Structural flexibility versus rigidity of the aromatic unit of DNA ligands: binding of aza- and azoniastilbene derivatives to duplex and quadruplex DNA†
Received
8th April 2019
, Accepted 14th June 2019
First published on 14th June 2019
Abstract
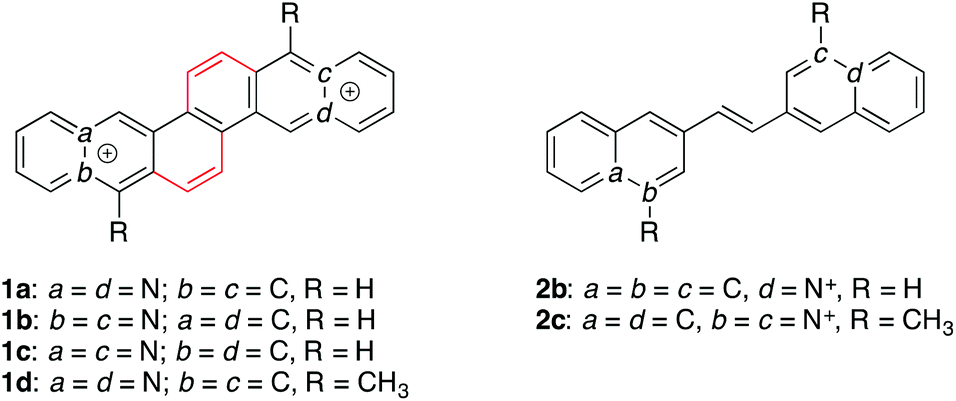
The known azastilbene (E)-1,2-di(quinolin-3-yl)ethane (2a) and the novel azoniastilbene derivatives (E)-2-(2-(naphthalen-2-yl)vinyl)quinolizinium (2b) and (E)-3,3′-(ethane-1,2-diyl)bis(1-methylquinolinin-1-ium) (2c) were synthesized. Their interactions with duplex and quadruplex DNA (G4-DNA) were studied by photometric, fluorimetric, polarimetric and flow-LD analysis, and by thermal DNA denaturation studies, as well as by 1H-NMR spectroscopy. The main goal of this study was a comparison of these conformationally flexible compounds with the known G4-DNA-binding diazoniadibenzo[b,k]chrysenes, that have a comparable π-system extent, but a rigid structure. We have observed that the aza- and azoniastilbene derivatives 2a–c, i.e. compounds with almost the same spatial dimensions and steric demand, bind to DNA with an affinity and selectivity that depends significantly on the number of positive charges. Whereas the charge neutral derivative 2a binds unspecifically to the DNA backbone of duplex DNA, the ionic compounds 2b and 2c are typical DNA intercalators. Notably, the bis-quinolinium derivative 2c binds to G4-DNA with moderate affinity (Kb = 4.8 × 105 M−1) and also stabilizes the G4-DNA towards thermal denaturation (ΔTm = 11 °C at ligand–DNA ratio = 5.0). Strikingly, the corresponding rigid counterpart, 4a,12a-diazonia-8,16-dimethyldibenzo[b,k]chrysene, stabilizes the G4-DNA to an even greater extent under identical conditions (ΔTm = 27 °C). These results indicate that the increased flexibility of a G4-DNA ligand does not necessarily lead to stronger interactions with the G4-DNA as compared with rigid ligands that have essentially the same size and π system extent.
Introduction
Among the non-canonical DNA forms, the quadruplex DNA (G4-DNA), that is formed in G-rich DNA sequences upon stacking of at least two guanine quartets, is currently attracting most attention.1 Thus, several G-rich DNA sequences with the propensity to fold into quadruplex structures have been identified in genomic nucleic acids,2 for example in telomeric DNA and some promoter regions of oncogenes.3 Moreover, it has been shown that some biologically relevant processes are directly related to quadruplex-DNA formation, such as the suppression of gene expression,4 or the induction of the cellular response to DNA damage.5 As a result of the essential biological functionality of quadruplex DNA, the association of an exogenous ligand with G4-DNA structures may have a significant effect on the biological function of G-rich DNA sequences, either by simply blocking the binding site of enzymes, which leads to their inhibition, or by increasing the thermodynamic stability and the lifetime of the G4-DNA.1,6 In the latter case, enzyme inhibition may also occur when the enzyme requires the unwound form of the particular DNA sequence. Based on this principle, numerous G4-DNA-targeting molecules have been developed that may affect the biological activity of the DNA.7 Along these lines, traditional DNA intercalators, i.e. cationic, planar, polycyclic (het)arenes, figure as a promising basis for the development of G4-DNA ligands. Thus, it has been shown that such intercalators have the propensity for a terminal π-stacking at the ends of G4-DNA structures.1,7 Such as intercalation of a ligand between base pairs in duplex DNA,8 the terminal π-stacking is driven by dispersion interactions between the aromatic ligand and the guanine quartet and by an additional hydrophobic effect as the lipophilic ligand migrates from the aqueous solution into the hydrophobic binding site.9 Because of the thermodynamically unfavorable process of the unwinding and disassembly of the quadruplex structure, planar aromatic ligands do usually not intercalate into G4-DNA. In this context, highly selective aromatic ligands have been developed that bind to the terminal end of the quadruplex due to their extended π-system, but whose structure does no longer allow a thermodynamically favorable intercalation into duplex DNA.1a,b,7 In this context, we have shown that polycyclic azoniahetarenes, for example the diazoniadibenzo[b,k]chrysenes (1a–c), bind to G4-DNA even in the absence of additional functional side chains, thus enabling the assessment of the intrinsic ligand properties.10 The binding modes of 1a–c were studied in detail and all experimental data indicate the terminal π-stacking of the diazoniachrysenes to quadruplex DNA as the major binding mode. Thus, the size and topology of the π-systems of the cationic hetarenes 1a–c enable sufficient overlap with the G-quartet mainly based on attractive π-stacking interactions.10 It should be noted, however, that this class of quadruplex ligands consists of highly rigid compounds that have essentially no conformational flexibility. At the same time, some of the most promising and efficient G4-DNA ligands, such as e.g. the bis(quinolinium)pyridine dicarboxamides,11 contain structural elements that provide a significant degree of conformational freedom, as the aromatic units are not completely annelated but connected by flexible linkers. It may be assumed that this structural flexibility facilitates the access of the ligand as well as the fit of the aromatic scaffold to the steric demand of the binding site. Along these lines, the influence of the structural flexibility of G4-DNA ligands has been shown recently by a comparison of structurally resembling flexible and rigid bis(quinolinium)-bisindole dicarboxamide derivatives.12 In these ligands, the bisindole core unit, that is involved in the terminal π stacking to G4-DNA, is either connected in a flexible biaryl structure or kept rigid in a completely fused structure. It has been shown that these flexible derivatives have a slightly higher affinity to G4-DNA than the rigid ones because of the better ability of the flexible ligands to adapt to the G4-DNA structure. It should be noted, however, that even the rigid ligands in this study still contained flexible elements as the two quinolinium units were attached to the bisindole core through amide bonds that allow torsional movement. Before this background, we proposed that ligands with a similarly extended π-system as the dibenzochrysene derivatives 1a–c, but with an increased conformational flexibility may have an increased affinity to G4-DNA. We identified the aza- and azoniastilbene derivatives 2a–c as a promising starting point to check this proposal because they have a similarly extended π-system as 1a–c, but more conformational flexibility due to the possible rotation about the aryl–alkene single bond. We proposed that these compounds bind to DNA as it has been shown already that pyridinium-based azoniastilbenes bind to DNA.13 And more recently, it has been demonstrated that substituted styryl-quinolinium derivatives bind to G4-DNA.14 Herein, we present the synthesis of stilbene derivatives 2a–c and the investigation of their DNA-binding properties. For better comparison with respect to size and shape, we also synthesized and investigated the dimethyldiazoniadibenzo–chrysene 1d in this study, because this derivative has methyl substituents at positions that correspond to the ones in the dimethyl-bis(quinolinium) derivative 2c.
