, 1.2 Å).37 The dioxygen species was deleted and the protein was prepared with Schrödinger's Protein Preparation Wizard;38 missing hydrogen-atoms were added, protonation states were assigned and the H-atom network was optimized. Two models of enzyme complexes were developed for the mixed quantum mechanics/molecular mechanics calculations; one with D-alanine and oxidized flavin and another containing the reduced FAD with the 2-iminopropanoate species. Both models were solvated in a TIP3P39 water box and neutralised with Na+ ions. (∼29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 water molecules and 1 Na+ ion were added.) Molecular dynamics simulations were carried out with GROMACS40 using Amber14 force field41 and GAFF42 to assign parameters to the protein and to the ligands, respectively. Periodic boundary conditions with particle mesh Ewald electrostatics43 were applied. The system was first minimized in 5000 steepest descent steps, and then it was heated to 300 K in three 100 ps equilibration steps in an NVT ensemble applying velocity-rescaling temperature coupling and using positional restraints of 1000 kJ mol−1 nm−2 for the protein backbone and ligand atoms. The final equilibration step contained 1 ns run in an NPT ensemble with 2 fs integration step at 300 K and 1.013 bar pressure applying a Parrinello Rahman barostat with restraints only on the ligand atoms. Molecular dynamics simulations were then performed for 50 ns at 300 K in a canonical ensemble. Due to the instability of the oxidized FAD–D-alanine complex in our test simulations we added restraints on the isoalloxazine part of the FAD and the substrate heavy atoms to avoid significant changes compared to the X-ray structure. The observed instability is attributed to the close distance between the FAD and substrate and, in particular, its Cα–H moiety. It is to be noted that our model is derived from an X-ray structure containing simultaneously the pre- and post-reaction species, and the substrate is not expected to form a stable complex in the pre-reaction phase. The reduced flavin iminopropanoate complex remained stable during the equilibration and in the production run, thus no constraints were applied in that case.
000 water molecules and 1 Na+ ion were added.) Molecular dynamics simulations were carried out with GROMACS40 using Amber14 force field41 and GAFF42 to assign parameters to the protein and to the ligands, respectively. Periodic boundary conditions with particle mesh Ewald electrostatics43 were applied. The system was first minimized in 5000 steepest descent steps, and then it was heated to 300 K in three 100 ps equilibration steps in an NVT ensemble applying velocity-rescaling temperature coupling and using positional restraints of 1000 kJ mol−1 nm−2 for the protein backbone and ligand atoms. The final equilibration step contained 1 ns run in an NPT ensemble with 2 fs integration step at 300 K and 1.013 bar pressure applying a Parrinello Rahman barostat with restraints only on the ligand atoms. Molecular dynamics simulations were then performed for 50 ns at 300 K in a canonical ensemble. Due to the instability of the oxidized FAD–D-alanine complex in our test simulations we added restraints on the isoalloxazine part of the FAD and the substrate heavy atoms to avoid significant changes compared to the X-ray structure. The observed instability is attributed to the close distance between the FAD and substrate and, in particular, its Cα–H moiety. It is to be noted that our model is derived from an X-ray structure containing simultaneously the pre- and post-reaction species, and the substrate is not expected to form a stable complex in the pre-reaction phase. The reduced flavin iminopropanoate complex remained stable during the equilibration and in the production run, thus no constraints were applied in that case.
QM/MM calculations
Starting structures for the QM/MM calculations were selected from the MD simulations. For the oxidative half-reaction, we selected a frame, where a water network was observed between the substrate and four water molecules, one of them occupying the previously proposed high affinity oxygen site.32 This water was replaced by an O2 molecule for further calculations and the structure was fully minimized at the QM/MM level. A 10 Å water shell (∼4800 water molecules) was kept in the QM/MM calculations. The neutral QM region consisted of the isoalloxazine ring system of the FAD with a capping methyl group, the substrate and the Arg285 residue in calculations for the reductive half-reaction. The oxygen with the three water molecules connecting it to the substrate was also part of the QM system for the oxidative half-reaction. The link atom approach was applied to separate the QM and MM regions that were interacting with electronic embedding. Atoms in a shell of 15 Å around the QM region were allowed to move, while the rest of the model was kept frozen.Relaxed coordinate scans were carried out applying sander44,45 combined with Gaussian0946 at the B3LYP/6-31G*47,48 level of theory, which was successfully applied previously in complex systems.29,49,50 The step size used to scan the reaction coordinates was 0.1 Å. The transition states were identified as the highest energy points of the scans. The reactant, intermediate and final states were further minimized without constraints. For each minimum, derivatives were evaluated at small displacements in both directions along each QM coordinate to verify the positive diagonal elements of the Hessian. Minimizations starting from both sides of the transition states and reaching the neighbouring minima were performed to confirm that each transition state connects the minima at the endpoints of the scan. For the optimized geometries B3LYP/6-31+G** energies were calculated. The unrestricted formalism (UB3LYP) was applied both in the triplet and open-shell singlet calculations.
