
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLacto-N-tetraose synthesis by wild-type and glycosynthase variants of the β-N-hexosaminidase from Bifidobacterium bifidum†
Katharina
Schmölzer
 a,
Melanie
Weingarten
b,
Kai
Baldenius
b and
Bernd
Nidetzky
*ac
a,
Melanie
Weingarten
b,
Kai
Baldenius
b and
Bernd
Nidetzky
*ac
aAustrian Centre of Industrial Biotechnology (acib), Petersgasse 14, 8010 Graz, Austria. E-mail: bernd.nidetzky@tugraz.at
bBASF SE, Carl-Bosch-Straße 38, 67056 Ludwigshafen, Germany
cInstitute of Biotechnology and Biochemical Engineering, Graz University of Technology, NAWI Graz, Petersgasse 12, 8010 Graz, Austria
First published on 16th May 2019
Abstract
Lacto-N-biose 1,2-oxazoline was prepared chemo-enzymatically and shown to be a donor substrate for β-1,3-glycosylation of lactose by the wild-type and glycosynthase variants (D320E, D320A, Y419F) of Bifidobacterium bifidum β-N-hexosaminidase. Lacto-N-tetraose, a core structure of human milk oligosaccharides, was formed in 20–60% yield of donor substrate (up to 8 mM product titre), depending on the degree of selectivity control by the enzyme used.
Complex oligosaccharides, like those present in human milk, are promising ingredients for health-related nutrition.1 To advance their applications, these oligosaccharides must be made available as structurally defined products. An oligosaccharide synthesis that exploits the high selectivity and reaction control of enzymes is therefore highly desirable.2 A promising strategy of enzyme development for target-oriented synthesis builds upon a glycoside hydrolase that is able to hydrolyse the required oligosaccharide product with high specificity. Converting that glycoside hydrolase into a “glycosynthase”3 by protein engineering would enable a biocatalytic synthesis in which the target oligosaccharide is assembled essentially from the structures of the hydrolytic fragments. The glycosynthase approach entails mechanistic repurposing of the glycoside hydrolase active site, so that the re-designed enzyme can still utilise a suitably activated donor substrate for glycosylation, but is unable to hydrolyse the oligosaccharide product thus formed.4
Here, we report different glycosynthase designs for the β-D-hexosaminidase LnbB from Bifidobacterium bifidum JCM 1254![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5 and explore the resulting enzyme variants, together with wild-type LnbB, in the synthesis of lacto-N-tetraose (LNT; Scheme 1).5b LNT represents a core structure of the human milk oligosaccharides (HMOs)1b and efficient route of its synthesis is of considerable interest.2,5b,6 LNT is hydrolysed by LnbB into lacto-N-biose (LNB) and lactose, as shown in earlier studies (Scheme 1a).5 LnbB is a member of glycoside hydrolase family GH-20 and had its crystal structure determined in complex with LNB (Fig. 1).5a LnbB is an exo-type enzyme that cleaves LNB from the substrate's non-reducing end (Fig. 1).5a,b The proposed enzymatic mechanism involves neighbouring group participation from the 2-acetamido group during glycosidic bond cleavage.5a The catalytic reaction proceeds through an oxazolinium ion intermediate (Scheme 1b) and has an overall retaining (β → β) stereochemical course. As suggested from studies of the mechanistically related β-D-glycosaminidases of glycoside hydrolase families GH-18,7 GH-208 and GH-85,7b,9 a promising glycosynthase strategy for LnbB involved site-directed substitution of the protein residues facilitating the attack of the 2-acetamido group, that is, Asp320 and Tyr419 (Fig. 1). Note: The LNB-thiazoline binds to LnbB very similarly as LNB does (Fig. S1, ESI†).5a To perform the glycosylation reactions, therefore, the resulting LnbB variants are to be combined with an oxazoline-activated LNB donor substrate (Scheme 1c).