Results
Synthesis
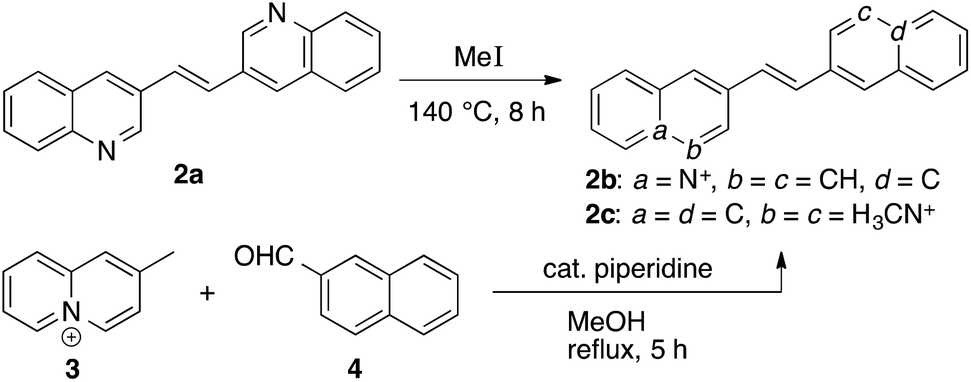
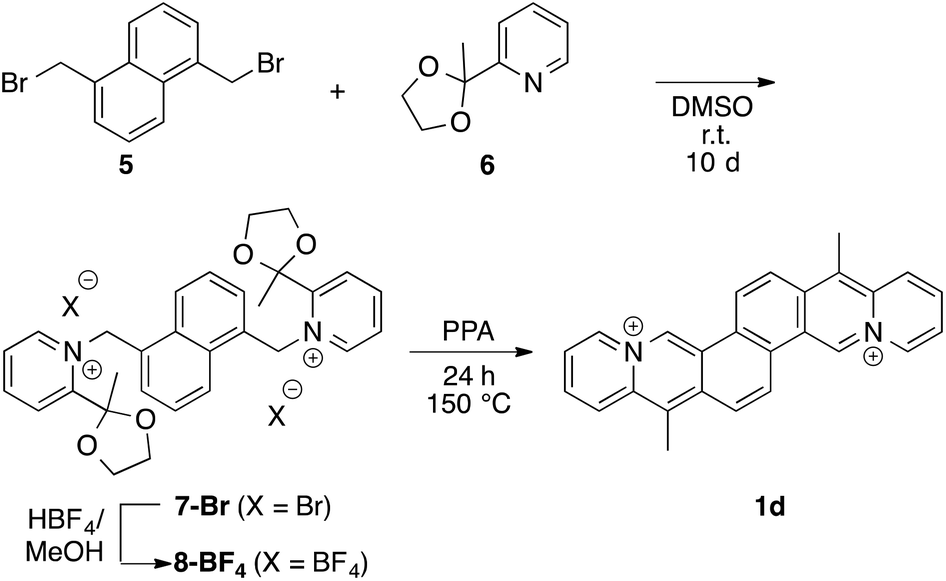
The (E)-1,2-di(quinolin-3-yl)ethane (2a) was prepared according to published procedure15 and subsequently quaternized by reaction with methyl iodide at high pressure10a to give the (E)-3,3′-(ethane-1,2-diyl)bis(1-methyl quinolinin-1-ium) (2c) in 70% yield (Scheme 1). The (E)-2-(2-(naphthalen-2-yl)vinyl)quinolizinium (2b) was synthesized in 62% yield by the base-catalyzed Knoevenagel-type reaction of 2-methylquinolizinium (3)10b with 2-naphthaldehyde (4) (Scheme 1).10b,c The dibenzochrysene derivative 1d was synthesized starting from the reaction of 1,5-di(bromomethyl)naphthalene (5)11 with 2-(2-methyl-(1,3)-dioxolan-2-yl)pyridine (6)13 and subsequent ion metathesis to give bis(pyridiniummethyl)naphthalene 7. The latter was treated with polyphosphoric acid (PPA) at 150 °C to give the cyclodehydration product 1d in 75% yield (Scheme 2).
 |
| | Scheme 1 Synthesis of stilbene derivatives 2b and 2c. | |
 |
| | Scheme 2 Synthesis of 4a,12a-diazonia-8,16-dimethyldibenzo[b,k]chrysene (1d). | |
Absorption and emission properties
The absorption and emission spectra of the aza- and azoniastilbene derivatives 2a–c were recorded in DMSO, MeCN, MeOH, water, and BPE buffer solution (Fig. 1, Table S1 in ESI†). The corresponding shifts and the band structures do not depend strongly on the solvent properties. Thus, the long-wavelength absorption maxima are lying in a rather small range of 325–333 nm (2a), 379–396 nm (2b) and 366–369 nm (2c), respectively. Likewise, most of the emission bands of each compound cover the same wavelength range with small Stokes shifts and low to moderate emission quantum yields (2a: λfl = 397–420 nm, Φfl = 0.2–0.5; 2b: λfl = 491–499 nm, Φfl = 0.2; 2c: λfl = 454–462 nm Φfl = <0.01–0.3). As the only exception, the emission spectra of derivative 2a in water and MeOH deviate from the ones in the other solvents; namely the bands are significantly broader with a very pronounced red-shifted shoulder (Fig. 1A), presumably indicating aggregation.
 |
| | Fig. 1 Absorption (dashed lines; c = 25 μM) and emission spectra [continuous lines; 2a: λex = 334 nm, 2b: 390 nm, 2c: 320 nm of compounds 2a (A), 2b (B), and 2c (C) in DMSO (red), MeCN (blue), water (green 2a: with 5% DMSO), MeOH (black), and phosphate buffer (orange, 2a: with 5% DMSO)]. | |
Spectrometric titrations with ct DNA
The interactions of the stilbene derivatives 2a–c with double-stranded calf thymus (ct) DNA were monitored by photometric and fluorimetric titrations (Fig. 2). The absorption of derivative 2a decreased during titration with no significant shift of the absorption maximum at 329 nm (Fig. 2A1). In the case of ligands 2b and 2c, the absorption maxima at 382 nm (2b) and 366 nm (2c) also decreased on addition of up to 2.8 (2b) and 6.1 (2c) molar equivalents of ct DNA, respectively, without shift of the maxima (Fig. 2A2 and A3). On further addition of DNA the absorption increased with a bathochromic shift of approx. 7 nm in each case. It should be noted that samples of 2b were occasionally contaminated with traces of the photodimer whose formation is indicated by a weak absorption at 333 nm. In general, the characteristic emission of the stilbene derivatives 2a–c was quenched on addition of ct DNA, which was accompanied in some cases by slight shifts of the emission maxima (Fig. 2B1–B3). In the case of ligand 2b, a very small fluorescence light-up effect was observed in the first titration steps (molar equivalent <1) before the quenching occurs. The data from the photometric or fluorimetric titrations were plotted as binding isotherms and analyzed based on the theoretical binding model (cf. ESI†).16 The resulting binding constants are 6.2 × 104 M−1 (2a), 5.1 × 104 M−1 (2b), and 2.0 × 105 M−1 (2c).
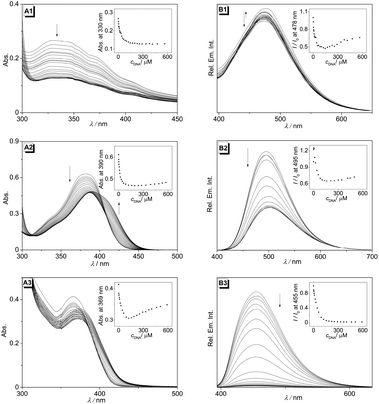 |
| | Fig. 2 Photometric (A) and fluorimetric titration (B); 2a: λex = 384 nm, 2b: λex = 342 nm, 2c: λex = 383 nm of ligands 2a (1), 2b (2) and 2c (3) (c = 20 μM) with ct DNA (c = 1.0 mM in base pairs) in BPE buffer (pH = 7.0; 2a–b: with 5% v/v DMSO); T = 20 °C. The arrows indicate the development of the absorption or emission bands during titration. Inset: Plot of absorption Abs/Abs0 and relative emission I/I0, resp., versus cDNA. | |
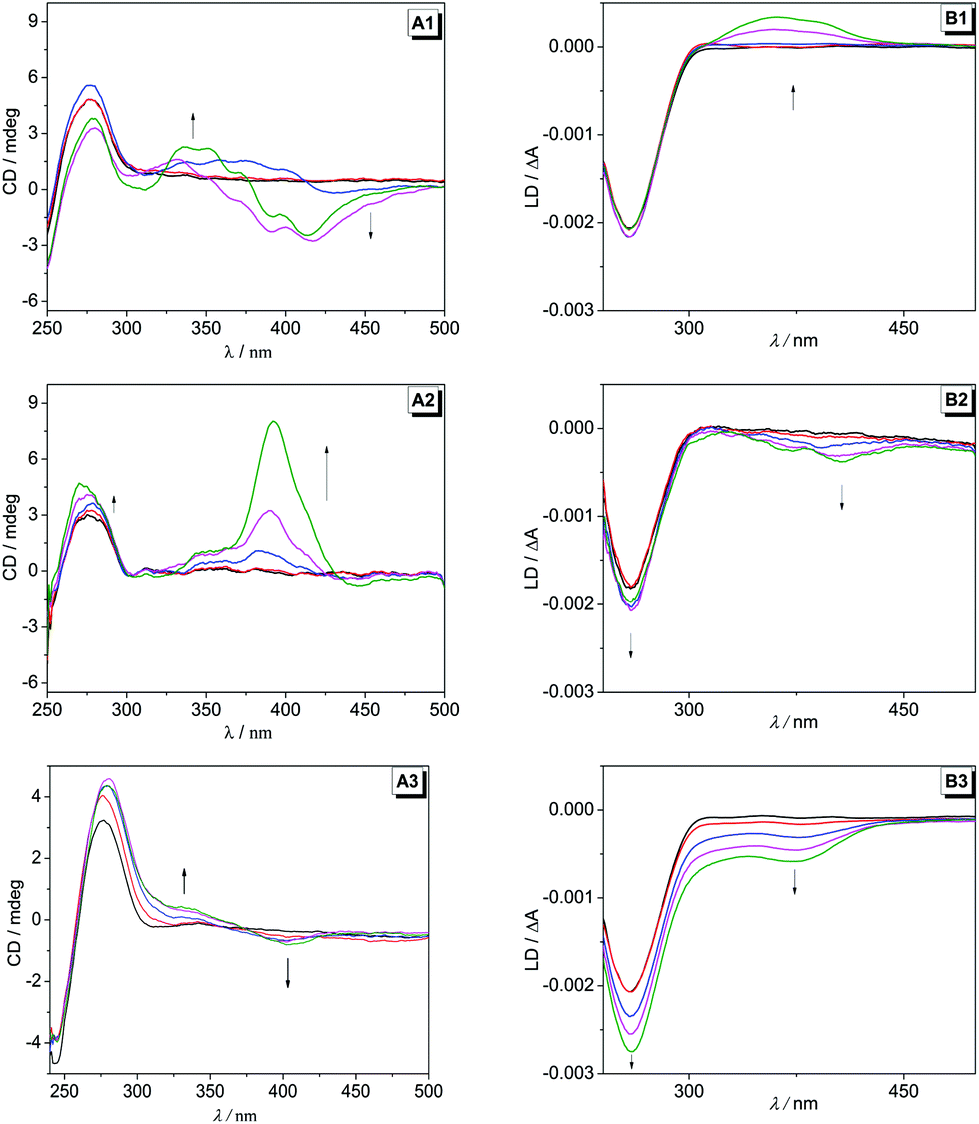
Polarimetric titrations with ct DNA
To gain further insight into the DNA-binding modes the interactions between the ligands 2a–c with ct DNA were examined by circular dichroism (CD) and flow linear dichroism (LD) spectroscopy (Fig. 3). Upon addition of DNA the compound 2a developed a significant positive induced CD (ICD) band at 344 nm and negative ICD bands at 417 nm and 307 nm, but the latter was only observed at ligand–DNA-ratio (LDR) = 0.5 and 1.0. At the same time, the characteristic positive CD band of the DNA at 277 nm fluctuated slightly with increasing LDR values. In addition, flow-LD measurements revealed a developing positive signal in the absorption range of the ligand 2a with a maximum at 361 nm (Fig. 3B1). The addition of DNA to a solution of compound 2b induced the formation of positive ICD bands at 386 nm and 340 nm with relatively high intensity at large LDR. The complementary LD-spectroscopic experiments showed a weak negative LD band at 403 and 369 nm whose intensity increased slightly with increasing LDR. The addition of DNA to compound 2c led to the formation of relatively weak positive and negative ICD bands at 332 nm and 405 nm (Fig. 3A3). The flow-LD measurements showed a developing negative LD band in the absorption range of the ligand with a maximum at 372 nm. Notably, the intensity of the characteristic negative band of the DNA at 260 nm increased significantly with increasing LDR values (Fig. 3B3).