Due to spin-contamination in the unrestricted singlet calculations, we applied spin projection to obtain the energy of the open shell singlet state (OSSSPE) using eqn (1) and (2):29,51,52
| OSSSPE = EOSS + CSC(EOSS − ET) | (1) |
| CSC = 〈SOSS〉/(〈ST〉 − 〈SOSS〉) | (2) |
The open shell singlet diradical character (Y%) was estimated using eqn (3).53
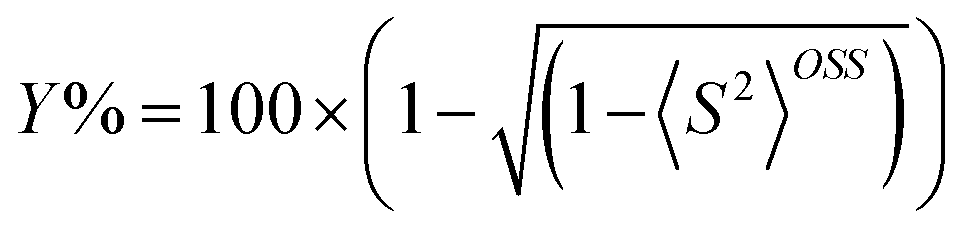 | (3) |
Natural bond orbital (NBO) analysis54 as implemented in Gaussain09 was performed.
Minimum energy crossing point (MECP) estimation
The MECP was located by applying a special minimization procedure.55,56 The main idea is to follow the effective gradient calculated as α∇(V1 − V2)2 + P(∇V1) during the minimization, where V1 and V2 are the potential energies on the two surfaces and P(∇V1) projects out the direction corresponding to the difference in the gradient of the two surfaces from the gradient on surface 1. The original program code55 written for Gaussian by Prof. Jeremy Harvey was adapted to estimate the MECP in AMBER-Gaussian QM/MM calculations. The MECP was located at the UBLYP/6-31+G** level of theory due to the severe instability of the open shell singlet diradical with the hybrid UB3LYP functional. The UB3LYP/6-31+G** energies for the open shell singlet and triplet states differ by less than 0.3 kcal mol−1 at the geometry corresponding to the UBLYP/6-31+G** MECP and, therefore, this geometry and the UB3LYP/6-31+G** energies were accepted as the MECP.Results and discussion
Reductive half-reaction
The first step of the catalytic cycle of DAAO starts with a hydride transfer from the bound substrate to the FAD cofactor.37 As the details of this reaction step are well established,15,37 it provides a good basis for validating our model system and the method applied.The B3LYP/6-31+G** QM/MM energies calculated with the relaxed coordinate scan along the substrate α-CH FAD-N5 distance give a 15.4 kcal mol−1 barrier for the reductive half-reaction of DAAO with D-alanine as a substrate (Fig. 2, red). This is in reasonable agreement with the experimental value of 13–15 kcal mol−1 (1.8 × 103–2.4 × 105 M−1 min−1)57 calculated from stopped flow rate constant measurements at 25 °C,18,58 and also with ab initio QM MD simulations performed for a smaller model system.15 The slight overestimation might be explained by the lack of dynamical and entropic effects in the calculations as well as the lack of tunnelling, which might contribute to the observed faster kinetics rates. NBO analysis of the optimized geometries (Fig. S1†) shows charge transfer from the substrate to the cofactor in line with the previously proposed hydride transfer mechanism (Table 1). Charges of the transition state (R1TS) are in between those of states R1 and R2 being slightly closer to the former.
 | ||
| Fig. 2 Energy diagrams of the reductive half-reactions for DAAO with D-alanine (red) and 3,3,3-trifluoro-D-alanine (CF3–D-alanine) (blue). See Scheme 1 for the meaning of R1 and R2. | ||
| D-Alanine | ||||
|---|---|---|---|---|
| R1 | R1TS | R2 | Change | |
| FAD | −0.128 | −0.568 | −1.114 | −0.987 |
| Substrate | −0.967 | −0.635 | −0.195 | 0.772 |
| H | 0.253 | 0.346 | 0.437 | 0.185 |
| Arg285 | 0.842 | 0.857 | 0.872 | 0.030 |
Despite the structural similarity of the bound form of 3,3,3-trifluoro-D-alanine (subsequently will be denoted as CF3–D-alanine)37 compared to the substrate D-alanine,37 the trifluoromethyl derivative inhibits the enzyme likely due to the strong electron withdrawing nature of the –CF3 group. To further test the capabilities of our model, we calculated the reductive half-reaction with this inhibitor ligand, as well. The reaction barrier was slightly higher (17.6 kcal mol−1) compared to the native substrate; however, the energy of the product is 10.3 kcal mol−1 higher than that of the reactants suggesting a thermodynamically controlled inhibition by CF3–D-alanine (Fig. 2, blue). NBO analysis showed significantly less charge transfer from the ligand to the cofactor (Table 2) in line with the strong electron withdrawing nature of the trifluoromethyl group.
| CF3–D-alanine | ||||
|---|---|---|---|---|
| R1 | R1TS | R2 | Change | |
| FAD | −0.099 | −0.540 | −0.901 | −0.805 |
| Substrate | −1.033 | −0.701 | −0.422 | 0.611 |
| H | 0.274 | 0.374 | 0.454 | 0.180 |
| Arg285 | 0.858 | 0.867 | 0.872 | 0.014 |
The above results are in accordance with both experimental data18,58 and Car-Parinello QM MD simulations15 performed for the active site model without considering the effect of the protein environment. These findings support that our model system and the applied method are suitable to study the reaction mechanism of D-amino acid oxidase.
