
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solid-phase synthesis of biocompatible N-heterocyclic carbene–Pd catalysts using a sub-monomer approach†
Durgadas
Cherukaraveedu‡
 ,
Paul T.
Cowling‡
,
Gavin P.
Birch
,
Mark
Bradley
* and
Annamaria
Lilienkampf
*
,
Paul T.
Cowling‡
,
Gavin P.
Birch
,
Mark
Bradley
* and
Annamaria
Lilienkampf
*
EaStCHEM School of Chemistry, Joseph Black Building, University of Edinburgh, EH9 3FJ, UK. E-mail: mark.bradley@ed.ac.uk; annamaria.lilienkampf@ed.ac.uk
First published on 8th May 2019
Abstract
Taking inspiration from the assembly of so-called peptoids (N-alkylglycine oligomers) we present a new synthetic methodology whereby N-heterocyclic carbene (NHC) based Pd ligands were assembled using a sub-monomer approach and loaded with Pd via solid-phase synthesis. This allowed the rapid generation a library of NHC–palladium catalysts that were readily functionalised to allow bioconjugation. These catalysts were able to rapidly activate a caged fluorophore and ‘switch-on’ an anticancer prodrug in 3D cell culture.
Introduction
Bioorthogonal reactions enable the selective visualisation and manipulation of biological processes in living systems and have been widely used in a number of applications.1–4 Transition metal mediated bioorthogonal reactions are of particular interest as they enable an array of non-natural chemical transformations that can be used to modulate living systems.5,6 Reactions mediated by copper,7,8 iron,9 gold,10–12 ruthenium,12–16 and iridium17 have all found applications in living systems, although palladium is perhaps the most utilised metal in a biological setting.18Palladium has gained popularity in bioorthogonal chemistry due its ability to perform catalytic cross-coupling reactions, enabling the generation of carbon–carbon and carbon–heteroatom bonds under mild, biological conditions,19–21 and more recently in vivo.22,23 Palladium catalysts have been used to initiate a range of intracellular reactions including dealkylation,24 decaging of propargyloxycarbonyl groups,19,24,25 as well as Suzuki–Miyaura cross-couplings.20,26–28 Thus, palladium mediated reactions have been used to selectively activate enzymes through deprotection of modified amino acids within proteins,21,29 to synthesise anticancer agents in cellulo from two benign precursors,19 as well as activate produgs;20–22,24 however, to date, the majority of the examples have used palladium nanoparticles entrapped within a polymeric support,19,20,22–24,26,27,30 simple palladium salts such as Pd(OAc)2,31–33 or designed targeted palladium ligands.34
This is in contrast to the highly active palladium catalysts used in conventional organic synthesis that use a variety of stabilising ligands (e.g. phosphines) or N-heterocyclic carbenes (NHC),35,36 but few have been used in a biological setting. Chen used an NHC–Pd catalyst (with imidazolium-based ligands bearing hydrophilic quaternary ammonium salts) to mediate the Suzuki–Miyaura coupling reaction between a boronic acid functionalised biotin and 4-iodophenylalanine modified cell surface proteins, enabling subsequent imaging of the cell surface with fluorescently labelled streptavidin.33 Recently, we reported a water soluble NHC–Pd catalyst coupled to a cell penetrating peptide, which was able to remove a propargyloxycarbonyl group from a pro-fluorophore in cells, thus demonstrating the first intracellular application of NHC–Pd chemistry.37 While catalyst loading was carried out on the solid-phase, the synthesis of the NHC ligand required multistep synthesis in solution, purification, and conjugation to a solid support.
Here, we report an efficient microwave assisted solid-phase synthesis of a series of biocompatible NHC–Pd catalysts and their chemistry in cells. To the best of our knowledge, a solid phase approach to the generation of NHC–Pd catalysts has not been reported and, with our approach, these stable NHC–Pd catalysts were prepared in good yields with the majority of the catalysts showing good activity in a biological setting.
