
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuPPL: Tub-tag mediated C-terminal protein–protein-ligation using complementary click-chemistry handles†
Andreas
Stengl
a,
Marcus
Gerlach
a,
Marc-André
Kasper
bc,
Christian P. R.
Hackenberger
 bc,
Heinrich
Leonhardt
a,
Dominik
Schumacher
*a and
Jonas
Helma
*a
bc,
Heinrich
Leonhardt
a,
Dominik
Schumacher
*a and
Jonas
Helma
*a
aDepartment of Biology II and Center for Integrated Protein Science Munich (CIPSM), Ludwig-Maximilians-Universität München, 82152 Planegg, Martinsried, Germany. E-mail: schumacher@biologie.uni-muenchen.de; helma@biologie.uni-muenchen.de
bLeibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP), Chemical Biology Department, Robert-Rössle-Strasse 10, 13125 Berlin, Germany
cHumboldt Universität zu Berlin, Department of Chemistry, Brook-Taylor-Str. 2, 12489 Berlin, Germany
First published on 1st April 2019
Abstract
We introduce a chemoenzymatic strategy for straightforward in vitro generation of C-terminally linked fusion proteins. Tubulin tyrosine ligase is used for the incorporation of complementary click chemistry handles facilitating subsequent formation of functional bispecific antibody-fragments. This simple strategy may serve as central conjugation hub for a modular protein ligation platform.
Recombinant fusion proteins have recently emerged as highly promising biopharmaceuticals.1,2 A prominent example are therapeutic antibody formats, such as Bispecific T-cell Engagers (BiTEs), a novel class of therapeutics, combining the antigen binding domains of two different antibodies to redirect immune cells to tumor cells.3,4 Individual proteins and their functionalities can be combined into a single biomolecule, however, how these proteins are connected is crucial but not trivial.1,5 Commonly, proteins are genetically fused as a single polypeptide. Even though genetic fusions are straightforward to construct, this approach comes with limitations such as strict C- to N-terminal linkage or the necessity of a mutual expression and purification strategy for both fusion partners.6 To circumvent these limitations alternative strategies have been described for in vitro conjugation. Although, amine or thiol reactive crosslinking reagents can serve as adaptors to covalently link proteins,7 occurring heterogeneity in conjugation site and stoichiometry can be detrimental to protein function. Chemical ligation methods like native chemical ligation (NCL) or expressed protein ligation (EPL) form native amide bonds and are established methods to increase conjugate homogeneity.8,9 However, like genetic fusions, they are also limited to N- to C-terminal linkage. The site of linkage and the relative orientation of the fusion partners can influence conjugation efficiency and functional activity.10 Therefore, different connections, such as N-to-N, C-to-C or AAx-to-AAy fusions are of high interest and became available by the development of several site-specific, bioorthogonal methods,11 for example, the incorporation