Oxidative half-reaction
The oxygen reaction with flavin is rather slow in solution, mainly due to the poorly catalysed first single-electron transfer step. Flavoenzymes accelerate this reaction up to several orders of magnitude showing that the protein environment effectively controls the reactivity of the partners. Positively charged residues (histidines, lysines) play a crucial role in many flavoprotein oxidases e.g. in glucose oxidase26 and sarcosine oxidase27,59 leading to a general consensus on the importance of the positive charge around the oxygen locus.60 Its proposed role is the stabilization of the superoxide anion O2˙− and providing a transferable proton. However, in choline oxidase a positively charged ammonium moiety plays the same key role in the catalysis61 showing that not only protein side chains can provide the electrostatic stabilization and the proton.25 Similarly, the active centre of the DAAO enzyme lacks acidic and basic residues; thus the protein is unable to provide the positive charge or the necessary proton for the catalytic cycle to conclude. The significant increase of the reaction rate measured by stopped flow spectrometry in the presence of the substrate suggests the involvement of the ligand in the reaction and supports a ternary complex mechanism.18,32,58 The oxygen and the substrate binding site are in the opposite sides of the FAD isoalloxazine ring thus prohibiting a direct proton transfer between the two species. MD simulations proposing the high affinity O2 binding site also revealed a water network connecting the O2 binding cavity on the FAD Si side to the Re side,32 where the substrate is bound.
Our MD simulations of the RgDAAO – reduced FAD – 2-iminopropanoate complex also revealed a water network between the Re and Si side of the flavin moiety providing a route to protonate the oxygen. A water molecule occupying the previously proposed high affinity O2 binding site was transformed into an O2 molecule for the further calculations of the oxidative-half reaction.
The charge distribution of the system with and without the O2 molecule was calculated by NBO analysis. The negative charge of the reduced FAD is localized on C4a, N5 and the O4 carbonyl oxygen atom before the oxygen binding (see Scheme 1 for atom numbering). These findings are supported by X-ray37 and 13C-NMR data65 and also by Car-Parinello QM MD results15 proposing high electron density on C4a. These atoms are all close to the binding site of the oxygen molecule and this facilitates the electron transfer between the FAD and the oxygen. Indeed, the NBO analysis of the oxygen bound structure both in the O1T triplet, and the O1OSS singlet states (see Scheme 2 for the designation of intermediate states and Table S1† for a complete list of QM atomic charges in the O1T, O1OSS, O2aCSS, and O3CSS states) showed that the majority of the negative charge is on the oxygen molecule (Table 3) and the largest change in the partial charges upon oxygen binding is observed for these atoms (Table S1†). These results suggest that the first step is a single electron transfer from FADH− to O2 resulting in the semiquinone-superoxide anion containing system.
 | ||
| Scheme 2 Reaction pathways investigated for the oxidative half-reaction of DAAO. T denotes triplet, OSS open shell singlet and CSS closed shell singlet states. The protonated imino compound in O2aCSS and the FAD semiquinone in the O2bOSS systems are not shown as they do not change with respect to the preceding state. Purple arrows indicate proton transfer from the FAD, while green arrows indicate proton transfer from the substrate. The transfer of electrons as obtained from NBO analysis is also shown. The pathway with covalent adduct formation was also investigated as shown in Scheme S1.† | ||
| O1T | O1OSS | O2aCSS | O3CSS | |
|---|---|---|---|---|
| FADH/FAD | −0.104 | −0.097 | −0.187 | −0.025 |
| O2/O2H/H2O2 | −0.846 | −0.850 | −0.713 | −0.089 |
| Substrate | 0.068 | 0.068 | 0.066 | −0.736 |
 | ||
| Fig. 3 (a) SOMO (mainly located on FAD) and (b) SOMO−1 (mainly located on O2˙−) orbitals for the semiquinone-superoxide open-shell singlet system. | ||
The high similarity of the triplet and open shell singlet states of the semiquinone-superoxide anion system suggests that a minimum energy crossing point (MECP) can be found that is similar both in geometry and energy. Therefore, we initiated a MECP search from the triplet state at the UB3LYP/6-31+G*, level; however, we were only able to locate a MECP at a geometry, in which the H-atom attached to N5 in the semiquinone-superoxide anion complex was largely transferred to the oxygen species. The singlet state of the MECP was essentially a closed shell and the corresponding energy is 9 kcal mol−1 higher than that of the semiquinone-superoxide anion. Although this is a valid MECP, we assumed that another one exists near the semiquinone-superoxide anion system and we were unable to identify it owing to the known spin contamination of the unrestricted DFT with hybrid functionals.66 Therefore, we repeated the MECP search at the UBLYP/6-31+G** level, and a MECP was located at a geometry close to the optimized semiquinone-superoxide anion system. The UB3LYP/6-31+G** energies of the singlet and triplet state at this MECP geometry agree within 0.3 kcal mol−1 and they are higher by 3.8 kcal mol−1 than the triplet energy at the corresponding optimized geometry. The open shell character of the singlet state at the MECP is 95%. These results show that the system can undergo spin-transition after the first electron transfer and the subsequent reaction steps start from an open shell singlet state (O1OSS, see Scheme 2). Therefore, the optimized open shell singlet geometry was used as the starting point in analysing further steps of the reaction.
First, we investigated the proton transfer from the FAD semiquinone to the superoxide anion. During the relaxed scan of the FAD-N5H–O distance to simulate the FAD deprotonation step, the obtained transition state is characterized by 1.2 Å O–H distance and 2.2 kcal mol−1 energy (Fig. 4). The optimized geometry of the intermediate state with the proton transferred to the oxygen species (O2aCSS) is characterized by an O–H bond length of 0.99 Å, and with an energy lower by 10.4 kcal mol−1 than that of the reactants in the triplet state.
