
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent-dependent photophysics of a red-shifted, biocompatible coumarin photocage†
Daniel
Offenbartl-Stiegert
a,
Tracey M.
Clarke
 *b,
Hugo
Bronstein
c,
Ha Phuong
Nguyen
a and
Stefan
Howorka
*a
*b,
Hugo
Bronstein
c,
Ha Phuong
Nguyen
a and
Stefan
Howorka
*a
aDepartment of Chemistry, Institute of Structural Molecular Biology, University College London, London WC1H 0AJ, UK. E-mail: s.howorka@ucl.ac.uk
bDepartment of Chemistry, University College London, London WC1H 0AJ, UK. E-mail: tracey.clarke@ucl.ac.uk
cDepartment of Chemistry, University of Cambridge, Cambridge CB2 1EW, UK
First published on 6th June 2019
Abstract
Controlling the activity of biomolecules with light-triggered photocages is an important research tool in the life sciences. We describe here a coumarin photocage that unusually combines the biocompatible optical properties of strong absorption at a long wavelength close to 500 nm and high photolysis quantum yields. The favourable properties are achieved by synthetically installing on the photocage scaffold a diethyl amino styryl moiety and a thionoester group rather than the lactone typical for coumarins. The photocage's photophysics are analysed with microsecond transient absorption spectroscopy to reveal the nature of the excited state in the photolysis pathway. The excited state is found to be strongly dependent on solvent polarity with a triplet state formed in DMSO and a charge-separated state in water that is likely due to aggregation. A long triplet lifetime is also correlated with a high photolysis quantum yield. Our study on the biocompatible photocage reveals fundamental insight for designing advanced photocages such as longer wavelengths in different solvent conditions tailored for applications in basic and applied research.
Introduction
Photocaging is a powerful approach used in basic chemistry, biology and biomedicine to control the activity of molecules with the non-invasive trigger of light under high spatial and temporal control.1–10 Photocages tune a molecule's activity by (i) forming a covalent linkage to the molecule, (ii) thereby blocking its activity, and (iii) absorbing light to photolyse the linkage in order to restore the molecule's activity.1To be compatible with advanced biomedical research, efficient photocages should feature high-yielding photolysis, which implies high extinction coefficients and photolysis quantum yields. In addition, absorption wavelengths at 500 nm or higher are increasingly demanded. Longer wavelengths are less mutagenic to biological cells,5 reach deeper into biological tissues,11 and help create scope for using a second orthologous photocage.11 Synthetic strategies to increase the absorption wavelength are to add auxochromes,12,20 extend the conjugated system,13 substitute oxygen in carbonyls or lactones,14 and combinations thereof.13 Yet, achieving long wavelength absorption and a high photolysis quantum yield within one photocage is usually difficult.11,13,15,16 Developing a photocage integrating both parameters is hence key for biological applications.11
A second demand is to understand the photocages’ fundamental photolytic process (Fig. 1). Insight is key to help design longer wavelength chromophores15 and may maximize yield of release.5 Yet, to establish design rules for efficient photocages several questions about fundamental steps after photo-induced excitation of photocages need to answered.17 For example, what is the detailed nature of the electronically excited state that induces photolysis?15 Furthermore, what is the lifetime of the relevant excited state and how does this affect the photolysis efficiency?18 In addition, to which extent do excited state and photolysis depend on the solvent in which the chromophore is dissolved? Organic solvents are often used in photophysical studies18,19 but biological use requires understanding from water-based data. A final question relates to the photolysis product as the expected release of both uncaged molecule and reformed photocage can be experimentally confirmed,20 while sometimes only the uncaged molecule is traced.13
 | ||
| Fig. 1 Schematic representation of photolysis pathways of photocage (1b) in DMSO (blue) and water (green). Note that the S1 and T1 energies (black) are approximately the same in water and in DMSO. The charge separated state (CS) is shown in blue for DMSO and green for water. The charge separated state (CS) in water (green) is lowered below the triplet state (shown by the grey arrow) and the photolysis thus proceeds via the CS state. Bottom: chemical structure of the photocage developed and investigated in this study. | ||
Here we pioneer a new coumarin photocage with long maximum wavelength of absorption and high photolysis quantum yield. Our photophysical analysis also advances the understanding of coumarin photocages (Fig. 1, 1a). Coumarins were selected as they are a widely used class of photocages2,11,13,14,21 that absorb at around 400 nm and efficiently photodissociate; they are hence compatible with cell biological research.11,13 Furthermore, they do not produce cytotoxic side-products observed for nitroaryl-based photocages that are popular for non-cellular biomolecular applications.22,23 The coumarin-based photocage presented in our study possesses an absorption maximum at 479 nm in water – longer than any previous coumarin scaffold11,13,14,24,25 – and high photolysis quantum yield.
