
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and biophysical properties of carbamate-locked nucleic acid (LNA) oligonucleotides with potential antisense applications†
Cameron
Thorpe
 a,
Sven
Epple
a,
Benjamin
Woods
a,
Afaf H.
El-Sagheer
ab and
Tom
Brown
*a
a,
Sven
Epple
a,
Benjamin
Woods
a,
Afaf H.
El-Sagheer
ab and
Tom
Brown
*a
aDepartment of Chemistry, University of Oxford, 12 Mansfield Road, Oxford OX1 3TA, UK. E-mail: tom.brown@chem.ox.ac.uk
bChemistry Branch, Department of Science and Mathematics, Faculty of Petroleum and Mining Engineering, Suez University, Suez 43721, Egypt
First published on 17th May 2019
Abstract
Antisense oligonucleotides (ASOs) are becoming important drugs for hard to treat diseases. Modifications to their DNA backbones are essential to inhibit degradation in vivo, but they can reduce binding affinity to RNA targets. To address this problem we have combined the enzymatic resistance of carbamate (CBM) DNA backbone analogues with the thermodynamic stability conferred by locked nucleic acid sugars (LNA). Using a dinucleotide phosphoramidite strategy and automated solid phase synthesis, we have synthesised a set of oligonucleotides modified with multiple LNA-CBM units. The LNA sugars restore binding affinity to RNA targets, and in this respect LNA position with respect to the CBM linkage is important. Oligonucleotides containing carbamate flanked on its 5′and 3′-sides by LNA form stable duplexes with RNA and unstable duplexes with DNA, which is desirable for antisense applications. Carbamate-LNA modified oligonucleotides also show increased stability in the presence of snake venom and foetal bovine serum compared to LNA or CBM backbones alone.
Introduction
Antisense oligonucleotides (ASOs) are short ∼20 mer sequences that bind to complementary ribonucleic nucleic acid (RNA) targets through Watson–Crick base pairing to promote enzymatic degradation (RNase H), induce RNA interference (RNAi) or regulate protein expression (splice modulation).1 Enabled by target specific binding, there are now several FDA approved ASO-based drugs in the clinic treating a range of genetic and hereditary diseases.2 Eteplirsen (Exondys 51)3 and Nusinersen (Spinraza)4 have been developed for the treatment of Duchenne muscular dystrophy and spinal muscular atrophy respectively. Antisense therapeutics are also making headway against severe hypercholesterolemia following the release of Mipomersen.5 Furthermore, with the recent approval in 2018 of Patisiran6 and Inotersen7 treating fatal hereditary transthyretin-mediated amyloidosis, the ASO field demonstrates its versatility for treatment of diseases that could be described as “undruggable” by small molecule approaches. Unfortunately, due to poor cell penetration8 combined with fast cellular degradation, unmodified sequences are less than ideal candidates for therapeutic applications. To overcome this, much research has focused on the incorporation of backbone modifications9,10 into ASOs. In regard to degradation of the phosphodiester backbone of ASOs by nuclease enzymes, chemical modification of sugar-phosphate backbones can significantly improve biological stability. Several classes of antisense modifications have emerged, and have been used in FDA approved ASOs. Common modifications include 2′-O-methoxy or 2′-O-(2-methoxyethyl) (MOE) ribose sugars, backbone modifications such as phosphorothioates11 (Mipomersen and Nusinersen) or phosphorodithioates and sugar alternatives such as morpholino (Eteplirsen) or threose nucleic acid (TNA).12,13 Amide14–18 and triazole19–21 artificial DNA backbones also have potential, but are yet to reach maturity in the antisense field. Further improvements to the design of chemically modified oligonucleotides (ONs), specifically those that increase hybridisation strength (RNA-target affinity) and stability in vivo are sought after, particularly if they also influence other properties such as cellular uptake.22Locked nucleic acid (LNA), also known as bridged nucleic acid (BNA), has been a transformative development in the nucleic acid field. LNA is a bicyclic ribose analogue with a 2′-O-4′-methylene sugar bridge. The C3′-endo conformation is preferred and consequently LNA displays unparalleled binding affinity to complementary RNA strands; LNA modifications can stabilise duplexes by up to +7 °C per modification.23,24 Building on the seminal work of Wengel23,25 and Obika,26,27 LNA has been used in a variety of applications including siRNA-mediated gene silencing, CRISPR-Cas9,28 and triplex formation.29,30 A combination of LNA with a DNA backbone mimic comprised of a six-atom triazole linkage improves duplex stability compared to the unmodified counterpart.20 In contrast, Watts et al.31 found that addition of LNA to a four-atom triazole linkage caused duplex destabilisation. This indicates that the contributions of linkage length and flexibility of DNA backbone analogues when combined with LNA as modulators of duplex stability is still poorly understood. It is thus important to expand the range of artificial backbones-LNA combinations beyond the triazole and amide systems.32,33 The carbamate backbone is a four-atom, flexible, charge-neutral linkage with two constitutional isomers CBM1 (Fig. 1A) and CBM2 (Fig. 1E). Carbamate backbone incorporation into DNA has previously been reported by Waldner and De Mesmaeker et al.34 They found that duplex destabilisation occurs, but good stability against enzymatic degradation is achieved. Hence, in isolation carbamates are less than ideal candidates for antisense applications because of reduced binding affinity. In pursuit of new modified oligonucleotide constructs, we sought to combine the enzymatic stability of carbamate backbones with the favourable thermodynamic properties of LNA. We now report the synthesis of a set of dinucleotide phosphoramidites designed to introduce the carbamate-LNA modifications in Fig. 1 into oligonucleotides sequences using standard solid phase synthesis. The modified oligomers were synthesised and evaluated by ultraviolet melting (hybridisation) studies in order to determine the most favourable carbamate-LNA combinations as modified DNA backbones. Enzymatic digestion assays were carried out using snake venom and foetal bovine serum.