 |
| | Fig. 3 CD (A) and flow-LD (B) spectra of mixtures of ligands 2a–c and ct DNA in BPE buffer (pH = 7.00; 2a,b: with 5% v/v DMSO) and at LDR 0.0 (black), 0.1 (red), 0.2 (blue), 0.5 (magenta) and 1.0 (green); cDNA = 20 μM; T = 20 °C. Arrows indicate the development of CD and LD bands with increasing LDR value. | |
Fluorescence-monitored quadruplex-DNA melting
The interactions of the ligands 1d and 2a–c with quadruplex DNA were investigated with representative quadruplex-forming oligonucleotide sequences. Firstly, the established FRET melting assay17 was employed to assess the stabilization of the quadruplex structure upon association with the ligands. With this method, the propensity of a ligand to stabilize quadruplex-DNA towards unfolding was determined by monitoring the temperature-dependent emission of the dye-labeled quadruplex-forming oligonucleotide F21T, fluo-d(G3TTA)3G3-tamra (fluo: fluorescein; TAMRA: tetramethyl-rhodamine), because only in the quadruplex form a Förster resonance energy transfer (FRET) between the two dyes is possible. The quadruplex-forming oligonucleotide was chosen for first principal studies, as it is among the most commonly used substrates for this well-established assay.17 The FRET melting experiments revealed that the ligand 2a does not stabilize the G4-DNA F21T, even at high LDR = 5.0 (Table 1), while the ligands 1d, 2b and 2c stabilize the G4-DNA F21T towards unfolding, as clearly indicated by the increase of the melting temperature, ΔTm, of 27 °C (1d), 3 °C (2b) and 11 °C (2c) at LDR = 5.0 (Table 1). To assess whether this stabilization of G4-DNA is a general feature of the ligands 2b and 2c the same experiments were also performed with the representative quadruplex-forming oligonucleotides FmycT [fluo-d(TGAG3TG3TAG3TG3TA2)-tamra], FkrasT [fluo-d(AG3CG2TGTG3A2GAG2A)-tamra] and Fa2T [fluo-d(ACAG4TGTG4)2-tamra] (Table 1), all of which have been used already with this assay.7,18 In all cases, the quadruplex structure is also stabilized to significantly more extent by ligand 2c as compared with 2b. Nevertheless, it was also observed that the degree of G4-DNA stabilization upon binding of ligand 2c is not the same for the different oligonucleotides. Hence, the extent of stabilization decreases in the order F21T (ΔTm = 11 °C), FkrasT (ΔTm = 9.9 °C), FmycT (ΔTm = 6.0 °C), and Fa2T (ΔTm = 3.1 °C; all values at LDR = 5). To further assess the selectivity of the quadruplex stabilization by 2c, the melting experiments were also performed in the presence of excess of the duplex-DNA forming oligonucleotide ds26.17 Under these conditions, the ligand 2c shows a thermal stabilization of the F21T by 7.5 °C at LDR = 5.0 (Table 1).
Table 1 Shift of melting temperature, ΔTm, of the G4-DNA F21T, FmycT, FkrasT and Fa2T in the presence of the ligands 1d and 2a–c
| LDR |
ΔTm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a/°C a/°C |
|
1d
|
2a
|
2b
|
2c
|
2b
|
2c
|
2b
|
2c
|
2b
|
2c
|
|
F21T
|
Fa2T
|
FmycT
|
FkrasT
|
|
c
DNA = 0.2 μM in Na-cacodylate buffer (pH = 7.2); estimated error: ±0.5 °C. Determined fluorimetrically based on the temperature-dependent change of FRET in F21T, FmycT, FkrasT and Fa2T.
With 1% v/v DMSO. ΔTm values in parentheses determined in the presence of ds26 d(CA2TCG2ATCGA2T2CGATC2GAT2G) (c = 3.0 μM).
|
| 1.3 |
+9.3 |
−0.3 |
+0.7 |
+4.3 (2.9) |
−0.2 |
<0.1 |
0.5 |
2.4 |
1.3 |
4.6 |
| 2.5 |
+21.6 |
+0.2 |
+1.6 |
+7.6 (5.0) |
0.1 |
−0.2 |
−0.3 |
2.9 |
1.3 |
7.2 |
| 5.0 |
+27.1 |
−0.4 |
+3.3 |
+10.6 (7.5) |
0.3 |
3.1 |
0.3 |
6.0 |
2.2 |
9.9 |
Spectrometric titrations with quadruplex-DNA
The interactions of the ligands 1d, 2b and 2c with the quadruplex-forming oligonucleotides 22AG dA(G3TTA)3G3 and a2 d(ACAG4TGTG4)2 were investigated by photometric and fluorimetric titrations (Fig. 4 and 5). Upon the addition of 22AG or a2, the absorption maxima of 2b (381 nm) and 2c (366 nm) decreased with the development of red-shifted absorption maxima at 401 nm (22AG) and 406 nm (a2) for 2b (Fig. 4A1 and B1), and 379 nm (22AG) and 382 nm (a2) for 2c (Fig. 4A2 and B2). In both titrations of the ligand 2c an isosbestic point was observed.
 |
| | Fig. 4 Photometric titration of 2b (1) and 2c (2) (c = 20 μM) with 22AG (A) and a2 (B) (cDNA = 200 μM) in K-phosphate buffer (pH = 7.0, 5% v/v DMSO), T = 20 °C. The arrows indicate the development of the absorption bands during the titration. Inset: Plot of absorption Abs./Abs.0versus cDNA/cligand. | |
 |
| | Fig. 5 Fluorimetric titration of 1d (1), 2b (2) and 2c (3) (c = 20 μM) with 22AG (A) and a2 (B) (cDNA = 200 μM) in K-phosphate buffer (pH = 7.0, 5% v/v DMSO), T = 20 °C; 1d: λex = 423 nm (22AG); 2b: λex = 390 nm (22AG and a2); 2c: λex = 385 nm (22AG) and 389 nm (a2). The arrows indicate the development of the emission bands during titration. Inset: Plot of relative emission intensity I/I0versus cDNA/cligand. | |
At the same time, the emission of derivatives 1d, 2b and 2c is significantly quenched upon binding to 22AG and a2 (Fig. 5). The analysis of the resulting binding isotherms from the fluorimetric titrations of the ligands 1d, 2b and 2c revealed binding constants of 1.5 × 106 M−1 (1d), 2.1 × 105 M−1 (2b) and 4.8 × 105 M−1 (2c) with 22AG and 5.8 × 105 M−1 (2b) and 4.0 × 105 M−1 (2c) with a2.