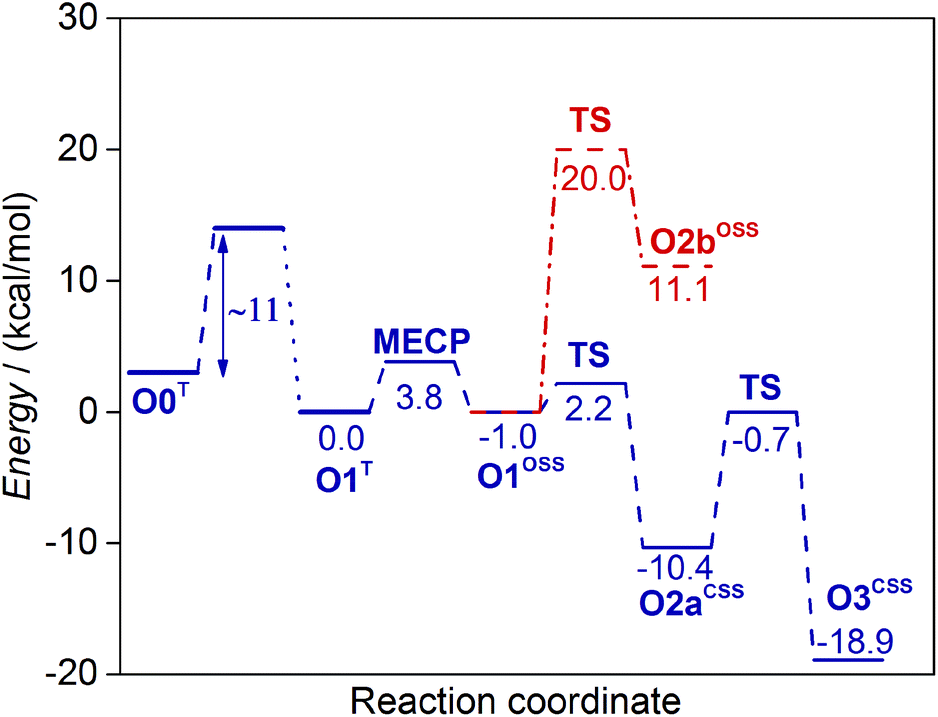 | ||
| Fig. 4 Energy diagrams of the non-covalent reaction pathways investigated in this study for the oxidative half-reaction of DAAO. See Scheme 2 for the designation of the intermediate states. The barrier of the first electron transfer step (O0T → O1T) corresponds to the experimental rate constant.18,58 | ||
NBO analysis of the intermediate state (O2aCSS) clearly shows that the proton transfer is accompanied by the transfer of an electron, since the resulting HOO− species has a charge close to that of the O2˙− superoxide anion (Table 3).
We followed the open-shell singlet diradical character (cf.eqn (3)) during the scan and its continuous decrease was observed while approaching the O2aCSS state containing HOO−. The reactant state (O1OSS) includes 89% open shell diradical character that decreases to 70% in the transition state, while the O2aCSS state is a pure closed shell singlet with an S2 value of 0. The singly occupied orbitals in the transition state (Fig. 5) are similar to those before the proton transfer (see Fig. 3 for the orbitals of the O1OSS state); the SOMO is mainly located on the FAD and the SOMO−1 resembles an O2 non-bonding orbital. The HOMO of the closed shell singlet state (O2aCSS) after the proton transfer also looks like an O2 non-bonding orbital (not shown). This suggests that the electron is transferred from a high energy FAD orbital to a non-bonding orbital of the O2 molecule and neither of them include orbitals assigned to proton binding. The proton moves along a line where no appreciable electron density of the singly occupied orbitals appears. Indeed, the orbitals bonding the proton to FAD-N5 (O1OSS state) and O (O2aCSS state) are lower energy orbitals. These observations suggest that the electron and the proton are exchanged between different sets of orbitals and the process can be classified as a proton-coupled electron transfer rather than as a H-atom transfer.67
 | ||
| Fig. 5 (a) SOMO and (b) SOMO−1 orbitals of the open-shell singlet transition state for the proton-coupled electron transfer step (O1OSS → O2aCSS). | ||
Following the proton-coupled electron transfer from the semiquinone to the superoxide anion, the formed HOO− abstracts a proton from the substrate to complete the reaction. The proton is transferred via a chain of water molecules (see above). The calculated energy of the transition state of the proton transfer compared to the energy of the reactants (O1T) is −0.7 kcal mol−1 and the energy drops to −18.9 kcal mol−1 in the final product state (Fig. 4). The optimized geometries and some characteristic geometric parameters are shown in Fig. 6. The reasonable barrier estimated for the O2aCSS → O3CSS step supports that in the absence of a proton source in the vicinity of the protein active site the proton can be transferred from the positively charged imino group of the substrate via a chain of water molecules.
 | ||
| Fig. 6 Characteristic geometrical parameters of selected intermediates and transition states. (a) FAD semiquinone-superoxide anion open shell singlet: O1OSS. The oxygen species is stabilized by further H-bonds to a water molecule and to the backbone NH of Gly54 not shown for the sake of clarity. (b) Transition state of proton-coupled electron transfer: O1OSS → O2aCSS. (c) Oxidised FAD and HOO−: O2aCSS. (d) Transition state of proton transfer: O2aCSS → O3CSS. (e) Oxidised FAD and H2O2: O3CSS. | ||
The alternative route, where the proton transfer from the substrate to the O2˙− superoxide anion precedes the proton transfer from the FAD, was also investigated. The calculated 20.0 kcal mol−1 barrier, however, is significantly higher than 2.2 kcal mol−1 obtained when the reaction starts with proton transfer from the FAD semiquinone (Fig. 4). The obtained open shell singlet intermediate with protonated oxygen species has an energy of 11.1 kcal mol−1, significantly higher than the highest energy transition state of the other route. Therefore, the route with proton transfer from the substrate (O1OSS → O2bOSS) preceding proton transfer from the FAD (O2bOSS → O3CSS) is highly unfavourable.