of unnatural amino acids (UAAs) by amber suppression.12 Alternatively, peptide-tag based systems, have been developed for enzyme catalyzed, site-specific incorporation of bioorthogonal handles13 and applied for protein–protein-ligation.14–17 In contrast to amber suppression, tag-based systems do not require engineered expression systems and often allow the incorporation of bioorthogonal reporters of choice after expression, resulting in highly modular ligation platforms.18,19 We have recently developed the Tub-tag technology for site-specific modification of recombinant proteins.20,21 The Tub-tag system utilizes the tubulin tyrosin ligase (TTL) and an alpha-tubulin derived C-terminal recognition sequence for the incorporation of unnatural tyrosine derivatives for chemoselective conjugation.22 For example, 3-azido-L-tyrosine was efficiently ligated to several proteins and enabled their modification by strain promoted click chemistry. Moreover, we were able to show that TTL is promiscuous towards a number of different bioorthogonal handles including O-propargyl-L-tyrosine allowing the ligation of compounds complementary for copper catalyzed click chemistry (copper(I)-catalyzed alkyne–azide cycloaddition (CuAAC)) (Fig. 1A).23 Whereas incorporation efficiency of 3-azido-L-tyrosine to a fluorescently labeled 14-mer C-terminal tubulin peptide (Tub tag peptide) is reported as >90%, under identical reaction conditions the incorporation of O-propargyl-L-tyrosine is less efficient.23 To improve incorporation on the Tub-tag peptide we investigated its dependence of the tyrosine derivative concentration. While the already high incorporation efficiency of 3-azido-L-tyrosine is only slightly improved, we observe a pronounced enhancement for O-propargyl-L-tyrosine from 25% to 95% when increasing the concentration from 1 mM to 16 mM (Fig. 1B and C). In a subsequent CuAAC reaction, O-propargyl-L-tyrosine is efficiently conjugated to 3-azido-L-tyrosine-modified Tub-tag peptide. After 32 minutes full conversion was achieved as monitored by RP-UPLC (Fig. S2†). Based on these observations, we decided to examine 3-azido- and O-propargyl-L-tyrosine as orthogonal handles for the generation of C-terminal fusion proteins. Therefore, we evaluated the TTL catalyzed incorporation of 3-azido- and O-propargyl-L-tyrosine on C-terminally Tub-tagged GFP-Binding Protein (GBP)24–26 and the reactivity of the installed handles in subsequent CuAAC reactions. We observed improved incorporation at 4 mM 3-azido-L-tyrosine, whereas 1 mM is sufficient to achieve full conversion after 3 h at 37 °C. Again, we found a strong concentration dependence on the incorporation efficiency of O-propargyl-L-tyrosine as monitored by SDS-PAGE and validated using MS analysis (Fig. S3 and S4†). Based on these findings we decided to perform all subsequent experiments with 1 mM 3-azido- and 10 mM O-propargyl-L-tyrosine for 3 h at 37 °C.
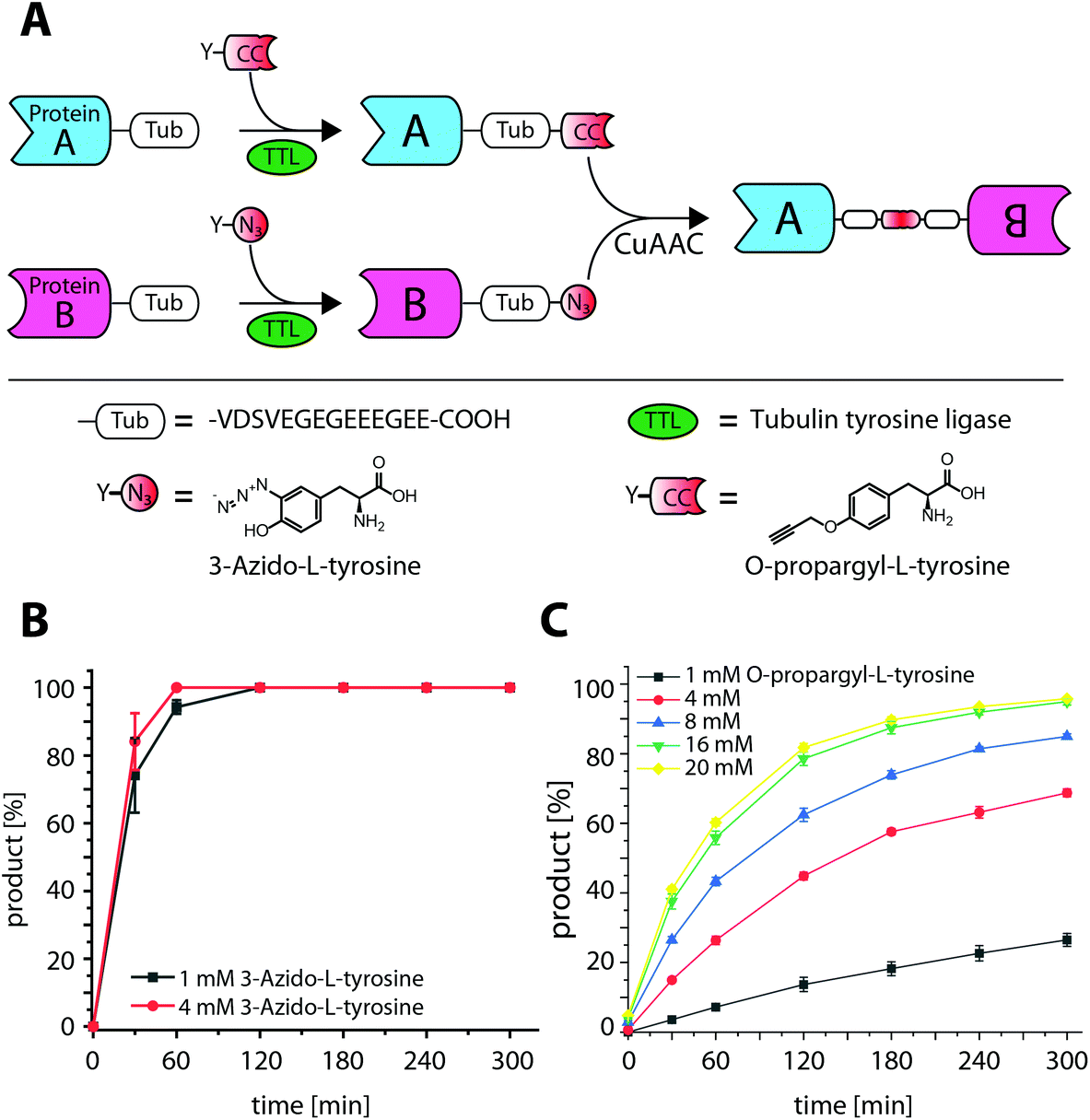 | ||