Results and discussion
A solid-phase synthetic route was developed for the NHC–Pd catalysts by adopting the ‘sub-monomer’ approach, originally developed by Zuckermann for the synthesis of peptoids (N-alkyl glycine oligomers), with sequential acylation and alkylation reactions on a resin.38,39 The NHC ligand chosen here was first reported by Meldal who showed that a resin-bound NHC–Pd complex (conjugated to a hydrophobic dipeptide) showed good catalytic efficiency in Suzuki–Miyaura cross-couplings in water.40 Here, this pyridine-derivatised imidazolium ligand was attached to a range of different amino acid spacers, with the aim of providing biocompatibility, aqueous stability, as well as a handle for biomolecule conjugation.Solid-phase synthetic route for the NHC–Pd catalysts
Fmoc-Ahx-OH was coupled to a 2-chlorotrityl chloride linker on a polystyrene resin (mesh 100–200), with subsequent removal of the Fmoc group giving 1, which was acylated with 2-bromoacetic acid (2 M in DMF) using DIC as a coupling reagent at 60 °C under μw irradiation for 20 min (Scheme 1). The bromide in 2 was substituted by imidazole to give 3, with optimised reaction conditions (ESI, Table S1†) allowing quantitative N-alkylation via an excess of imidazole (2 M) at 60 °C (μw heating for 40 min) in anhydrous DMSO with 0.5 M AgNO3. The addition of AgNO3 was required for full conversion (based on HPLC analysis, ESI, Fig. S1†). The imidazole in 3 was N-alkylated with 2-(bromomethyl)pyridine (1 M) to give 4 with >95% conversion using Et3N (1 M) and AgNO3 (0.5 M) at 60 °C in anhydrous DMF (for optimisation see ESI, Table S2†) giving the NHC ligand in >95% purity and 65% overall yield over 3 steps (ESI, Fig. S2 and S3†). Palladium loading of the NHC-ligand 4 on solid-phase was carried out as reported by Meldal40 with minor modifications. In brief, the carbene was generated on the resin from the imidazolium ion with the phosphazene base 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine (BEMP) in anhydrous DMF. Pd(COD)Cl2 was added subsequently to generate the NHC–Pd catalyst 5 (Scheme 1), which was cleaved off the resin with 30% HFIP in DCM and purified by semi-preparative RP-HPLC. This allowed the rapid and efficient generation of >100 mg quantities of the catalyst 5 that could be freely stored.§ | ||