Our report also provides a step-change in characterizing the identity of the photocage's excited state and its kinetics in organic solvent DMSO and water. We reveal that the nature of the excited state is strongly solvent dependent. A triplet state is formed in DMSO and a charge-separated state in water. The nature of the excited state is important as it influences the photolysis rate. The insight into key steps of the photolysis pathway can help design further improved photocages to feature efficient photodissociation at wavelength approaching the optical window.
Results and discussion
To achieve long-wavelength absorption for (1a), coumarin was equipped with two new moieties. A diethyl amino styryl moiety,13 and a thionoester group rather than the lactone typical for coumarins (Fig. 1).11 The synthetic route to styryl thiocoumarin (1a) is shown in Scheme 1. Iodine-containing analogue (3) was prepared via the initial diazotisation13 of amine derivative (2) and halogen substitution. The methyl group of (3) was then subjected to Riley oxidation and sodium borohydride reduction to yield alcohol (4). The hydroxyl group was subsequently protected with TIPS to give (5). TIPS was preferred over alternative protecting groups such as acetyl or tetrahydropyranyl which did not sufficiently ward off side reactions during the following synthetic steps. Using a ligand-free Heck-coupling, protected iodinated coumarin (5) was furnished with styrene (6) which had been prepared by a standard Wittig reaction. The resulting styrene coumarin (7) was then thiated using Lawesson's reagent to yield TIPS-protected thionoester (8). Following deprotection with triethylamine trihydrofluoride, coumarin (1a) was obtained. Alternative deprotecting routes such as TASF, TBAF and acyl chloride/methanol were either too harsh or inefficient. Unprotected (1a) was used for the spectroscopic analysis of the chromophore's ground state. However, (1a) was also modified with an acetyl group to produce (1b) in order to characterize the excited state of the photocage and to determine the rate of photolysis.15 Three batches of (1a) and (1b) were synthesised over the course of this study ensuring reproducible spectroscopic data.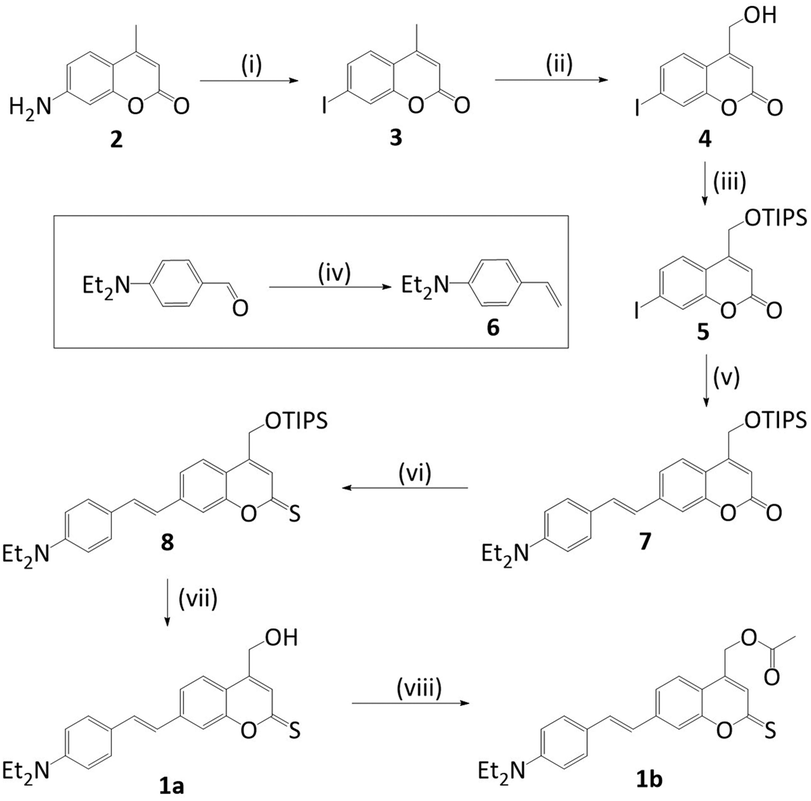 | ||
| Scheme 1 Synthesis of styryl thiocoumarin photocages (1a) and (1b). (i) H2SO4, H2O; NaNO2, KI, 0 °C; 56% (ii) SeO2, reflux; NaBH4, EtOH/THF; 45% (iii) DMAP, DIPEA, TIPS·Cl, DMF; 58% (iv) PPH3CH3Br, KOtBu, THF; 66% (v) 6, Pd(OAc)2, TEA; dry DMF; 47% (vi) Lawesson's reagent, dry toluene, reflux; 33% (vii) Et3N·3HF, THF; 39% (viii) acetic acid, DMAP, DCC, dry DCM; 76%. | ||