| Fig. 1 Tub-tag mediated protein–protein ligation (TuPPL) process and TTL catalyzed ligation of 3-azido- and O-propargyl-L-tyrosine with a Tub-tag peptide. (A) Schematic depiction of TuPPL. Tubulin tyrosine ligase (TTL) catalyzes the site-specific incorporation of tyrosine derivatives on a C-terminal peptide tag (Tub-tag). Incorporation of derivatives carrying complementary click chemistry handles on Tub-tagged proteins enables their C-terminal ligation by CuAAC, (B) ligation of 3-azido- and (C) O-propargyl-L-tyrosine to the Tub-tag peptide (CF-VDSVEGEGEEEGEE). Samples were taken at different time points of the TTL reaction and analyzed with RP-UPLC. Quantitation of substrate and product was performed through peak integration as described before.17 The mean values and standard deviations of three replicate reactions are shown. | ||
To validate reactivity of installed handles, we used CuAAC to conjugate 6-Carboxyfluorescein-azide (N3-6FAM) to O-propargyl-L-tyrosine modified GBP (alkynyl-GBP). Conversion from unlabeled to 6FAM labeled GBP was determined by band shifts in Coomassie stained SDS-gels and in-gel fluorescence (Fig. 2A). Quantitative formation of GBP-6FAM in 60 min demonstrated that O-propargyl-L-tyrosine can efficiently be ligated to Tub-tagged proteins and that the installed alkyne-handle is reactive for the subsequent CuAAC conjugation. In the same manner, efficient incorporation of 3-azido-L-tyrosine and reactivity was demonstrated by generating 3-azido-L-tyrosine modified GBP (azido-GBP) and subsequent labeling with biotin-PEG4-alkyne at >99% efficiency in ≤10 min (Fig. 2B). These results show that C-terminally Tub-tagged proteins can be equipped with highly CuAAC-reactive alkyne- and azide-handles by incorporation of respective tyrosine derivatives.
 | ||
| Fig. 2 CuAAC reactivity of alkynyl-GBP and azido-GBP with complementary small molecules 6FAM-azide or biotin-PEG4-alkyne, respectively. (A) Alkynyl-GBP reactivity in CuAAC reactions is demonstrated by conjugation of 6FAM-azide. The increase in molecular weight upon conjugation is observed as a band shift in SDS-PAGE analysis and detection of 6FAM fluorescence. Over 95% conversion is reached after 60 min estimated by densitometric analysis, (B) azido-GBP reactivity in CuAAC reactions is demonstrated by the conjugation of biotin-PEG4-alkyne with over 99% efficacy in ≤10 min. | ||
Encouraged by these observations we set out to generate a homodimeric fusion protein consisting of two GBP entities. In this regard, Tub-tagged GBP was functionalized either with 3-azido- or O-propargyl-L-tyrosine in two individual, but procedural similar, TTL-catalyzed reactions. In a second step, CuAAC of alkynyl-GBP and azido-GBP generated the C-terminally linked GBP-homodimer, as illustrated in Fig. 3A. We followed the click reaction over time by SDS-PAGE analysis and observed the formation of the homodimeric product GBP-GBP (Fig. 3B). Since our initial coupling conditions (2 h, 30 °C in PBS) gave only moderate conversion (∼20%) we set out to optimize the