Scheme 1 The microwave assisted solid-phase synthesis of NHC–Pd catalyst 5. (a) 0.3 M Fmoc-Ahx-OH, 0.5 M DIPEA, anhydrous DCM–DMF (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 1 h; (b) 20% piperidine in DMF, 2 × 10 min; (c) 2 M BrCH2CO2H, 1 M DIC, anhydrous DMF, 20 min, 60 °C, μw; (d) 2 M imidazole, 0.5 M AgNO3 in anhydrous DMSO, 40 min, 60 °C, μw; (e) 1 M 2-(bromomethyl)pyridine, 1 M EtN3, 0.5 M AgNO3, anhydrous DMF, 90 min, 60 °C, μw; (f) BEMP, anhydrous DMF, N2, 45 min, then Pd(COD)Cl2 overnight; (g) 30% HFIP–DCM, 1 h. All conversions were monitored by cleavage of a small sample from the resin and characterisation by HPLC and NMR. As drawn, the catalyst is Pd(II) with Y most likely formate (from the HPLC purification buffer). :1), 1 h; (b) 20% piperidine in DMF, 2 × 10 min; (c) 2 M BrCH2CO2H, 1 M DIC, anhydrous DMF, 20 min, 60 °C, μw; (d) 2 M imidazole, 0.5 M AgNO3 in anhydrous DMSO, 40 min, 60 °C, μw; (e) 1 M 2-(bromomethyl)pyridine, 1 M EtN3, 0.5 M AgNO3, anhydrous DMF, 90 min, 60 °C, μw; (f) BEMP, anhydrous DMF, N2, 45 min, then Pd(COD)Cl2 overnight; (g) 30% HFIP–DCM, 1 h. All conversions were monitored by cleavage of a small sample from the resin and characterisation by HPLC and NMR. As drawn, the catalyst is Pd(II) with Y most likely formate (from the HPLC purification buffer). | ||
Synthesis of NHC–Pd catalyst library
This solid-phase synthesis route was used to generate an NHC–Pd catalyst library (5–12). Using the NHC moiety with different amino acid spacers gave a range of both hydrophilic and hydrophobic groups to compare the potential effect on catalytic activity, as well as the robustness of the synthetic method (Fig. 1). The catalysts were synthesised using the 2-chlorotrityl linker, but the methodology was also compatible with the Rink-amide linker (ESI, catalyst 13). The ligands for catalysts 5–12 were fully characterised using NMR, HPLC, and HRMS, and after palladium loading, the catalysts were cleaved off the resin, purified by semi-preparative RP-HPLC, and characterised by ESI-MS and HPLC (ESI, Table S3†). The purity of the catalysts was confirmed by analytical HPLC (ESI, Fig. S4 and S5† – the “naked ligands” and the Pd-loaded ligands displayed different retention times) and the presence of palladium also established by HRMS (ESI, Fig. S6†) (the presence of the Pd meant that the 1H NMR spectra were highly broadened). These catalysts were stable for two weeks at room temperature in ACN/H2O, and ≥8 weeks at 4 °C. | ||