It was investigated if the proton transfer from the FAD and from the substrate can proceed in a concerted way. Two-dimensional relaxed scans for the transfer of the two protons were performed; however they do not reveal a lower energy concerted pathway. These results show that in the lowest barrier pathway a proton transfer from the FAD essentially precedes that from the substrate.
The formation of a covalent adduct between the O2 species and FAD was also investigated (Scheme S1†). The oxidative half-reaction with the formation of the C4a-(hydro)peroxy adduct is characteristic of monooxygenases and it is also observed for pyranose-2-oxidase. The calculated reaction barrier for the adduct formation in DAAO is over 11 kcal mol−1 and thus it is highly unfavoured compared to the non-covalent pathway (Fig. S2†).
The first electron transfer appears to be rate limiting also in the oxidative half-reaction of DAAO as no intermediates were observed.18,36 The experimental rate constant of the oxidative half-reaction in yeast DAAO is in the 106 M−1 min−1 range18,58 corresponding to ∼11 kcal mol−1 barrier.57 Assigning this barrier to the electron transfer is also consistent with the calculated transition states if we make the reasonable assumption of exothermic electron transfer. Then the energies of the transition states of the subsequent reaction steps exceed the energy of the reactants by less than 4 kcal mol−1 with barriers lower than 10 kcal mol−1 (Fig. 4), and a mechanism with the rate limiting first step emerges.
The oxidative half-reaction has to include spin transition since the system is in the triplet state before the reaction owing to the presence of the O2 molecule, and it is in a closed shell singlet state with oxidized FAD and H2O2 molecules at the completion of the reaction. Our calculations show that the FAD semiquinone-superoxide anion system has similar geometries and energies in triplet and open shell singlet states suggesting that the minimum energy crossing point (MECP) is in the vicinity. Indeed, a MECP was identified that has an energy higher by 3.8 kcal mol−1 than that of the triplet minimum. The actual rate constant may be lower than that for a spin-allowed reaction with the same barrier since the surface hopping probability is typically lower than 1. The reaction rate lowering was estimated to be equivalent with 1.4–5.5 kcal mol−1 barrier height increase at room temperature for a wide range of reactions.56 These considerations suggest that the spin transition can occur with a substantially lower barrier than 11 kcal mol−1 assigned to the electron transfer. Taking into account that the subsequent proton-coupled electron transfer occurs via a 2.2 kcal mol−1 barrier and is significantly exothermic (10.4 kcal mol−1 lower in energy than the starting O1T state) no accumulation of the intermediates is predicted in accordance with the experimental observations.18,36
The proton-coupled electron transfer from the FAD semiquinone to the oxygen species proceeds with a barrier (2.2 kcal mol−1) that is significantly lower than the barrier of the proton transfer from the substrate to the oxygen species (20.0 kcal mol−1). Therefore, the spin transition step is followed by a proton-coupled electron transfer and this completes the reoxidation of the FAD. The oxidized FAD–HOO− system adopts a closed shell singlet configuration and this does not change as the system is transformed to the product by a proton transfer to the oxygen species discussed below.
The oxidative half-reaction is completed by a proton transfer from the substrate to the oxygen species via a water chain. In contrast to other oxidases where the proton source is an amino acid side chain26 there is no amino-acid in the close environment of the oxygen species that could be a candidate for proton donation. However, the substrate was found to remain in the complex and to facilitate the electron transfer rate limiting to the oxidative half-reaction.18,33 In addition, the substrate that is in a zwitterionic state after oxidation is able to donate a proton when it forms a neutral imine group.9,58 Although the positions of the oxygen species and the substrate do not allow a direct proton transfer, it is feasible via a water chain observed in previous molecular dynamics simulations32 and also in the present study.
The reactions of flavoenzymes are typically monitored spectrophotometrically by measuring the absorbance depending on the oxidation state of the FAD. The rate constant of ∼106 M−1 min−1 (∼11 kcal mol−1 barrier57) for the oxidative half-reaction of DAAO was also determined spectrophotometrically18,58 and thus it refers to the reaction up to the proton-coupled electron transfer accompanied by a change of the electronic configuration of the FAD from the open shell to closed shell singlet. The last step of the reaction, namely the proton transfer from the substrate to the oxygen species via the water network, does not change the electronic structure of the FAD and is not included in the measured reaction rate. However, the calculated energy of the transition state of the last step does not exceed those of the previous steps. Similarly, the calculated barrier of the last step does not exceed the barrier (∼11 kcal mol−1) corresponding to the experimentally determined rate constant of the previous steps. These findings show that the proton transfer does not affect the overall reaction rate of the oxidative half-reaction.
It has to be noted that the quantum tunnelling effect was not considered in our calculations; thus, the above conclusions are valid within the transition state theory with the assumption that calculated enthalpy differences well approximate free-energy differences.
The results are obtained for yeast (Rhodotorula gracilis) DAAO, where the rate limiting step of the whole catalytic reaction includes the hydride transfer in the reductive half-reaction.12,20,70 The corresponding barrier is 13–15 kcal mol−1 (ref. 57) (1.8 × 103–2.4 × 105 M−1 min−1)18,58 and our calculated barrier of 15.4 kcal mol−1 for the hydride transfer is in reasonable agreement with the experimental values. It has to be also noted that the turn-over of human DAAO is lower than that of yeast DAAO71 since the rate limiting step in the former is the conformational change of the loop covering the active site and this loop is not present in the yeast enzyme. However, the flavin oxidation rates in DAAO from various species are between 1.9 × 106 and 1.0 × 107 M−1 min−1 (ref. 9, 18 and 58) corresponding to barriers within 1 kcal mol−1 (ref. 57) variation thus suggesting that these orthologs share the same mechanism for the oxidative half-reaction.
