
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot ortho-amination of aryl C–H bonds using consecutive iron and copper catalysis†
Martyn C.
Henry
 ,
Rochelle
McGrory
,
Réka J.
Faggyas
,
Mohamed A. B.
Mostafa
and
Andrew
Sutherland
*
,
Rochelle
McGrory
,
Réka J.
Faggyas
,
Mohamed A. B.
Mostafa
and
Andrew
Sutherland
*
WestCHEM, School of Chemistry, The Joseph Black Building, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: Andrew.Sutherland@glasgow.ac.uk
First published on 18th April 2019
Abstract
A one-pot approach for ortho-coupling of arenes with non-actived N-nucleophiles has been developed using sequential iron and copper catalysis. Regioselective ortho-activation of anisoles, anilines and phenols was achieved through iron(III) triflimide catalysed iodination, followed by a copper(I)-catalysed, ligand-assisted coupling reaction with N-heterocycle, amide and sulfonamide-based nucleophiles. The synthetic utility of this one-pot, two-step method for the direct amination of ortho-aryl C–H bonds was demonstrated with the late-stage functionalisation of 3,4-dihydroquinolin-2-ones. This allowed the preparation of a TRIM24 bromodomain inhibitor and a series of novel analogues.
Introduction
In the last few decades, significant progress in the development of transition metal catalysed reactions has resulted in regioselective formation of aryl C–N bonds and the use of these processes for the general synthesis of pharmaceutically active compounds, organic materials and natural products.1 Early methods involved the copper- or palladium-catalysed amination of aryl (pseudo)halides using Ullmann–Goldberg, Chan–Evans–Lam or Buchwald–Hartwig methods (Fig. 1a).2,3 More recently, procedures have also been developed for the amination of aryl halides using a combination of iron and copper catalysis.4 While extensive reaction optimisation has resulted in highly efficient transformations, a drawback to this approach is the requirement of pre-functionalised arenes. | ||
| Fig. 1 Selected methods for ortho C–H aryl amination. | ||
To avoid arene ring pre-functionalisation, more recent strategies have focused on ortho-directed dehydrogenative coupling of aryl C–H bonds with non-activated amines and amides using chelating auxiliary groups in the presence of oxidants such as oxygen or hypervalent iodine compounds.5 This method has found application for the regioselective substitution of a wide range of arenes, including the ortho-amination of anilines.5,6 For example, Rodríguez and co-workers described the efficient copper-catalysed amination of anilines using the 2-picolinic acid directing group and (diacetoxyiodo)benzene as the oxidant (Fig. 1b).7,8 The auxiliary group could be removed under basic conditions to access the ortho-aminated aniline in high overall yield. Mechanistically distinct, palladium-catalysed ortho-amination of para-substituted anilides with N-fluorobenzenesulfonimide (NFSI) has also been described (Fig. 1c).9 In this transformation, it was proposed that the in situ generated FPdN(SO2Ph)2 formed a dearomatised spiro-cyclopalladium intermediate that underwent an ortho-nucleophilic amination with benzenesulfonimide. Alternatively, with ortho-substituted anilides, this approach enabled the preparation of para-benzenesulfonimide functionalised arenes.
We recently reported a one-pot para-amination and amidation of activated arenes using sequential iron and copper catalysis.10 The process permitted the reaction of para-aryl C–H bonds with non-activated amines and amides via iron(III)-triflimide catalysed arene bromination, followed by a copper(I)-catalysed coupling reaction. However, attempted ortho-amination of para-substituted anilines using this one-pot, two-step approach led to inseparable mixtures of the coupled product and the aniline starting material. Further studies revealed this was due to competing reduction of the bromide intermediate during the relatively slow copper-catalysed Goldberg-type11 amination. As current methods for transition metal catalysed ortho-amination of arenes requires either aryl ring pre-functionalisation or the use of chelating auxiliary groups with strong oxidising conditions and often precious metals, we were interested in overcoming the issues of the one-pot iron and copper catalysed method for direct access to ortho-aminated arenes. Herein, we describe a one-pot intermolecular ortho-amination of arenes using an iron-catalysed iodination, followed by a copper-catalysed, ligand-assisted coupling reaction (Fig. 1d). As well as exploring the scope of this method for the ortho-amination or amidation of anilines, anisoles and phenols, we also describe the use of the procedure for the late-stage amidation of quinolin-2-ones and the structural diversification of a TRIM24 bromodomain inhibitor.
Results and discussion
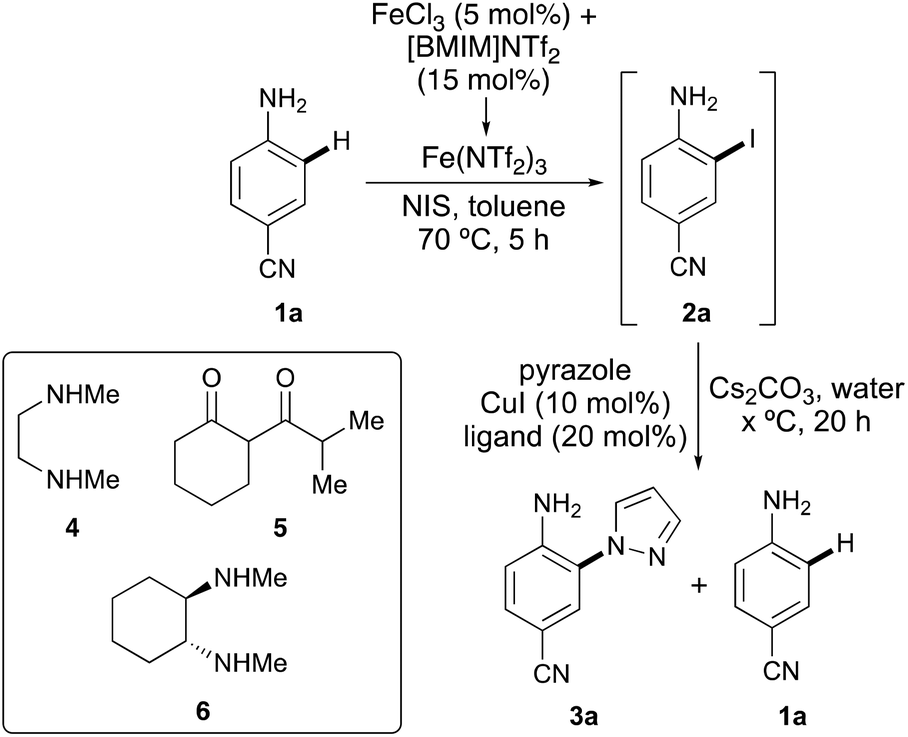
The study began by the investigation of the one-pot ortho-amination of para-aminobenzonitrile (1a) with pyrazole (Table 1). Iron(III) chloride and the ionic liquid, [BMIM]NTf2 were used for the in situ formation of the super Lewis acid, iron triflimide and the activation of N-bromosuccinimide (NBS) for ortho-bromination of 1a (entry 1).10,12 Following full conversion to the bromide (4 h), the copper(I)-catalysed coupling reaction with pyrazole, using N,N′-dimethylethylenediamine (4) (DMEDA) as the copper-chelating ligand was investigated. This gave a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of coupled product 3a and reduced compound 1a.13,14 It was proposed that a more reactive aryl halide bond may facilitate the coupling reaction over the reduction pathway. Therefore, the process was repeated using N-iodosuccinimide (NIS) during the halogenation step (entries 2 and 3). While this initially resulted in only a modest improvement (entry 2), the combination of using a lower reaction temperature (130 °C) during the coupling step gave a 6:1 ratio of 3a and 1a (entry 3). To further refine the process, alternative ligands were considered.15 Problems associated with challenging Goldberg-type coupling reactions are generally due to catalyst deactivation via competitive N-arylation of the ligand.16 To overcome this issue, ligands such as diketone 5 that exist in a delocalised enolate form and are less likely to undergo arylation have been utilised. The use of diketone 5 during the one-pot iodination and pyrazole coupling of 1a did lead to coupled product 3a, but there was no improvement in the product ratio (entry 4). On screening other ligands that facilitate a wide range of N-arylation reactions, racemic trans-N,N′-dimethylcyclohexane-1,2-diamine (6) gave the highest selectivity of 12:1 for 3aversus1a from the one-pot process (entry 5).17 Further optimisation revealed that increasing the amount of pyrazole led to complete suppression of the reductive pathway and generation of only coupled product 3a (entry 6). It should be noted that the involvement of Fe3+ during the first step is crucial for the success of the one-pot process. When iodination of 1a is performed under the same conditions as in Table 1 (70 °C), but without the presence of iron(III) chloride, only 40% conversion is observed after 20 h.
:1 mixture of coupled product 3a and reduced compound 1a.13,14 It was proposed that a more reactive aryl halide bond may facilitate the coupling reaction over the reduction pathway. Therefore, the process was repeated using N-iodosuccinimide (NIS) during the halogenation step (entries 2 and 3). While this initially resulted in only a modest improvement (entry 2), the combination of using a lower reaction temperature (130 °C) during the coupling step gave a 6:1 ratio of 3a and 1a (entry 3). To further refine the process, alternative ligands were considered.15 Problems associated with challenging Goldberg-type coupling reactions are generally due to catalyst deactivation via competitive N-arylation of the ligand.16 To overcome this issue, ligands such as diketone 5 that exist in a delocalised enolate form and are less likely to undergo arylation have been utilised. The use of diketone 5 during the one-pot iodination and pyrazole coupling of 1a did lead to coupled product 3a, but there was no improvement in the product ratio (entry 4). On screening other ligands that facilitate a wide range of N-arylation reactions, racemic trans-N,N′-dimethylcyclohexane-1,2-diamine (6) gave the highest selectivity of 12:1 for 3aversus1a from the one-pot process (entry 5).17 Further optimisation revealed that increasing the amount of pyrazole led to complete suppression of the reductive pathway and generation of only coupled product 3a (entry 6). It should be noted that the involvement of Fe3+ during the first step is crucial for the success of the one-pot process. When iodination of 1a is performed under the same conditions as in Table 1 (70 °C), but without the presence of iron(III) chloride, only 40% conversion is observed after 20 h.
Using the optimised conditions, the scope of the one-pot ortho-amination of a range of activated para-substituted arenes with pyrazole was explored (Scheme 1). Various anilines were subjected to the standard one-pot iron-catalysed activation and copper-catalysed coupling process and gave the ortho-substituted adducts (3a–3e) cleanly, in good yields (58–71%). This method allows direct ortho-substitution of unprotected anilines and despite the use of nucleophilic substrates, only N-arylation by pyrazole was observed. Extension of the one-pot process for the ortho N-arylation of anisoles, gave the corresponding ortho-substituted compounds (3f–3k) in 48–71% yield. Again, use of the highly regioselective iron-catalysed mono-iodination reaction led to formation of the ortho-coupled adducts as the sole products, even using highly reactive arenes with multiple activating groups (e.g.1i, 1k).