1H-NMR spectroscopic studies
Titrations of the diazoniastilbene 2c to the quadruplex-forming oligonucleotide Tel6 d(T2AG3) was monitored by 1H-NMR spectroscopy (Fig. 6). This hexanucleotide forms an equilibrium between monomeric and a terminally π-stacked dimeric G4-DNA [d(T2AG3)]4 in aqueous solution16,19 Although Tel6 is only a very simplified quadruplex model as it does not contain the loop structures, such as e.g.22AG, this oligonucleotide or variations thereof are often employed for the NMR spectroscopic detection of terminally stacking G4-DNA ligands,20 because the interpretation of the NMR data is straightforward. It should be noted that due to limited solubility at the employed concentration range only up to 1.5 molar equivalents of the ligand 2c could be added. The titration of ligand 2c to Tel6 led to significant shifts of the 1H NMR signals of the G4-DNA Tel6 (Fig. 6). Thus, NMR signals of the guanine imino protons of the dimeric quadruplex broadened and shifted to high-field by 0.10–0.12 ppm on addition of up to 0.4 molar equivalents of 2c and eventually disappeared during the course of titration. At the same time, the imino protons of the monomeric quadruplex were shifted by 0.27–0.32 ppm to higher field. Except for the proton signals of A3H2 and T1H6, most of the bands of the aromatic protons in the range of 6–8 ppm remained relatively sharp up to 0.8 molar equivalents of 2c (cf. ESI, Fig. S2†). In addition, the 1H-NMR signals of ligand 2c also developed and shifted during the titration. As a general trend, the signals of the protons H2, H4, H6, and H7 were shifted to lower field by up to 0.4 ppm upon association of the ligand with the DNA, whereas the proton H8 is slightly shifted to higher field and H5 does not experience a significant shift (cf. ESI, Fig. S2†).
 |
| | Fig. 6 The 1H NMR spectra (600 MHz) of the imino protons of Tel6 (2 mM in bases) in K-phosphate buffer (H2O:D2O = 9:1; 95 mM, pH 7.0; T = 25 °C) with increasing amount of 2c; * = monomeric quadruplex. | |
Discussion
The results from the spectrometric DNA titrations with ct DNA clearly indicate the binding interactions between duplex DNA and the aza- and azoniastilbene derivatives 2a–c; however, with significantly different affinity and binding mode. Thus, the association of the charge neutral derivative 2a with ct DNA is rather weak, as shown by the lack of a shift of absorption band during the photometric DNA titration and by the inefficient fluorescence quenching by DNA. Moreover, the positive LD signal of the DNA-bound ligand indicates groove binding. As the development and band structure of the ICD bands does not reveal a clear trend and depends strongly on the ligand concentration, it is assumed that this ligand forms only loosely bound aggregates along the DNA grooves whose actual structure depends on the ligand–DNA ratio. This assumption is supported by the observation that the development of absorption bands of 2a during titration is not characteristic of a DNA binder, but rather indicates the random association of the hardly soluble compound along the DNA backbone, as shown by the plain decrease of absorption band with no significant shift. Therefore the determined binding constant is rather an aggregation constant of the ligand. This undirectional binding mode of this ligand to DNA is explained by the lack of a positive charge, which is usually required for high affinity of DNA ligands. In contrast, the ligands 2b and 2c exhibit the characteristic spectroscopic features of DNA intercalators, specifically polycyclic azoniahetarenes,10c,21 upon complex formation22 namely a hypochromic effect and red shift of the absorption band, fluorescence quenching, the development of a weak positive or negative ICD band, typical binding constants (2b: 5.1 × 104 M−1, 2c: 2.0 × 105 M−1) and – mostly indicative of the binding mode – a negative LD band in the ligand absorption range. It is tempting to conclude that the ligands 2b and 2c have different alignments in the intercalation site as the ICD signals develop with different phase. However, the sign of the ICD signals depends on the relative orientation of the transition dipole moments of the ligand and the DNA base pairs. Considering dipole moments of the ligands that are aligned along the long molecular axis in 2b and along the short molecular axis in 2c, as deduced from the substitution pattern, both ligands intercalate into DNA with the long molecule axis perpendicular to the long axis of the binding site.
In addition, it was demonstrated that the ligands 2b and 2c bind to quadruplex DNA also, as shown exemplarily with the two representative DNA forms 22AG and a2 as well as with the dye labeled quadruplex-forming oligonucleotides F21T, FmycT, FkrasT and Fa2T. With regard to the association with quadruplex DNA, the azo- and azoniastilbene derivatives 2a–c show a similar trend of binding constants as with duplex DNA. It should be noted that the binding constants of the ligands with ct DNA and quadruplex DNA appear to be the same (e.g. for 2c: KctDNAB = 2.0 × 105 M−1 ), however; they were determined in solutions with different ionic strength (10 mM BPE buffer versus 95 mM K-phosphate buffer). Therefore, they cannot be directly compared because it is known that the binding constants, especially the ones of cationic ligands, decrease with increasing ionic strength. In fact, a control experiment showed that 2c has a significantly smaller binding constant with ct DNA of KB = 6.7 × 104 M−1 at higher ionic strength (cf. ESI†). In general, with the increasing number of positive charges in the molecule the binding interaction with the DNA is getting stronger; however, this effect is more pronounced in the case of quadruplex DNA 22AG. Thus, the uncharged derivative 2a does not have a stabilizing effect on quadruplex DNA which indicates a very weak binding interaction. In contrast, the positively charged stilbene derivatives 2b and 2c induce a significant stabilization of the quadruplex DNA F21T, FmycT, FkrasT and Fa2T toward unfolding, and this effect is much stronger in the case of the dicationic derivative 2c than with the monocationic one. This difference may be explained by the effect of the positive charge, namely attractive Coulomb interactions with the phosphate backbone as well as thermodynamically favorable release of counter ions from the grooves and their subsequent solvation in the aqueous medium.9 The ligands 2b and 2c also bind to the ILPR DNA a2 as indicated by the spectrometric titrations and binding constants that are in the same range as the ones observed with 22AG. However, the stabilization of the ILPR quadruplex toward unfolding is not very pronounced according to the relatively low shifts of melting temperature of Fa2T upon ligand binding. This difference between binding constants and ΔTm values may be explained by the different buffer solutions that are used in each experiment.18 Moreover, even the rather moderate increase of the DNA melting temperature of Fa2T is indicative of ligand binding, because similar results were obtained with the well-established quadruplex ligands thiazole orange (ΔTm = 3.1 °C), porphyrine TMPyP4 (ΔTm = 6.3 °C) and coralyne (ΔTm = 1.8 °C) under identical conditions.18
), however; they were determined in solutions with different ionic strength (10 mM BPE buffer versus 95 mM K-phosphate buffer). Therefore, they cannot be directly compared because it is known that the binding constants, especially the ones of cationic ligands, decrease with increasing ionic strength. In fact, a control experiment showed that 2c has a significantly smaller binding constant with ct DNA of KB = 6.7 × 104 M−1 at higher ionic strength (cf. ESI†). In general, with the increasing number of positive charges in the molecule the binding interaction with the DNA is getting stronger; however, this effect is more pronounced in the case of quadruplex DNA 22AG. Thus, the uncharged derivative 2a does not have a stabilizing effect on quadruplex DNA which indicates a very weak binding interaction. In contrast, the positively charged stilbene derivatives 2b and 2c induce a significant stabilization of the quadruplex DNA F21T, FmycT, FkrasT and Fa2T toward unfolding, and this effect is much stronger in the case of the dicationic derivative 2c than with the monocationic one. This difference may be explained by the effect of the positive charge, namely attractive Coulomb interactions with the phosphate backbone as well as thermodynamically favorable release of counter ions from the grooves and their subsequent solvation in the aqueous medium.9 The ligands 2b and 2c also bind to the ILPR DNA a2 as indicated by the spectrometric titrations and binding constants that are in the same range as the ones observed with 22AG. However, the stabilization of the ILPR quadruplex toward unfolding is not very pronounced according to the relatively low shifts of melting temperature of Fa2T upon ligand binding. This difference between binding constants and ΔTm values may be explained by the different buffer solutions that are used in each experiment.18 Moreover, even the rather moderate increase of the DNA melting temperature of Fa2T is indicative of ligand binding, because similar results were obtained with the well-established quadruplex ligands thiazole orange (ΔTm = 3.1 °C), porphyrine TMPyP4 (ΔTm = 6.3 °C) and coralyne (ΔTm = 1.8 °C) under identical conditions.18
In the case of ligand 2c, the association with the quadruplex-forming oligonucleotide Tel6 as simple model was further confirmed by NMR-spectroscopic analysis. The broadening and significant upfield-shift of the imino protons of the guanine residues of the quadruplex usually indicate a terminal π-stacking of the ligand. Although it may be tempting to assume intercalation, this mode of binding is usually not favorable in quadruplex structures.23 Moreover, the significant shifts of the ligand protons during complex formation may be explained by the π-stacking to a terminal quartet and the positioning of the ligand protons in the anisotropic cones of the guanine quartet.