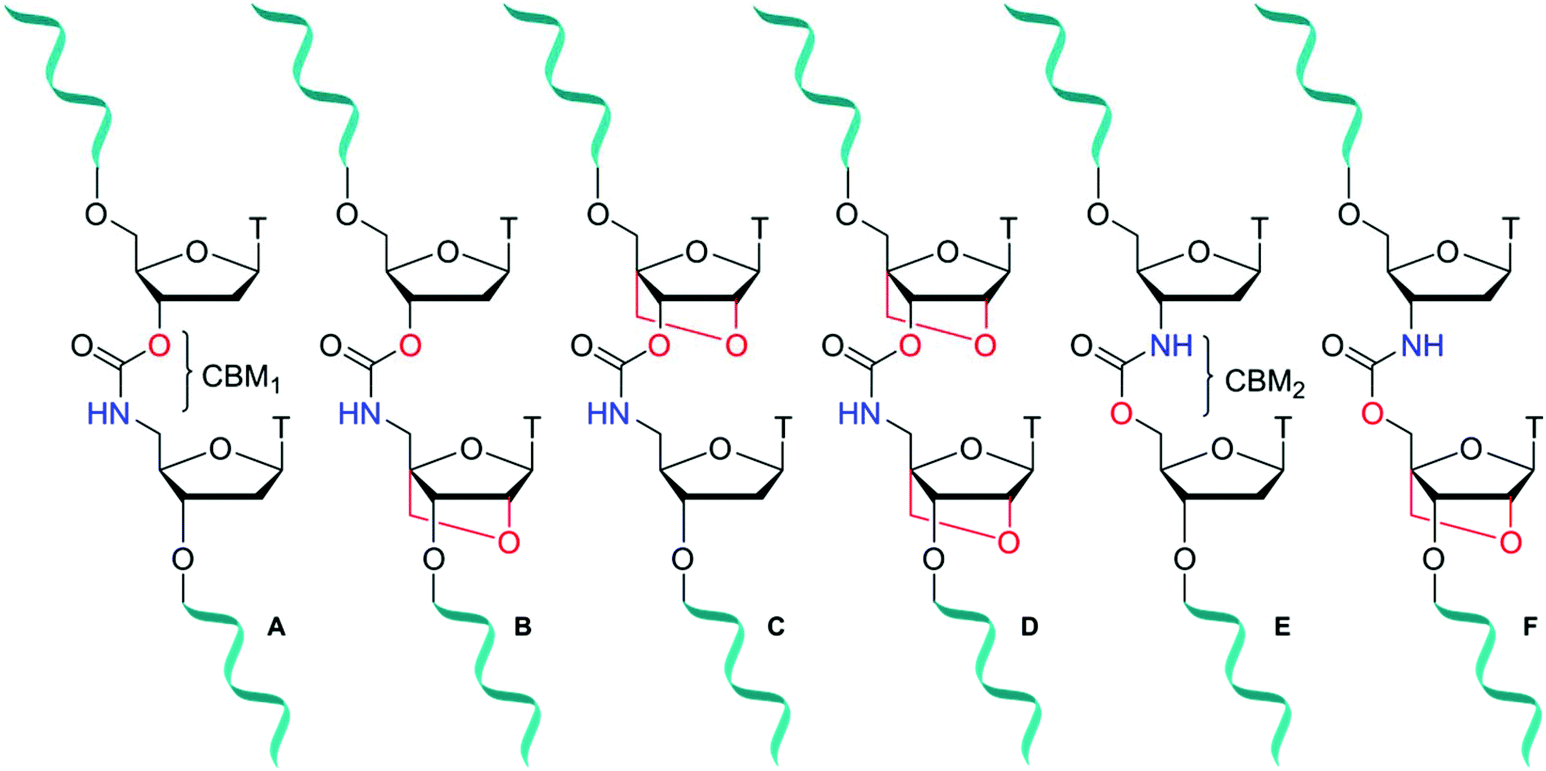 | ||
| Fig. 1 Carbamate-modified DNA backbones containing LNA sugars. Set 1 (A–D) and set 2 (E, F) comprise of dimers with the constitutional carbamate isomers CBM1 and CBM2 respectively. A = DNA-CBM1-DNA, B = DNA-CBM1-LNA, C = LNA-CBM1-DNA, D = LNA-CBM1-LNA, E = DNA-CBM2-DNA, F = DNA-CBM2-LNA. | ||
Results and discussion
Synthesis of LNA-CBM1 dinucleotides
Compounds 13–16 (Scheme 1) were synthesised by ligation of 3′-activated carbonyls 2 and 4 with 5′-nucleophilic amines 5 and 8 following a previously reported synthesis by Waldner et al.34 Starting material 1 was reacted with p-nitrophenyl (PNP) chloroformate to give the activated nucleoside 2. Subsequent reaction with amine 5 under basic conditions gave dinucleotide 9 which was converted to phosphoramidite 13 using standard phosphitylating conditions. For the LNA monomers, a divergent synthetic strategy was used. Compounds 3 and 6 were synthesised35 and compound 3 was converted to an activated imidazole carbamate 4. Unlike DNA analogue 2, activation of the 3′-OH by p-nitrophenyl (PNP) chloroformate was slow and resulted in poor yields. It has been previously suggested that PNP carbonates could be susceptible to degradation in basic solvent,36 preventing efficient activation of the 3′-position. To overcome this, we opted for a one pot strategy directly reacting alcohol 3 with CDI to form imidazole carbamate 4 which was then used without further purification.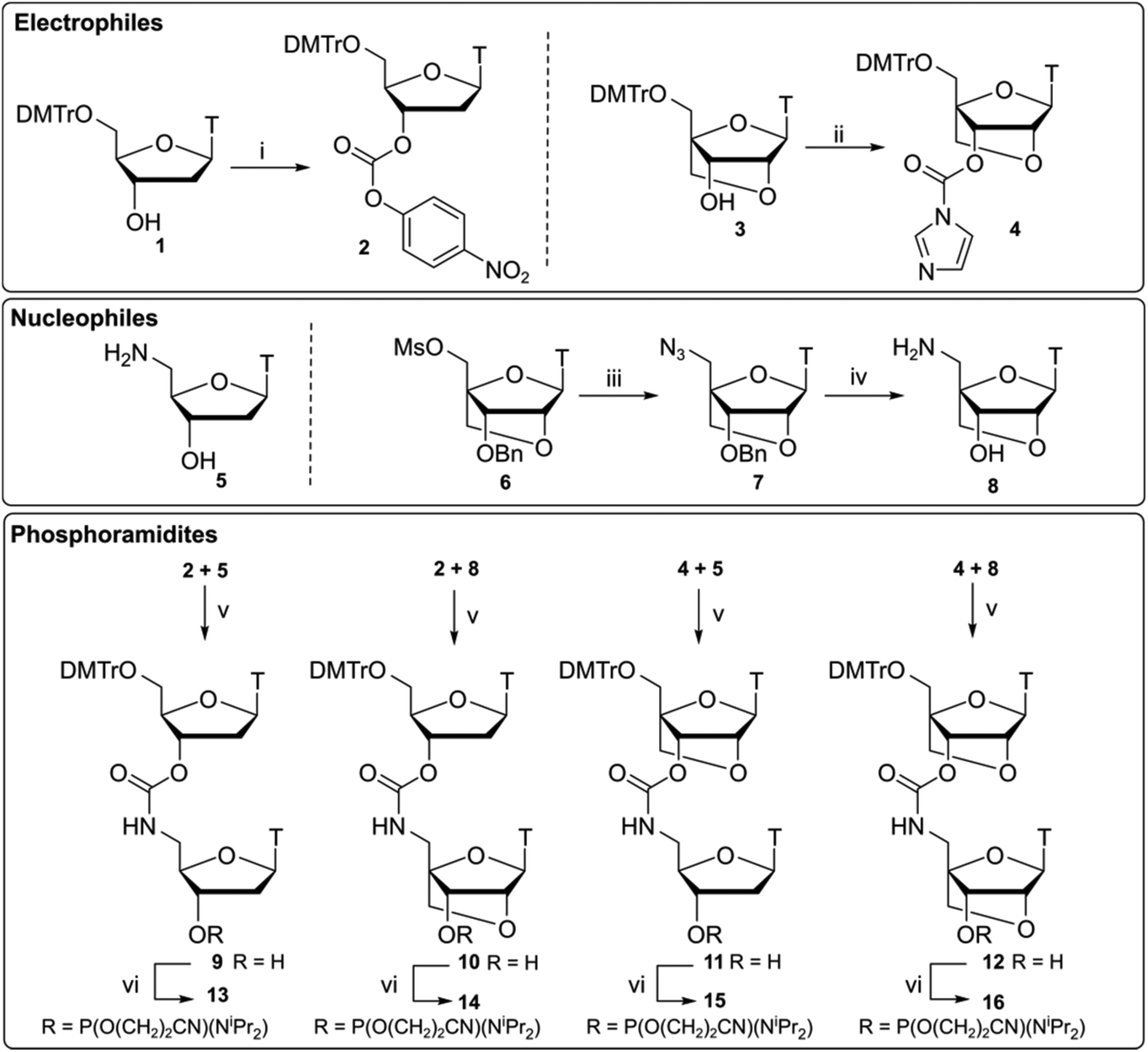 | ||
| Scheme 1 Synthesis of CBM1-linked dinucleotides 13–16 using different combinations of 3′-activated carbonates 2, 4 and 5′-amines 5, 8. Reagents and conditions (i) p-nitrophenyl chloroformate, pyridine, 80 °C, 20 h, crude 2 60%; (ii) CDI, THF, rt, 20 h, crude 4 84%; (iii) NaN3, DMF, 65 °C, 20 h, 7 90%; (iv) H2, Pd(OH)2 (20%)/C MeOH, rt, 3 h; NH4HCO2, reflux, 4 h, 8 87%; (v) DMAP, pyridine, 80 °C, 20 h, 9 62%, 10 51%, 11 63%, 12 70%; (vi) Chloro(diisopropylamino)-β-cyanoethoxyphosphine, DIPEA, CH2Cl2, rt, 1 h, 13 54%, 14 57%, 15 36%, 16 88%. | ||
Following work by Obika,37 LNA nucleoside 6 was reacted with sodium azide to form its 5′-azide 7. Reduction of the azido group was performed as a one pot procedure by palladium-catalysed hydrogenation under H2 gas. Subsequent deprotection of the 3′-benzyl group was then facilitated by the addition of NH4HCO2 to give amino nucleoside 8. Combinations of the required DNA and LNA monomers 2, 4, 5, 8 were then used to give CBM1-linked dinucleotides analogues 10–12. As to be expected, coupling of the more sterically hindered 3′-activated LNA monomer 4 was found to be slower but proceeded with comparable yields to the DNA analogues. These compounds were then converted to their respective phosphoramidites 14–16.
Synthesis of LNA-CBM2 dinucleotide analogues