5 and explore the resulting enzyme variants, together with wild-type LnbB, in the synthesis of lacto-N-tetraose (LNT; Scheme 1).5b LNT represents a core structure of the human milk oligosaccharides (HMOs)1b and efficient route of its synthesis is of considerable interest.2,5b,6 LNT is hydrolysed by LnbB into lacto-N-biose (LNB) and lactose, as shown in earlier studies (Scheme 1a).5 LnbB is a member of glycoside hydrolase family GH-20 and had its crystal structure determined in complex with LNB (Fig. 1).5a LnbB is an exo-type enzyme that cleaves LNB from the substrate's non-reducing end (Fig. 1).5a,b The proposed enzymatic mechanism involves neighbouring group participation from the 2-acetamido group during glycosidic bond cleavage.5a The catalytic reaction proceeds through an oxazolinium ion intermediate (Scheme 1b) and has an overall retaining (β → β) stereochemical course. As suggested from studies of the mechanistically related β-D-glycosaminidases of glycoside hydrolase families GH-18,7 GH-208 and GH-85,7b,9 a promising glycosynthase strategy for LnbB involved site-directed substitution of the protein residues facilitating the attack of the 2-acetamido group, that is, Asp320 and Tyr419 (Fig. 1). Note: The LNB-thiazoline binds to LnbB very similarly as LNB does (Fig. S1, ESI†).5a To perform the glycosylation reactions, therefore, the resulting LnbB variants are to be combined with an oxazoline-activated LNB donor substrate (Scheme 1c).
 | ||
| Scheme 1 Strategy of lacto-N-tetraose (LNT) synthesis from lacto-N-biose 1,2-oxazoline (LNB-oxa) by LnbB from B. bifidum. (a) Hydrolysis of LNT by wild-type LnbB. (b) Proposed catalytic mechanism of LnbB. (c) LNB 1,2-oxazoline as possible donor substrate for synthesis of LNT via glycosylation of lactose. | ||
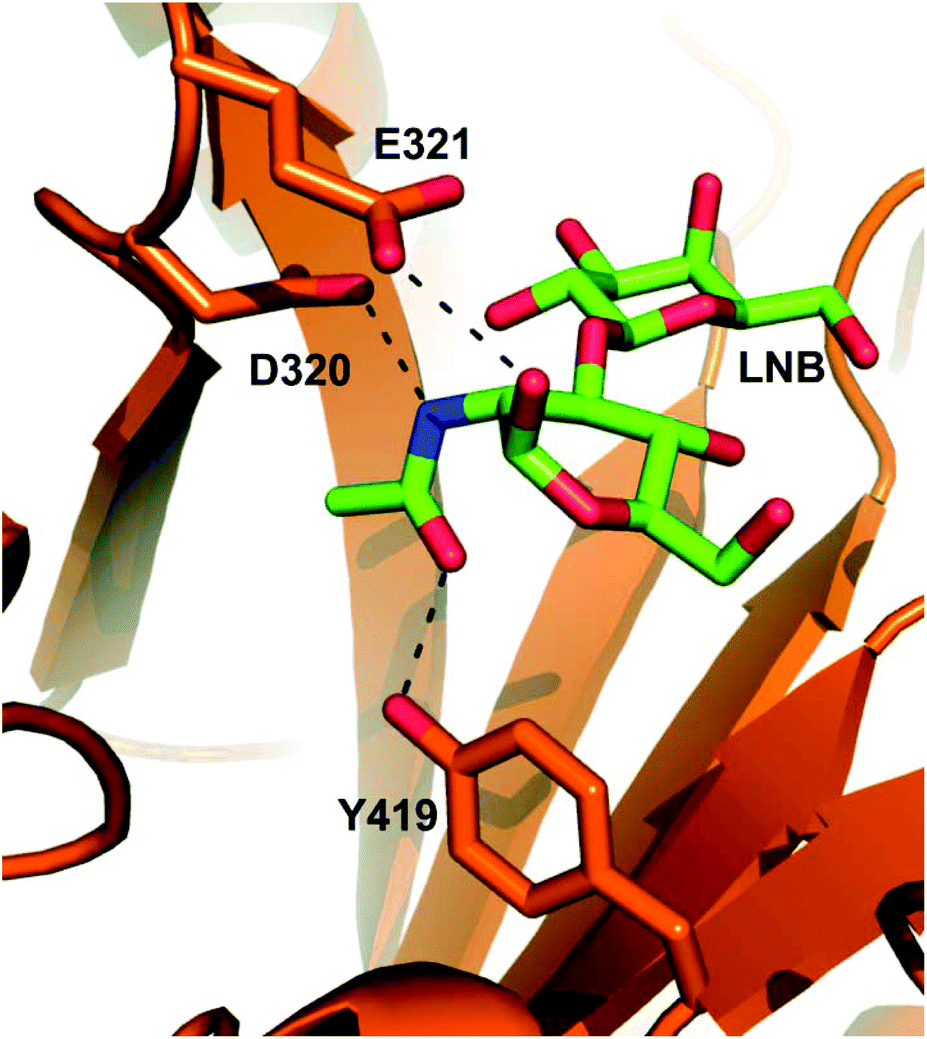 | ||
| Fig. 1 Close-up view of the active site in the experimental structure of LnbB complexed with LNB (PDB code 4H04). Key active-site residues (Asp320, polarising residue; Glu321, acid/base; Tyr419, stabilisation of oxazolinium reaction intermediate) are drawn in sticks. LNB (drawn with green-colored carbon atoms) is shown. The D320A and Y419F variants involve removal of stabilising interactions due to deletion of functional groups from the original amino acid residues. The D320E variant may involve weakened electrostatic stabilisation due to increased steric demand for the side chain of Glu as compared to the side chain of Asp. | ||
We prepared three LnbB variants (D320E, D320A, Y419F; for methods used and for protein sequence information; see ESI†) expected to show a considerably perturbed anchimeric assistance from the 2-acetamido group in their catalytic reactions (Fig. 1). The D320Q variant was also considered, but its expression in E. coli failed in our hands. Using a 4-nitro-phenyl-β-LNB (LNB-β-pNP) substrate (20 mM) for trans-glycosylation, indeed, these LnbB variants exhibited drastically (102–104-fold) decreased LNT hydrolase activity as compared to the wild-type enzyme (∼1 U mg−1; 37 °C; 15% DMSO). Previous evaluation of D320A and Y419F variants in the hydrolysis of LNB-β-pNP is consistent with this activity decrease.5a Enzymatic reactions in the