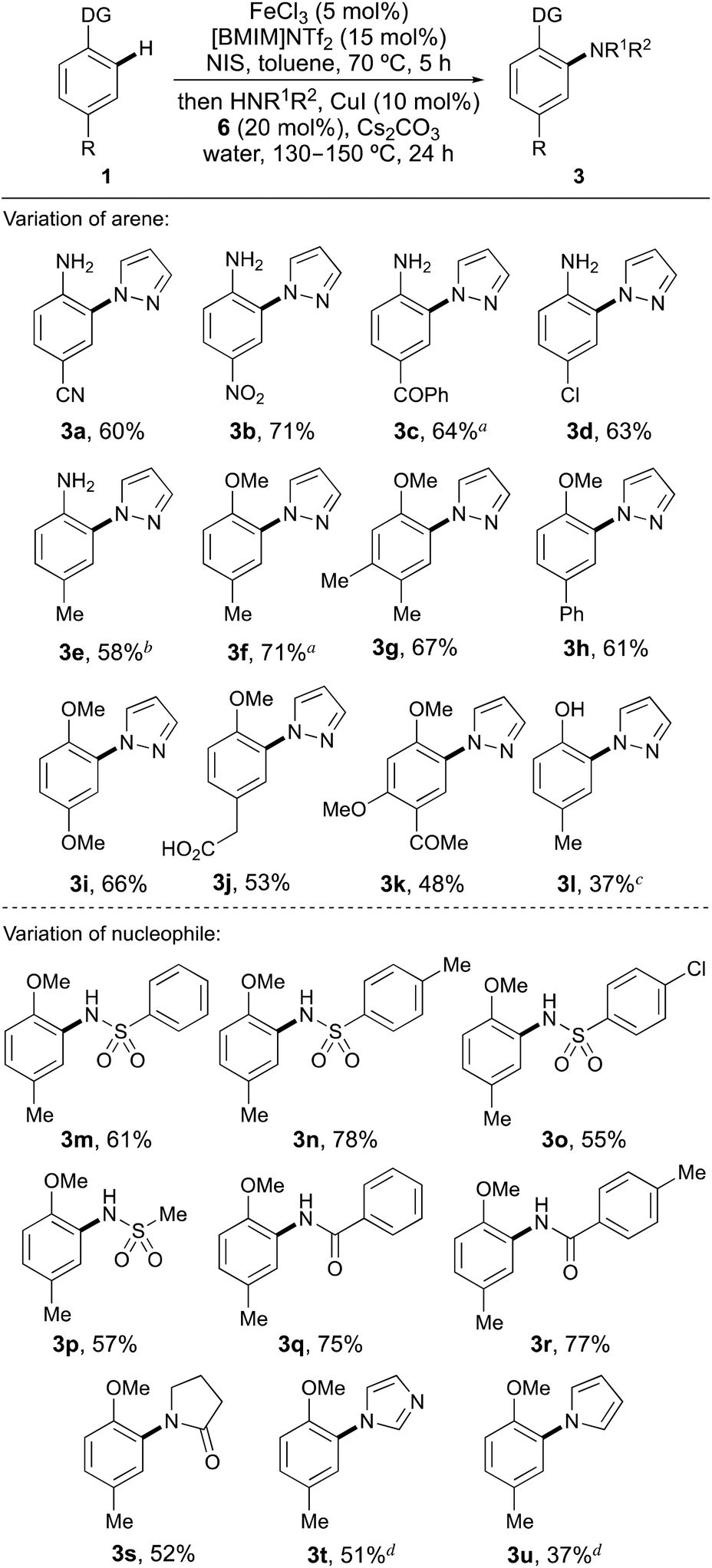 | ||
| Scheme 1 One-pot ortho-amination and amidation of arenes. aIodination step was complete after 20 h. bIodination step was done at 40 °C. cIodination step was performed using AgNTf2 (7.5 mol%) at 40 °C. dThe second step required 36 h. | ||
A limitation of this method was found in the attempted ortho-substitution of phenols (Scheme 1). Investigation of p-cresol (1l) as a substrate for the one-pot process yielded less than 20% of the coupled product with low conversion observed for each step. We previously reported that iodination of phenols could be improved using the softer Lewis acid, silver triflimide to activate NIS.18 The use of silver triflimide (7.5 mol%) was more effective for ortho-iodination of p-cresol (1l); however, the one-pot process yielded the coupled product 3l in a moderate 37% yield.
Using para-methylanisole (1f), the scope of the nucleophile was next explored (Scheme 1). With the standard one-pot conditions for anisoles, various sulfonamides and amides were smoothly coupled, allowing isolation of the ortho-substituted adducts (3m–3s) in good yields (52–78%). Using weaker, less nucleophilic N-heterocycles such as imidazole and pyrrole required a longer reaction time for the copper-catalysed N-substitution step (36 h), but still gave the ortho-substituted products 3t and 3u, cleanly. Two limitations were found when examining the scope of the nucleophile for the one-pot process. Electron-rich alkylamine nucleophiles such as morpholine gave low conversion (<10%) to the coupled product. The activity of the copper catalyst in these processes is likely compromised by competitive binding with the alkylamine nucleophiles, which are present in significantly higher concentration than the diamine ligand. Hindered heterocyclic nucleophiles such as 3,5-dimethylpyrazole also gave no coupled product. In this case, coupling at the ortho-position of an arene with an N-heterocyclic nucleophile also bearing an adjacent substituent is too hindered under these conditions for this transformation to proceed.
A proposed mechanism for the one-pot, two-step process is shown in Scheme 2. The combination of iron(III) chloride and the [BMIM]NTf2 ionic liquid leads to the formation of the super Lewis acid, Fe(NTf2)37.19,20 The highly delocalised nature of the triflimide counterion allows effective activation of NIS by Fe3+, resulting in rapid regioselective, electrophilic aromatic iodination of the arene ring system. In the second step, addition of copper(I) iodide and the chelating diamine ligand, trans-N,N′-dimethylcyclohexane-1,2-diamine (6) results in formation of 1,2-diamine-ligated copper(I) intermediate 8. Mechanistic studies by the Buchwald and Hartwig groups have shown subsequent reaction of copper(I) intermediate 8 with the base and nucleophile generates 1,2-diamine-ligated copper(I) amidate 9.21 While radical and nonradical pathways have been proposed for reaction of copper(I) amidate 9 with haloarenes, the same mechanistic studies using experimental tests for radical intermediates and DFT calculations have shown that oxidative addition to 10 occurs without free aryl radical intermediates.22 Despite this insight, the precise mechanism of aryl halide activation is still not well-established but Cu(III)-intermediates such as 10 are calculated to be kinetically accessible by a concerted oxidative addition or by internal electron transfer within the coordination sphere of the metal.21a Fast reductive elimination of 10 then forms the C–N coupled product and regenerates 1,2-diamine-ligated copper(I) intermediate 8. As shown by the results above (Scheme 1), the sequential transformations are highly compatible within the one-pot halogenation–amination process, leading to the clean formation of C–N coupled products. It should also be noted that during the course of this study, by-products due to an Ullmann-type, bi-aryl coupling were never observed.
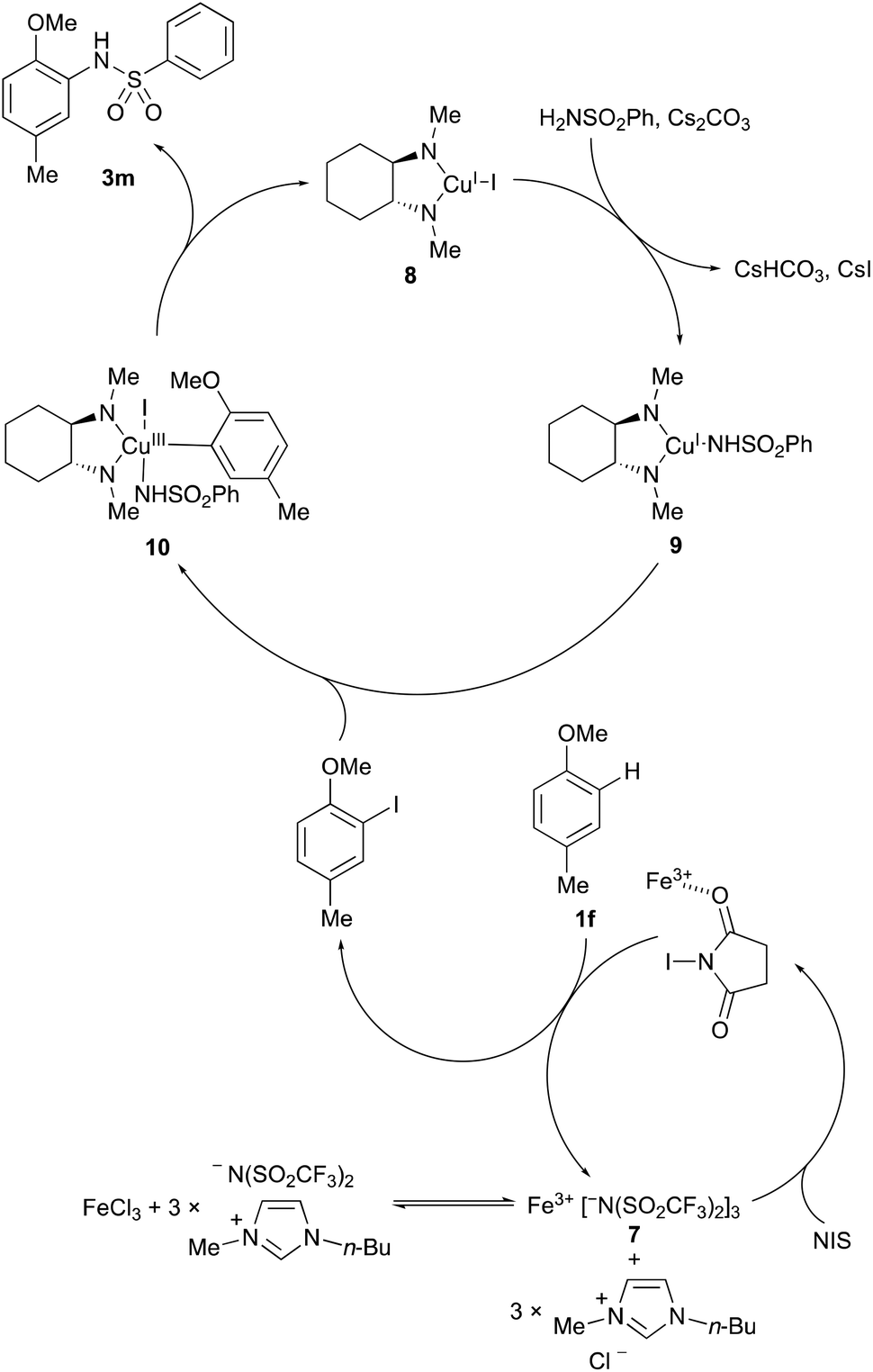 | ||
| Scheme 2 Proposed one-pot Fe(III)- and Cu(I)-catalysed ortho-amidation of 1f. | ||
Having examined the scope and limitations of the one-pot ortho-amination process, we wanted to demonstrate the synthetic utility of this approach for the preparation of pharmaceutically relevant targets. Palmer and co-workers have shown that sulfonamide substituted 3,4-dihydroquinolin-2-ones, such as 16a (Scheme 3) are inhibitors of the bromodomain containing protein TRIM24, which is implicated in human cancers.23 It was proposed that use of the one-pot activation and ortho-amidation process with N-methyl 3,4-dihydroquinoline-2-one 14 would allow rapid, late-stage access to 16a and a range of novel analogues. 3,4-Dihydroquinolin-2-one 14 was prepared via a two pot-process. Initially, 7-methoxy-3,4-dihydro-1H-quinolin-2-one (13) was prepared via a one-pot multistep process, that involved formation of the diazonium salt intermediate of aniline 11 under mild conditions, followed by a base-free Heck–Matsuda reaction with methyl acrylate to give cinnamate intermediate 12.24 In the second part of the one-pot process, re-utilisation of the palladium catalyst during a hydrogenation reaction resulted in reduction and cyclisation to give 3,4-dihydroquinolin-2-one 13 in 79% overall yield. N-Methylation under standard conditions then gave 14 in 92% yield. Iodination of 14 was found to proceed with lower catalyst loadings of both iron(III) chloride (2.5 mol%) and [BMIM]NTf2 (7.5 mol%), under milder conditions (40 °C) than the general process. In addition, despite the potential substitution of either ortho-site, only iodination of the 6-position was observed by 1H NMR analysis of the reaction mixture. Subsequent copper(I)-catalysed coupling with benzenesulfonamide completed the one-pot transformation and gave TRIM24 inhibitor, 16a in 56% yield. Application of the one-pot ortho-amidation of 14 for the late-stage synthesis of novel analogues of 16a was then explored. Using other sulfonamides, benzamides and heterocyclic nucleophiles gave coupled products 16b–16f in good overall yields (51–61%). Overall, the utilisation of both one-pot processes allowed rapid access to a range of novel pharmaceutically relevant targets.