CuAAC conditions (Fig. S5†). From this screen two conditions turned out to be beneficial; firstly, lowering the pH to 5.5 with MES as buffer component and, secondly, the addition of 10% DMSO when using PBS, pH 7.4 as buffer component. At 30 °C GBP-dimer formation reaches ∼60% conversion (pH 5.5, MES) or ∼50% conversion (PBS/10% DMSO) after 10 min (Fig. 3B). We observed a slight loss of protein over time in MES, pH 5.5 presumably due to protein aggregation; however, this effect was not observed in PBS/10% DMSO. At 4 °C we also observe efficient coupling with ∼50% conversion after 90 min without any significant loss of protein. Control reactions confirm that dimer formation is dependent on the presence of both complementary functionalized GBP monomers and Cu(I) as a catalyst. These results show that by using the TuPPL workflow C-terminally linked homodimers can be generated efficiently at equimolar protein concentrations. We confirmed homodimer formation by MS-analysis (Fig. S6†). To evaluate the preserved binding of the GBP-dimers to their antigen eGFP, GBP-dimers were purified by preparative size exclusion chromatography (Fig. 4A). Due to the C-terminal linkage, both N-terminal eGFP binding sites of the GBP-dimer face opposite directions, thus, functional GBP-dimers bind two eGFP molecules.15 To test this bivalent eGFP-binding we performed analytic size exclusion chromatography of GBP-dimer incubated with eGFP at different molar ratios. At a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of eGFP to GBP-dimer we observed three products: (i) GBP-dimer, (ii) GBP-dimer bound to a single eGFP and (iii) to two eGFP molecules (Fig. 4B). A molar excess of 4:1 eGFP over GBP-dimer leads to saturation of all binding sites, confirming functional bivalency of the purified GBP-homodimer. Furthermore, we would like to emphasize the near-quantitative eGFP binding of almost all GBP-dimer molecules. This highlights that the C-terminal linkage allows quantitative bivalent binding of two molecules which has been reported to be problematic in genetic N-to-C fusions of antibodies27 and that the mild conjugation procedure fully preserves the antibodies’ antigen binding properties.
:1 molar ratio of eGFP to GBP-dimer we observed three products: (i) GBP-dimer, (ii) GBP-dimer bound to a single eGFP and (iii) to two eGFP molecules (Fig. 4B). A molar excess of 4:1 eGFP over GBP-dimer leads to saturation of all binding sites, confirming functional bivalency of the purified GBP-homodimer. Furthermore, we would like to emphasize the near-quantitative eGFP binding of almost all GBP-dimer molecules. This highlights that the C-terminal linkage allows quantitative bivalent binding of two molecules which has been reported to be problematic in genetic N-to-C fusions of antibodies27 and that the mild conjugation procedure fully preserves the antibodies’ antigen binding properties.
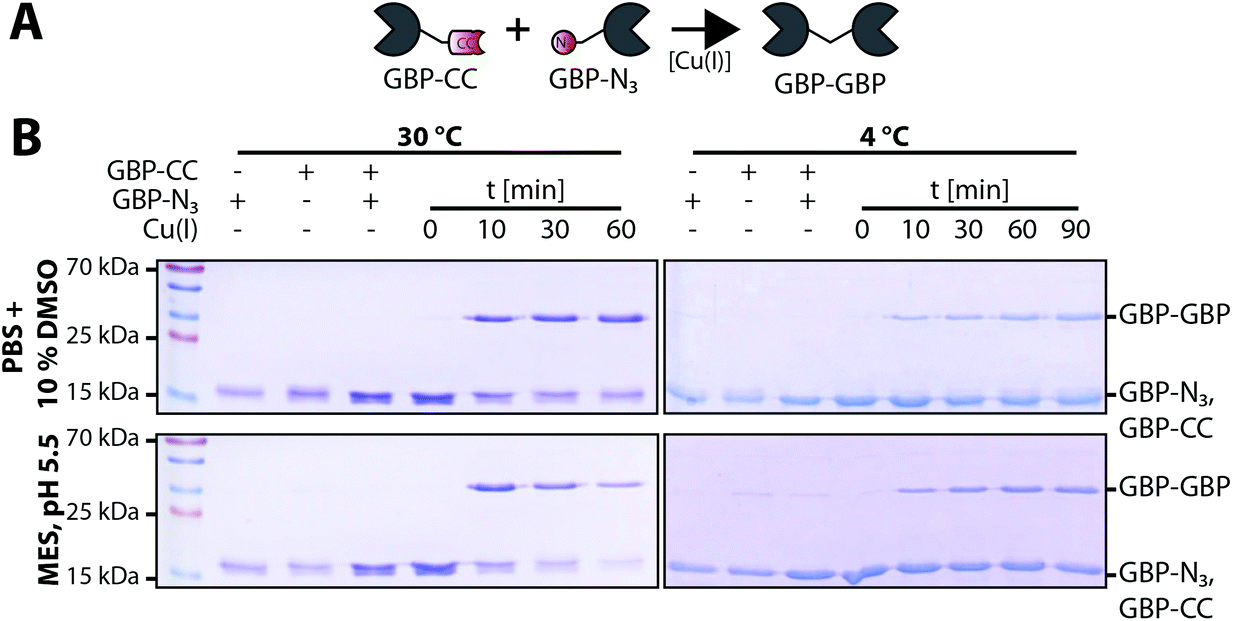 | ||