UV-Vis spectroscopic analysis of photocage (1a) determined the increase of absorption wavelength, λmax. Indeed, the absorption spectrum for (1a) in water (Fig. 2) reveals absorption maximum λmax at 479 nm. This is longer than for styryl coumarin (λmax = 408 nm (ref. 13)) and all other coumarin photocages reported to date.11,13,14,24,25 The broad absorption peaks along with the low solubility in water potentially indicates the presence of aggregation which is investigated later in the manuscript. The extinction coefficient ε of (1a) in water is 1.04 × 104 M−1 cm−1 (Table 1) which is in the range observed for coumarins.20 Furthermore, the absorption characteristics of (1a) are strongly dependent on the solvent (ESI Fig. S1,†Table 1).26 As an overall trend, increasing solvent polarity from hexane to water leads to a red-shift of absorption wavelength. Acetonitrile is an exception indicating that factors other than solvent polarity also play a role such as additional subtle solvent–molecule interactions. The solvatochromism may be explained by the more polar nature of the excited state compared to the ground state, and a stabilisation of the excited state by polar solvents.
 | ||
| Fig. 2 UV-Vis absorption profile of styryl thiocoumarin (1a). 50 μM in water. | ||
| Hexane | DCM | ACN | DMSO | Water | |
|---|---|---|---|---|---|
| λ max (nm) | 440 | 463 | 454 | 475 | 479 |
| ε (×104 M−1 cm−1) | 1.26 | 2.46 | 2.18 | 1.54 | 1.04 |
The detailed photophysical reason for the solvatochromism was established with density functional theory (DFT) and time-dependent (TD-DFT) calculations. The DFT calculations (CAM-B3LYP/6-31G(d), ESI Tables S2–S5†) determined the ground state geometries and molecular orbitals of (1a). The HOMO and LUMO were found to be partially spatially separated which implies a partial charge transfer upon excitation to the π–π* singlet state. To confirm stabilisation of the charge-transfer state by a more polar solvent, the energy levels of the excited state in different solvents were calculated with TD-DFT using the SCRF/PCM model. The results supported the hypothesis that the more polar solvent water stabilises the π–π* excited singlet state more effectively, with a progressive decrease in the energy of the π–π* state calculated with increasing solvent polarity. Furthermore, these calculations indicate that the lowest energy excited singlet state changes with solvent: a π–π* state in water, but an n–π* state in the non-polar solvent heptane.