The proposed mechanism of FAD oxidation in DAAO differs from the FAD oxidation put forward for other oxidases. In glucose oxidase, although the reaction was proposed to start similarly with a rate determining single electron transfer,27,60 the subsequent steps as suggested by calculations on model systems63 are different; a proton transfer from the FAD to the oxygen species is followed by a proton coupled electron transfer where the proton comes from a charged HIS residue and the electron from the FAD. In contrast, a synchronous H-atom transfer from FAD N5 and proton transfer from a solvent exchangeable site is proposed in choline oxidase where the positive charge of the oxidized substrate contributes to O2 activation.34 The proton transfer from a charged His residue is possibly coupled with the first electron transfer from the FAD and precedes the slow H-atom transfer from the FAD in aryl-alcohol oxidase.35 The first electron transfer was postulated to be coupled with the proton transfer from a charged His residue to form a hydroperoxy-flavin intermediate in pyranose 2-oxidase.30 The reaction is completed by the rate determining proton abstraction from FAD N5 leading to H2O2 elimination.31 This versatile mechanism of FAD oxidation in oxidases is in line with the structural differences on the one hand, and with the modest energy differences among the various mechanisms, on the other hand. A characteristic structural variation among oxidases is the presence or absence of a charged residue capable of facilitating electron transfer to O2 and donating proton to the oxygen species. Concerning the energy differences among the potential routes for FAD oxidation, the calculated small energy variations for the open shell singlet and triplet states of the FAD semiquinone-superoxide containing system, just as the appearance of another MECP for the proton transfer suggest that small structural variations may produce changes in the dominating reaction route of the oxidative half-reaction. This argument is corroborated by the highly varying reaction rates of reduced flavin with oxygen among flavin dependent enzymes23,72 and the different routes followed by the two classes of flavoenzymes, oxidases and monooxygenases, the latter typically forming quasi-stable C4a(hydro)peroxyflavin.
Conclusions
The mechanism of the oxidative half-reaction of the D-amino acid oxidase enzyme was elucidated by combining the results of DFT level QM/MM calculations with experimental data from the literature. The oxidative half-reaction starts with a single electron transfer from the reduced FAD to the O2 molecule. The resulting FAD semiquinone-superoxide anion system is a stable intermediate. The system is in the triplet state at the start of the oxidative half-reaction and the triplet–singlet transition occurs in the semiquinone-superoxide anion system with a low barrier. The next step is a proton-coupled electron transfer between the FAD and the oxygen species, and this is accompanied by a change in the electronic configuration from an open shell to closed shell singlet. The reaction completes with proton abstraction from the oxidized substrate via a chain of water molecules. The experimentally observed reaction barrier for the oxidative half-reaction is ∼11 kcal mol−1 (ref. 57) (106 M−1 min−1 range)18,58 and it is assigned to the first electron transfer as no intermediates of the oxidative half-reaction were experimentally observed.18,36 This is consistent with the calculated energy surface of the subsequent low barrier steps. The oxidized substrate contributes to O2 activation in the rate-determining single-electron transfer as its presence lowers the barrier of the oxidative half-reaction.18 Our calculations confirm its additional role of providing a proton in the last step of H2O2 formation as suggested in ref. 32. The proposed detailed mechanism of the oxidative half-reaction of DAAO does not agree with the mechanism of other oxidases22,27,29–31,34,35,60,63 and provides another example for the versatility of oxidation in flavin-dependent enzymes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Jeremy Harvey for providing us with the MECP searching QM computer code that was transformed into a QM/MM MECP searching program. We are indebted to Profs Imre Pápai and György M. Keserű for valuable comments. The computing time granted on the Hungarian HPC Infrastructure at KIFÜ-NIIF Institute is gratefully acknowledged. The research was supported by the National Brain Research Program (2017-1.2.1-NKP-2017-00002) of the National Research, Development and Innovation Office, Hungary, the Hungarian Scientific Research Fund (OTKA, Grant No. K111862 and K116305) and the bilateral Hungarian-Slovenian science and technology cooperation under Grant Number TÉT 16-1-2016-0069 and BI-HU/17-18-009.Notes and references
- L. Pollegioni and S. Sacchi, Cell. Mol. Life Sci., 2010, 67, 2387–2404 CrossRef CAS PubMed.
- H. Wolosker, Mol. Neurobiol., 2007, 36, 152–164 CrossRef CAS PubMed.
- J. W. Newcomer and J. H. Krystal, Hippocampus, 2001, 11, 529–542 CrossRef CAS PubMed.
- P. W. J. Burnet, S. L. Eastwood, G. C. Bristow, B. R. Godlewska, P. Sikka, M. Walker and P. J. Harrison, Mol. Psychiatry, 2008, 13, 658–660 CrossRef CAS PubMed.
- I. Chumakov, M. Blumenfeld, O. Guerassimenko, L. Cavarec, M. Palicio, H. Abderrahim, L. Bougueleret, C. Barry, H. Tanaka, P. La Rosa, A. Puech, N. Tahri, A. Cohen-Akenine, S. Delabrosse, S. Lissarrague, F.-P. Picard, K. Maurice, L. Essioux, P. Millasseau, P. Grel, V. Debailleul, A.-M. Simon, D. Caterina, I. Dufaure, K. Malekzadeh, M. Belova, J.-J. Luan, M. Bouillot, J.-L. Sambucy, G. Primas, M. Saumier, N. Boubkiri, S. Martin-Saumier, M. Nasroune, H. Peixoto, A. Delaye, V. Pinchot, M. Bastucci, S. Guillou, M. Chevillon, R. Sainz-Fuertes, S. Meguenni, J. Aurich-Costa, D. Cherif, A. Gimalac, C. Van Duijn, D. Gauvreau, G. Ouellette, I. Fortier, J. Raelson, T. Sherbatich, N. Riazanskaia, E. Rogaev, P. Raeymaekers, J. Aerssens, F. Konings, W. Luyten, F. Macciardi, P. C. Sham, R. E. Straub, D. R. Weinberger, N. Cohen, D. Cohen, G. Ouelette and J. Realson, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 13675–13680 CrossRef CAS PubMed.