| Fig. 1 The NHC–Pd catalysts 5–12 synthesised by the optimised solid-phase synthesis protocol (Scheme 1). As drawn, the catalyst is Pd(II). In the biological experiments Y can be exchanged to various species with different coordination/charge states possible. | ||
Screening of catalytic activity
The fluorogenic probe based on 2,7-dichlorofluorescein (DCF) was used to evaluate the activity of the catalysts.41 The profluorophore O-propargylated DCF-1 was converted to the fluorescent molecule DCF-2 (λEx/Em 480/520 nm) upon Pd-catalysed cleavage of the propargyl group (Fig. 2A). Catalyst screening was carried out at 37 °C in PBS and in MCF-7 cell lysate to evaluate catalytic activity under biologically relevant conditions. Catalysts 5–12 (0.8 mol%) were incubated with DCF-1 (10 μM) and the increase in fluorescence measured over 4 h with an additional measurement at 20 h (ESI, Fig. S7†). With the exception of 7 and 11, all the catalysts were active in PBS with 8 and 9 showing comparable activity to 0.8 mol% Pd(OAc)2 (Fig. 2B). The decaging reactions were notably slower in cell lysate with 8 and 9 again showing the best catalytic efficiency under these conditions and outperforming Pd(OAc)2 (ESI, Fig. S8A†). Although shorter hydrophobic spacers seemed to be preferred in PBS (8 > 9 > 10), clear structure–activity relationships could not be established for the catalytic activity, particularly in the cell lysate, highlighting the need for methods to rapidly generate combinatorial catalyst libraries.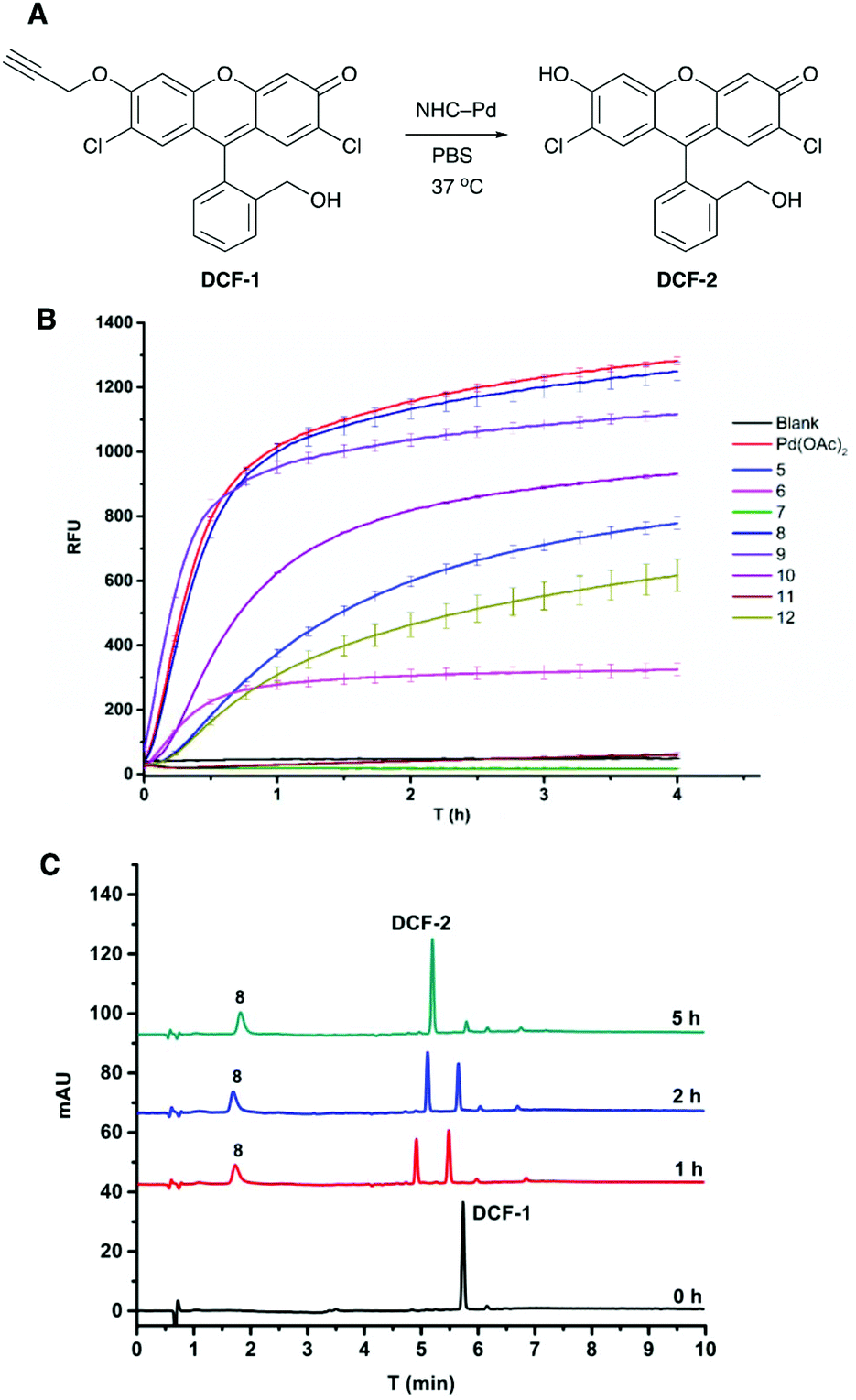 | ||
| Fig. 2 (A) Pd catalysed depropargylation reaction of profluorophore DCF-1 to give fluorescent DCF-2 (λEx/Em 480/520 nm). (B) Screening of catalysts 5–12 (0.8 mol%) for the activation of DCF-1 (10 μM) in PBS (n = 3). The reactions were monitored over 4 h and the increase in fluorescence recorded over time and compared to blank (no catalyst) and 0.8 mol% Pd(OAc)2. (C) The catalytic decaging of DCF-1 (50 μM) with catalyst 8 (2 mol%) monitored by HPLC (detection at 282 nm) over 5 h with the reaction carried out in PBS, showing >92% of DCF-2. | ||
Catalyst 8 was further evaluated by monitoring the decaging of DCF-1 (50 μM) by HPLC, with the experiments performed in PBS and in human plasma to mirror a cellular environment rich in proteins. In PBS, 8 (2 mol%) gave 92% conversion to DCF-2 after 5 h (Fig. 2C), whereas in plasma 97% conversion was observed (ESI, Fig. S8B†).