After examining coumarin chromophore (1a), we investigated the photocaged compound (1b). The spectroscopic properties, including solvatochromism, of (1b) are very similar to (1a) (ESI Fig. S1 and S2†). The excited state of (1b) was experimentally elucidated with microsecond transient absorption spectroscopy (TAS).27,28 In this technique, the chromophore is excited with a beam from a pulsed laser of wavelength λpump. A probe beam with λprobe is then applied to determine the change in absorption as a function of wavelength and time delay between excitation and probing. The experimental set-up for TAS is shown in ESI Fig. S3.†
The transient absorption spectrum of the excited state of (1b) in DMSO was determined using λpump = 475 nm. As shown in Fig. 3A, the excited state of (1b) absorbs at 1090 nm and features vibronic structure with a shoulder at approximately 970 nm. The decay kinetics of the spectral response were monoexponential, and the decay lifetime, τ, invariant of pump excitation density (Fig. 3B and ESI Fig. S4†): both key characteristics of a triplet state. Fitting to a monoexponential function yielded a lifetime of τ = 63 ± 2 μs (n = 5). The presence of solvated electrons can be discounted since they absorb above 1500 nm in DMSO and have a lifetime on the order of a few nanoseconds.29,30 Our TAS analysis is the first to experimentally confirm a triplet excited state in a coumarin photocage.
 | ||
| Fig. 3 Characterisation of the excited state of (1b) with TAS. (A) Absorption spectrum of the excited triplet state showing its decay over time. Pump intensity = 37 μJ cm−2, 5 μM in DMSO, λpump = 475 nm. (B) Change in optical density versus time at different pump intensities (μJ cm−2) in DMSO. (C) Quenching and successful recovery of the transient triplet state of (1b) in nitrogen and oxygen. λprobe = 1090 nm; λpump = 475 nm, 5 μM in DMSO. | ||
To further corroborate the presence of a triplet state, the oxygen sensitivity of the transient species was investigated. Triplet excited states are known to decay faster in oxygen due to quenching via triplet–triplet annihilation with triplet oxygen.31,32 Oxygen addition should hence strongly reduce lifetime and signal amplitude of the excited state of (1b). Indeed, both parameters were considerably lower in the presence of O2 and the triplet now decayed fully within 2 μs (Fig. 3C). In further support, the absorption and lifetime of the triplet could be fully recovered when the analysis solution was degassed with N2 which is inert against triplet quenching.
Formation of a triplet state was corroborated by minimal observation of the alternative pathway of fluorescence. As illustrated in a Jablonski diagram, an optically excited chromophore in the S1 state can convert via intersystem crossing to a triplet state, or decay via fluorescence to the ground state (ESI Fig. S5†). In support of a strongly populated triplet state, the fluorescence quantum yield, ϕf, of (1b) in DMSO was so low that it could not be detected (ESI Fig. S6 and S7†); a minimal ϕf of 1% was found in DCM. The proposed efficient intersystem crossing to the triplet state is supported by this low fluorescence quantum yield, even in polar solvents such as water and DMSO where the 1π–π* state is stabilised below the non-fluorescent 1n–π* state (Tables S3 and S5–S7†). π–π* states are expected to be substantially more fluorescent than n–π* excited states because their fluorescence back to the ground state is symmetry-allowed. However, a contribution from H-aggregate formation quenching the fluorescence is also a possibility.33
After establishing the triplet nature of the excited state in DMSO, we set out to determine whether the photolysis of (1b) proceeds via the triplet state. Photolysis was investigated in O2 and in argon (O2-free) atmosphere to probe how a change in triplet lifetime affects the photolysis rate in DMSO. Photolysis was achieved using a quartz tungsten halogen lamp to irradiate the sample. The beam was passed through a bandpass filter with a centre wavelength of 475 nm. The power density at 475 nm was measured to be 6.7 mW cm−2 (ESI Fig. S8†). The progress of photolysis was determined by tracking the residual content of (1b) by HPLC, followed by normalization to the amount of starting material.13 NMR spectra of the photolysis products suggested formation of smaller fragments (ESI Fig. S9†).