presence of lactose (600 mM) showed the formation of a single trans-glycosylation product, identified as LNT by reference to an authentic standard in HPLC and TLC (Fig. S2, ESI†). However, the LNT was released in less than 13% yield of donor substrate that was converted primarily via hydrolysis. Reaction time course analysis (Fig. S3, ESI†) revealed marked differences between the individual enzymes as regards their ability to degrade the LNT formed. The wild-type enzyme hydrolysed the LNT quickly. The specific hydrolysis rate of LNT was about 100-fold lower than that of LNB-β-pNP. The Y419F variant also hydrolysed the LNT, albeit at a 25-fold slower rate than the wild-type enzyme. With both Asp320 variants, the LNT hydrolysis was barely detectable. In the D320E variant, the hydrolysis of LNT was ∼3000-fold slower than the hydrolysis of LNB-β-pNP. Reaction parameters for enzymatic conversions of LNB-β-pNP in the presence of lactose are summarised in Table 1.
| LnbB | LNB-oxaa | LNB-β-pNPb | ||||
|---|---|---|---|---|---|---|
| trans-Glycosylationc (μmol mg−1 min−1) | R TH | LNT hydrolysis (μmol mg−1 min−1) | trans-Glycosylationc (μmol mg−1 min−1) | R TH | LNT hydrolysis (μmol mg−1 min−1) | |
| a 12 mM LNB-oxa, 600 mM lactose, 37 °C, pH 7.5. b 20 mM LNB-β-pNP, 600 mM lactose, 15% DMSO, 37 °C, pH 5.8. c Hydrolase activity toward the donor substrate is given in brackets (=trans-glycosylation/RTH ratio). d R TH is the ratio of LNT to LNB concentration at maximum LNT yield. e R TH is the ratio of the enzyme activities for trans-glycosylation and LNB-β-pNP hydrolysis. f Not detectable. g Calculated based on 22 h values. Significant contribution of spontaneous LNB-oxa hydrolysis. | ||||||
| WT | 25 (∼15) | 1.7 | 9.5 × 10−1 | 4.0 (∼100) | 0.04 | 1.0 |
| D320E | 2.2 × 10−1 (∼6 × 10−1) | 0.4 | n.d.f | 2.1 × 10−1 (∼7 × 10−1) | 0.3 | 2.3 × 10−4 |
| D320A | 8.9 × 10−4 (∼4 × 10−3) | 0.2g | n.d.f | 8.0 × 10−4 (∼5 × 10−3) | 0.2 | n.d.f |
| Y419F | 7.5 × 10−2 (∼9 × 10−2) | 0.8 | 4.8 × 10−3 | 3.3 × 10−2 (∼3 × 10−1) | 0.1 | 3.8 × 10−2 |
To synthesise the LNB 1,2-oxazoline (LNB-oxa), we first prepared LNB from α-galactose 1-phosphate (5.4 mmol) and GlcNAc (1.8 mmol) using LNB phosphorylase10 (50 μg mL−1; 0.6 μM) in pH-controlled reaction (pH 6.8; 37 °C; 40 mL total volume). The LNB was recovered in an overall isolated yield of 80% by anion exchange chromatography for removal of excess α-galactose 1-phosphate. The LNB thus purified contained less than 5% GlcNAc (Fig. S4, ESI†). The 1,2-oxazoline was prepared according to a modified protocol of Shoda and co-workers (ESI†),11 applying 2-chloro-1,3-dimethyl-1H-benzimidazol-3-ium chloride (CDMBI) in 2-fold molar excess over LNB (0.7 mmol) at 3 °C. The CDMBI was added in portions over 15 min and the reaction continued for 1 h. The reaction mixture was filtered, washed and freeze-dried as described in the ESI.† Solid product was obtained at 0.5 mmol scale in about 79% yield from LNB (quantitative 1H NMR spectroscopy, Fig. S5, S6a, ESI;†13C NMR spectroscopy, Fig. S6b, ESI†) and could be stored at −20 °C for several weeks. Extraction into acetonitrile was used to desalt the product before applying it to the enzymatic reactions (ESI†).