 | ||
| Scheme 3 Synthesis and late-stage functionalisation of 3,4-dihydroquinolin-2-one 14. aThe second step required 72 h. | ||
Conclusions
In summary, a one-pot method for the ortho-amination and amidation of arenes has been developed using sequential iron and copper catalysis. Regioselective activation of arenes via iodination, using NIS and the super Lewis acid, iron(III) triflimide, followed by copper(I)-catalysed, ligand-assisted coupling with a range of N-nucleophiles gave the ortho-substituted products in good yields. As well as allowing access to a range of anisole adducts, this approach also permits direct ortho-amination of unprotected anilines, without the requirement of a directing auxiliary group. The selective and mild nature of this method was further demonstrated by the late-stage synthesis of a quinolin-2-one inhibitor of the bromodomain containing protein TRIM24, as well as a series of novel analogues. Further applications of one-pot iron and copper-catalysed arene substitution reactions are currently being investigated.Experimental
All reagents and starting materials were obtained from commercial sources and used as received. The synthesis of compounds 1324 and 1425 were previously described in the literature. All dry solvents were purified using a solvent purification system. All reactions were performed in oven-dried glassware under an atmosphere of argon unless otherwise stated. Brine refers to a saturated solution of sodium chloride. Flash column chromatography was performed using silica gel 60 (40–63 μm) and neutral aluminium oxide (50–200 μm). Aluminium-backed plates pre-coated with silica gel 60F254 were used for thin layer chromatography and were visualised with a UV lamp or by staining with potassium permanganate. 1H NMR spectra were recorded on a NMR spectrometer at either 400 or 500 MHz and data are reported as follows: chemical shift in ppm relative to tetramethylsilane or the solvent as the internal standard (CDCl3, δ 7.26 ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet or overlap of nonequivalent resonances, integration). 13C NMR spectra were recorded on an NMR spectrometer at either 101 or 126 MHz and data are reported as follows: chemical shift in ppm relative to tetramethylsilane or the solvent as internal standard (CDCl3, δ 77.0 ppm), multiplicity with respect to hydrogen (deduced from DEPT experiments, C, CH, CH2 or CH3). IR spectra were recorded on a FTIR spectrometer; wavenumbers are indicated in cm−1. Mass spectra were recorded using electrospray or electron impact techniques. HRMS spectra were recorded using a dual-focusing magnetic analyser mass spectrometer. Melting points are uncorrected.
N-(2-Amino-5-cyanophenyl)-1H-pyrazole (3a)26
Iron(III) chloride (4.0 mg, 0.025 mmol) was dissolved in 1-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide (22 μL, 0.075 mmol) and stirred for 0.5 h at room temperature and then added to a suspension of N-iodosuccinimide (0.11 g, 0.50 mmol) in toluene (0.5 mL). 4-Aminobenzonitrile (1a) (0.059 g, 0.50 mmol) was added and the mixture was stirred at 70 °C for 5 h. Upon completion of the iodination step, the reaction mixture was cooled to room temperature and pyrazole (0.10 g, 1.5 mmol), copper(I) iodide (0.0095 g, 0.050 mmol), caesium carbonate (0.33 g, 1.0 mmol), trans-N,N′-dimethylcyclohexane-1,2-diamine (6) (16 μL, 0.10 mmol) and water (0.25 mL) were added. The reaction mixture was degassed under argon for 0.1 h and then heated to 130 °C for 24 h. The reaction mixture was cooled to room temperature, diluted with ethyl acetate (10 mL), washed with a 1 M aqueous sodium thiosulfate solution (10 mL). The aqueous layer was extracted with ethyl acetate (3 × 10 mL) and the combined organic layers were washed with brine (10 mL). The organic phase was dried (MgSO4), filtered and concentrated in vacuo. Purification by flash column chromatography (petroleum ether/ethyl acetate, 7:3) gave N-(2-amino-5-cyanophenyl)-1H-pyrazole (3a) (0.055 g, 60%) as a colourless oil. Spectroscopic data were consistent with the literature.26δH (500 MHz, CDCl3) 5.45 (2H, br s, NH2), 6.50 (1H, t, J 2.2 Hz, 4′-H), 6.81 (1H, d, J 8.4 Hz, 3-H), 7.39 (1H, dd, J 8.4, 1.9 Hz, 4-H), 7.47 (1H, d, J 1.9 Hz, 6-H), 7.75 (1H, d, J 2.2 Hz, ArH), 7.77 (1H, d, J 2.2 Hz, ArH); δC (126 MHz, CDCl3) 99.7 (C), 107.2 (CH), 117.0 (CH), 119.2 (C), 125.5 (C), 127.3 (CH), 129.7 (CH), 132.2 (CH), 141.2 (CH), 145.0 (C); m/z (ESI) 207 (MNa+. 100%).
N-(2-Amino-5-nitrophenyl)-1H-pyrazole (3b)
N-(2-Amino-5-nitrophenyl)-1H-pyrazole (3b) was synthesised as described for N-(2-amino-5-cyanophenyl)-1H-pyrazole (3a) using 4-nitroaniline (1b) (0.070 g, 0.50 mmol) and pyrazole (0.10 g, 1.5 mmol). The iodination step was carried out at 70 °C for 5 h and the N-arylation step at 130 °C for 24 h. Purification by flash column chromatography (petroleum ether/ethyl acetate, 4:1) gave N-(2-amino-5-nitrophenyl)-1H-pyrazole (3b) (0.073 g, 71%) as a yellow solid. Mp 137–139 °C; νmax/cm−1 (neat) 3319 (NH), 3080 (CH), 1633, 1614, 1527 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1494, 1333, 1316, 707; δH (500 MHz, CDCl3) 5.84 (2H, br s, NH2), 6.52 (1H, t, J 2.0 Hz, 4′-H), 6.80 (1H, d, J 8.9 Hz, 3-H), 7.78 (1H, d, J 2.0 Hz, ArH), 7.86 (1H, d, J 2.0 Hz, ArH), 8.05 (1H, dd, J 8.9, 2.5 Hz, 4-H), 8.17 (1H, d, J 2.5 Hz, 6-H); δC (126 MHz, CDCl3) 107.4 (CH), 115.8 (CH), 119.4 (CH), 124.2 (C), 124.4 (CH), 129.8 (CH), 138.1 (C), 141.2 (CH), 146.8 (C); m/z (EI) 204.0654 (M+. C9H8N4O2 requires 204.0647), 174 (20%), 159 (19), 149 (20), 131 (15), 83 (100), 77 (23), 69 (28), 57 (46).
C), 1494, 1333, 1316, 707; δH (500 MHz, CDCl3) 5.84 (2H, br s, NH2), 6.52 (1H, t, J 2.0 Hz, 4′-H), 6.80 (1H, d, J 8.9 Hz, 3-H), 7.78 (1H, d, J 2.0 Hz, ArH), 7.86 (1H, d, J 2.0 Hz, ArH), 8.05 (1H, dd, J 8.9, 2.5 Hz, 4-H), 8.17 (1H, d, J 2.5 Hz, 6-H); δC (126 MHz, CDCl3) 107.4 (CH), 115.8 (CH), 119.4 (CH), 124.2 (C), 124.4 (CH), 129.8 (CH), 138.1 (C), 141.2 (CH), 146.8 (C); m/z (EI) 204.0654 (M+. C9H8N4O2 requires 204.0647), 174 (20%), 159 (19), 149 (20), 131 (15), 83 (100), 77 (23), 69 (28), 57 (46).
3-Pyrazol-1′-yl-4-aminobenzophenone (3c)
3-Pyrazol-1′-yl-4-aminobenzophenone (3c) was synthesised as described for N-(2-amino-5-cyanophenyl)-1H-pyrazole (3a) using 4-aminobenzophenone (1c) (0.059 g, 0.50 mmol) and pyrazole (0.10 g, 1.5 mmol). The iodination step was carried out at 70 °C for 20 h and the N-arylation step at 130 °C for 24 h. Purification by flash column chromatography (dichloromethane/petroleum ether, 9:1 to dichloromethane/petroleum ether, 4:1) gave 3-pyrazol-1′-yl-4-aminobenzophenone (3c) (0.076 g, 64%) as a colourless oil. νmax/cm−1 (neat) 3360 (NH), 1611 (CO), 1518, 1396, 1269, 1148; δH (500 MHz, CDCl3) 5.44 (2H, br s, NH2), 6.47 (1H, t, J 2.2 Hz, 4′-H), 6.82 (1H, d, J 8.4 Hz, 5-H), 7.44–7.50 (2H, m, 3′′-H and 5′′-H), 7.56 (1H, tt, J 7.4, 1.3 Hz, 4′′-H), 7.65 (1H, dd, J 8.4, 1.9 Hz, 6-H), 7.72–7.77 (3H, m, 2-H, 2′′-H and 6′′-H), 7.79 (1H, d, J 2.2 Hz, ArH), 7.81 (1H, d, J 2.2 Hz, ArH); δC (126 MHz, CDCl3) 106.8 (CH), 115.9 (CH), 125.3 (C), 126.1 (CH), 126.9 (C), 128.3 (2 × CH), 129.5 (2 × CH), 129.9 (CH), 131.5 (CH), 131.7 (CH), 138.4 (C), 140.8 (CH), 145.3 (C), 194.6 (C); m/z (ESI) 286.0944 (MNa+. C16H13N3NaO requires 286.0951).