Compounds 24 and 27 (Scheme 2) were synthesised using a 3′-nucleophilic amine and activated 5′-carbonate (reverse connectivity to CBM1). Compound 17 was obtained by reduction of commercially available 3′-azidothymidine (AZT) over palladium on activated charcoal in good yield. In order to synthesise compound 19 we activated the 5′-OH of unprotected thymidine but this led to an intramolecular reaction via 3′-OH nucleophilic attack on the PNP carbonate. Thus, a 5′/3′ protection strategy was employed similar to that of Utagawa et al.,38 where 5′-O-DMTr/3′-O-TBDMS protected thymidine was used in the generation of the 5′-activated PNP carbonate (compound 19) via compound 18. Compound 22 was formed under basic conditions and 3′-O-TBDMS protection was removed to form alcohol 23. This was subsequently phosphitylated to give the phosphoramidite DNA–DNA dimer 24.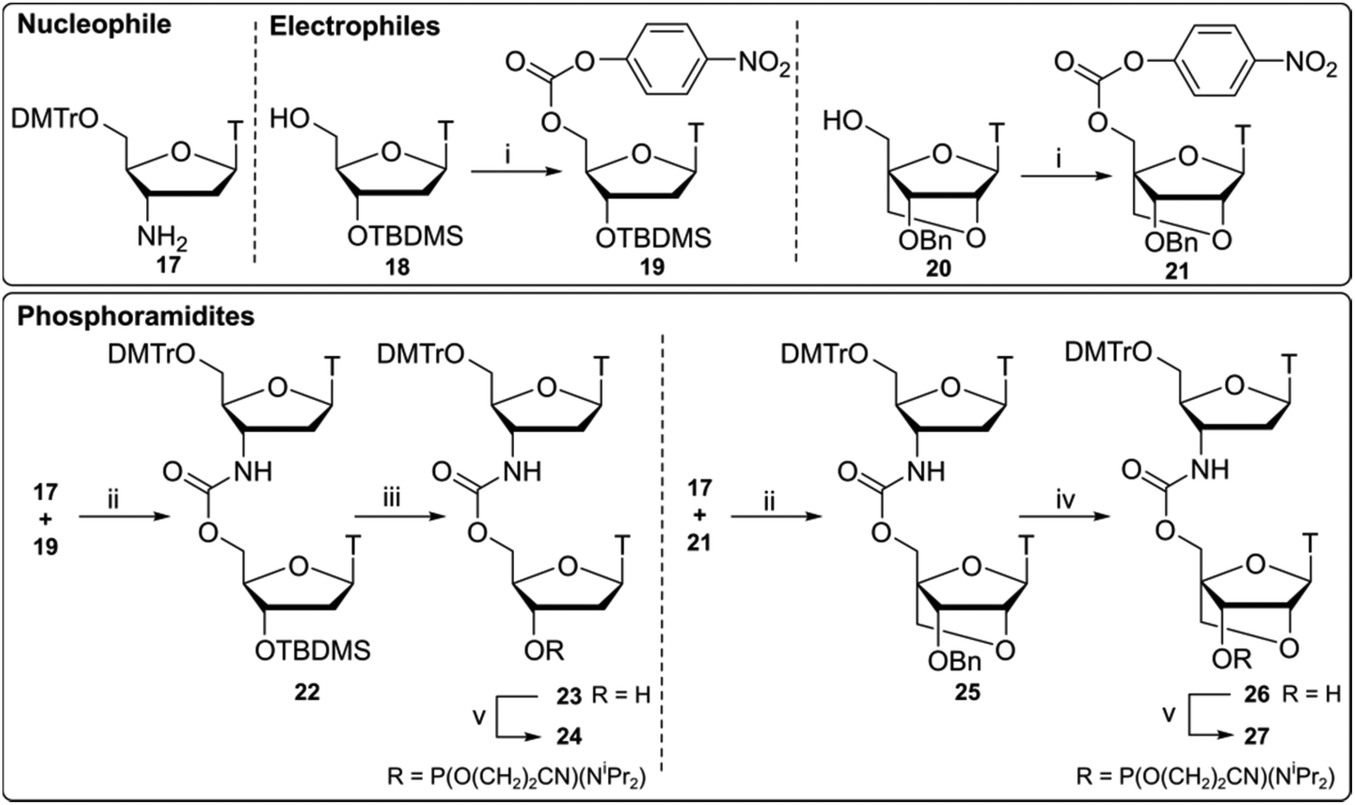 | ||
| Scheme 2 Synthesis of CBM2 dinucleotides 24, 27 using activated 3′-amine 17 and 5′-activated carbonates 19, 21. Reagents and conditions (i) p-nitrophenyl chloroformate, pyridine, 80 °C, 20 h, 19 76%, 21 crude 73%, (ii) HOBt, pyridine, 80 °C, 20 h, 22 67%, 25 63%; (iii) TBAF, THF, 0 °C, 2 h, 23 77%; (iv) Pd(OH)2 (20%)/C, NH4HCO2, 65 °C, 2 h, 26 97%; (v) Chloro(diisopropylamino)-β-cyanoethoxyphosphine, DIPEA, CH2Cl2, rt, 1 h, 24 80%, 27 43%. | ||
Following a protocol from Koshkin et al.,35 5′-activated 21 (Scheme 2) was synthesised. Dimer 25 was formed by coupling of amine 17 with LNA monomer 21 and the 3′-benzyl ether was deprotected by reduction over a Pd(OH)2-catalyst to yield alcohol 26 followed by conversion to the phosphoramidite DNA-LNA building block 27. Previous DNA carbamate duplex melting experiments34 indicate that the CBM2 linkage is more destabilising than CBM1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22 and recent biophysical studies with LNA-triazole backbones also showed that 5′-LNA adjacent to the modified linkage is highly destabilising in both DNA and RNA duplexes.20 Based on these observations we did not pursue the synthesis of this 5′-LNA carbamate monomer and subsequent 5′-LNA carbamate dimers.39–41
22 and recent biophysical studies with LNA-triazole backbones also showed that 5′-LNA adjacent to the modified linkage is highly destabilising in both DNA and RNA duplexes.20 Based on these observations we did not pursue the synthesis of this 5′-LNA carbamate monomer and subsequent 5′-LNA carbamate dimers.39–41
Duplex stability (hybridisation) studies
:DNA duplexes than within DNA:RNA hybrids, causing smaller reductions in melting temperature (additional data, Fig. S1 and S2†).
| ON | ON sequence (5′-3′) | DNA target | RNA target | ||
|---|---|---|---|---|---|
| T m | ΔTm | T m | ΔTm | ||
| ON1 | GCTTGCTTCGTTCC | 60.2 | — | 63.6 | — |
| ON2 | GCTTGCTxTCGTTCC | 57.5 | −2.7 | 57.2 | −6.4 |
| ON3 | GCTTGCTLxTCGTTCC | 50.5 | −9.7 | 56.1 | −7.5 |
| ON4 | GCTTGCTxTLCGTTCC | 60.3 | 0.1 | 61.3 | −2.3 |
| ON5 | GCTTGCTLxTLCGTTCC | 53.4 | −6.8 | 62.1 | −1.5 |
| ON6 | GCTTGCTyTCGTTCC | 53.6 | −6.6 | 55.0 | −8.6 |
| ON7 | GCTTGCTyTLCGTTCC | 51.7 | −8.5 | 57.1 | −6.5 |