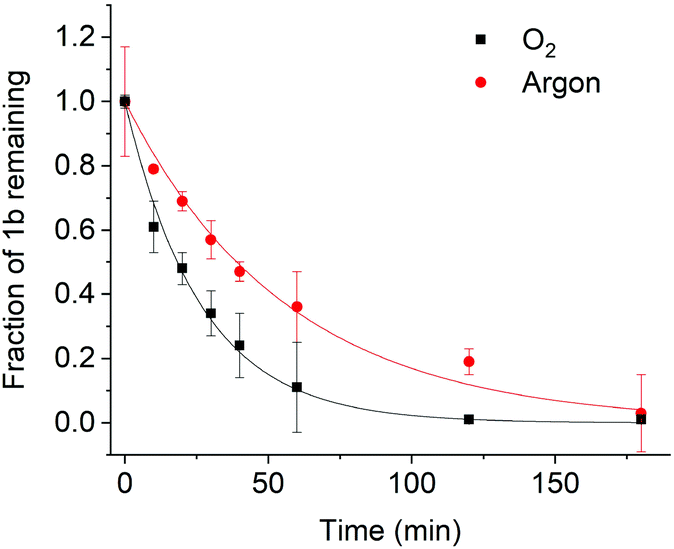
As shown in Fig. 4, (1b) uncaging in O2 has a half-life of 54 ± 6 min. This is similar to other coumarin photocages considering that a much lower light intensity of 6.7 mW cm−2 was used for (1b) compared to high intensities of up to 28 mW cm−2 for other cages13 which can be damaging to cells.15 When the photolysis reaction was studied in the absence of O2 – corresponding to a longer triplet lifetime – faster photolysis was observed (Fig. 4). The fast kinetics with a half-life of 10 ± 2 min correspond to a higher photolysis quantum yield. The strong influence of the presence or absence of oxygen on the photolysis kinetics validate the proposed relationship between a long-lived triplet state and high photolysis quantum yield.15 As further insight into the photolysis mechanism, the continued presence of photolysis under oxygen implies that photolysis must proceed very quickly once the triplet state is generated. Shortening the triplet lifetime therefore does not fully quench photolysis.
 | ||
| Fig. 4 Photolysis of (1b) in oxygen and argon using a concentration of 50 μM in DMSO. The solid lines are a fit to a monoexponential decay function. The photocage is stable in the dark even after an extended incubation time of 6 h. | ||
After demonstrating the presence of a triplet state and its link to photolysis in solvent DMSO, we analysed the excited state in water, the biologically relevant solvent. In stark contrast to our previous findings, the optical TAS analysis of (1b) could not detect any long-lived triplet excited state in water (Fig. 5). Instead, the excited state kinetics showed a power law behaviour, markedly different from the mono-exponential behaviour in DMSO that is characteristic of triplets. As another striking difference, TAS kinetics did not depend on the absence or presence of oxygen (Fig. 5). The power law kinetics and lack of oxygen sensitivity in water are consistent with a charge-separated water-stabilized state rather than a long-lived triplet state.
 | ||
| Fig. 5 Characterization of the excited state kinetics of (1b) using TAS in water and DMSO using different atmospheres. Pump intensity = 32 μJ cm−2, λpump = 475 nm, λprobe = 1090 nm. The concentration of (1b) was 5 μM. | ||
The observation of triplets in DMSO but a charge-separated state in water was investigated further using solvent-dependent TD-DFT calculations. The aim was to identify any differences in the energy levels of the singlet and triplet excited states that could account for the different photophysics in water and DMSO. However, these calculations (CAM-B3LYP/6-31G(d), ESI Tables S6 and S7†) showed that the singlet and triplet excited states had virtually identical energies and transition contributions in both solvents. Consequently, an intrinsic energetic origin to the differing behaviour is unlikely. An alternative explanation is that the energy levels of the excited state could be altered by aggregate formation, which is not accounted for in our DFT calculations.