Prodrug activation in cancer cells and in cancer cell spheroids
The catalyst 8 was evaluated for its efficiency to activate the caged anticancer drug N-propargyl protected 5-fluorouracil24 (Pro-5-FU) (Fig. 3A). In PBS, 5 mol% of 8 was able to convert Pro-5-FU (100 μM) into the active drug 5-fluorouracil (5-FU) within 48 h, as monitored by HPLC (Fig. 3B).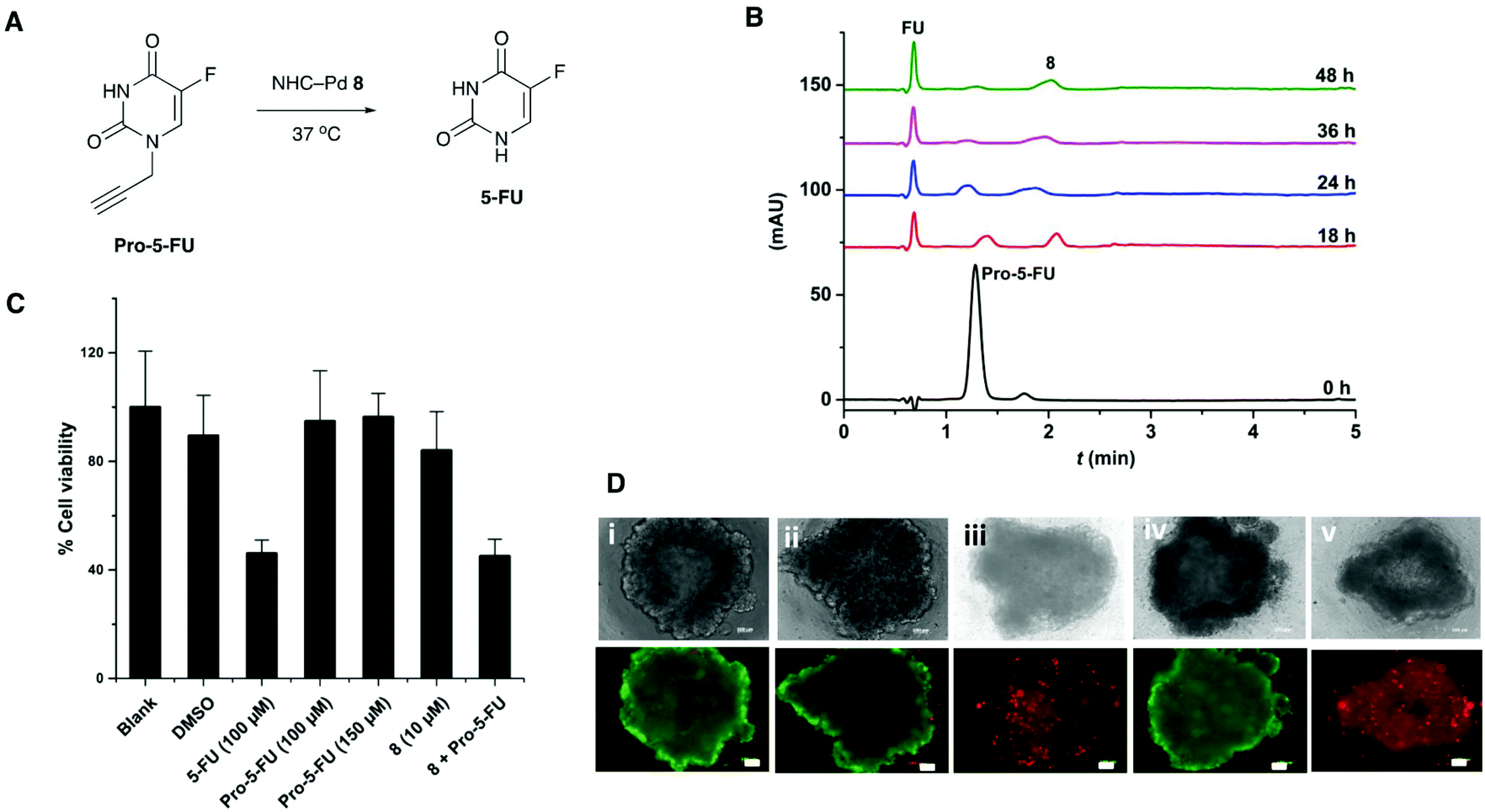 | ||
| Fig. 3 (A) The Pd-catalysed decaging of prodrug 5-fluoro-1-propargyluracil Pro-5-FU into the active anticancer drug 5-fluorouracil 5-FU. (B) Decaging of Pro-5-FU (100 μM) with catalyst 8 (5 mol%) in PBS (pH 7.4, 37 °C) monitored by HPLC with detection at 282 nm. (C) MTT cytotoxicity assay for prodrug activation in MCF-7 cells. The prodrug Pro-5-FU (100 μM with 1% DMSO) did not induce cytotoxicity and the NHC–Pd catalyst 8 (10 μM) only showed minor reduction in cell viability (84% viability) after 5 days incubation (untreated control cells were defined as 100% viable). Co-treatment with catalyst 8 (10 mol%, 10 μM) and Pro-5-FU (100 μM) for 5 days resulted in comparable cytotoxicity to 5-FU. (D) In situ activation of prodrug Pro-5-FU by catalyst 8 in 3D MCF-7 spheroids (drug activation most likely happening extracellularly). The spheroids were imaged for live/dead status, Green cells (λEx/Em 495/520 nm) are live whilst the red cells (λEx/Em 595/615 nm) are dead. (i) Untreated spheroid (control); (ii) Spheroid treated with (10 μM) of catalyst 8; (iii) Spheroid treated with 5-FU (100 μM) resulting in cell death; (iv) Spheroid treated with prodrug Pro-5-FU (100 μM) showing good viability; (v) Spheroid co-treated with the Pro-5-FU (100 μM) and catalyst 8 (10 μM) suggesting cell death equivalent to that seen with 100 μM of 5-FU. Scale bar 100 μm. | ||
The co-treatment of MCF-7 cell first with Pro-5-FU (100 μM) for 24 h followed by catalyst 8 (10 mol%) for 4 days resulted in comparable cytotoxicity (MTT assay) to 5-FU (Fig. 3C), with the drug activation likely taking place both intra- and extracellularly (Pro-5-FU is readily taken up by cells30). Catalyst 8 did not show notable cytotoxicity with 84% cell viability at 10 μM concentration.