| ON | ON sequence (5′-3′) | DNA target | RNA target | ||
|---|---|---|---|---|---|
| T m | ΔTm | T m | ΔTm | ||
| ON1 | GCTTGCTTCGTTCC | 60.2 | — | 63.6 | — |
| ON8 | GCTxTGCTxTCGTxTCC | 52.1 | −8.1 | 44.8 | −18.8 |
| ON9 | GCTLxTGCTLxTCGTLxTCC | <30 | <−30.2 | 40.6 | −23.0 |
| ON10 | GCTxTLGCTxTLCGTxTLCC | 60.1 | −0.1 | 59.6 | −4.0 |
| ON11 | GCTLxTLGCTLxTLCGTLxTLCC | 41.2 | −19.0 | 61.6 | −2.0 |
| ON12 | GCTyTGCTyTCGTyTCC | 37.9 | −22.3 | 36.5 | −27.1 |
| ON13 | GCTyTLGCTyTLCGTyTLCC | 34.7 | −25.5 | 46.8 | −16.8 |
LNA has been shown to increase duplex stability by up to +7 °C per modification in unmodified phosphodiester duplexes.42 To evaluate the positional effects of LNA on carbamate modified duplexes, LNA-containing dinucleotides 14–16 were used to incorporate LNA sugars adjacent to the modified linkages. Unlike with phosphodiester backbones, the presence of 5′-LNA (Fig. 2A and B, ON9) caused further destabilisation to the duplexes compared to the CBM1 backbone with unmodified sugars (ON8). Introduction of an LNA modification to the 3′ side (Table 1 ON4, Table 2 ON10, Fig. 2A and B, ON10) had the inverse effect, stabilising duplexes against both DNA and RNA targets by +2.6 and +4.1 °C (ON2 vs. ON4) per modification respectively. Addition of LNA to both sides of the linkage against DNA targets resulted in moderate destabilisation, seemingly combining the two effects of 3′ and 5′ LNA (Fig. 2A, ON11). In contrast, LNA addition to both sides of the carbamate linkage against RNA targets (Fig. 2B, ON11) resulted in the most stable carbamate duplexes, outperforming the 3′ addition alone. This is likely to be a consequence of the conformational influence of the LNA sugar. In unmodified DNA duplexes the deoxyribose sugar is C2′-endo, leading to a B-form helix. Conversely, LNA prefers a C3′-endo conformation characteristic of A-form RNA. Hence, as the LNA content of the DNA strand increases, it can more efficiently hybridise with RNA. As a result, increased LNA content improves hybridisation of modified DNA to complementary RNA sequences. These results also demonstrate that the LNA sugar influences the conformation of the backbone linkage directly on its 3′-side, as observed by Petersen et al.,43 even if this is not a canonical phosphodiester. However, its influence on a phosphodiester is much greater than on the carbamate analogue. The closer an analogue is to a pure phosphodiester, the greater will be the positive influence of LNA. For example, ON4 and ON10 display high levels of stability because influence of LNA is directed to the phosphodiester linkage at its 3′-side, as in normal LNA:RNA duplexes. When 5′-LNA modifications are introduced (e.g. in ON9), LNA-induced conformational change is now directed to the 3′-carbamate linkage which clearly cannot accommodate the necessary structural differences to the same degree as the phosphodiester. This may be due to the relative conformational rigidity of the carbamate group.