Aggregate formation was examined via concentration-dependent steady-state absorption spectroscopy. A concentration dependence of the absorption profile was indeed observed in water but not DMSO (ESI Fig. S10 and S11†). In particular, increased absorption at the shorter-wavelength peak at higher concentrations suggest H-aggregates in water.33 The absorption of these aggregates therefore coincides with higher-lying electronic absorption bands of the compound. As further characteristic of H-aggregates, both (1a) and (1b) showed large Stoke's shifts and low fluorescence quantum yields (ESI Fig. S6 and S7†). These observations imply that aggregation stabilises the charge-separated state below that of the triplet in water, presumably by allowing a lower energy intermolecular charge-separated state to form.
To complement our analysis, we investigated the photolysis of (1b) in water in both O2 and argon. The kinetics of photolysis were in a similar time range as DMSO (Fig. 6). By contrast, the difference between O2 and argon atmospheres, with half-lives of 18 and 39 minutes, respectively, was much smaller than in DMSO. This smaller difference is consistent with the presence of charge-separated states, which relax back to the ground state independently of the presence of oxygen. The photolytic properties including quantum yield are summarised in Table 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20 and Table S8† indicating that our new dye compares favourably to other coumarin11,13,14 as well as BODIPY photocages.15
20 and Table S8† indicating that our new dye compares favourably to other coumarin11,13,14 as well as BODIPY photocages.15
 | ||
| Fig. 6 Photolysis of (1b) in oxygen and argon using a concentration of 50 μM in water, showing a much smaller difference in rate of consumption between argon and oxygen atmosphere than in DMSO. The solid lines are a fit to a monoexponential decay function. | ||
| Water (argon) | Water (O2) | DMSO (argon) | DMSO (O2) | |
|---|---|---|---|---|
| a Photolysis rate constant for irradiation at 475 nm. b Quantum yield of photolysis at λ = 475 nm. c Product of molar absorption coefficient and quantum yield of photolysis at λ = 475 nm. | ||||
| k (×10−3 s−1) | 17.8 | 37.9 | 71.4 | 12.8 |
| t 1/2 (min) | 39 | 18 | 54 | 10 |
|
Φ
475b |
0.034 | 0.071 | 0.133 | 0.024 |
|
ε
475 × Φ475c (M−1 cm−1) |
337 | 712 | 2049 | 375 |
Conclusions
This study has described a new coumarin-based photocage that overcomes two pertinent hurdles in the field. In terms of optical properties, the photocage favourably combines long wavelength and a high photolysis quantum yield not previously attained. It is therefore highly suitable for applications in biology such as to optically control the activity of nucleotides3,10 and DNA nanostructures34–37 in cellular tissues. In addition, the report has revealed the electronic nature of the excited state active in the photolysis mechanism, and its solvent dependency, something which has not been done before. In new insight, photolysis in water was shown to involve a charge-separated state while the pathway in organic solvent proceeds via a triplet state. A long triplet lifetime was correlated with higher photolysis quantum yields.The new fundamental understanding can help guide the future design of photocages with enhanced triplet population and lifetime in organic solvents and water. Routes to increase the triplet yield are to promote intersystem crossing to the triplet state, e.g. by incorporating heavy atoms or by reducing the S1 − T1 energy gap.15,38 However, these strategies do not guarantee a long triplet lifetime. Longer triplet lifetimes could be engineered by an energetically high T1 triplet state provided it is not above any charged-separated state. Alternatively, there should be no change in the orbital angular momentum between the T1 and S0 states.39,40 The future design will also have to consider the possible formation of the charge-separated state due to aggregation. The interaction may not occur when the photocage is attached to the caged compound that is large or polar enough to minimise dimerisation of photocage moieties. Another simple strategy to avoid aggregation is to improve the solubility of the photocage by introducing polar moieties. In conclusion, our study has provided a photocage with advanced bio-relevant optical properties and offers fundamental insight to establish design rules for efficient photocage that promote basic and applied research.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. H. acknowledges support by the UK EPSRC grant EP/N009282/1, BBSRC grants BB/M025373/1 and BB/N017331/1, and the Leverhulme Trust RPG-2017-015. TMC would like to acknowledge support from EPSRC project EP/N026411/1. We thank Dr Abil Aliev and Helena Philpott for helping with NMR analysis and Dr Kersti Karu for HRMS measurements.References
- G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2006, 45, 4900–4921 CrossRef CAS PubMed.