| Fig. 3 GBP-homodimer formation by CuAAC mediated ligation of alkynyl-GBP and azido-GBP. (A) Schematic of Cu(I) catalyzed C-terminal ligation of alkynyl-GBP (GBP-CC) and azido-GBP (GBP-N3) to form a covalently linked GBP-homodimer. (B) SDS-PAGE time course analysis of GBP-homodimer formation at 30 °C or 4 °C. The monomeric substrates, alkynyl-GBP and azido-GBP(∼17 kDa) form a dimeric product (GBP–GBP, ∼35 kDa). At 30 °C the reactions reach ∼60% conversion (pH 5.5, MES) or ∼50% conversion (PBS/10% DMSO) after 10 min with a loss of total protein observable in MES, pH 5.5 but not in PBS/10% DMSO. At 4 °C we also observe efficient coupling with ∼50% conversion after 90 min without any significant loss of protein. Control reactions confirm that dimer formation is dependent on the presence of (i) both complementary functionalized GBP monomers and (ii) Cu(I) as a catalyst. | ||
 | ||
| Fig. 4 Purification of GBP-homodimer and confirmation of bivalent antigen binding. (a) Size exclusion chromatogram of GBP-homodimer preparation after TuPPL and SDS-PAGE analysis of pooled peak fractions. (b) Size exclusion chromatogram of eGFP incubated with GBP-homodimer in a molar ratio of 0:1 (black), 1:1 (cyan) or 4:1 (magenta). At a 1:1 molar ratio the following species can be observed: GBP–GBP bound to (i) no, (ii) one or (iii) two eGFP molecules, whereas, complexes containing eGFP can be identified by following the absorbance at 488 nm (dashed line). A molar excess of 4:1 eGFP over GBP–GBP leads to saturation of all binding sites, confirming functional bivalency of the purified GBP-homodimer. Impurities of monomeric GBP (‡) and GBP:eGFP complex (‡‡) are present. | ||
In vitro ligation is especially beneficial for fusing proteins that require different production strategies. Thus, we finally set out to ligate two different antibody fragments isolated from different bacterial compartments. On the one hand we purified GBP from whole cell lysate and on the other hand a trastuzumab derived single chain Fragment variable (TscFv) from the periplasm. Subsequently, we functionalized both proteins with O-propargyl- or 3-azido-L-tyrosine, respectively, to generate alkynyl-GBP and azido-TscFv (MS analysis see Fig. S4†). Ligation of alkynyl-GBP and azido-TscFv by TuPPL was achieved by CuAAC (Fig. 5B) with a conjugation efficacy of 62% after 30 min and generated heterodimers (GBP-TscFv) were purified by SEC. Removal of residual copper ions was achieved by dialysis against EDTA containing buffer and confirmed by ICP-OES (Table S1†). A functional GBP-TscFv heterodimer is bispecific, whereas one paratope binds eGFP and the other the extracellular domain of the Her2-receptor. Fluorescence microscopy, shows that GBP-TscFv heterodimers recruit eGFP to the plasma membrane of Her2 overexpressing cells (SKBR3), but not to Her2 low expressing cells (MDAMB468) (Fig. 5C). These results demonstrate the functional integrity of both fusion partners after ligation, thus, confirming the applicability of TuPPL for the generation of C-terminally linked bispecific fusion proteins.