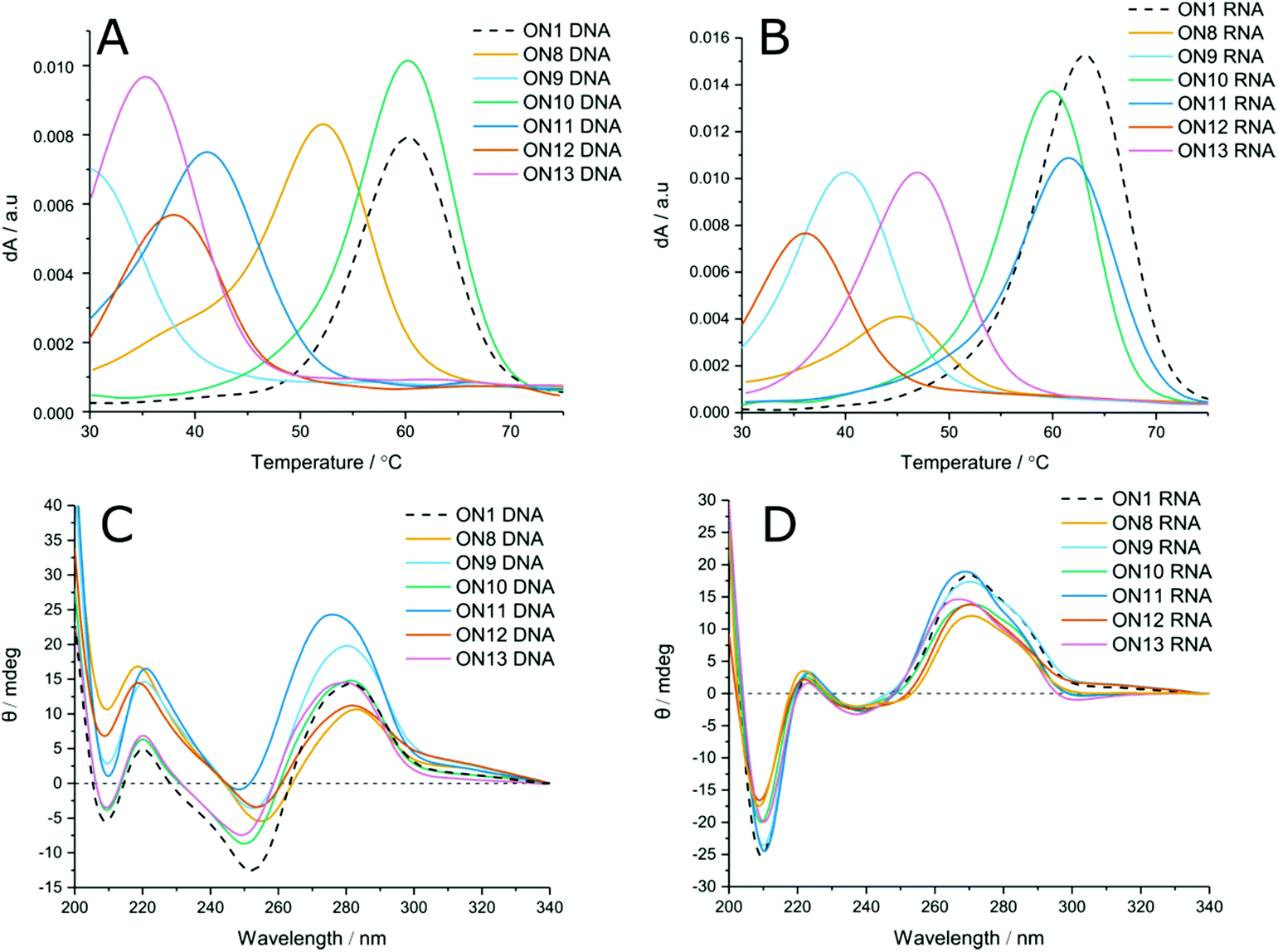 | ||
| Fig. 2 (Top) UV-melting data, 3 μM of oligonucleotides were added to 10 mM phosphate, 200 mM NaCl buffer, pH 7.0 and heated from 20–85 °C. Tm values were determined from an average of six ramps from a smoothed plot of dA/t. (A) First derivative Tm curves against DNA targets containing triple modifications of CBM-LNA, (B) First derivative Tm curves against RNA targets containing triple modifications of CBM-LNA. (Bottom) Circular dichroism spectra of 3 μM oligonucleotides in 10 mM phosphate, 200 nM NaCl buffer, pH 7.0. Spectra were averaged from four scans and smoothed to 20 points using a third order polynomial. (C) Triple incorporation of CBM-LNA modifications against DNA targets, (D) Triple incorporation of CBM-LNA modifications against RNA targets. Overall duplex conformation against both targets remained largely unchanged. Against DNA targets hypsochromic shifts were observed with addition of LNA. | ||
To evaluate the effect of increased numbers of modifications, LNA-CBM1 dimers were introduced at three distinct sites within the ON sequences (ON8–13, Table 2, and Fig. 2). In these templates the same destabilising and stabilising effects were seen as with the single addition. In most cases, the change in Tm per modification was observed to be additive. 5′-LNA modifications were shown to be highly destabilising, reducing Tm by >30 °C and 23 °C against DNA and RNA respectively. However, the reduction in stability caused by the CBM1 backbone in a DNA oligonucleotide hybridised to a DNA target can be reversed by the presence of an adjacent 3′-LNA (ON10) or by a 5′/3′-LNA sandwich in DNA:RNA hybrids (ON11). Combining the results from single and triple incorporations of LNA-CBM1 combinations, the trends in duplex stability against DNA targets is as follows: TxTL > TxT > TLxTL > TLxT and against RNA targets TLxTL > TxTL > TxT > TLxT.
:RNA hybrids (ON13 vs. ON12 = +10.3 °C). This again highlights possible structural and conformational differences between CBM1 (x) and CBM2 (y) isomers. In summary, for DNA targets we find duplex stability TyT > TyTL whereas against RNA targets TyTL > TyT. Summarising Tm results it is clear that the CBM1-linkage has better stabilising properties compared to CBM2.