The wild-type LnbB showed high hydrolase activity toward LNB-oxa (≥15 U mg−1). All LnbB variants were substantially (≥100-fold) less active (Table 1). Time courses of LNT formation from LNB-oxa (12 mM) and lactose (600 mM) are summarised in Fig. 2. trans-Glycosylation was selective for β-1,3-glycosylation of lactose, giving LNT (Fig. S7–S9, ESI†). Reaction of the wild-type enzyme (Fig. 2a) involved an almost instantaneous release of LNT in ∼60% yield of the LNB-oxa donor present. However, the LNT was degraded rapidly afterwards and almost none of it was left after 23 h. The specific activity of wild-type LnbB for hydrolysis of LNT (Table 1) was ∼5% that of the enzyme for the hydrolysis of LNB-oxa under the conditions used. The initial “burst” of LNT formation was used to determine the selectivity of LnbB for utilising the LNB-oxa donor substrate for glycosylation of lactose as compared to utilising it for hydrolysis (RTH). The RTH value of ∼1.7 thus obtained was considerably higher than the RTH value of 0.04 obtained for LnbB utilising LNB-β-pNP as the donor substrate.
 | ||
| Fig. 2 LNT synthesis from LNB-oxa (12 mM) and lactose (600 mM) by wild-type LnbB and mutants thereof. (a) Wild-type, 0.5 μM; (b) D320E, 4 μM; (c) D320A, 20 μM; (d) Y419F, 4 μM. LNT, black circles; LNB, green reverse triangle. The LNT and LNB concentrations used for RTH calculation are indicated by an arrow. | ||
The D320E variant was employed in 8-fold higher concentration than the wild-type LnbB to compensate its lower activity. The reaction time course for this variant showed a fast release of LNT in a yield of ∼30% of the LNB-oxa donor used (Fig. 2b). The initially formed product was stable over extended reaction times, thus demonstrating the absence of LNT hydrolase activity, and therefore the successful glycosynthase design, for this LnbB variant. However, limitation on the LNT yield in reactions of the D320E variant arose from the enzyme's relatively high activity for hydrolysis of the LNB-oxa donor substrate under the conditions used. We performed the enzymatic reaction under exactly the same conditions as in Fig. 2b but used a 5-fold higher concentration of the D320E variant (20 μM) (Fig. S10, ESI†). The LNT yield was still ∼30%. Therefore, this result showed that that the product yield was limited by enzyme selectivity (RTH). The spontaneous hydrolysis of LNB-oxa could be excluded as a relevant factor. The RTH value of ∼0.4 determined for the D320E variant from the time course data in Fig. 2b revealed the significant (∼4-fold) decrease in selectivity brought about by the site-directed substitution of Asp320 by a glutamic acid. Decreasing the activity for LNB-oxa hydrolysis is a clear task for further engineering of the D320E variant to optimise the synthetic utility of this glycosynthase for LNT production.