 | ||
| Fig. 5 C-Terminally linked GBP-TscFv heterodimer recruits eGFP to the plasma membrane of Her2 overexpressing (SKBR3) but not Her2 low expressing cells (MDAMB468). (A) Schematic of Cu(I)-catalyzed C-terminal ligation of alkynyl-GBP and azido-TscFv (single-chain fragment variable derived from the Her2 binding antibody trastuzumab) to form a bispecific heterodimer. (b) SDS-PAGE time course analysis of GBP-TscFv heterodimer formation at 30 °c. (C) eGFP recruitment of GBP-TscFv heterodimer to fixed Her2 overexpressing cells is demonstrated by fluorescence microscopy and verifies the functional integrity of both paratopes of the heterodimer. No recruitment can be observed when either (i) the heterodimer is excluded or (ii) Her2 low expressing cells (MDAMB468) are used. Scalebars represent 10 μm. | ||
Herein, we describe TuPPL as a modular site-specific conjugation approach for the C-terminal ligation of proteins post expression. The modularity of TuPPL allows parallel generation of azido- and alkynyl-functionalized proteins as well as straightforward ligation of proteins that are produced in different expression systems. We showed that TTL catalyzed incorporation of 3-azido-L- and O-propargyl-L-tyrosine in combination with CuAAC chemistry enables the convenient generation of homodimeric and heterodimeric antibody-fragments without diminishing the protein's function and no residual copper contaminants. Since the relative orientation of binding sites in a multimeric complex has been shown to influence their binding ability,10 fusion proteins in general might benefit from C-to-C-terminal linkage generated by TuPPL. Especially for antibodies this C-to-C linkage will be beneficial since N-terminal fusion, as generated by standard genetic fusion or EPL, can impair antigen binding due to steric obstruction of the paratope.27 In addition, our approach may readily be combined with other site-specific protein modification techniques due to the use of universal bioorthogonal handles. Future work may include the use of alternative bioorthogonal handles and conjugation reactions to expand TuPPL's modularity. Moreover, the use of TuPPL can be expanded to many more proteins such as antibodies, enzymes or proteinaceous toxins. In this case, a set of pre-functionalized proteins could serve as building blocks for a modular and scalable protein ligation platform in which TuPPL serves as a central conjugation hub.
Conflicts of interest
The Tub-tag technology is part of a patent application filed by D.S., J.H, C.P.R.H and H.L. and subject to the spin-off project Tubulis.Acknowledgements
We thank J. Koch and H. Flaswinkel for providing recombinant eGFP and H. Mescheder and T. Stoschek for critical scientific discussions. A. S. was trained and supported by the graduate school GRK1721 of the Deutsche Forschungsgemeinschaft (DFG). This work was further supported by the DFG with grants to H. L. (SFB1243, SPP1623) and C. P. R. H. (SPP1623), the Einstein Stiftung Berlin (Leibniz-Humboldt Professorship) to C. P. R. H., the Boeringer Ingelheim Fond (Plus 3 award) to C. P. R. H., by the Leibniz Association with the Leibniz Wettbewerb to C. P. R. H. and H. L. and by the German Federal Ministry for Economic Affairs and Energy with grants to D. S. and J. H. (EXIST FT I).Notes and references
- U. Brinkmann and R. E. Kontermann, mAbs, 2017, 9, 182–212 CrossRef CAS PubMed.
- K. Garber, Nat. Rev. Drug Discovery, 2014, 13, 799–801 CrossRef CAS PubMed.
- P. A. Baeuerle and C. Reinhardt, Cancer Res., 2009, 69, 4941–4944 CrossRef CAS PubMed.
- M. Mack, G. Riethmuller and P. Kufer, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7021–7025 CrossRef CAS.
- S. Berger, P. Lowe and M. Tesar, Centre d'Immunologie Pierre Fabre, in MAbs, Taylor & Francis Group, LLC, 2015, vol. 7, pp. 456–460 Search PubMed.
- M. Schmittnaegel, E. Hoffmann, S. Imhof-Jung, C. Fischer, G. Drabner, G. Georges, C. Klein and H. Knoetgen, Mol. Cancer Ther., 2016, 15, 2130–2142 CrossRef CAS PubMed.
- S. Panowski, S. Bhakta, H. Raab, P. Polakis and J. R. Junutula, mAbs, 2014, 6, 34–45 CrossRef PubMed.