The initial goal of this study was the comparison of the DNA-binding properties of the stilbene derivatives 2a–c with the ones of the structurally resembling, quadruplex-binding dibenzochrysenes 1a–c. Specifically, it should be tested whether the increased structural flexibility of the stilbenes – while maintaining a similar longitudinal extension of the π-system – increases the affinity of the former ligands to DNA. Indeed, it was observed that the structurally flexible diazoniastilbene 2c has a stabilizing effect on quadruplex DNA F21T (ΔTm = 11 °C; LDR = 5.0); but the diazoniabenzochrysene derivatives 1a–c have been shown to induce an even larger increase of the melting temperature of quadruplex DNA F21T under almost identical conditions (ΔTm = 6–19 °C, LDR = 5.0).10b In the case of 1a10b and 2c (Table 1), the thermal stabilization of the quadruplex is only marginally influenced by the presence of duplex DNA ds26, indicating a high selectivity of these ligands for quadruplex DNA relative to duplex. Nevertheless, this selectivity appears to be slightly more pronounced for the rigid diazoniadibenzochrysene 1a because the decrease of the melting temperature is smaller (ΔΔTm = −2.5 °C, LDR = 5.0) than the one observed with 2c (ΔΔTm = −3.1 °C). Moreover, the binding constant of 2c with quadruplex DNA 22AG is about half as large as the ones of the ligands 1a–c (2c: 4.8 × 105 M−1versus1a–c: 2.5–3 × 106 M−110b). These observations imply that the gain in flexibility in ligand 2c as compared with the structurally rigid dibenzochrysenes 1a–c does not compensate the loss of overall π-surface. Thus, these results show in a direct comparison of two ligands with resembling extent and shape of the π system that at least under equilibrium conditions the π-stacking or dispersion interactions of a ligand contribute more to the affinity of a ligand to terminal binding sites of the quadruplex structure than its flexibility, although the latter would allow the ligand to adjust its conformation within the binding site in an induced-fit process. Although it may be obvious from literature data1,7 that both rigid and flexible ligands can bind to quadruplex DNA with reasonable affinity and selectivity, to the best of our knowledge there is only one direct and explicit comparison reported between two types of ligands with closely resembling ligand structures that mainly differ in terms of rigidity of the π system.12 However, it should be emphasized that the latter study is not directly comparable with the results presented herein, because even the “rigid” ligands contain flexible substituents that may influence the overall binding affinity to G4-DNA. Complementary to those findings, our results could be helpful for ligand design, because we present the counterintuitive observation that the affinity and selectivity of a given flexible ligand may be even increased by the introduction of more rigidity. This small but significant effect may be explained by thermodynamic factors, because the rigid ligand does not loose conformational freedom, thus causing no additional entropic penalty, upon transfer from the solution to the constrained binding site.
Notably, the differences of the ligand-induced shifts of quadruplex melting temperatures on addition of 2b and 2c (ΔΔTm = 7 °C (F21T) and ΔΔTm = 3 °C (Fa2T) at LDR = 5) appear to be too large to be solely caused by the different charges and may need further attention. Considering the known, but still rather unexplored “methyl effect” of DNA ligands, the increased affinity of 2c may also be caused by its methyl substituents, i.e. supported by additional dispersion interactions between the methyl groups and the hydrophobic binding site.24 To assess whether this methyl effect also affects the quadruplex-stabilizing properties of the diazoniadibenzo[b,k]chrysene scaffold and to have a better comparison with 2c, the dimethyl-substituted derivative 1d was also investigated in this study as it resembles the methyl-substitution pattern of 2c. Moreover, it was assumed that due to its close structural resemblance with the parent compounds 1a–c the derivative 1d also binds to quadruplex DNA through terminal π stacking. In fact, the methyl-substituted derivative 1d also induces a much better stabilization of the quadruplex DNA F21T toward thermal unfolding as the parent compounds 1a–c (ΔTm = 27 °C; LDR = 5.0), although with slightly smaller binding constant. Unfortunately, the bad solubility of some 1d-DNA complexes hampered its complete investigation. But at least the obtained results are in agreement with the ones with ligand 2c, which indicates that a methyl effect may operate in quadruplex ligands, presumably based on attractive dispersion interactions of the methyl substituents with the hydrophobic binding site. Nevertheless, this assumption has to be verified in a systematic study with a larger series of methyl-substituted ligand derivatives.
Conclusions
We have shown that aza- and azoniastilbene derivatives 2a–c, i.e. compounds with almost the same spatial dimensions and steric demand, bind to DNA with an affinity and selectivity that depends significantly on the number of positive charges. Whereas the charge neutral derivative 2a only binds non-specifically to the DNA backbone of duplex DNA, the ionic compounds 2b and 2c are typical DNA intercalators. Most notably, the bis-quinolinium derivative 2c binds to quadruplex DNA with moderate affinity and also induces a pronounced stabilization of the quadruplex DNA towards thermal denaturation, presumably caused by an additional methyl effect. In contrast to the proposed properties of the stilbene derivatives 2b and 2c, these ligands have a significantly weaker stabilizing effect on quadruplex DNA than the dibenzochrysene derivatives 1a–d. From this observation we carefully conclude that the increased flexibility of a quadruplex-DNA ligand does not lead to stronger interactions with the quadruplex DNA as compared with rigid ligands that have essentially the same size and extent of π-system. Certainly, this finding has to be further substantiated with a larger series of ligands and quadruplex forms; however, the present study already reveals that structural rigidity and lack of conformational freedom, as in the diazoniadibenzochrysene series, is not a disadvantage with respect to quadruplex-DNA binding.
Experimental
Equipment
Melting points were determined with a BÜCHI 545 (Büchi, Flawil, CH) and are uncorrected. NMR spectra were recorded on a Bruker AV 400 (1H: 400 MHz and 13C: 100 MHz) or on a Varian VNMR-S 600 (1H: 600 MHz, 13C: 150 MHz) at 20 °C; chemical shifts are given in ppm (δ) values relative to tetramethylsilane (TMS) as internal standard (δ = 0.0). Combustion analyses were carried out by Mr Rochus Breuer (Organic Chemistry I, University of Siegen). Mass spectra were recorded on a Finnigan LCQ deca (driving voltage: 6 kV, impingement gas: argon, capillary temperature: 200 °C, auxiliary gas: nitrogen). Photometric titrations were recorded with a Varian Cary 100 Bio. A Varian Cary Eclipse spectrometer was used for the spectrofluorimetric analyses. The CD and flow-LD measurements were performed on a Chirascan spectrometer (Applied Photophysics).
Materials
Commercially purchased reagents were used without further purification. Chemicals were obtained from Alfa Aesar GmbH & Co. KG, Karlsruhe, Germany (N-bromosuccinimide, 3-bromoquinoline, ethenyltriethylsilane), Acros Organics [n-butyllithium solution (2.5 M in hexane), 2-butenoic acid, 4-pyridinecarbonitrile]. 1,5-di(bromomethyl)naphthalene (5)11 2-(2-methyl-(1,3)-dioxolan-2-yl)pyridine (6),13 2-methylquinolizinium bromide (3)25 and (E)-1,2-di(quinolin-3-yl)ethane (2a)15 were prepared according to literature protocols. The oligonucleotides fluo-d(G3T2AG3T2AG3T2AG3)–tamra (F21T, fluo = fluoresceine; tamra = tetramethylrhodamine), FmycT [fluo-d(TGAG3TG3TAG3TG3TA2)-tamra], FkrasT [fluo-d(AG3CG2TGTG3A2GAG2A)-tamra], Fa2T fluo-d(ACAG4TGTG4)2-tamra, d(AG3T2AG3T2AG3T2AG3) (22AG), a2 d(ACAG4TGTG4)2 and d(T2AG3) (Tel6) were purchased from Metabion or Biomers and ct DNA was purchased from Merck and used without any further purification. DNA solutions in buffer were prepared according to published procedures;26 BPE buffer (pH 7.0): 6.0 mM Na2HPO4, 2.0 mM NaH2PO4, 1.0 mM Na2EDTA; Na-cacodylate buffer (pH 7.2–7.3): 10 mM Na(CH3)2AsO2·3 H2O, 10 mM KCl, 90 mM LiCl; K-phosphate buffer (pH 7.0): 25 mM K2HPO4, 70 mM KCl.