Next, MCF-7 spheroids were treated with the prodrug Pro-5-FU (100 μM) and catalyst 8 (10 mol%) for 5 days, stained with the LIVE/DEAD™ Cell Imaging Kit (488/570), and analysed by fluorescence microscopy. Spheroids treated with both the prodrug and catalyst resulted in cell death comparable to cells treated with 5-FU, whereas treatment with only the catalyst or the prodrug Pro-5-FU had no effect on the viability of the 3D spheroids (Fig. 3D). These results demonstrate that the catalyst was able to decage a protected prodrug in a more representative 3D cancer model, which may have applications in future anticancer prodrug therapies.
Conclusions
A highly efficient microwave assisted solid-phase synthesis of NHC–Pd catalysts, based on the ‘sub monomer’ approach was developed and used to generate biocompatible catalysts on scale. Catalyst 8, comprising of the NHC–Pd moiety linked to a glycine, was the most robust catalyst in the series and was able to activate a fluorogenic probe in a biological setting and decage the protected anticancer drug (5-fluoro-1-propalgyluracil) in a 3D cancer cell culture resulting in comparable cell death to 5-fluorouracil. These robust NHC-Pd catalysts have a carboxylic acid that can be readily converted to a stable active ester thus providing a handle for bioconjugation and offering applications for both specific cell targeting ligands and bioorthogonal prodrug activations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the European commission for the Marie Skłodowska-Curie Individual Fellowship for D. C. (MSCA-IF-2014-EF, MetalCell 659489), and the Engineering and Physical Sciences Research Council and Medical Research Council CDT Optima (grant number EP/L016559/1) and the Rosetrees Trust for funding. We also thank the SIRCAMS at University of Edinburgh for the HRMS analysis and the School of Chemistry NMR facility.Notes and references
- J. Li and P. R. Chen, Nat. Chem. Biol., 2016, 12, 129–137 CrossRef CAS PubMed.
- J. G. Rebelein and T. R. Ward, Curr. Opin. Biotechnol., 2018, 53, 106–114 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- J. J. Soldevila-Barreda and N. Metzler-Nolte, Chem. Rev., 2019, 119, 829–869 CrossRef CAS PubMed.