Circular dichroism
Circular dichroism (CD) was used to evaluate changes in global duplex structure by measurement of polarised light absorption from 200–340 nm (Fig. 2C and D). From these results it was found that the helical conformation remains essentially unperturbed and that structural changes compared to the unmodified control are modest. DNA:DNA duplexes adopt B-form structures characterised by transitions at 220, 255 and 280 nm. Likewise, DNA:RNA hybrids adopt A-like duplexes with negative and positive peaks at 210 and 270 nm. Increasing the proportion of LNA in both duplexes (ON11) results in a hypsochromic shift consistent with reports in the literature.20 Comparison of DNA:DNA to DNA:RNA hybrids suggests that LNA has greater structural effects on DNA:DNA B-form duplexes. High levels of LNA within the DNA duplex favour a B → A transition, indicated by hypsochromic shift from 280 nm towards 270 nm and greater deviation from the unmodified control. In DNA:RNA hybrids, the duplex is already in or close to the A-form. Therefore, no significant transition is observed, and the CD spectrum deviates to a lesser extent from the unmodified sequence (additional CD data Fig. S3†).
Enzymatic stability assays
Enzymatic stability of modified ONs is important to ensure optimal biological half-life and therapeutic efficacy. To evaluate if CBM1-LNA modifications enhance enzymatic stability, oligonucleotides were incubated with phosphodiesterase 1 from C. adamanteus and foetal bovine serum to determine the stability of the modified templates towards a range of nucleases.Triply modified ONs were incubated at 37 °C in a phosphate buffer containing snake venom phosphodiesterase 1 and frozen at set time points up to 1 hour (Fig. 3 top). Unmodified control (ON1) was fully degraded within five minutes, along with 3′-LNA control (ON14), indicated by loss of all full-length bands. Triple addition of 5′/3′-LNA showed substantial resistance to enzymatic degradation; only the end nucleotides were excised (Fig. 3A, lane 11). Isolated CBM1-linkages were found to offer little resistance towards degradation (Fig. 3B, ON8). However, a combination of 5′/3′-LNA and CBM1-linkages (ON11) produced the best resistance, even reducing degradation of the oligonucleotide compared to 5′/3′-LNA (ON11 vs. ON15, additional data Fig. S4†). Initial degradation of the 3′-end in LNA/CBM-modified oligonucleotide ON11 leads to a degradation product with a single nucleotide removed. Due to the presence of the neutral carbamate modifications, the overall mass to charge ratio increases, making the oligonucleotide run slower on the gel (Fig. 3B and D, lane 25–29). In contrast, degradation from the 3′-end of the all phosphodiester LNA-modified oligonucleotide ON15 generates a shorter fragment with the expected faster migration (Fig. 3A and C, lane 10–14).
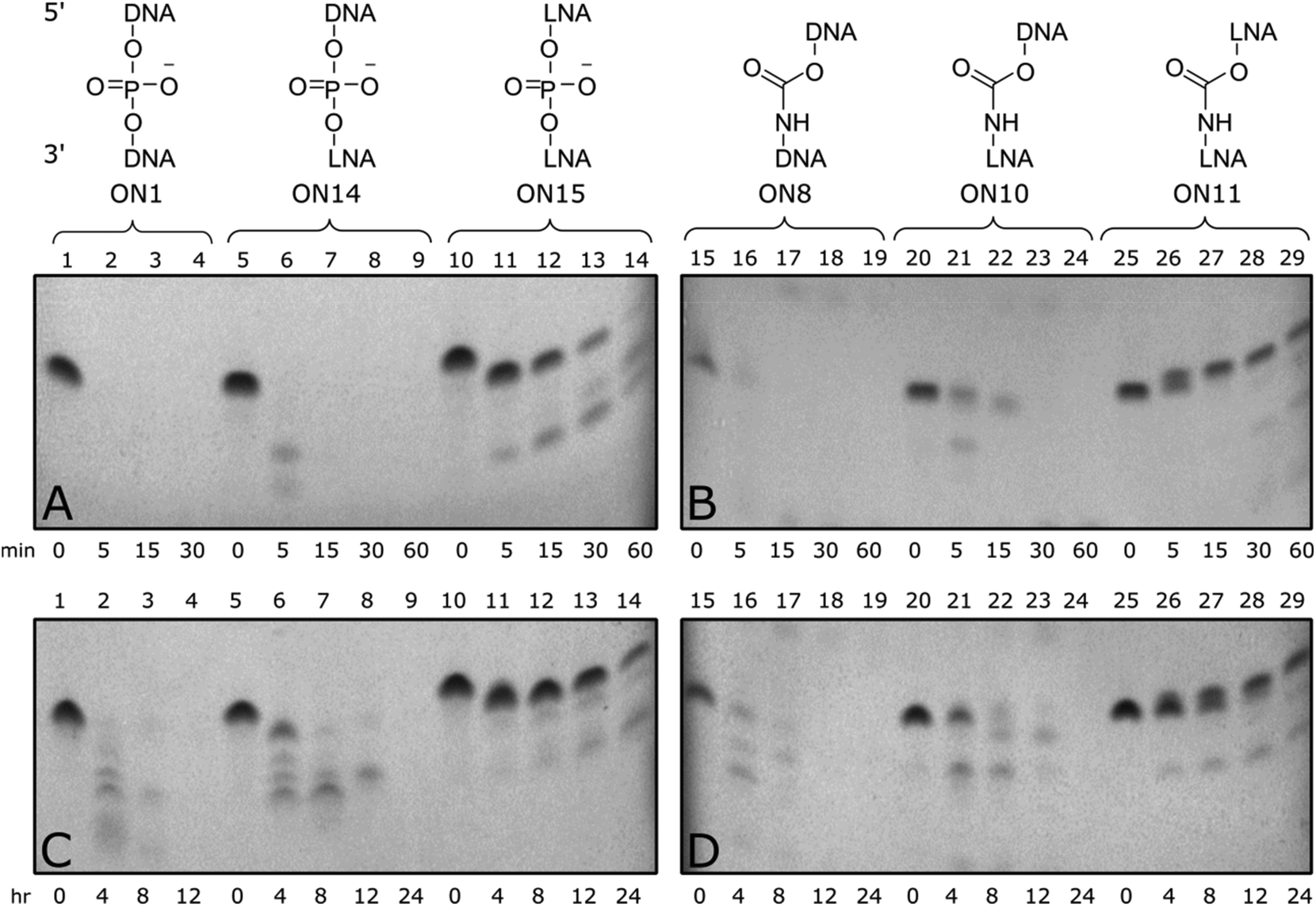 | ||
| Fig. 3 (Top) 20% denatured polyacrylamide gel electrophoresis (PAGE) results from snake venom 3′-exonuclease assay. Samples were incubated in 50 mM Tris-HCl, 10 mM MgCl2 buffer pH 9.0 at 37 °C. Aliquots were removed at t = 0, 5, 15, 30 and 60 min and frozen before analysis. (A) Gel showing unmodified and LNA controls, (B) gel showing oligonucleotides with carbamate and combined LNA-carbamate modification. ON14 GCTTLGCTTLCGTTLCC, ON15 GCTLTLGCTLTLCGTLTLCC, L = LNA. Results show increased nuclease resistance when introducing three separate 5′/3′-LNA-CBM1 modifications (ON11). (Bottom) 20% denatured polyacrylamide gel electrophoresis (PAGE) results from FBS assay of modified templates containing triple incorporation of CBM1-LNA dinucleotides. Samples were incubated in Dulbecco's PBS buffer at 37 °C. Aliquots were removed at t = 0, 4, 8, 12 and 24 h and frozen before analysis. (C) Gel showing unmodified and LNA controls, (D) gel showing oligonucleotides with carbamate and combined LNA-carbamate modification. Combined 5′/3′-LNA and CBM1 modifications (ON11) showed increased serum resistance compared to unmodified control. | ||
Triply modified ONs were also incubated at 37 °C in foetal bovine serum (FBS) for up to 24 hours and analysed by 20% denatured polyacrylamide gel electrophoresis (PAGE) (Fig. 3 bottom). Much like the snake venom assay, addition of DNA-CBM1-linkages or isolated LNA sugars was found to be less effective towards enzymatic resistance (Fig. 3C, ON14, Fig. 3D, ON8) only stable for up to 4–8 hours. When two consecutive LNAs were added (Fig. 3C, ON15), serum enzymes were unable to fully digest the ONs cleaving only terminal regions. Enzymatic resistance can be further enhanced by combining multiple LNA sugars with multiple CBM1 linkages (Fig. 3D, ON11).