- C. Rojas, J. Alt, N. A. Ator, A. G. Thomas, Y. Wu, N. Hin, K. Wozniak, D. Ferraris, R. Rais, T. Tsukamoto and B. S. Slusher, Neuropsychopharmacology, 2016, 41, 1610–1619 CrossRef CAS PubMed.
- B. Szilágyi, G. G. Ferenczy and G. M. Keserű, Expert Opin. Drug Discovery, 2018, 13, 973–982 CrossRef PubMed.
- H.-Y. Lane, C.-H. Lin, M. F. Green, G. Hellemann, C.-C. Huang, P.-W. Chen, R. Tun, Y.-C. Chang and G. E. Tsai, JAMA Psychiatry, 2013, 70, 1267 CrossRef CAS PubMed.
- G. Molla, S. Sacchi, M. Bernasconi, M. S. Pilone, K. Fukui and L. Pollegioni, FEBS Lett., 2006, 580, 2358–2364 CrossRef CAS PubMed.
- T. Kohiki, Y. Kato, Y. Nishikawa, K. Yorita, I. Sagawa, M. Denda, T. Inokuma, A. Shigenaga, K. Fukui and A. Otaka, Org. Biomol. Chem., 2017, 15, 5289–5297 RSC.
- A. A. Raibekas, K. Fukui and V. Massey, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3089–3093 CrossRef CAS.
- K. A. Kurtz, M. A. Rishavy, W. W. Cleland and P. F. Fitzpatrick, J. Am. Chem. Soc., 2000, 122, 12896–12897 CrossRef CAS.
- R. T. Terry-Lorenzo, L. E. Chun, S. P. Brown, M. L. R. Heffernan, Q. K. Fang, M. A. Orsini, L. Pollegioni, L. W. Hardy, K. L. Spear and T. H. Large, Biosci. Rep., 2014, 34, 487–499 CrossRef CAS PubMed.
- M. A. Vanoni, A. Cosma, D. Mazzeo, A. Mattevi, F. Flavia Todone and B. Curti, Biochemistry, 1997, 36, 5624–5632 CrossRef CAS PubMed.
- A. Tilocca, A. Gamba, M. A. Vanoni and E. Fois, Biochemistry, 2002, 41, 14111–14121 CrossRef CAS PubMed.
- T. Kawazoe, H. K. Park, S. Iwana, H. Tsuge and K. Fukui, Chem. Rec., 2007, 7, 305–315 CrossRef CAS PubMed.
- V. Massey and Q. H. Gibson, Fed. Proc., 1964, 23, 18–29 CAS.
- L. Pollegioni, B. Langkau, W. Tischer, S. Ghisla and M. S. Pilone, J. Biol. Chem., 1993, 268, 13850–13857 CAS.
- M. S. Pilone, Cell. Mol. Life Sci, 2000, 57, 1732–1747 CrossRef CAS PubMed.
- L. Pollegioni, W. Blodig and S. Ghisla, J. Biol. Chem., 1997, 272, 4924–4934 CrossRef CAS PubMed.
- M. Delgado, S. Görlich, J. E. Longbotham, N. S. Scrutton, S. Hay, V. Moliner and I. Tuñón, ACS Catal., 2017, 7, 3190–3198 CrossRef CAS PubMed.
- E. Romero, J. R. Gómez Castellanos, G. Gadda, M. W. Fraaije and A. Mattevi, Chem. Rev., 2018, 118, 1742–1769 CrossRef CAS PubMed.
- V. Massey, J. Biol. Chem., 1994, 269, 22459–22462 CAS.
- P. Chaiyen, M. W. Fraaije and A. Mattevi, Trends Biochem. Sci., 2012, 37, 373–380 CrossRef CAS PubMed.
- G. Gadda, Biochemistry, 2012, 51, 2662–2669 CrossRef CAS PubMed.
- J. P. Roth, R. Wincek, G. Nodet, D. E. Edmondson, W. S. McIntire and J. P. Klinman, J. Am. Chem. Soc., 2004, 126, 15120–15131 CrossRef CAS PubMed.
- J. P. Roth and J. P. Klinman, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 62–67 CrossRef CAS PubMed.
- Q. Su and J. P. Klinman, Biochemistry, 1999, 38, 8572–8581 CrossRef CAS PubMed.
- S. Visitsatthawong, P. Chenprakhon, P. Chaiyen and P. Surawatanawong, J. Am. Chem. Soc., 2015, 137, 9363–9374 CrossRef CAS PubMed.
- T. Wongnate, P. Surawatanawong, S. Visitsatthawong, J. Sucharitakul, N. S. Scrutton and P. Chaiyen, J. Am. Chem. Soc., 2014, 136, 241–253 CrossRef CAS PubMed.
- J. Sucharitakul, T. Wongnate and P. Chaiyen, J. Biol. Chem., 2011, 286, 16900–16909 CrossRef CAS PubMed.
- J. Saam, E. Rosini, G. Molla, K. Schulten, L. Pollegioni and S. Ghisla, J. Biol. Chem., 2010, 285, 24439–24446 CrossRef CAS PubMed.