- M. Martinez-Calvo and J. L. Mascarenas, Chimia, 2018, 72, 791–801 CrossRef CAS PubMed.
- Y. Bai, J. Chen and S. C. Zimmerman, Chem. Soc. Rev., 2018, 47, 1811–1821 RSC.
- J. Clavadetscher, S. Hoffmann, A. Lilienkampf, L. Mackay, R. M. Yusop, S. A. Rider, J. J. Mullins and M. Bradley, Angew. Chem., Int. Ed., 2016, 55, 15662–15666 CrossRef CAS PubMed.
- Y. Bai, X. Feng, H. Xing, Y. Xu, B. K. Kim, N. Baig, T. Zhou, A. A. Gewirth, Y. Lu, E. Oldfield and S. C. Zimmerman, J. Am. Chem. Soc., 2016, 138, 11077–11080 CrossRef CAS PubMed.
- P. K. Sasmal, S. Carregal-Romero, A. A. Han, C. N. Streu, Z. Lin, K. Namikawa, S. L. Elliott, R. W. Koster, W. J. Parak and E. Meggers, ChemBioChem, 2012, 13, 1116–1120 CrossRef CAS PubMed.
- K. Tsubokura, K. K. H. Vong, A. R. Pradipta, A. Ogura, S. Urano, T. Tahara, S. Nozaki, H. Onoe, Y. Nakao, R. Sibgatullina, A. Kurbangalieva, Y. Watanabe and K. Tanaka, Angew. Chem., Int. Ed., 2017, 56, 3579–3584 CrossRef CAS PubMed.
- A. M. Perez-Lopez, B. Rubio-Ruiz, V. Sebastian, L. Hamilton, C. Adam, T. L. Bray, S. Irusta, P. M. Brennan, G. C. Lloyd-Jones, D. Sieger, J. Santamaria and A. Unciti-Broceta, Angew. Chem., Int. Ed., 2017, 56, 12548–12552 CrossRef CAS PubMed.
- C. Vidal, M. Tomas-Gamasa, P. Destito, F. Lopez and J. L. Mascarenas, Nat. Commun., 2018, 9, 1913 CrossRef PubMed.
- C. Streu and E. Meggers, Angew. Chem., Int. Ed., 2006, 45, 5645–5648 CrossRef CAS PubMed.
- M. Tomas-Gamasa, M. Martinez-Calvo, J. R. Couceiro and J. L. Mascarenas, Nat. Commun., 2016, 7, 12538 CrossRef PubMed.
- T. Volker, F. Dempwolff, P. L. Graumann and E. Meggers, Angew. Chem., Int. Ed., 2014, 53, 10536–10540 CrossRef PubMed.
- M. I. Sanchez, C. Penas, M. E. Vazquez and J. L. Mascarenas, Chem. Sci., 2014, 5, 1901–1907 RSC.
- S. Bose, A. H. Ngo and L. H. Do, J. Am. Chem. Soc., 2017, 139, 8792–8795 CrossRef CAS PubMed.
- S. V. Chankeshwara, E. Indrigo and M. Bradley, Curr. Opin. Chem. Biol., 2014, 21, 128–135 CrossRef CAS PubMed.
- J. Clavadetscher, E. Indrigo, S. V. Chankeshwara, A. Lilienkampf and M. Bradley, Angew. Chem., Int. Ed., 2017, 56, 6864–6868 CrossRef CAS PubMed.
- R. M. Yusop, A. Unciti-Broceta, E. M. Johansson, R. M. Sanchez-Martin and M. Bradley, Nat. Chem., 2011, 3, 239–243 CrossRef CAS PubMed.
- J. Li, J. Yu, J. Zhao, J. Wang, S. Zheng, S. Lin, L. Chen, M. Yang, S. Jia, X. Zhang and P. R. Chen, Nat. Chem., 2014, 6, 352–361 CrossRef CAS PubMed.
- M. A. Miller, B. Askevold, H. Mikula, R. H. Kohler, D. Pirovich and R. Weissleder, Nat. Commun., 2017, 8, 15906 CrossRef CAS PubMed.