N-(2-Amino-5-chlorophenyl)-1H-pyrazole (3d)26
N-(2-Amino-5-chlorophenyl)-1H-pyrazole (3d) was synthesised as described for N-(2-amino-5-cyanophenyl)-1H-pyrazole (3a) using 4-chloroaniline (1d) (0.064 g, 0.50 mmol) and pyrazole (0.10 g, 1.5 mmol). The iodination step was carried out at 70 °C for 5 h and the N-arylation step at 130 °C for 24 h. Purification by flash column chromatography (dichloromethane) gave N-(2-amino-5-chlorophenyl)-1H-pyrazole (3d) (0.061 g, 63%) as a brown oil. Spectroscopic data were consistent with the literature.26δH (500 MHz, CDCl3) 4.75 (2H, br s, NH2), 6.44 (1H, t, J 2.2 Hz, 4′-H), 6.74 (1H, d, J 8.5 Hz, 3-H), 7.08 (1H, dd, J 8.5, 2.4 Hz, 4-H), 7.18 (1H, d, J 2.4 Hz, 6-H), 7.70 (1H, d, J 2.2 Hz, ArH), 7.73 (1H, d, J 2.2 Hz, ArH); δC (126 MHz, CDCl3) 106.8 (CH), 118.2 (CH), 122.2 (C), 123.7 (CH), 126.8 (C), 128.2 (CH), 129.8 (CH), 139.7 (C), 140.9 (CH); m/z (ESI) 216 (MNa+. 100%).N-(2-Amino-5-methylphenyl)-1H-pyrazole (3e)26
N-(2-Amino-5-methylphenyl)-1H-pyrazole (3e) was synthesised as described for N-(2-amino-5-cyanophenyl)-1H-pyrazole (3a) using 4-methylaniline (1e) (0.054 g, 0.50 mmol) and pyrazole (0.10 g, 1.5 mmol). The iodination step was carried out at 40 °C for 5 h and the N-arylation step at 130 °C for 24 h. Purification by flash column chromatography (petroleum ether/ethyl acetate, 7:3) gave N-(2-amino-5-methylphenyl)-1H-pyrazole (3e) (0.051 g, 58%) as an orange oil. Spectroscopic data were consistent with the literature.26δH (400 MHz, CDCl3) 2.27 (3H, s, 5-CH3), 4.48 (2H, br s, NH2), 6.42 (1H, t, J 2.1 Hz, 4′-H), 6.74 (1H, d, J 8.1 Hz, 3-H), 6.96 (1H, dd, J 8.1, 1.3 Hz, 4-H), 7.00 (1H, d, J 1.3 Hz, 6-H), 7.70 (1H, d, J 2.1 Hz, ArH), 7.73 (1H, d, J 2.1 Hz, ArH); δC (101 MHz, CDCl3) 20.3 (CH3), 106.3 (CH), 117.4 (CH), 124.7 (CH), 126.6 (C), 127.6 (C), 129.1 (CH), 129.8 (CH), 138.5 (C), 140.5 (CH); m/z (ESI) 174 (MNa+. 100%).
N-(2-Methoxy-5-methylphenyl)-1H-pyrazole (3f)
N-(2-Methoxy-5-methylphenyl)-1H-pyrazole (3f) was synthesised as described for N-(2-amino-5-cyanophenyl)-1H-pyrazole (3a) using 4-methylanisole (1f) (0.063 mL, 0.50 mmol) and pyrazole (0.10 g, 1.5 mmol). The iodination step was carried out at 70 °C for 20 h and the N-arylation step at 130 °C for 24 h. Purification by flash column chromatography (hexane/diethyl ether, 4:1) gave N-(2-methoxy-5-methylphenyl)-1H-pyrazole (3f) (0.067 g, 71%) as a colourless oil. νmax/cm−1 (neat) 2936 (CH), 1524 (CC), 1504, 1462, 1283, 1242, 1036, 804, 744; δH (400 MHz, CDCl3) 2.33 (3H, br s, 5-CH3), 3.83 (3H, s, OCH3), 6.41 (1H, dd, J 2.4, 1.9 Hz, 4′-H), 6.92 (1H, d, J 8.4 Hz, 3-H), 7.05–7.10 (1H, m, 4-H), 7.55 (1H, d, J 2.3 Hz, 6-H), 7.69 (1H, dd, J 2.4, 0.4 Hz, ArH), 8.03 (1H, dd, J 1.9, 0.4 Hz, ArH); δC (101 MHz, CDCl3) 20.4 (CH3), 56.1 (CH3), 106.1 (CH), 112.3 (CH), 125.6 (CH), 128.3 (CH), 129.4 (C), 130.8 (C), 131.5 (CH), 139.9 (CH), 149.1 (C); m/z (EI) 188.0942 (M+. C11H12N2O requires 188.0950), 159 (55%), 144 (34).
N-(2-Methoxy-4,5-dimethylphenyl)-1H-pyrazole (3g)
N-(2-Methoxy-4,5-dimethylphenyl)-1H-pyrazole (3g) was synthesised as described for N-(2-amino-5-cyanophenyl)-1H-pyrazole (3a) using 3,4-dimethylanisole (1g) (0.070 mL, 0.50 mmol) and pyrazole (0.10 g, 1.5 mmol). The iodination step was carried out at 70 °C for 5 h and the N-arylation step at 130 °C for 24 h. Purification by flash column chromatography (hexane/diethyl ether, 9:1) gave N-(2-methoxy-4,5-dimethylphenyl)-1H-pyrazole (3g) (0.068 g, 67%) as a colourless oil. νmax/cm−1 (neat) 2932 (CH), 1520, 1466, 1396, 1242, 1188, 1034, 748; δH (400 MHz, CDCl3) 2.23 (3H, s, CH3), 2.29 (3H, s, CH3), 3.82 (3H, s, OCH3), 6.39 (1H, dd, J 2.3, 1.7 Hz, 4′-H), 6.81 (1H, s, 3-H), 7.47 (1H, s, 6-H), 7.67 (1H, d, J 1.7 Hz, ArH), 7.97 (1H, d, J 2.3 Hz, ArH); δC (101 MHz, CDCl3) 18.7 (CH3), 19.9 (CH3), 56.1 (CH3), 105.9 (CH), 113.9 (CH), 126.1 (CH), 127.2 (C), 129.2 (C), 131.4 (CH), 136.4 (C), 139.7 (CH), 149.1 (C); m/z (ESI) 225.0994 (MNa+. C12H14N2NaO requires 225.0998).
3-Pyrazol-1′-yl-4-methoxybiphenyl (3h)
3-Pyrazol-1′-yl-4-methoxybiphenyl (3h) was synthesised as described for N-(2-amino-5-cyanophenyl)-1H-pyrazole (3a) using 4-methoxybiphenyl (1h) (0.050 g, 0.19 mmol) and pyrazole (0.051 g, 0.75 mmol). The iodination step was carried out at 70 °C for 5 h and the N-arylation step at 130 °C for 24 h. Purification by flash column chromatography (hexane/diethyl ether, 7:3) gave 3-pyrazol-1′-yl-4-methoxybiphenyl (3h) (0.077 g, 61%) as a white solid. Mp 52–54 °C; νmax/cm−1 (neat) 2940 (CH), 1528 (CC), 1489, 1404, 1281, 1250, 1018, 756; δH (500 MHz, CDCl3) 3.92 (3H, s, OCH3), 6.45 (1H, dd, J 2.3, 1.8 Hz, 4′-H), 7.11 (1H, d, J 8.6 Hz, 5-H), 7.32 (1H, tt, J 7.4, 1.2 Hz, 4′′-H), 7.38–7.44 (2H, m, 3′′-H and 5′′-H), 7.52 (1H, dd, J 8.6, 2.4 Hz, 6-H), 7.58–7.63 (2H, m, 2′′-H and 6′′-H), 7.73 (1H, d, J 1.8 Hz, ArH), 7.99 (1H, d, J 2.4 Hz, 2-H), 8.08 (1H, d, J 2.3 Hz, ArH); δC (126 MHz, CDCl3) 56.1 (CH3), 106.3 (CH), 112.7 (CH), 123.9 (CH), 126.3 (CH), 126.8 (2 × CH), 127.1 (CH), 128.8 (2 × CH), 129.9 (C), 131.6 (CH), 134.5 (C), 139.8 (C), 140.2 (CH), 150.7 (C); m/z (ESI) 273.0992 (MNa+. C16H14N2NaO requires 273.0998).
N-(2,5-Dimethoxyphenyl)-1H-pyrazole (3i)
N-(2,5-Dimethoxyphenyl)-1H-pyrazole (3i) was synthesised as described for N-(2-amino-5-cyanophenyl)-1H-pyrazole (3a) using 1,4-dimethoxybenzene (1i) (0.069 g, 0.50 mmol) and pyrazole (0.10 g, 1.5 mmol). The iodination step was carried out at 70 °C for 5 h and the N-arylation step at 150 °C for 24 h. Purification by flash column chromatography (petroleum ether/ethyl acetate, 9:1) gave N-(2,5-dimethoxyphenyl)-1H-pyrazole (3i) (0.067 g, 66%) as a colourless oil. νmax/cm−1 (neat) 2940 (CH), 1597, 1520, 1504, 1211, 1042, 748; δH (400 MHz, CDCl3) 3.82 (6H, br s, 2 × OCH3), 6.42 (1H, dd, J 2.4, 1.9 Hz, 4′-H), 6.83 (1H, dd, J 9.0, 3.1 Hz, 4-H), 6.98 (1H, d, J 9.0 Hz, 3-H), 7.36 (1H, d, J 3.1 Hz, 6-H), 7.70 (1H, d, J 1.9 Hz, ArH), 8.10 (1H, d, J 2.4 Hz, ArH); δC (101 MHz, CDCl3) 55.9 (CH3), 56.7 (CH3), 106.3 (CH), 110.0 (CH), 113.6 (CH), 114.0 (CH), 130.2 (C), 131.6 (CH), 140.1 (CH), 145.1 (C), 154.1 (C); m/z (ESI) 227.0785 (MNa+. C11H12N2NaO2 requires 227.0791).