- G. C. R. Ellis-Davies, Nat. Methods, 2007, 4, 619–628 CrossRef CAS PubMed.
- N. Ankenbruck, T. Courtney, Y. Naro and A. Deiters, Angew. Chem., Int. Ed., 2018, 57, 2768–2798 CrossRef CAS PubMed.
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS PubMed.
- P. Klan, T. Solomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef CAS PubMed.
- D. Woll, S. Laimgruber, M. Galetskaya, J. Smirnova, W. Pfleiderer, B. Heinz, P. Gilch and U. E. Steiner, J. Am. Chem. Soc., 2007, 129, 12148–12158 CrossRef PubMed.
- R. H. Pawle, V. Eastman and S. W. Thomas, J. Mater. Chem., 2011, 21, 14041–14047 RSC.
- A. Patchorn, B. Amit and R. B. Woodward, J. Am. Chem. Soc., 1970, 92, 6333–6335 CrossRef.
- V. Gatterdam, T. Stoess, C. Menge, A. Heckel and R. Tampé, Angew. Chem., Int. Ed., 2012, 51, 3960–3963 CrossRef CAS PubMed.
- Q. Y. Liu and A. Deiters, Acc. Chem. Res., 2014, 47, 45–55 CrossRef CAS PubMed.
- L. Fournier, C. Gauron, L. J. Xu, I. Aujard, T. Le Saux, N. Gagey-Eilstein, S. Maurin, S. Dubruille, J. B. Baudin, D. Bensimon, M. Volovitch, S. Vriz and L. Jullien, ACS Chem. Biol., 2013, 8, 1528–1536 CrossRef CAS PubMed.
- C. M. Krauter, J. Mohring, T. Buckup, M. Pernpointner and M. Motzkus, Phys. Chem. Chem. Phys., 2013, 15, 17846–17861 RSC.
- C. Bao, G. Fan, Q. Lin, B. Li, S. Cheng, Q. Huang and L. Zhu, Org. Lett., 2012, 14, 572–575 CrossRef CAS PubMed.
- L. Fournier, I. Aujard, T. Le Saux, S. Maurin, S. Beaupierre, J. B. Baudin and L. Jullien, Chem. – Eur. J., 2013, 19, 17494–17507 CrossRef CAS PubMed.
- P. P. Goswami, A. Syed, C. L. Beck, T. R. Albright, K. M. Mahoney, R. Unash, E. A. Smith and A. H. Winter, J. Am. Chem. Soc., 2015, 137, 3783–3786 CrossRef CAS PubMed.
- I. Aujard, C. Benbrahim, M. Gouget, O. Ruel, J. B. Baudin, P. Neveu and L. Jullien, Chemistry, 2006, 12, 6865–6879 CrossRef CAS PubMed.
- Y. Kajii, T. Nakagawa, S. Suzuki, Y. Achiba, K. Obi and K. Shibuya, Chem. Phys. Lett., 1991, 181, 100–104 CrossRef CAS.
- T. Slanina, P. Shrestha, E. Palao, D. Kand, J. A. Peterson, A. S. Dutton, N. Rubinstein, R. Weinstain, A. H. Winter and P. Klan, J. Am. Chem. Soc., 2017, 139, 15168–15175 CrossRef CAS PubMed.
- A. Gandioso, S. Contreras, I. Melnyk, J. Oliva, S. Nonell, D. Velasco, J. Garcia-Amoros and V. Marchan, J. Org. Chem., 2017, 82, 5398–5408 CrossRef CAS PubMed.