- T. L. Bray, M. Salji, A. Brombin, A. M. Perez-Lopez, B. Rubio-Ruiz, L. C. A. Galbraith, E. E. Patton, H. Y. Leung and A. Unciti-Broceta, Chem. Sci., 2018, 9, 7354–7361 RSC.
- J. T. Weiss, J. C. Dawson, K. G. Macleod, W. Rybski, C. Fraser, C. Torres-Sanchez, E. E. Patton, M. Bradley, N. O. Carragher and A. Unciti-Broceta, Nat. Commun., 2014, 5, 3277 CrossRef PubMed.
- J. T. Weiss, J. C. Dawson, C. Fraser, W. Rybski, C. Torres-Sanchez, M. Bradley, E. E. Patton, N. O. Carragher and A. Unciti-Broceta, J. Med. Chem., 2014, 57, 5395–5404 CrossRef CAS PubMed.
- E. Indrigo, J. Clavadetscher, S. V. Chankeshwara, A. Lilienkampf and M. Bradley, Chem. Commun., 2016, 52, 14212–14214 RSC.
- F. Wang, Y. Zhang, Z. Du, J. Ren and X. Qu, Nat. Commun., 2018, 9, 1209 CrossRef PubMed.
- P. Destito, A. Sousa-Castillo, J. R. Couceiro, F. Lopez, M. A. Correa-Duarte and J. L. Mascarenas, Chem. Sci., 2019, 10, 2598–2603 RSC.
- J. Wang, S. Zheng, Y. Liu, Z. Zhang, Z. Lin, J. Li, G. Zhang, X. Wang, J. Li and P. R. Chen, J. Am. Chem. Soc., 2016, 138, 15118–15121 CrossRef CAS PubMed.
- G. Y. Tonga, Y. D. Jeong, B. Duncan, T. Mizuhara, R. Mout, R. Das, S. T. Kim, Y. C. Yeh, B. Yan, S. Hou and V. M. Rotello, Nat. Chem., 2015, 7, 597–603 CrossRef CAS PubMed.
- J. Li, S. Lin, J. Wang, S. Jia, M. Yang, Z. Hao, X. Zhang and P. R. Chen, J. Am. Chem. Soc., 2013, 135, 7330–7338 CrossRef CAS PubMed.
- C. D. Spicer and B. G. Davis, Chem. Commun., 2011, 47, 1698–1700 RSC.
- X. J. Ma, H. X. Wang and W. Z. Chen, J. Org. Chem., 2014, 79, 8652–8658 CrossRef CAS PubMed.
- M. Martinez-Calvo, J. R. Couceiro, P. Destito, J. Rodriguez, J. Mosquera and J. L. Mascarenas, ACS Catal., 2018, 8, 6055–6061 CrossRef CAS PubMed.
- G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151–5169 RSC.
- J. F. Jensen, K. Worm-Leonhard and M. Meldal, Eur. J. Org. Chem., 2008, 3785–3797 CAS.
- E. Indrigo, J. Clavadetscher, S. V. Chankeshwara, A. Megia-Fernandez, A. Lilienkampf and M. Bradley, Chem. Commun., 2017, 53, 6712–6715 RSC.
- R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS.
- T. S. Burkoth, A. T. Fafarman, D. H. Charych, M. D. Connolly and R. N. Zuckermann, J. Am. Chem. Soc., 2003, 125, 8841–8845 CrossRef CAS PubMed.
- K. Worm-Leonhard and M. Meldal, Eur. J. Org. Chem., 2008, 5244–5253 CrossRef CAS.
- M. Santra, S. K. Ko, I. Shin and K. H. Ahn, Chem. Commun., 2010, 46, 3964–3966 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Supporting figures and tables, and experimental procedures. See DOI: 10.1039/c9ob00716d |
| ‡ Equal contribution. |
| § The solid-phase approach used here offers advantages over more traditional solution-phase methods, namely the use of mass-action to drive the chemistries and the removal of intermediary purification steps. The ligand was previously synthesised in three steps (in solution) with a combined reaction time of 7.5 h (compared to 3 h in this work) requiring both silica column chromatography and preparative RP-HPLC purification.40 |
| This journal is © The Royal Society of Chemistry 2019 |