2′′-(4-Methoxy-3-pyrazol-1′-ylphenyl)acetic acid (3j)
2′′-(4-Methoxy-3-pyrazol-1′-ylphenyl)acetic acid (3j) was synthesised as described for N-(2-amino-5-cyanophenyl)-1H-pyrazole (3a) using 4-methoxyphenylacetic acid (1j) (0.083 g, 0.50 mmol) and pyrazole (0.10 g, 1.5 mmol). The iodination step was carried out at 70 °C for 5 h and the N-arylation step at 150 °C for 24 h. The reaction mixture was cooled to room temperature, diluted with dichloromethane (10 mL) and extracted with 1 M aqueous sodium hydroxide solution (10 mL). The aqueous layer was separated and acidified with 1 M aqueous hydrochloric acid and extracted into dichloromethane (3 × 30 mL). The combined organic layers were dried (MgSO4) and concentrated in vacuo. Trituration with hexane gave 2′′-(4-methoxy-3-pyrazol-1′-ylphenyl)acetic acid (3j) (0.062 g, 53%) as a white solid. Mp 94–96 °C; νmax/cm−1 (neat) 2940 (CH), 1713 (CO), 1526 (CC), 1462, 1410, 1287, 1248, 1180, 1152, 1022, 760; δH (400 MHz, CDCl3) 3.62 (2H, s, CH2), 3.86 (3H, s, OCH3), 6.42 (1H, dd, J 2.3, 1.8 Hz, 4′-H), 6.99 (1H, d, J 8.5 Hz, 5-H), 7.21 (1H, dd, J 8.5, 2.1 Hz, 6-H), 7.67 (1H, d, J 2.1 Hz, 2-H), 7.73 (1H, d, J 1.8 Hz, ArH), 8.02 (1H, d, J 2.3 Hz, ArH); δC (101 MHz, CDCl3) 40.1 (CH2), 56.0 (CH3), 106.3 (CH), 112.4 (CH), 126.4 (CH), 126.9 (C), 129.1 (CH), 129.2 (C), 131.9 (CH), 139.9 (CH), 150.2 (C), 175.5 (C); m/z (ESI) 255.0738 (MNa+. C12H12N2NaO3 requires 255.0740).
N-(5-Acetyl-2,4-dimethoxyphenyl)-1H-pyrazole (3k)
N-(5-Acetyl-2,4-dimethoxyphenyl)-1H-pyrazole (3k) was synthesised as described for N-(2-amino-5-cyanophenyl)-1H-pyrazole (3a) using 2,4-dimethoxyacetophenone (1k) (0.090 g, 0.50 mmol) and pyrazole (0.10 g, 1.5 mmol). The iodination step was carried out at 70 °C for 5 h and the N-arylation step at 150 °C for 24 h. Purification by flash column chromatography (petroleum ether/ethyl acetate, 4:1) followed by trituration with diethyl ether gave N-(5-acetyl-2,4-dimethoxyphenyl)-1H-pyrazole (3k) (0.059 g, 48%) as a white solid. Mp 122–124 °C; νmax/cm−1 (neat) 2931 (CH), 1659 (CO), 1605, 1520, 1265, 1227, 1026, 741; δH (500 MHz, CDCl3) 2.60 (3H, s, COMe), 3.93 (3H, s, OCH3), 3.99 (3H, s, OCH3), 6.41 (1H, dd, J 2.3, 1.8 Hz, 4′-H), 6.57 (1H, s, 3-H), 7.69 (1H, d, J 1.8 Hz, ArH), 7.78 (1H, d, J 2.3 Hz, ArH), 8.08 (1H, s, 6-H); δC (126 MHz, CDCl3) 31.7 (CH3), 56.0 (CH3), 56.2 (CH3), 95.8 (CH), 106.1 (CH), 120.6 (C), 123.4 (C), 128.8 (CH), 131.3 (CH), 140.2 (CH), 156.7 (C), 159.9 (C), 196.8 (C); m/z (ESI) 269.0889 (MNa+. C13H14N2NaO3 requires 269.0897).
N-(2-Hydroxy-5-methylphenyl)-1H-pyrazole (3l)
An oven-dried microwave vial was flushed with argon and charged with N-iodosuccinimide (0.11 g, 0.50 mmol) and dry toluene (3 mL). To this suspension was added silver bis(trifluoromethanesulfonyl)imide (0.015 g, 0.038 mmol) and p-cresol (1l) (0.053 mL, 0.50 mmol). The reaction mixture was stirred at 40 °C for 5 h in the dark. The reaction mixture was cooled to room temperature and pyrazole (0.10 g, 1.5 mmol), copper(I) iodide (0.0095 g, 0.050 mmol), caesium carbonate (0.33 g, 1.0 mmol), trans-N,N′-dimethylcyclohexane-1,2-diamine (6) (0.16 μL, 0.10 mmol) and water (0.5 mL) were added. The reaction mixture was degassed under argon for 0.1 h and then heated to 130 °C for 24 h. The reaction mixture was then cooled to room temperature, diluted with ethyl acetate (10 mL), washed with a 1 M aqueous sodium thiosulfate solution (10 mL). The aqueous layer was extracted with ethyl acetate (3 × 10 mL) and the combined organic layers were washed with brine (10 mL). The organic phase was dried (MgSO4), filtered and concentrated in vacuo. Purification by flash column chromatography (hexane/diethyl ether, 4:1) gave N-(2-hydroxy-5-methylphenyl)-1H-pyrazole (3l) (0.032 g, 37%) as a colourless oil. νmax/cm−1 (neat) 3036 (OH), 2920 (CH), 1518 (CC), 1333, 1279, 1250, 750; δH (400 MHz, CDCl3) 2.33 (3H, s, 5-CH3), 6.49 (1H, dd, J 2.4, 2.1 Hz, 4′-H), 6.95–7.01 (2H, m, 3-H and 4-H), 7.18 (1H, br s, 6-H), 7.72 (1H, d, J 2.1 Hz, ArH), 7.99 (1H, d, J 2.4 Hz, ArH), 11.11 (1H, s, OH); δC (101 MHz, CDCl3) 20.6 (CH3), 106.7 (CH), 118.3 (CH), 118.7 (CH), 124.5 (C), 126.6 (CH), 128.2 (CH), 128.9 (C), 138.9 (CH), 147.0 (C); m/z (ESI) 197.0684 (MNa+. C10H10N2NaO requires 197.0685).
N-(2-Methoxy-5-methylphenyl)benzenesulfonamide (3m)
N-(2-Methoxy-5-methylphenyl)benzenesulfonamide (3m) was synthesised as described for N-(2-amino-5-cyanophenyl)-1H-pyrazole (3a) using 4-methylanisole (1f) (0.063 mL, 0.50 mmol) and benzenesulfonamide (0.24 g, 1.5 mmol). The iodination step was carried out at 70 °C for 5 h and the N-arylation step at 150 °C for 24 h. Purification by flash column chromatography (hexane/diethyl ether, 1:1) gave N-(2-methoxy-5-methylphenyl)benzenesulfonamide (3m) (0.084 g, 61%) as a white solid. Mp 134–136 °C; νmax/cm−1 (neat) 3011 (CH), 1506, 1389, 1327, 1256, 1165, 1090, 1030, 907, 727; δH (400 MHz, CDCl3) 2.25 (3H, s, 5-CH3), 3.54 (3H, s, OCH3), 6.59 (1H, d, J 8.3 Hz, 3-H), 6.82 (1H, dd, J 8.3, 1.6 Hz, 4-H), 6.98 (1H, br s, NH), 7.34 (1H, d, J 1.6 Hz, 6-H), 7.36–7.43 (2H, m, 3′-H and 5′-H), 7.48 (1H, t, J 7.4 Hz, 4′-H), 7.71–7.77 (2H, m, 2′-H and 6′-H); δC (101 MHz, CDCl3) 20.7 (CH3), 55.7 (CH3), 110.5 (CH), 122.4 (CH), 125.4 (C), 125.9 (CH), 127.2 (2 × CH), 128.7 (2 × CH), 130.6 (C), 132.8 (CH), 139.2 (C), 147.7 (C); m/z (ESI) 300.0653 (MNa+. C14H15NNaO3S requires 300.0665).
N-(2-Methoxy-5-methylphenyl)-4′-methylbenzenesulfonamide (3n)
N-(2-Methoxy-5-methylphenyl)-4′-methylbenzenesulfonamide (3n) was synthesised as described for N-(2-amino-5-cyanophenyl)-1H-pyrazole (3a) using 4-methylanisole (1f) (0.063 mL, 0.50 mmol) and p-toluenesulfonamide (0.26 g, 1.5 mmol). The iodination step was carried out at 70 °C for 5 h and the N-arylation step at 150 °C for 24 h. Purification by flash column chromatography (hexane/diethyl ether, 1:1) gave N-(2-methoxy-5-methylphenyl)-4′-methylbenzenesulfonamide (3n) (0.113 g, 78%) as a white solid. Mp 68–70 °C; νmax/cm−1 (neat) 3400 (NH), 2936 (CH), 1595 (CC), 1508, 1389, 1330, 1252, 1163, 1123, 1090, 808; δH (400 MHz, CDCl3) 2.25 (3H, s, 4′-CH3), 2.35 (3H, s, 5-CH3), 3.58 (3H, s, OCH3), 6.60 (1H, d, J 8.4 Hz, 3-H), 6.81 (1H, dd, J 8.4, 2.1 Hz, 4-H), 6.95 (1H, br s, NH), 7.18 (2H, d, J 8.2 Hz, 3′-H and 5′-H), 7.34 (1H, d, J 2.1 Hz, 6-H), 7.63 (2H, d, J 8.2 Hz, 2′-H and 6′-H); δC (101 MHz, CDCl3) 20.8 (CH3), 21.5 (CH3), 55.7 (CH3), 110.5 (CH), 121.8 (CH), 125.6 (CH), 125.7 (C), 127.2 (2 × CH), 129.3 (2 × CH), 130.6 (C), 136.4 (C), 143.5 (C), 147.5 (C); m/z (ESI) 314.0809 (MNa+. C15H17NNaO3S requires 314.0821).