- H. P. Nguyen, S. Stewart, M. N. Kukwikila, S. F. Jones, D. Offenbartl-Stiegert, S. Mao, S. Balasubramanian, S. Beck and S. Howorka, Angew. Chem., Int. Ed., 2019, 58, 6620–6624 CrossRef CAS PubMed.
- R. O. Schonleber, J. Bendig, V. Hagen and B. Giese, Bioorg. Med. Chem., 2002, 10, 97–101 CrossRef CAS PubMed.
- M. Beier and J. D. Hoheisel, Nucleic Acids Res., 1999, 27, 1970–1977 CrossRef CAS PubMed.
- H. Lusic, D. D. Young, M. O. Lively and A. Deiters, Org. Lett., 2007, 9, 1903–1906 CrossRef CAS PubMed.
- V. Hagen, B. Dekowski, V. Nache, R. Schmidt, D. Geissler, D. Lorenz, J. Eichhorst, S. Keller, H. Kaneko, K. Benndorf and B. Wiesner, Angew. Chem., Int. Ed., 2005, 44, 7887–7891 CrossRef CAS PubMed.
- V. Hagen, F. Kilic, J. Schaal, B. Dekowski, R. Schmidt and N. Kotzur, J. Org. Chem., 2010, 75, 2790–2797 CrossRef CAS PubMed.
- I. Lopez-Duarte, P. Chairatana, Y. Wu, J. Perez-Moreno, P. M. Bennett, J. E. Reeve, I. Boczarow, W. Kaluza, N. A. Hosny, S. D. Stranks, R. J. Nicholas, K. Clays, M. K. Kuimova and H. L. Anderson, Org. Biomol. Chem., 2015, 13, 3792–3802 RSC.
- R. Godin, Y. Wang, M. A. Zwijnenburg, J. Tang and J. R. Durrant, J. Am. Chem. Soc., 2017, 139, 5216–5224 CrossRef CAS PubMed.
- H. Ohkita, S. Cook, Y. Astuti, W. Duffy, S. Tierney, W. Zhang, M. Heeney, I. McCulloch, J. Nelson, D. D. Bradley and J. R. Durrant, J. Am. Chem. Soc., 2008, 130, 3030–3042 CrossRef CAS PubMed.
- U. Schindewolf, Angew. Chem., Int. Ed. Engl., 1968, 7, 190–203 CrossRef CAS.
- T. K. Cooper, D. C. Walker, H. A. Gillis and N. V. Klassen, Can. J. Chem., 1973, 51, 2195–2206 CrossRef CAS.
- C. Grewer and H. D. Brauer, J. Phys. Chem., 1994, 98, 4230–4235 CrossRef CAS.
- F. Wilkinson and A. A. AbdelShafi, J. Phys. Chem. A, 1997, 101, 5509–5516 CrossRef CAS.
- F. C. Spano and C. Silva, Annu. Rev. Phys. Chem., 2014, 65, 477–500 CrossRef CAS PubMed.
- N. C. Seeman and H. F. Sleiman, Nat. Rev. Mater., 2017, 3, 17068 CrossRef.
- J. R. Burns, A. Seifert, N. Fertig and S. Howorka, Nat. Nanotechnol., 2016, 11, 152–156 CrossRef CAS PubMed.
- S. Howorka, Science, 2016, 352, 890–891 CrossRef CAS PubMed.
- P. Chidchob, D. Offenbartl-Stiegert, D. McCarthy, X. Luo, J. N. Li, S. Howorka and H. F. Sleiman, J. Am. Chem. Soc., 2019, 141, 1100–1108 CrossRef CAS PubMed.
- M. El-Sayed, Acc. Chem. Res., 1968, 1, 8–16 CrossRef CAS.
- S. M. King, R. Matheson, F. B. Dias and A. P. Monkman, J. Phys. Chem. B, 2008, 112, 8010–8016 CrossRef CAS PubMed.
- F. B. Dias, J. Santos, D. R. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. Berberan-Santos, M. R. Bryce and A. P. Monkman, Adv. Sci., 2016, 3, 1600080 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ob00632j |
| This journal is © The Royal Society of Chemistry 2019 |