Methods
The spectrophotometric, spectrofluorimetric and CD-spectroscopic titrations with DNA, and the thermal DNA denaturation studies were performed according to reported protocols (cf. ESI†).10a,b To ensure a sufficient solubility of the ligands during the titrations, all experiments were performed with 5%v/v DMSO as cosolvent. Binding constants of ligand–DNA complexes were determined by fitting of the experimental data from the photometric and fluorimetric DNA titrations to the theoretical model according to the published procedure (cf. ESI†).10a,27
Synthesis of 1,5-bis{[1-(2-methyl-1,3-dioxolan-2 yl)pyridinium]methyl}naphtalenebis(tetrafluoroborate) (7).
A solution of 1,5-di(bromomethyl)naphthalene (5)11 (0.75 g, 2.39 mmol) and 2-(2-methyl-(1,3)-dioxolan-2-yl)pyridine (6)13 (1.09 g, 6.60 mmol) in DMSO (15 mL) were stirred at r.t. for 10 d. The yellow brown solution was added into EtOAc (100 mL). The precipitated crude solid was filtered and washed with EtOAc and Et2O. The solid was recrystalized from MeOH (0.5 mL, with added HBF4) to obtain product 7 (600 mg, 0.92 mmol, 36%); mp = 252–254 °C (dec.). – 1H NMR (400 MHz, DMSO-d6): δ = 1.84 (6 H, s, CH3), 3.73–3.76 (4 H, m, CH2), 4.02–4.08 (4 H, m, CH2), 6.25 (4 H, s, CH2N+), 7.43 (1 H, d, 3J = 7 Hz, CH), 7.63 (2 H, dd, 3J = 8 Hz, CH), 7.99 (2 H, d, 3J = 9 Hz, CH), 8.20 (2 H, ddd, 3J = 8 Hz, 3J = 6 Hz, 4J = 1 Hz, CH), 8.39 (2 H, d, 3J = 8 Hz, CH), 8.74 (2 H, dd, 3J = 9 Hz, 3J = 8 Hz, CH), 9.05 (2 H, d, 3J = 6 Hz, CH). – 13C-NMR (DMSO-d6, 100 MHz): 26.0, 61.1, 65.3, 106.1, 109.1, 125.5, 125.7, 126.9, 128.3, 128.9, 132.2, 133.9, 147.6, 148.8, 156.3. – El. anal. for C30H32N2B2F8O4 × H2O (676.21) calcd (%): C 53.29, H 5.07, N 4.14, found: C 52.93; H 4.13; N 4.21.
Synthesis of 4a,12a-diazonia-8,16-dimethyldibenzo[b,k]chrysenebis(tetrafluoroborate) (1d).
The bispyridinium 7 (295 mg, 0.45 mmol) was stirred in PPA (3.06 g) at 150 °C for 24 h. The mixture was cooled to 100 °C and saturated aqueous solution of NaBF4 (10 mL) was added. After cooling slowly to r.t. the aquesous solution was extracted with MeNO2. The organic layer was separated, dried with Na2SO4, and the solvent was evaporated to obtain the product 1d as yellow needles (179 mg, 0.34 mmol, 75%); mp >300 °C. – 1H NMR (400 MHz, DMF-d7): δ = 3.49 (6 H, s, CH3), 8.34 (2 H, dd, 3J = 7 Hz, CH), 8.49 (2 H, dd, 3J = 8 Hz, CH), 9.14 (2 H, d, 3J = 9 Hz, CH), 9.17 (2 H, d, 3J = 9.4 Hz, CH), 9.69 (2 H, d, 3J = 9 Hz, CH), 9.77 (2 H, d, 3J = 7 Hz, CH), 11.62 (2 H, s, CH). – 13C-NMR (DMF-d7, 100 MHz): δ = 13.4 (CH3), 122.8 (Cq), 123.0 (CH), 124.1 (CH), 125.5 (CH), 128.2 (CH), 128.8 (Cq), 132.4 (Cq), 132.9 (CH), 133.9 (Cq), 135.8 (CH). – El. anal. for C26H20N2B2F8·2 H2O (563.33) calcd (%): C 54.78, H 4.24, N 4.91; found C 55.37, H 4.01, N 5.44.
Synthesis of (E)-2-(2-(naphthalene-2-yl)vinyl)quinolizinium bromide (2b).
A solution of 2-methylquinolizinium bromide (3)25 (448 mg, 2.00 mmol), 2-naphthaldehyde (4) (469 mg, 3.00 mmol) and piperidine (0.20 mL) in MeOH (5 mL) was stirred under reflux for 5 h. The reaction mixture was cooled to r.t. and filtered. The filter cake was washed with cold MeOH and then with Et2O. The obtained orange-colored solid was recrystalized from MeOH to give the product 2b (450 mg, 1.24 mmol, 62%) as orange crystals (note: due to its photoreactivity this compound should be handled in the dark); mp = 272–274 °C (dec.). 1H NMR (600 MHz, DMSO-d6): δ = 7.55–7.60 (2 H, m, 1′-H, 2′-H), 7.70 (1 H, d, 3J = 16 Hz, 4′-H), 7.92–8.04 (5 H, m, 3-H, 9-H, 7′-H, 8′-H, 9′-H), 8.12 (1 H, d, 3J = 16 Hz, 5′-H), 8.19 (1 H, s, 10′-H), 8.31 (1 H, t, 3J = 8 Hz, 8-H), 8.43–8.50 (2 H, m, 7-H, 6′-H), 8.60 (1 H, s, 1-H), 9.26 (1 H, d, 3J = 8 Hz, 4-H), 9.33 (1 H, d, 3J = 7 Hz, 6-H). – 13C NMR (150 MHz, DMSO-d6): δ = 120.3, 122.8, 123.0, 123.6, 124.3, 126.9, 126.9, 127.3, 127.7, 128.4, 128.7, 129.1, 133.0, 133.1, 133.5, 136.6, 136.9, 138.4, 142.8, 144.9. – MS (ESI+): m/z (rel. intensity) = 282 (100) [M+]. – El. anal. for C21H16BrN × H2O (368.28) calcd (%): C 66.33, H 4.77, N 3.68, found: C 66.41; H 4.71; N 3.64.
Synthesis of (E)-3,3′-(ethane-1,2-diyl)bis(1-methylquinolin-1-ium) (2c).
In a 200 mL sealed tube, a mixture of 2a15 (141 mg, 0.50 mmol) in MeI (5.0 mL) was stirred at 140 °C for 5 h. The reaction mixture was cooled to r.t. and Et2O (30 mL) was added. The yellow precipitate was filtered and washed with Et2O. The remaining solid was dissolved in a small amount of MeOH (100 ml) and the solid was passed through a bromide-saturated ion-exchange column (DOWEX®1 × 8) three times. The solvent was evaporated and the solid was re-crystalized from MeOH/MeCN to obtain product 2c as a yellow solid (note: due to its photoreactivity this compound was handled in the dark); mp = 292–294 °C. 1H NMR (600 MHz, DMSO-d6): δ = 4.71 (6 H, s, 2 × CH3), 7.94 (2 H, s, 1′-H, 2′-H), 8.11 (2 H, t, 3J = 8 Hz, 6-H, 6′′-H), 8.28–8.32 (2 H, m, 7-H, 7′′-H), 8.53 (2 H, d, 3J = 8 Hz, 5-H, 5′′-H), 8.55 (2 H, d, 3J = 9 Hz, 8-H, 8′′-H), 9.40 (2 H, s, 4-H, 4′′-H), 9.92 (2 H, s, 2-H, 2′′-H). – 13C NMR (150 MHz, DMSO-d6): δ = 45.8, 119.3, 127.8, 129.1, 129.9, 130.5, 130.6, 135.5, 137.5, 142.9, 149.2. – El. anal. for C22H20Br2·0.5 H2O (508.25) calcd (%): C 51.99, H 4.76, N 5.51, found: C 52.28; H 4.78; N 5.55.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Generous support by the Deutsche Forschungsgemeinschaft is gratefully acknowledged. We thank Ms Jennifer Hermann, Ms Sandra Uebach and Dr Stefanie Müller for technical assistance.
Notes and references
-
(a) C. K. Kwok and C. J. Merrick, Trends Biotechnol., 2017, 35, 997 CrossRef CAS PubMed;
(b) S. Neidle, Nat. Rev. Chem., 2017, 1, 0041 CrossRef CAS;
(c) S. Neidle, J. Med. Chem., 2016, 59, 5987 CrossRef CAS PubMed;
(d) D. Rhodes and H. J. Lipps, Nucleic Acids Res., 2015, 43, 8627 CrossRef CAS PubMed;
(e) S. Balasubramanian, L. H. Hurley and S. Neidle, Nat. Rev. Drug Discovery, 2011, 10, 261 CrossRef CAS PubMed;
(f) G. W. Collie and G. N. Parkinson, Chem. Soc. Rev., 2011, 40, 5867 RSC.
-
(a) T. Caneque, S. Müller and R. Rodriguez, Nat. Rev. Chem., 2018, 2, 202 CrossRef;
(b) A. Laguerre, J. M. Y. Wong and D. Monchaud, Sci. Rep., 2016, 6, 32141 CrossRef CAS;
(c) V. S. Chambers, G. Marsico, J. M. Boutell, M. Di Antonio, G. P. Smith and S. Balasubramanian, Nat. Biotechnol., 2015, 33, 877 CrossRef.