N-(2-Methoxy-5-methylphenyl)-4′-chlorobenzenesulfonamide (3o)
N-(2-Methoxy-5-methylphenyl)-4′-chlorobenzenesulfonamide (3o) was synthesised as described for N-(2-amino-5-cyanophenyl)-1H-pyrazole (3a) using 4-methylanisole (1f) (0.063 mL, 0.50 mmol) and p-chlorobenzenesulfonamide (0.29 g, 1.5 mmol). The iodination step was carried out at 70 °C for 5 h and the N-arylation step at 150 °C for 24 h. Purification by flash column chromatography (hexane/diethyl ether, 1:1) gave N-(2-methoxy-5-methylphenyl)-4′-chlorobenzenesulfonamide (3o) (0.089 g, 55%) as a yellow oil. νmax/cm−1 (neat) 3273 (NH), 2932 (CH), 1585 (CC), 1508, 1335, 1252, 1165, 1123, 1086, 752; δH (400 MHz, CDCl3) 2.27 (3H, s, 5-CH3), 3.58 (3H, s, OCH3), 6.61 (1H, d, J 8.3 Hz, 3-H), 6.85 (1H, dd, J 8.3, 2.0 Hz, 4-H), 6.97 (1H, br s, NH), 7.32–7.38 (3H, m, 6-H, 3′-H and 5′-H), 7.67 (2H, d, J 8.9 Hz, 2′-H and 6′-H); δC (101 MHz, CDCl3) 20.7 (CH3), 55.7 (CH3), 110.5 (CH), 122.6 (CH), 125.0 (C), 126.3 (CH), 128.7 (2 × CH), 129.0 (2 × CH), 130.7 (C), 137.7 (C), 139.2 (C), 147.8 (C); m/z (ESI) 334.0265 (MNa+. C14H1435ClNNaO3S requires 334.0275).
N-(2-Methoxy-5-methylphenyl)methanesulfonamide (3p)
N-(2-Methoxy-5-methylphenyl)methanesulfonamide (3p) was synthesised as described for N-(2-amino-5-cyanophenyl)-1H-pyrazole (3a) using 4-methylanisole (1f) (0.063 mL, 0.50 mmol) and methanesulfonamide (0.14 g, 1.5 mmol). The iodination step was carried out at 70 °C for 5 h and the N-arylation step at 150 °C for 24 h. Purification by flash column chromatography (petroleum ether/ethyl acetate, 7:3) gave N-(2-methoxy-5-methylphenyl)methanesulfonamide (3p) (0.061 g, 57%) as a colourless oil. νmax/cm−1 (neat) 3267 (NH), 2934 (CH), 1508 (CC), 1387, 1323, 1252, 1161, 1121, 1028, 970, 760; δH (500 MHz, CDCl3) 2.30 (3H, s, 5-CH3), 2.94 (3H, s, SO2CH3), 3.85 (3H, s, OCH3), 6.74 (1H, br s, NH), 6.80 (1H, d, J 8.3 Hz, 3-H), 6.92 (1H, dd, J 8.3, 1.9 Hz, 4-H), 7.33 (1H, d, J 1.9 Hz, 6-H); δC (126 MHz, CDCl3) 20.8 (CH3), 39.0 (CH3), 55.9 (CH3), 110.6 (CH), 121.7 (CH), 125.7 (C), 125.9 (CH), 131.0 (C), 147.5 (C); m/z (ESI) 238.0502 (MNa+. C9H13NNaO3S requires 238.0508).
N-(2-Methoxy-5-methylphenyl)benzamide (3q)27
N-(2-Methoxy-5-methylphenyl)benzamide (3q) was synthesised as described for N-(2-amino-5-cyanophenyl)-1H-pyrazole (3a) using 4-methylanisole (1f) (0.063 mL, 0.50 mmol) and benzamide (0.18 g, 1.5 mmol). The iodination step was carried out at 70 °C for 5 h and the N-arylation step at 150 °C for 24 h. Purification by flash column chromatography (hexane/diethyl ether, 4:1) gave N-(2-methoxy-5-methylphenyl)benzamide (3q) (0.091 g, 75%) as a colourless oil. Spectroscopic data were consistent with the literature.27δH (500 MHz, CDCl3) 2.33 (3H, s, 5-CH3), 3.87 (3H, s, OCH3), 6.78 (1H, d, J 8.3 Hz, 3-H), 6.86 (1H, dd, J 8.3 Hz, 2.1 Hz, 4-H), 7.44–7.55 (3H, m, 3′-H, 4′-H and 5′-H), 7.86–7.90 (2H, m, 2′-H and 6′-H), 8.38 (1H, d, J 2.1 Hz, 6-H), 8.52 (1H, br s, NH); δC (126 MHz, CDCl3) 21.0 (CH3), 55.9 (CH3), 109.8 (CH), 120.5 (CH), 124.1 (CH), 127.0 (2 × CH), 127.5 (C), 128.7 (2 × CH), 130.6 (C), 131.6 (CH), 135.3 (C), 146.1 (C), 165.1 (C); m/z (ESI) 264 (MNa+. 100%).
N-(2-Methoxy-5-methylphenyl)-4′-methylbenzamide (3r)
N-(2-Methoxy-5-methylphenyl)-4′-methylbenzamide (3r) was synthesised as described for N-(2-amino-5-cyanophenyl)-1H-pyrazole (3a) using 4-methylanisole (1f) (0.063 mL, 0.50 mmol) and p-toluamide (0.10 g, 1.5 mmol). The iodination step was carried out at 70 °C for 5 h and the N-arylation step at 150 °C for 24 h. Purification by flash column chromatography (hexane/diethyl ether, 4:1) gave N-(2-methoxy-5-methylphenyl)-4′-methylbenzamide (3r) (0.098 g, 77%) as a white solid. Mp 56–58 °C; νmax/cm−1 (neat) 3429 (NH), 2920 (CH), 1670 (CO), 1530 (CC), 1477, 1423, 1248, 1223, 1138, 1030, 799; δH (400 MHz, CDCl3) 2.33 (3H, s, 5-CH3), 2.41 (3H, s, 4′-CH3), 3.87 (3H, s, OCH3), 6.78 (1H, d, J 8.2 Hz, 3-H), 6.85 (1H, dd, J 8.2, 2.0 Hz, 4-H), 7.27 (2H, d, J 8.1 Hz, 3′-H and 5′-H), 7.78 (2H, d, J 8.1 Hz, 2′-H and 6′-H), 8.38 (1H, d, J 2.0 Hz, 6-H), 8.50 (1H, br s, NH); δC (101 MHz, CDCl3) 21.0 (CH3), 21.5 (CH3), 55.9 (CH3), 109.8 (CH), 120.4 (CH), 123.9 (CH), 127.0 (2 × CH), 127.6 (C), 129.4 (2 × CH), 130.6 (C), 132.5 (C), 142.1 (C), 146.1 (C), 165.1 (C); m/z (ESI) 278.1146 (MNa+. C16H17NNaO2 requires 278.1151).
N-(2-Methoxy-5-methylphenyl)pyrrolidin-2′-one (3s)28
N-(2-Methoxy-5-methylphenyl)pyrrolidin-2′-one (3s) was synthesised as described for N-(2-amino-5-nitrophenyl)-1H-pyrazole (3a) using 4-methylanisole (1f) (0.063 mL, 0.50 mmol) and pyrrolidin-2-one (0.11 mL, 1.5 mmol). The iodination step was carried out at 70 °C for 5 h and the N-arylation step at 150 °C for 24 h. Purification by flash column chromatography (ethyl acetate) gave N-(2-methoxy-5-methylphenyl)pyrrolidin-2′-one (3s) (0.053 g, 52%) as a colourless oil. Spectroscopic data were consistent with the literature.28δH (500 MHz, CDCl3) 2.12–2.20 (2H, m, 4′-H2), 2.28 (3H, s, 5-CH3), 2.54 (2H, t, J 7.8 Hz, 3′-H2) 3.73 (2H, t, J 7.0 Hz, 5′-H2), 3.79 (3H, s, OCH3), 6.84 (1H, d, J 8.9 Hz, 3-H), 7.04–7.07 (2H, m, 4-H and 6-H); δC (126 MHz, CDCl3) 18.9 (CH2), 20.4 (CH3), 31.2 (CH2), 50.0 (CH2), 55.8 (CH3), 112.1 (CH), 126.9 (C), 129.1 (CH), 129.2 (CH), 130.4 (C), 152.7 (C), 175.2 (C); m/z (ESI) 228 (MNa+. 100%).N-(2-Methoxy-5-methylphenyl)imidazole (3t)29
N-(2-Methoxy-5-methylphenyl)imidazole (3t) was synthesised as described for N-(2-amino-5-cyanophenyl)-1H-pyrazole (3a) using 4-methylanisole (1f) (0.063 mL, 0.50 mmol) and imidazole (0.10 g, 1.5 mmol). The iodination step was carried out at 70 °C for 5 h and the N-arylation step at 150 °C for 36 h. Purification by flash column chromatography (ethyl acetate/methanol, 9:1) gave N-(2-methoxy-5-methylphenyl)imidazole (3t) (0.049 g, 51%) as a colourless oil. Spectroscopic data were consistent with the literature.29δH (500 MHz, CDCl3) 2.33 (3H, s, 5-CH3), 3.80 (3H, s, OCH3), 6.94 (1H, d, J 8.4 Hz, 3-H), 7.09 (1H, d, J 1.8 Hz, 6-H), 7.14 (1H, dd, J 8.4, 1.8 Hz, 4-H), 7.15–7.24 (2H, m, 5′-H and 4′-H), 7.79 (1H, br s, 2′-H); δC (126 MHz, CDCl3) 20.4 (CH3), 55.9 (CH3), 112.4 (CH), 120.3 (CH), 126.1 (CH), 126.2 (C), 128.7 (CH), 129.2 (CH), 130.7 (C), 137.8 (CH), 150.4 (C); m/z (ESI) 189 (MH+. 100%).
N-(2-Methoxy-5-methylphenyl)pyrrole (3u)
N-(2-Methoxy-5-methylphenyl)pyrrole (3u) was synthesised as described for N-(2-amino-5-nitrophenyl)-1H-pyrazole (3a) using 4-methylanisole (1f) (0.063 mL, 0.50 mmol) and pyrrole (0.10 mL, 1.5 mmol). The iodination step was carried out at 70 °C for 5 h and the N-arylation step at 150 °C for 36 h. Purification by flash column chromatography (hexane/diethyl ether, 9:1) gave N-(2-methoxy-5-methylphenyl)pyrrole (3u) (0.035 g, 37%) as a colourless oil. νmax/cm−1 (neat) 2931 (CH), 1512 (CC), 1483, 1327, 1242, 1070, 1024, 723; δH (400 MHz, CDCl3) 2.32 (3H, s, 5-CH3), 3.79 (3H, s, OCH3), 6.30 (2H, t, J 2.2 Hz, 3′-H and 4′-H), 6.91 (1H, d, J 8.2 Hz, 3-H), 6.97 (2H, t, J 2.2 Hz, 2′-H and 5′-H), 7.05 (1H, dd, J 8.2, 2.1 Hz, 4-H), 7.10 (1H, d, J 2.1 Hz, 6-H); δC (101 MHz, CDCl3) 20.4 (CH3), 56.0 (CH3), 108.7 (2 × CH), 112.4 (CH), 122.0 (2 × CH), 126.4 (CH), 127.7 (CH), 130.0 (C), 130.5 (C), 150.6 (C); m/z (ESI) 210.0892 (MNa+. C12H13NNaO requires 210.0889).