The above results suggest that nuclease enzymes can digest oligonucleotides that contain single backbone/sugar modifications; hence there is a requirement for multiple modifications to achieve stability. Carbamates themselves are not expected to be especially labile to enzymatic hydrolysis in cells, but many nucleases have large footprints holding several DNA residues close to the catalytic site.44 As a result, unmodified phosphodiester linkages within this location could be susceptible to hydrolysis. Multiple LNA/CBM1 linkages (as in ON11) presents a region with very little unmodified DNA, such that few phosphodiesters will be within range of the nuclease catalytic site. This greatly inhibits strand cleavage. Overall the enzymatic cleavage experiments show that a combination of multiple 5′/3′-LNA-CBMs provides a high level of stability to enzymatic degradation, while maintaining affinity for RNA targets.45
Conclusions
The combined effects of LNA sugars and DNA backbone analogues on DNA:DNA and DNA:RNA duplex stability are poorly understood. In this paper we have used simple and efficient automated solid-phase methods to synthesise oligonucleotides containing several carbamate-LNA and carbamate-DNA backbone combinations to shed light on this. With or without LNA, the CBM1 linkage possesses more favourable duplex stabilising properties than CBM2 and was therefore the main focus of this study. LNA either stabilises or destabilises duplexes containing carbamate backbones depending on the location of the LNA sugar relative to the CBM moiety. In DNA:DNA duplexes, and also when the modified DNA strand is paired to an RNA target (DNA:RNA hybrids) addition of LNA to the 3′-side of the CBM1 backbone partly compensates for reduced duplex stability resulting from the carbamate linkage, whereas in both DNA duplexes and DNA:RNA hybrids, addition of LNA to the 5′-side of the CBM1 backbone is strongly destabilising. The most interesting case is when a 5′/3′-LNA sandwich surrounds CBM1. This strongly destabilises DNA:DNA duplexes but the DNA:RNA hybrid is almost as stable as the unmodified duplex; for a 14-mer containing three such modified backbone linkages the DNA:RNA hybrid is a remarkable 20 °C more stable than the DNA duplex. This is significant because in general the biological target for antisense oligonucleotides is RNA, and differentiation between DNA and RNA targets is important. Another key factor to consider in a cellular context is stability to degradation. Nuclease digestion studies indicate that LNA-CBM1 modifications provide improved enzymatic resistance compared to LNA or CBM alone. Finally, LNA-CBM1 linkages cannot be degraded by phosphodiesterase enzymes into 5′-LNA mononucleotides; hence oligonucleotides containing this dual modification should not give rise to LNA fragments that could be incorporated into genomic DNA in vivo. Consequently they might have altered (potentially more favourable) toxicological profiles compared oligonucleotides which contain phosphodiester backbones or close analogues. Taking the above properties into account, LNA-carbamate oligonucleotides, and potentially other combinations of LNA with artificial DNA backbones, could be future candidates for use in diagnostic, therapeutic and in vivo imaging applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. T. is supported by funding from the Biotechnology and Biological Sciences Research Council (BBSRC) [grant number BB/M011224/1] in partnership with ATDBio Ltd. and A. H. E-S is supported by BBSRC grant BB/R008655/1, also in partnership with ATDBio Ltd. S. E. is grateful to the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine (EP/L015838/1) for a studentship, generously supported by AstraZeneca, Diamond Light Source, Defence Science and Technology Laboratory, Evotec, GlaxoSmithKline, Janssen, Novartis, Pfizer, Syngenta, Takeda, UCB and Vertex.Notes and references
- C. F. Bennett and E. E. Swayze, Annu. Rev. Pharmacol. Toxicol., 2010, 50, 259–293 CrossRef CAS PubMed.
- T. Koo and M. J. Wood, Hum. Gene Ther., 2013, 24, 479–488 CrossRef CAS PubMed.
- A. Aartsma-Rus and A. M. Krieg, Nucleic Acid Ther., 2017, 27, 1–3 CrossRef CAS PubMed.
- A. Aartsma-Rus, Nucleic Acid Ther., 2017, 27, 67–69 CrossRef CAS PubMed.
- S. T. Crooke and R. S. Geary, Br. J. Clin. Pharmacol., 2013, 76, 269–276 CrossRef CAS PubMed.
- S. M. Hoy, Drugs, 2018, 78, 1625–1631 CrossRef CAS PubMed.
- S. J. Keam, Drugs, 2018, 78, 1371–1376 CrossRef CAS PubMed.
- R. Kanasty, J. R. Dorkin, A. Vegas and D. Anderson, Nat. Mater., 2013, 12, 967–977 CrossRef CAS PubMed.