-
(a) J. L. Huppert and S. Balasubramanian, Nucleic Acids Res., 2007, 35, 406 CrossRef CAS;
(b) J. L. Huppert, A. Bugaut, S. Kumari and S. Balasubramanian, Nucleic Acids Res., 2008, 36, 6260 CrossRef CAS;
(c) A. Verma, V. K. Yadav, R. Basundra, A. Kumar and S. Chowdhury, Nucleic Acids Res., 2009, 37, 4194 CrossRef CAS PubMed.
-
(a) C. L. Grand, T. J. Powell, R. B. Nagle, D. J. Bearss, D. Tye, M. Gleason-Guzman and L. H. Hurley, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 6140 CrossRef CAS PubMed;
(b) A. Siddiqui-Jain, C. L. Grand, D. J. Bearss and L. H. Hurley, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11593 CrossRef CAS PubMed;
(c) S. Balasubramanian, L. H. Hurley and S. Neidle, Nat. Rev. Drug Discovery, 2011, 10, 261 CrossRef CAS PubMed;
(d) Y. Li, J. Syed, Y. Suzuki, S. Asamitsu, N. Shioda, T. Wada and H. Sugiyama, ChemBioChem, 2016, 17, 928 CrossRef CAS PubMed;
(e) G. W. Collie and G. N. Parkinson, Chem. Soc. Rev., 2011, 40, 5867 RSC;
(f) S. J. Adam, C. L. Grand, D. J. Bearss and L. H. Hurley, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11593 CrossRef PubMed;
(g) S. Kumari, A. Bugaut, J. L. Huppert and S. Balasubramanian, Nat. Chem. Biol., 2007, 3, 218 CrossRef CAS PubMed.
-
(a) S. Balasubramanian and S. Neidle, Curr. Opin. Chem. Biol., 2009, 13, 345 CrossRef CAS PubMed;
(b) R. Hänsel-Hertsch, M. Di Antonio and S. Balasubramanian, Nat. Rev. Mol. Cell Biol., 2017, 18, 279 CrossRef PubMed.
-
(a) R. C. Monsen and J. O. Trent, Biochimie, 2018, 152, 134 CrossRef CAS PubMed;
(b) H.-S. Lee, M. Carmena, M. Liskovykh, E. Peat, J.-H. Kim, M. Oshimura, H. Masumoto, M.-P. Teulade-Fichou, Y. Pommier, W. C. Earnshaw, V. Larionov and N. Kouprina, Cancer Res., 2018, 78, 6282 CrossRef CAS PubMed.
-
(a) S. Asamitsu, T. Bando and H. Sugiyama, Chem. – Eur. J., 2019, 25, 417 CrossRef CAS PubMed;
(b) E. Ruggiero and S. N. Richter, Nucleic Acids Res., 2018, 46, 3270 CrossRef CAS PubMed;
(c) T. Che, Y.-Q. Wang, Z.-L. Huang, J.-H. Tan, Z.-S. Huang and S.-B. Chen, Molecules, 2018, 23, 493 CrossRef PubMed;
(d) Y. Xu, Chem. Soc. Rev., 2011, 40, 2719 RSC;
(e) A. Ali and S. Bhattacharya, Bioorg. Med. Chem. Lett., 2014, 22, 4506 CrossRef CAS PubMed;
(f) P. Murat, Y. Singh and E. Defrancq, Chem. Soc. Rev., 2011, 40, 5293 RSC.
-
(a) S. Bhaduri, N. Ranjan and D. P. Arya, Beilstein J. Org. Chem., 2018, 14, 1051 CrossRef CAS PubMed;
(b) A. Rescifina, C. Zagni, M. G. Varrica, V. Pistarà and A. Corsaro, Eur. J. Med. Chem., 2014, 74, 95 CrossRef CAS PubMed.
-
H. Ihmels and L. Thomas, in Materials Science of DNA Chemistry, ed. J.-I. Jin, CRC Press, Boca Raton, 2011, p. 49 Search PubMed.
-
(a) A. Granzhan, H. Ihmels and K. Jäger, Chem. Commun., 2009, 1249 RSC;
(b) K. Jäger, J. W. Bats, H. Ihmels, A. Granzhan, S. Uebach and B. O. Patrick, Chem. – Eur. J., 2012, 18, 10903 CrossRef PubMed;
(c) A. Granzhan and H. Ihmels, Synlett, 2016, 1775 CAS;
(d) H. Ihmels, K. Löhl, T. Paululat and S. Uebach, New J. Chem., 2018, 42, 13813 RSC.
- A. de Cian, E. DeLemos, J.-L. Mergny, M.-P. Teulade-Fichou and D. Monchaud, J. Am. Chem. Soc., 2007, 129, 1856 CrossRef CAS PubMed.
- B. Prasad, J. Jamroskovic, S. Bhowmik, R. Kumar, T. Romell, N. Sabouri and E. Chorell, Chem. – Eur. J., 2018, 24, 7926 CrossRef CAS PubMed.
- W. Sbliwa, G. Matusiak and B. Bachowska, Croat. Chem. Acta, 2006, 79, 513 Search PubMed.
-
(a) M.-Q. Wang, Y. Wu, Z.-Y. Wang, Q.-Y. Chen, F.-Y. Xiao, Y.-C. Jiang and A. Sang, Dyes Pigm., 2017, 145, 1 CrossRef CAS;
(b) M.-Q. Wang, J. Xu, L. Zhang, Y. Liao, H. Wei, Y.-Y. Yin, Q. Liu, M.-Q. Wang, W.-X. Zhu, Z.-Z. Song, S. Li and Y.-Z. Zhang, Bioorg. Med. Chem. Lett., 2015, 25, 5672 CrossRef CAS PubMed;
(c) R. Chennoufi, H. Bougherara, N. Gagey-Eilstein, B. Dumat, E. Henry, F. Subra, S. Bury-Moné, F. Mahuteau-Betzer, P. Tauc, M.-P. Teulade-Fichou and E. Deprez, Sci. Rep., 2016, 6, 21458 CrossRef CAS PubMed;
(d) M.-Q. Wang, J. Xu, L. Zhang, Y. Liao, H. Wei, Y.-Y. Yin, Q. Liu and Y. Zhang, Bioorg. Med. Chem., 2019, 27, 552 CrossRef CAS PubMed.
- X. Zhang, E. L. Clennan, N. Arulsamy, R. Weber and J. Weber, J. Org. Chem., 2016, 81, 5474 CrossRef CAS PubMed.
- Y. Wang and D. J. Patel, Biochemistry, 1992, 31, 8112 CrossRef CAS PubMed.
-
(a) D. Renčiuk, J. Zhou, L. Beautepaire, A. Guédin, A. Bourdoncle and J.-L. Mergny, Methods, 2012, 57, 122 CrossRef PubMed;
(b) A. de Cian, L. Guittat, M. Kaiser, B. Saccà, S. Amrane, A. Bourdoncle, P. Alberti, M.-P. Teulade-Fichou, L. Lacroix and J.-L. Mergny, Methods, 2007, 42, 183 CrossRef CAS PubMed.
-
(a) D. Dzubiel, H. Ihmels, M. M. A. Mahmoud and L. Thomas, Beilstein J. Org. Chem., 2014, 10, 2963 CrossRef PubMed;
(b) J. W. Yan, S.-B. Chen, H.-Y. Liu, W.-J. Ye, T.-M. Ou, J.-H. Tan, D. Li, L.-Q. Gu and Z.-S. Huang, Chem. Commun., 2014, 50, 6927 RSC;
(c) S. Paramasivan and P. H. Bolton, Nucleic Acids Res., 2008, 36, e106 CrossRef PubMed;
(d) J. Carvalho, T. Quintela, N. M. Gueddouda, A. Bourdoncle, J. Mergny, G. F. Salgado, J. A. Queiroz and C. Cruz, Org. Biomol. Chem., 2018, 16, 2776 RSC;
(e) A. Chauhan, S. Paladhi, M. Debnath and J. Dash, Org. Biomol. Chem., 2016, 14, 5761 RSC;
(f) J. Lavrado, P. M. Borralho, S. A. Ohnmacht, R. E. Castro, C. M. P. Rodrigues, R. Moreira, D. J. V. A. dos Santos, S. Neidle and A. Paulo, ChemMedChem, 2013, 8, 1648 CAS.
- G. A. Michelotti, E. F. Michelotti, A. Pullner, R. C. Duncan, D. Eick and D. Levens, Mol. Cell. Biol., 1996, 16, 2656 CrossRef CAS PubMed.
- See e.g.