N-Methyl-6-benzenesulfonamide-7-methoxy-3,4-dihydro-1H-quinolin-2-one (16a)
N-Methyl-6-benzenesulfonamide-7-methoxy-3,4-dihydro-1H-quinolin-2-one (16a) was synthesised as described for N-(2-amino-5-cyanophenyl)-1H-pyrazole (3a) using N-methyl-7-methoxy-3,4-dihydro-1H-quinolin-2-one (14) (0.0500 g, 0.262 mmol), benzenesulfonamide (0.122 g, 0.780 mmol), iron(III) chloride (1.01 mg, 0.00625 mmol) and [BMIM]NTf2 (6.00 μL, 0.0196 mmol). The iodination step was carried out at 40 °C for 4 h and the N-arylation step at 150 °C for 20 h. Purification by flash column chromatography (ethyl acetate/hexane, 7:3) gave N-methyl-6-benzenesulfonamide-7-methoxy-3,4-dihydro-1H-quinolin-2-one (16a) (0.0510 g, 56%) as a white solid. Mp 185–188 °C; νmax/cm−1 (neat) 3252 (NH), 2967 (CH), 1665 (CO), 1616, 1518, 1332, 1171, 1146, 1115, 1092, 1061, 934, 763; δH (400 MHz, CDCl3) 2.61 (2H, dd, J 9.2, 6.8 Hz, 4-H2), 2.83 (2H, dd, J 7.6, 6.8 Hz, 3-H2), 3.29 (3H, s, NCH3), 3.55 (3H, s, OCH3), 6.33 (1H, s, 8-H), 6.76 (1H, br s, NH), 7.35 (1H, s, 5-H), 7.39–7.44 (2H, m, 3′-H and 5′-H), 7.52 (1H, tt, J 7.2, 1.2 Hz, 4′-H), 7.70–7.74 (2H, m, 2′-H and 6′-H); δc (101 MHz, CDCl3) 24.6 (CH2), 29.6 (CH3), 31.8 (CH2), 55.8 (CH3), 98.7 (CH), 118.6 (C), 120.0 (C), 122.5 (CH), 127.2 (2 × CH), 128.7 (2 × CH), 132.8 (CH), 138.8 (C), 139.3 (C), 149.8 (C), 170.4 (C); m/z (ESI) 369.0873 (MNa+. C17H18N2NaO4S requires 369.0879).
N-Methyl-6-(p-toluenesulfonamide)-7-methoxy-3,4-dihydro-1H-quinolin-2-one (16b)
N-Methyl-6-(p-toluenesulfonamide)-7-methoxy-3,4-dihydro-1H-quinolin-2-one (16b) was synthesised as described for N-methyl-6-benzenesulfonamide-7-methoxy-3,4-dihydro-1H-quinolin-2-one (16a) using N-methyl-7-methoxy-3,4-dihydro-1H-quinolin-2-one (14) (0.0500 g, 0.262 mmol) and p-toluenesulfonamide (0.134 g, 0.780 mmol). The iodination step was carried out at 40 °C for 4 h and the N-arylation step at 150 °C for 20 h. Purification by flash column chromatography (ethyl acetate/hexane, 7:3) gave N-methyl-6-(p-toluenesulfonamide)-7-methoxy-3,4-dihydro-1H-quinolin-2-one (16b) (0.0530 g, 56%) as a white solid. Mp 162–164 °C; νmax/cm−1 (neat) 3255 (NH), 2924 (C–H), 1661 (CO), 1512, 1342, 1150; δH (400 MHz, CDCl3) 2.38 (3H, s, CH3), 2.61 (2H, dd, J = 9.2, 7.2 Hz, 4-H2), 2.82 (2H, dd, J = 9.2, 6.8 Hz, 3-H2), 3.29 (3H, s, NCH3), 3.59 (3H, s, OCH3), 6.35 (1H, s, NH), 6.79 (1H, s, 8-H), 7.20 (2H, d, J = 8.5 Hz, 4′-H and 6′-H), 7.34 (1H, s, 5-H), 7.62 (2H, d, J = 8.5 Hz, 3′-H and 7′-H); δC (101 MHz, CDCl3) 21.5 (CH3), 24.6 (CH2), 29.6 (CH3), 31.8 (CH2), 55.9 (CH3), 98.8 (CH), 118.5 (C), 120.3 (C), 122.0 (CH), 127.2 (2 × CH), 129.4 (2 × CH), 136.4 (C), 138.5 (C), 143.6 (C), 149.5 (C), 170.4 (C); m/z (ESI) 383.1022 (MNa+. C18H20N2O4S requires 383.1036).
N-Methyl-6-benzamide-7-methoxy-3,4-dihydro-1H-quinolin-2-one (16c)
N-Methyl-6-benzamide-7-methoxy-3,4-dihydro-1H-quinolin-2-one (16c) was synthesised as described for N-methyl-6-benzenesulfonamide-7-methoxy-3,4-dihydro-1H-quinolin-2-one (16a) using N-methyl-7-methoxy-3,4-dihydro-1H-quinolin-2-one (14) (0.0500 g, 0.262 mmol) and benzamide (0.094 g, 0.780 mmol). The iodination step was carried out at 40 °C for 4 h and the N-arylation step at 150 °C for 20 h. Purification by flash column chromatography (dichloromethane/ethyl acetate, 4:1) gave N-methyl-6-benzamide-7-methoxy-3,4-dihydro-1H-quinolin-2-one (16c) (0.0480 g, 60%) as a white solid. Mp 168–170 °C; νmax/cm−1 (neat) 3428 (NH), 2941 (CH), 1661 (CO), 1530, 1124, 1059; δH (400 MHz, CDCl3) 2.65 (2H, dd, J = 9.2, 7.2 Hz, 4-H2), 2.89 (2H, dd, J = 9.2, 6.8 Hz, 3-H2), 3.37 (3H, s, NCH3), 3.95 (3H, s, OCH3), 6.57 (1H, s, NH), 7.47–7.59 (3H, m, 4′-H, 5′-H and 6′-H), 7.87–7.91 (2H, m, 3′-H and 7′-H), 8.37 (1H, s, 8-H), 8.42 (1H, s, 5-H); δC (101 MHz, CDCl3) 24.9 (CH2), 29.6 (CH3), 32.0 (CH2), 56.2 (CH3), 98.3 (CH), 118.4 (C), 119.5 (CH), 122.6 (C), 127.0 (2 × CH), 128.8 (2 × CH), 131.8 (CH), 135.1 (C), 136.8 (C), 147.6 (C), 165.1 (C), 170.4 (C); m/z (ESI) 333.1200 (MNa+. C18H18N2O3 requires 333.1210).
N-Methyl-6-(4′-methylbenzamide)-7-methoxy-3,4-dihydro-1H-quinolin-2-one (16d)
N-Methyl-6-(4′-methylbenzamide)-7-methoxy-3,4-dihydro-1H-quinolin-2-one (16d) was synthesised as described for N-methyl-6-benzenesulfonamide-7-methoxy-3,4-dihydro-1H-quinolin-2-one (16a) using N-methyl-7-methoxy-3,4-dihydro-1H-quinolin-2-one (14) (0.0500 g, 0.262 mmol) and p-toluamide (0.106 g, 0.783 mmol). The iodination step was carried out at 40 °C for 4 h and the N-arylation step at 150 °C for 20 h. Purification by flash column chromatography (dichloromethane/diethyl ether, 3:2) gave N-methyl-6-(4′-methylbenzamide)-7-methoxy-3,4-dihydro-1H-quinolin-2-one (16d) (0.0520 g, 61%) as a white solid. Mp 164–166 °C; νmax/cm−1 (neat) 3428 (NH), 2955 (CH), 1663 (CO), 1614, 1533, 1472, 1427, 1350, 1265, 1244, 1206, 1126, 1061, 745; δH (500 MHz, CDCl3) 2.43 (3H, s, 4′-CH3), 2.62–2.66 (2H, m, 4-H2), 2.86–2.90 (2H, m, 3-H2), 3.37 (3H, s, NCH3), 3.94 (3H, s, OCH3), 6.56 (1H, s, 8-H), 7.29 (2H, d, J 8.2 Hz, 3′-H and 5′-H), 7.78 (2H, d, J 8.2 Hz, 2′-H and 6′-H), 8.36 (1H, s, 5-H), 8.39 (1H, br s, NH); δc (126 MHz, CDCl3) 21.5 (CH3), 24.9 (CH2), 29.6 (CH3), 32.0 (CH2), 56.2 (CH3), 98.3 (CH), 118.3 (C), 119.4 (CH), 122.7 (C), 127.0 (2 × CH), 129.4 (2 × CH), 132.3 (C), 136.6 (C), 142.3 (C), 147.6 (C), 165.1 (C), 170.5 (C); m/z (ESI) 347.1364 (MNa+. C19H20N2NaO3 requires 347.1366).
N-Methyl-6-(2-pyrrolidinone)-7-methoxy-3,4-dihydro-1H-quinolin-2-one (16e)
N-Methyl-6-(2-pyrrolidinone)-7-methoxy-3,4-dihydro-1H-quinolin-2-one (16e) was synthesised as described for N-methyl-6-benzenesulfonamide-7-methoxy-3,4-dihydro-1H-quinolin-2-one (16a) using N-methyl-7-methoxy-3,4-dihydro-1H-quinolin-2-one (14) (0.0500 g, 0.262 mmol) and 2-pyrrolidinone (59.3 μL, 0.780 mmol). The iodination step was carried out at 40 °C for 4 h and the N-arylation step at 150 °C for 72 h. Purification by flash column chromatography (methanol/ethyl acetate, 1:20 to methanol/ethyl acetate, 1:10) gave N-methyl-6-(2-pyrrolidinone)-7-methoxy-3,4-dihydro-1H-quinolin-2-one (16e) (0.0360 g, 51%) as a white solid. Mp 188–190 °C; νmax/cm−1 (neat) 2970 (CH), 1667 (CO), 1612, 1520, 1450, 1420, 1350, 1281, 1057; δH (400 MHz, CDCl3) 2.15–2.23 (2H, m, 4′-H2), 2.55 (2H, t, J = 8.4 Hz, 5′-H2), 2.64 (2H, dd, J = 9.2, 6.8 Hz, 4-H2), 2.83 (2H, dd, J = 9.2, 6.8 Hz, 3-H2), 3.36 (3H, s, NCH3), 3.73 (2H, t, J = 7.2 Hz, 3′-H2), 3.86 (3H, s, OCH3), 6.57 (1H, s, 8-H), 7.05 (1H, s, 5-H); δC (101 MHz, CDCl3) 18.9 (CH2), 24.4 (CH2), 29.7 (CH3), 31.1 (CH2), 31.8 (CH2), 50.0 (CH2), 56.0 (CH3), 100.0 (CH), 118.4 (C), 121.6 (C), 127.6 (CH), 140.8 (C), 154.2 (C), 170.5 (C), 175.4 (C); m/z (ESI) 297.1214 (MNa+. C15H18N2NaO3 requires 297.1210).