- P. Cappelletti, L. Piubelli, G. Murtas, L. Caldinelli, M. Valentino, G. Molla, L. Pollegioni and S. Sacchi, Biochim. Biophys. Acta, Proteins Proteomics, 2015, 1854, 1150–1159 CrossRef CAS PubMed.
- S. Gannavaram and G. Gadda, Biochemistry, 2013, 52, 1221–1226 CrossRef CAS PubMed.
- J. Carro, P. Ferreira, A. T. Martínez and G. Gadda, Biochemistry, 2018, 57, 1790–1797 CrossRef CAS PubMed.
- V. Massey and G. Palmer, Biochemistry, 1966, 5, 3181–3189 CrossRef CAS PubMed.
- S. Umhau, L. Pollegioni, G. Molla, K. Diederichs, W. Welte, M. S. Pilone and S. Ghisla, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 12463–12468 CrossRef CAS PubMed.
- G. Madhavi Sastry, M. Adzhigirey, T. Day, R. Annabhimoju and W. Sherman, J. Comput.-Aided Mol. Des., 2013, 27, 221–234 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey, M. L. Klein, J. D. Chandrasekhar, J. Madura, W. R. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- S. Pronk, S. Páll, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson, D. van der Spoel, B. Hess and E. Lindahl, Bioinformatics, 2013, 29, 845–854 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- D. A. Case, V. Babin, J. T. Berryman, R. M. Betz, Q. Cai, D. S. Cerutti, T. E. Cheatham, T. A. Darden, R. E. Duke, H. Gohlke, A. W. Goetz, S. Gusarov, N. Homeyer, P. Janowski, J. Kaus, I. Kolossváry, A. Kovalenko, T. S. Lee, S. LeGrand, T. Luchko, R. Luo, B. Madej, K. M. Merz, F. Paesani, D. R. Roe, A. Roitberg, C. Sagui, R. Salomon-Ferrer, G. Seabra, C. L. Simmerling, W. Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu and P. A. Kollman, AMBER 14, University of San Francisco, California, CA, 2014 Search PubMed.
- A. W. Götz, M. A. Clark and R. C. Walker, J. Comput. Chem., 2014, 35, 95–108 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision A.01), Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- N. Jongkon, D. Gleeson and M. P. Gleeson, Org. Biomol. Chem., 2018, 16, 6239–6249 RSC.
- F. Claeyssens, K. E. Ranaghan, N. Lawan, S. J. Macrae, F. R. Manby, J. N. Harvey and A. J. Mulholland, Org. Biomol. Chem., 2011, 9, 1578 RSC.
- K. Yamaguchi, Y. Takahara, T. Fueno and K. N. Houk, Theor. Chim. Acta, 1988, 73, 337–364 CrossRef CAS.
- H. Isobe, S. Yamanaka, S. Kuramitsu and K. Yamaguchi, J. Am. Chem. Soc., 2008, 130, 132–149 CrossRef CAS PubMed.
- V. Bachler, G. Olbrich, F. Neese and K. Wieghardt, Inorg. Chem., 2002, 41, 4179–4193 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- J. N. Harvey, M. Aschi, H. Schwarz and W. Koch, Theor. Chim. Acta, 1998, 99, 95–99 CrossRef CAS.
- J. N. Harvey, Phys. Chem. Chem. Phys., 2007, 9, 331–343 RSC.
- Kinetic constants (k) were transformed to energy barriers (E) using the equation E = −RT*ln(k/Z) with RT = 0.59 kcal/mol and Z = 6.21 × 1012 s−1.
- D. J. Porter, J. G. Voet and H. J. Bright, J. Biol. Chem., 1977, 252, 4464–4473 CAS.
- G. Zhao, R. C. Bruckner and M. S. Jorns, Biochemistry, 2008, 47, 9124–9135 CrossRef CAS PubMed.
- J. P. Klinman, Acc. Chem. Res., 2007, 40, 325–333 CrossRef CAS PubMed.
- G. Gadda, F. Fan and J. V. Hoang, Arch. Biochem. Biophys., 2006, 451, 182–187 CrossRef CAS.
- A. Hernández-Ortega, F. Lucas, P. Ferreira, M. Medina, V. Guallar and A. T. Martínez, Biochemistry, 2012, 51, 6595–6608 CrossRef PubMed.
- R. Prabhakar, P. E. M. Siegbahn, B. F. Minaev and Å. Hans, J. Phys. Chem. B, 2002, 106, 3742–3750 CrossRef CAS.
- P. J. Silva, PeerJ, 2016, 4, e2805 CrossRef.
- R. Miura and Y. Miyake, J. Biochem., 1987, 102, 1345–1354 CrossRef CAS.
- A. S. Menon and L. Radom, J. Phys. Chem. A, 2008, 112, 13225–13230 CrossRef CAS PubMed.
- S. Hammes-Schiffer and A. A. Stuchebrukhov, Chem. Rev., 2010, 110, 6939–6960 CrossRef CAS PubMed.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, Rev. Bioenerg., 1985, 811, 265–322 CrossRef CAS.
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599–610 CrossRef CAS.
- C. M. Harris, L. Pollegioni and S. Ghisla, Eur. J. Biochem., 2001, 268, 5504–5520 CrossRef CAS PubMed.
- S. Sacchi, E. Rosini, L. Pollegioni and G. Molla, Curr. Pharm. Des., 2013, 19, 2499–2511 CrossRef CAS PubMed.
- A. Mattevi, Trends Biochem. Sci., 2006, 31, 276–283 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ob00975b |
| This journal is © The Royal Society of Chemistry 2019 |