(a) P. Kumar and R. Barthwal, Biochimie, 2018, 147, 153 CrossRef PubMed;
(b) K. Padmapriya and R. Barthwal, Biochim. Biophys. Acta, 2017, 1861, 37 CrossRef CAS PubMed;
(c) L. Scaglioni, R. Mondelli, R. Artali, F. R. Sirtori and S. Mazzini, Biochim. Biophys. Acta, Gen. Subj., 2016, 1860, 1129 CrossRef CAS PubMed;
(d) K. Padmapriya and R. Barthwal, Bioorg. Med. Chem. Lett., 2016, 26, 4915 CrossRef CAS PubMed;
(e) S. Tripathi, T. P. Pradeep and R. Barthwal, ChemBioChem, 2016, 17, 554 CrossRef CAS PubMed;
(f) T. P. Pradeep and R. Barthwal, Biochimie, 2016, 128, 59 CrossRef PubMed;
(g) W. Gai, Q. Yang, J. Xiang, W. Jiang, Q. Li, H. Sun, A. Guan, Q. Shang, H. Zhang and Y. Tang, Nucleic Acids Res., 2013, 41, 2709 CrossRef CAS PubMed;
(h) R. Ferreira, R. Artali, J. Farrera-Sinfreu, F. Albericio, M. Royo, R. Eritja and S. Mazzini, Biochim. Biophys. Acta, Gen. Subj., 2011, 1810, 769 CrossRef CAS PubMed;
(i) E. Gavathiotis, R. A. Heald, M. F. G. Stevens and M. S. Searle, Angew. Chem., Int. Ed., 2001, 40, 4749 CrossRef CAS.
-
(a) S. K. Manna, A. Mandal, S. K. Mondal, A. K. Adak, A. Jana, S. Das, S. Chattopadhyay, S. Roy, S. K. Ghorai, S. Samanta, M. Hossain and M. Baidya, Org. Biomol. Chem., 2015, 13, 8037 RSC;
(b) B. Abarca, R. Custodio, A. M. Cuadro, D. Sucunza, A. Domingo, F. Mendicuti, J. Alvarez-Builla and J. J. Vaquero, Org.
Lett., 2014, 16, 3464 CrossRef CAS PubMed;
(c) R. M. Suárez, P. Bosch, D. Sucunza, A. M. Cuadro, A. Domingo, F. Mendicuti and J. J. Vaquero, Org. Biomol. Chem., 2015, 13, 527 RSC;
(d) P. Bosch, D. Sucunza, F. Mendicuti, A. Domingo and J. J. Vaquero, Org. Chem. Front., 2018, 5, 1916 RSC.
- M. M. Aleksic and V. Kapetanovic, Acta Chim. Slov., 2014, 61, 555 CAS.
-
(a) E. Gavathiotis, R. A. Heald, M. F. G. Stevens and M. S. Searle, Angew. Chem., Int. Ed., 2001, 40, 4749 CrossRef CAS;
(b) H. Mita, T. Ohyama, Y. Tanaka and Y. Yamamoto, Biochemistry, 2006, 45, 6765 CrossRef CAS;
(c) d. S. M. Webba, Methods, 2007, 43, 264 CrossRef.
-
(a) A. K. F. Martensson and P. Lincoln, Phys. Chem. Chem. Phys., 2018, 20, 11336 RSC;
(b) E. J. Barreiro, A. E. Kümmerle and C. A. M. Fraga, Chem. Rev., 2011, 111, 5215 CrossRef CAS PubMed;
(c) C. S. Leung, S. S. F. Leung, J. Tirado-Rives and W. L. Jorgensen, J. Med. Chem., 2012, 55, 4489 CrossRef CAS PubMed;
(d) B. Rajendar, Y. Sato, S. Nishizawa and N. Teramae, Bioorg. Med. Chem. Lett., 2007, 17, 3682 CrossRef CAS PubMed;
(e) B. Rajendar, A. Rajendran, Y. Sato, S. Nishizawa and N. Teramae, Org. Biomol. Chem., 2009, 17, 351 CrossRef CAS PubMed;
(f) B. Rajendar, A. Rajendran, Z. Ye, E. Kanai, Y. Sato, S. Nishizawa, M. Sikorski and N. Teramae, Org. Biomol. Chem., 2010, 8, 4949 RSC;
(g) K. Benner, H. Ihmels, S. Kölsch and P. M. Pithan, Org. Biomol. Chem., 2014, 12, 1725 RSC.
- D. V. Berdnikova, O. A. Fedorova, E. V. Tulyakova, H. Li, S. Kölsch and H. Ihmels, Photochem. Photobiol., 2014, 91, 723 CrossRef PubMed.
-
(a) M. M. Paquette, R. A. Kopelman, E. Beitler and N. L. Frank, Chem. Commun., 2009, 5424 RSC;
(b) W. Paw and R. Eisenberg, Inorg. Chem., 1997, 36, 2287 CrossRef CAS PubMed;
(c) J.-L. Pozzo, A. Samat, R. Guglielmetti and D. D. Keukeleire, J. Chem. Soc., Perkin Trans. 2, 1993, 1327 RSC.
- A. Granzhan, H. Ihmels and G. Viola, J. Am. Chem. Soc., 2007, 129, 1254 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Absorption and emission data of 2a–c; thermal DNA denaturation analysis; NMR spectra. See DOI: 10.1039/c9ob00809h |
| ‡ Author names are in alphabetical order that does not reflect the specific contribution of each author. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
M.
Karbasiyoun
*,
M.
Karbasiyoun





 ), however; they were determined in solutions with different ionic strength (10 mM BPE buffer versus 95 mM K-phosphate buffer). Therefore, they cannot be directly compared because it is known that the binding constants, especially the ones of cationic ligands, decrease with increasing ionic strength. In fact, a control experiment showed that 2c has a significantly smaller binding constant with ct DNA of KB = 6.7 × 104 M−1 at higher ionic strength (cf. ESI†). In general, with the increasing number of positive charges in the molecule the binding interaction with the DNA is getting stronger; however, this effect is more pronounced in the case of quadruplex DNA 22AG. Thus, the uncharged derivative 2a does not have a stabilizing effect on quadruplex DNA which indicates a very weak binding interaction. In contrast, the positively charged stilbene derivatives 2b and 2c induce a significant stabilization of the quadruplex DNA F21T, FmycT, FkrasT and Fa2T toward unfolding, and this effect is much stronger in the case of the dicationic derivative 2c than with the monocationic one. This difference may be explained by the effect of the positive charge, namely attractive Coulomb interactions with the phosphate backbone as well as thermodynamically favorable release of counter ions from the grooves and their subsequent solvation in the aqueous medium.9 The ligands 2b and 2c also bind to the ILPR DNA a2 as indicated by the spectrometric titrations and binding constants that are in the same range as the ones observed with 22AG. However, the stabilization of the ILPR quadruplex toward unfolding is not very pronounced according to the relatively low shifts of melting temperature of Fa2T upon ligand binding. This difference between binding constants and ΔTm values may be explained by the different buffer solutions that are used in each experiment.18 Moreover, even the rather moderate increase of the DNA melting temperature of Fa2T is indicative of ligand binding, because similar results were obtained with the well-established quadruplex ligands thiazole orange (ΔTm = 3.1 °C), porphyrine TMPyP4 (ΔTm = 6.3 °C) and coralyne (ΔTm = 1.8 °C) under identical conditions.18
), however; they were determined in solutions with different ionic strength (10 mM BPE buffer versus 95 mM K-phosphate buffer). Therefore, they cannot be directly compared because it is known that the binding constants, especially the ones of cationic ligands, decrease with increasing ionic strength. In fact, a control experiment showed that 2c has a significantly smaller binding constant with ct DNA of KB = 6.7 × 104 M−1 at higher ionic strength (cf. ESI†). In general, with the increasing number of positive charges in the molecule the binding interaction with the DNA is getting stronger; however, this effect is more pronounced in the case of quadruplex DNA 22AG. Thus, the uncharged derivative 2a does not have a stabilizing effect on quadruplex DNA which indicates a very weak binding interaction. In contrast, the positively charged stilbene derivatives 2b and 2c induce a significant stabilization of the quadruplex DNA F21T, FmycT, FkrasT and Fa2T toward unfolding, and this effect is much stronger in the case of the dicationic derivative 2c than with the monocationic one. This difference may be explained by the effect of the positive charge, namely attractive Coulomb interactions with the phosphate backbone as well as thermodynamically favorable release of counter ions from the grooves and their subsequent solvation in the aqueous medium.9 The ligands 2b and 2c also bind to the ILPR DNA a2 as indicated by the spectrometric titrations and binding constants that are in the same range as the ones observed with 22AG. However, the stabilization of the ILPR quadruplex toward unfolding is not very pronounced according to the relatively low shifts of melting temperature of Fa2T upon ligand binding. This difference between binding constants and ΔTm values may be explained by the different buffer solutions that are used in each experiment.18 Moreover, even the rather moderate increase of the DNA melting temperature of Fa2T is indicative of ligand binding, because similar results were obtained with the well-established quadruplex ligands thiazole orange (ΔTm = 3.1 °C), porphyrine TMPyP4 (ΔTm = 6.3 °C) and coralyne (ΔTm = 1.8 °C) under identical conditions.18