- S. B. Kent, Chem. Soc. Rev., 2009, 38, 338–351 RSC.
- T. W. Muir, D. Sondhi and P. A. Cole, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6705–6710 CrossRef CAS.
- B. M. Hutchins, S. A. Kazane, K. Staflin, J. S. Forsyth, B. Felding-Habermann, P. G. Schultz and V. V. Smider, J. Mol. Biol., 2011, 406, 595–603 CrossRef CAS PubMed.
- D. Schumacher, C. P. Hackenberger, H. Leonhardt and J. Helma, J. Clin. Immunol., 2016, 36(Suppl. 1), 100–107 CrossRef CAS PubMed.
- K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS PubMed.
- D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592–605 CrossRef CAS PubMed.
- K. Wagner, M. J. Kwakkenbos, Y. B. Claassen, K. Maijoor, M. Bohne, K. F. van der Sluijs, M. D. Witte, D. J. van Zoelen, L. A. Cornelissen, T. Beaumont, A. Q. Bakker, H. L. Ploegh and H. Spits, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16820–16825 CrossRef CAS PubMed.
- M. D. Witte, J. J. Cragnolini, S. K. Dougan, N. C. Yoder, M. W. Popp and H. L. Ploegh, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11993–11998 CrossRef CAS PubMed.
- J. E. Hudak, R. M. Barfield, G. W. de Hart, P. Grob, E. Nogales, C. R. Bertozzi and D. Rabuka, Angew. Chem., Int. Ed., 2012, 51, 4161–4165 CrossRef CAS PubMed.
- D. A. Levary, R. Parthasarathy, E. T. Boder and M. E. Ackerman, PLoS One, 2011, 6, e18342 CrossRef CAS PubMed.
- T. Kruger, S. Weiland, G. Falck, M. Gerlach, M. Boschanski, S. Alam, K. M. Muller, T. Dierks and N. Sewald, Angew. Chem., Int. Ed., 2018, 57, 7245–7249 CrossRef.
- J. Lotze, U. Reinhardt, O. Seitz and A. G. Beck-Sickinger, Mol. BioSyst., 2016, 12, 1731–1745 RSC.
- D. Schumacher, J. Helma, F. A. Mann, G. Pichler, F. Natale, E. Krause, M. C. Cardoso, C. P. Hackenberger and H. Leonhardt, Angew. Chem., Int. Ed., 2015, 54, 13787–13791 CrossRef CAS PubMed.
- M. Effenberger, A. Stengl, K. Schober, M. Gerget, M. Kampick, T. R. Muller, D. Schumacher, J. Helma, H. Leonhardt and D. H. Busch, J. Immunol., 2019 DOI:10.4049/jimmunol.1801435.
- A. Banerjee, T. D. Panosian, K. Mukherjee, R. Ravindra, S. Gal, D. L. Sackett and S. Bane, ACS Chem. Biol., 2010, 5, 777–785 CrossRef CAS PubMed.
- D. Schumacher, O. Lemke, J. Helma, L. Gerszonowicz, V. Waller, T. Stoschek, P. M. Durkin, N. Budisa, H. Leonhardt, B. G. Keller and C. P. R. Hackenberger, Chem. Sci., 2017, 8, 3471–3478 RSC.
- A. Kirchhofer, J. Helma, K. Schmidthals, C. Frauer, S. Cui, A. Karcher, M. Pellis, S. Muyldermans, C. S. Casas-Delucchi, M. C. Cardoso, H. Leonhardt, K. P. Hopfner and U. Rothbauer, Nat. Struct. Mol. Biol., 2010, 17, 133–138 CrossRef CAS PubMed.
- J. Helma, M. C. Cardoso, S. Muyldermans and H. Leonhardt, J. Cell Biol., 2015, 209, 633–644 CrossRef CAS PubMed.
- U. Rothbauer, K. Zolghadr, S. Tillib, D. Nowak, L. Schermelleh, A. Gahl, N. Backmann, K. Conrath, S. Muyldermans, M. C. Cardoso and H. Leonhardt, Nat. Methods, 2006, 3, 887–889 CrossRef CAS PubMed.
- M. M. Harmsen and H. J. De Haard, Appl. Microbiol. Biotechnol., 2007, 77, 13–22 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ob00508k |
| This journal is © The Royal Society of Chemistry 2019 |