- P. E. Nielsen, Annu. Rev. Biophys. Biomol. Struct., 1995, 24, 167–183 CrossRef CAS PubMed.
- A. Shivalingam, A. E. S. Tyburn, A. H. El-Sagheer and T. Brown, J. Am. Chem. Soc., 2017, 139, 1575–1583 CrossRef CAS PubMed.
- N. Iwamoto, D. C. D. Butler, N. Svrzikapa, S. Mohapatra, I. Zlatev, D. W. Y. Sah, Meena, S. M. Standley, G. Lu, L. H. Apponi, M. Frank-Kamenetsky, J. J. Zhang, C. Vargeese and G. L. Verdine, Nat. Biotechnol., 2017, 35, 845–851 CrossRef CAS.
- W. Brad Wan and P. P. Seth, J. Med. Chem., 2016, 59, 9645–9667 CrossRef.
- A. Khvorova and J. K. Watts, Nat. Biotechnol., 2017, 35, 238–248 CrossRef CAS PubMed.
- I. Idziak, G. Just, M. J. Damha and P. A. Giannaris, Tetrahedron Lett., 1993, 34, 5417–5420 CrossRef CAS.
- A. Demesmaeker, A. Waldner, J. Lebreton, P. Hoffmann, V. Fritsch, R. M. Wolf and S. M. Freier, Angew. Chem., Int. Ed. Engl., 1994, 33, 226–229 CrossRef.
- A. De Mesmaeker, C. Lesueur, M.-O. Bévièrre, A. Waldner, V. Fritsch and R. M. Wolf, Angew. Chem., Int. Ed. Engl., 1996, 35, 2790–2794 CrossRef.
- D. Mutisya, C. Selvam, B. D. Lunstad, P. S. Pallan, A. Haas, D. Leake, M. Egli and E. Rozners, Nucleic Acids Res., 2014, 42, 6542–6551 CrossRef CAS PubMed.
- P. Tanui, S. D. Kennedy, B. D. Lunstad, A. Haas, D. Leake and E. Rozners, Org. Biomol. Chem., 2014, 12, 1207–1210 RSC.
- A. H. El-Sagheer and T. Brown, Chem. Soc. Rev., 2010, 39, 1388–1405 RSC.
- P. Kumar, A. H. El-Sagheer, L. Truong and T. Brown, Chem. Commun., 2017, 53, 8910–8913 RSC.
- P. Kumar, L. Truong, Y. R. Baker, A. H. El-Sagheer and T. Brown, ACS Omega, 2018, 3, 6976–6987 CrossRef CAS PubMed.
- S. M. Freier and K.-H. Altmann, Nucleic Acids Res., 1997, 25, 4429–4443 CrossRef CAS PubMed.
- S. K. Singh, P. Nielsen, A. A. Koshkin and J. Wengel, Chem. Commun., 1998, 455–456 RSC.
- S. K. Singh and J. Wengel, Chem. Commun., 1998, 1247–1248 RSC.
- A. A. Koshkin, S. K. Singh, P. Nielsen, V. K. Rajwanshi, R. Kumar, M. Meldgaard, C. E. Olsen and J. Wengel, Tetrahedron, 1998, 54, 3607–3630 CrossRef CAS.
- S. Obika, D. Nanbu, Y. Hari, K. Morio, Y. In, T. Ishida and T. Imanishi, Tetrahedron Lett., 1997, 38, 8735–8738 CrossRef CAS.
- S. Obika, M. Onoda, J. Andoh, T. Imanishi, K. Morita and M. Koizumi, Chem. Commun., 2001, 1992–1993 RSC.
- C. R. Cromwell, K. Sung, J. Park, A. R. Krysler, J. Jovel, S. K. Kim and B. P. Hubbard, Nat. Commun., 2018, 9, 1448 CrossRef PubMed.
- H. Kaur, B. R. Babu and S. Maiti, Chem. Rev., 2007, 107, 4672–4697 CrossRef CAS PubMed.
- J. Elmén, M. Lindow, S. Schütz, M. Lawrence, A. Petri, S. Obad, M. Lindholm, M. Hedtjärn, H. F. Hansen, U. Berger, S. Gullans, P. Kearney, P. Sarnow, E. M. Straarup and S. Kauppinen, Nature, 2008, 452, 896–899 CrossRef PubMed.
- V. Sharma, S. Singh, P. M. Krishnamurthy, J. Alterman, R. Haraszti, A. Khvorova, A. K. Prasad and J. Watts, Chem. Commun., 2017, 53, 8906–8909 RSC.
- M. A. Islam, A. Fujisaka, S. Mori, K. R. Ito, T. Yamaguchi and S. Obika, Bioorg. Med. Chem., 2018, 26, 3634–3638 CrossRef CAS PubMed.
- A. Lauritsen and J. Wengel, Chem. Commun., 2002, 5, 530–531 RSC.
- A. Waldner, A. De Mesmaeker and J. Lebreton, Bioorg. Med. Chem. Lett., 1994, 4, 405–408 CrossRef CAS.
- A. A. Koshkin, J. Fensholdt, H. M. Pfundheller and C. Lomholt, J. Org. Chem., 2001, 66, 8504–8512 CrossRef CAS PubMed.
- W. S. Mungall and J. K. Kaiser, J. Org. Chem., 1977, 42, 703–706 CrossRef CAS.
- S. Obika, O. Nakagawa, A. Hiroto, Y. Hari and T. Imanishi, Chem. Commun., 2003, 3, 2202–2203 RSC.
- E. Utagawa, M. Sekine and K. Seio, J. Org. Chem., 2006, 71, 7668–7677 CrossRef CAS PubMed.
- V. K. Sharma, S. K. Singh, K. Bohra, C. S. R. L., V. Khatri, C. E. Olsen and A. K. Prasad, Nucleosides, Nucleotides Nucleic Acids, 2013, 32, 256–272 CrossRef CAS PubMed.
- M. M. Matin, J. Appl. Sci. Res., 2008, 4, 1478–1482 CAS.
- G. S. G. De Carvalho, J.-L. Fourrey, R. H. Dodd and A. D. Da Silva, Tetrahedron Lett., 2008, 50, 463–466 CrossRef.
- P. M. McTigue, R. J. Peterson and J. D. Kahn, Biochemistry, 2004, 43, 5388–5405 CrossRef CAS PubMed.
- M. Petersen, K. Bondensgaard, J. Wengel and J. Peter Jacobsen, J. Am. Chem. Soc., 2002, 124, 5974–5982 CrossRef CAS PubMed.
- W. Yang, Q. Rev. Biophys., 2011, 44, 1–93 CrossRef CAS.
- J. Kurreck, Nucleic Acids Res., 2002, 30, 1911–1918 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ob00691e |
| This journal is © The Royal Society of Chemistry 2019 |