N-Methyl-6-pyrazole-7-methoxy-3,4-dihydro-1H-quinolin-2-one (16f)
N-Methyl-6-(1H-pyrazole)-7-methoxy-3,4-dihydro-1H-quinolin-2-one (16f) was synthesised as described for N-methyl-6-benzenesulfonamide-7-methoxy-3,4-dihydro-1H-quinolin-2-one (16a) using N-methyl-7-methoxy-3,4-dihydro-1H-quinolin-2-one (14) (0.0500 g, 0.262 mmol) and pyrazole (0.531 g, 0.780 mmol). The iodination step was carried out at 40 °C for 4 h and the N-arylation step at 150 °C for 20 h. Purification by flash column chromatography (ethyl acetate/hexane, 7:3) gave N-methyl-6-(1H-pyrazole)-7-methoxy-3,4-dihydro-1H-quinolin-2-one (16f) (0.0400 g, 60%) as a white solid. Mp 168–170 °C; νmax/cm−1 (neat) 2947 (CH), 1667 (CO), 1620, 1528, 1466, 1350, 1188, 1134, 1034; δH (400 MHz, CDCl3) 2.66 (2H, dd, J = 9.2, 6.8 Hz, 4-H2), 2.89 (2H, dd, J = 9.2, 6.8 Hz, 3-H2), 3.40 (3H, s, NCH3), 3.89 (3H, s, OCH3), 6.42 (1H, dd, J = 2.3, 1.7 Hz, 4′-H), 6.65 (1H, s, 8-H), 7.54 (1H, s, 5-H), 7.69 (1H, d, J = 1.7 Hz, 3′-H), 7.99 (1H, d, J = 2.3 Hz, 5′-H); δC (101 MHz, CDCl3) 24.4 (CH2), 29.7 (CH3), 31.8 (CH2), 56.4 (CH3), 100.1 (CH), 106.2 (CH), 118.8 (C), 124.3 (CH), 124.5 (C), 131.4 (CH), 140.0 (CH), 140.2 (C), 150.5 (C), 170.4 (C); m/z (ESI) 280.1052 (MNa+. C14H15N3NaO2 requires 280.1056).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the EPSRC (studentship to M. C. H., EP/M508056/1), the University of Glasgow (studentship to R. J. F) and the Ministry of Higher Education and Scientific Research, Omar Al-Mukhtar University, Libya (studentship to M. A. B. M.) is gratefully acknowledged.Notes and references
- (a) R. Hili and A. K. Yudin, Nat. Chem. Biol., 2006, 2, 284 CrossRef CAS PubMed; (b) Amino Group Chemistry: From Synthesis to the Life Sciences, ed. A. Ricci, Wiley-VCH, Weinheim, 2007 Search PubMed.
- For reviews of copper-catalysed aryl amination, see: (a) K. Kunz, U. Scholz and D. Ganzer, Synlett, 2003, 2428 CrossRef CAS; (b) S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400 CrossRef CAS PubMed; (c) C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525 RSC; (d) K. Okano, H. Tokuyama and T. Fukuyama, Chem. Commun., 2014, 50, 13650 RSC.
- For reviews of palladium-catalysed aryl amination, see: (a) M. Carril, R. SanMartin and E. Domínguez, Chem. Soc. Rev., 2008, 37, 639 RSC; (b) J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534 CrossRef CAS PubMed; (c) D. S. Surry and S. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338 CrossRef CAS PubMed; (d) C. Fischer and B. Koenig, Beilstein J. Org. Chem., 2011, 7, 59 CrossRef CAS PubMed.
- For example, see: (a) M. Taillefer, N. Xia and A. Ouali, Angew. Chem., Int. Ed., 2007, 46, 934 CrossRef CAS PubMed; (b) A. Correa and C. Bolm, Angew. Chem., Int. Ed., 2007, 46, 8862 CrossRef CAS PubMed; (c) Z. Wang, H. Fu, Y. Jiang and Y. Zhao, Synlett, 2008, 2540 CAS; (d) A. Correa, S. Elmore and C. Bolm, Chem. – Eur. J., 2008, 14, 3527 CrossRef CAS PubMed; (e) S. L. Buchwald and C. Bolm, Angew. Chem., Int. Ed., 2009, 48, 5586 CrossRef CAS; (f) D. Guo, H. Huang, Y. Zhou, J. Xu, H. Jiang, K. Chen and H. Liu, Green Chem., 2010, 12, 276 RSC; (g) N. Panda, A. K. Jena, S. Mohapatra and S. R. Rout, Tetrahedron Lett., 2011, 52, 1924 CrossRef CAS.
- For recent reviews, see: (a) M.-L. Louillat and F. W. Patureau, Chem. Soc. Rev., 2014, 43, 901 RSC; (b) J. Jiao, K. Murakami and K. Itami, ACS Catal., 2016, 6, 610 CrossRef CAS; (c) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247 CrossRef CAS PubMed; (d) M. C. Henry, M. A. B. Mostafa and A. Sutherland, Synthesis, 2017, 49, 4586 CrossRef CAS.
- For example, see: (a) C. Tang and N. Jiao, J. Am. Chem. Soc., 2012, 134, 18924 CrossRef CAS; (b) M.-L. Louillat, A. Biafora, F. Legros and F. W. Patureau, Angew. Chem., Int. Ed., 2014, 53, 3505 CrossRef CAS.
- Á. M. Martinez, N. Rodríguez, R. G. Arrayás and J. C. Carretero, Chem. Commun., 2014, 50, 2801 RSC.
- Q. Li, S.-Y. Zhang, G. He, Z. Ai, W. A. Nack and G. Chen, Org. Lett., 2014, 16, 1764 CrossRef CAS PubMed.
- K. Sun, Y. Li, T. Xiong, J. Zhang and Q. Zhang, J. Am. Chem. Soc., 2011, 133, 1694 CrossRef CAS.
- M. A. B. Mostafa, E. D. D. Calder, D. T. Racys and A. Sutherland, Chem. – Eur. J., 2017, 23, 1044 CrossRef CAS PubMed.
- I. Goldberg, Ber. Dtsch. Chem. Ges., 1906, 39, 1691 CrossRef.
- (a) D. T. Racys, C. E. Warrilow, S. L. Pimlott and A. Sutherland, Org. Lett., 2015, 17, 4782 CrossRef CAS PubMed; (b) M. A. B. Mostafa, R. M. Bowley, D. T. Racys, M. C. Henry and A. Sutherland, J. Org. Chem., 2017, 82, 7529 CrossRef CAS PubMed; (c) M. C. Henry, H. M. Senn and A. Sutherland, J. Org. Chem., 2019, 84, 346 CrossRef CAS PubMed.
- Analysis of the one-pot amination of 1a by 1H NMR spectroscopy showed that full conversion to the bromide intermediate was achieved during the first step. Hence, recovery of 1a at the end of the copper-catalysed amination step is due to reduction of the bromide intermediate and not returned unreacted aniline.
- In previous studies (see ref. 10), control experiments showed that the presence of the ionic liquid has no effect on the copper-catalysed C–N bond forming step.
- G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054 CrossRef CAS PubMed.
- (a) A. Shafir and S. L. Buchwald, J. Am. Chem. Soc., 2006, 128, 8742 CrossRef CAS PubMed; (b) A. Shafir, P. A. Lichtor and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 3490 CrossRef CAS PubMed.
- Buchwald and co-workers have suggested that the success of cyclic diamine ligands for Goldberg-type coupling reactions may be due to an increase of the stability constant of the catalytically active copper–nucleophile complex: (a) A. Klapars, J. C. Antilla, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2001, 123, 7727 CrossRef CAS PubMed; (b) A. Klapars, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 7421 CrossRef CAS PubMed.
- D. T. Racys, S. A. I. Sharif, S. L. Pimlott and A. Sutherland, J. Org. Chem., 2016, 81, 772 CrossRef CAS PubMed.
- (a) M. Earle, B. J. McAuley, A. Ramani, K. Seddon and J. Thompson, WO02072260, 2002; (b) M. J. Earle, U. Hakala, B. J. McAuley, M. Nieuwenhuyzen, A. Ramani and K. R. Seddon, Chem. Commun., 2004, 1368 RSC.
- S. Antoniotti, V. Dalla and E. Duñach, Angew. Chem., Int. Ed., 2010, 49, 7860 CrossRef CAS PubMed.
- For example, see: (a) J. W. Tye, Z. Weng, A. M. Johns, C. D. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 9971 CrossRef CAS PubMed; (b) E. R. Strieter, B. Bhayana and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 78 CrossRef CAS PubMed; (c) R. Giri, A. Brusoe, K. Troshin, J. Y. Wang, M. Font and J. F. Hartwig, J. Am. Chem. Soc., 2018, 140, 793 CrossRef CAS PubMed.
- E. Sperotto, G. P. M. van Klink, G. van Koten and J. G. de Vries, Dalton Trans., 2010, 39, 10338 RSC.
- W. S. Palmer, G. Poncet-Montange, G. Liu, A. Petrocchi, N. Reyna, G. Subramanian, J. Theroff, A. Yau, M. Kost-Alimova, J. P. Bardenhagen, E. Leo, H. E. Shepard, T. N. Tieu, X. Shi, Y. Zhan, S. Zhao, M. C. Barton, G. Draetta, C. Toniatti, P. Jones, M. G. Do and J. N. Andersen, J. Med. Chem., 2016, 59, 1440 CrossRef CAS PubMed.
- R. J. Faggyas, M. Grace, L. Williams and A. Sutherland, J. Org. Chem., 2018, 83, 12595 CrossRef CAS PubMed.
- Z. Kuang, B. Li and Q. Song, Chem. Commun., 2018, 54, 34 RSC.
- B. J. Liddle, R. M. Silva, T. J. Morin, F. P. Macedo, R. Shukla, S. V. Lindeman and J. R. Gardinier, J. Org. Chem., 2007, 72, 5637 CrossRef CAS PubMed.
- T.-S. Jiang and G.-W. Wang, J. Org. Chem., 2012, 77, 9504 CrossRef CAS PubMed.
- L. V. Desai, H. A. Malik and M. S. Sanford, Org. Lett., 2006, 8, 1141 CrossRef CAS PubMed.
- K. M. McCauley, J. M. Vrtis, J. Dupont and W. A. van der Donk, J. Am. Chem. Soc., 2000, 122, 2403 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra for all compounds. See DOI: 10.1039/c9ob00712a |
| This journal is © The Royal Society of Chemistry 2019 |