The D320A variant showed very low specific trans-glycosylation activity, about 105-fold lower than the specific activity of the wild-type enzyme (Table 1). It was no longer possible for this variant to compensate the low specific activity by the enzyme amount applied to the reaction. Due to the slow enzymatic reaction rate, spontaneous hydrolysis of the LNB-oxa became a significant path of the overall conversion of the donor substrate. The LNT formation by the D320A variant was therefore low, about 12% after 22 h (Fig. 2c). At this time of the reaction, all of the LNB-oxa substrate was used up. Degradation of LNT by the D320A variant was not observed.
Contrary to the D320E and D320A variants, the Y419F variant retained a substantial level of LNT hydrolase activity that was ∼6% of the activity for conversion of LNB-oxa (Table 1). The reaction time course (Fig. 2d) therefore involved characteristically the formation and degradation of LNT. The LNT accumulated to a yield of up to 30% but was completely hydrolysed at extended reaction times.
In summary, this study establishes LNB-oxa as donor substrate for trans-glycosylation by LnbB. The enzyme is of interest for target-oriented synthesis of complex oligosaccharides, but lacking a suitable donor its significant potential could hitherto not be realised.5b Here, the enzymatic LNB transfer from LNB-oxa to lactose yielded LNT in a single-step, regioselective transformation. Glycosynthase variants of LnbB, the D320E variant in particular, appear useful due to their diminished activity for product hydrolysis. However, optimized balance between LNT hydrolysis and hydrolysis of the LNB-oxa donor substrate remains a challenge for LnbB biocatalyst development. Generally, exo-acting β-glycosaminidases are less well studied for synthetic application than their endo-acting counterparts.4b,5b,7b,8a,b,12,13 For synthesis of free oligosaccharides, in contrast to synthesis of glycoconjugates, however, the exo-acting enzymes might offer better control over the product structure(s) obtained. In harnessing an exo-acting enzyme for disaccharide transfer, this study extends the scope of β-glycosaminidase-catalysed oligosaccharide synthesis.
Conflicts of interest
M. W. and K. B. are employees of BASF SE with an interest in the commercial production of chemicals. There are no conflicts to declare for K. S. and B. N.Acknowledgements
The authors thank Prof. Hansjörg Weber from the Graz University of Technology for NMR analysis and Margaretha Schiller for her excellent technical support.Notes and references
- (a) C. Kunz and S. Rudloff, Int. Dairy J., 2006, 16, 1341 CrossRef CAS; (b) L. Bode, Glycobiology, 2012, 22, 1147 CrossRef CAS PubMed.
- (a) N. S. Han, T.-J. Kim, Y.-C. Park, J. Kim and J.-H. Seo, Biotechnol. Adv., 2012, 30, 1268 CrossRef CAS PubMed; (b) X. Chen, Adv. Carbohydr. Chem. Biochem., 2015, 72, 113 CrossRef PubMed.
- (a) L. F. Mackenzie, Q. Wang, R. A. J. Warren and S. G. Withers, J. Am. Chem. Soc., 1998, 120, 5583 CrossRef CAS; (b) C. Malet and A. Planas, FEBS Lett., 1998, 440, 208 CrossRef CAS PubMed; (c) M. Moracci, A. Trincone, G. Perugino, M. Ciaramella and M. Rossi, Biochemistry, 1998, 37, 17262 CrossRef CAS PubMed; (d) Y. Honda and M. Kitaoka, J. Biol. Chem., 2006, 281, 1426 CrossRef CAS PubMed; (e) Y. Honda, S. Fushinobu, M. Hidaka, T. Wakagi, H. Shoun, H. Taniguchi and M. Kitaoka, Glycobiology, 2008, 18, 325 CrossRef CAS PubMed; (f) A. D'Almeida, M. Ionata, V. Tran, C. Tellier, M. Dion and C. Rabiller, Tetrahedron: Asymmetry, 2009, 20, 1243 CrossRef.
- (a) P. M. Danby and S. G. Withers, ACS Chem. Biol., 2016, 11, 1784 CrossRef CAS PubMed; (b) M. Faijes and A. Planas, Carbohydr. Res., 2007, 342, 1581 CrossRef CAS PubMed; (c) B. Cobucci-Ponzano, A. Strazzulli, M. Rossi and M. Moracci, Adv. Synth. Catal., 2011, 353, 2284 CrossRef CAS; (d) P. Bojarová and V. Křen, Trends Biotechnol., 2009, 27, 199 CrossRef PubMed; (e) M. R. Hayes and J. Pietruszka, Molecules, 2017, 22, 1434 CrossRef PubMed.
- (a) T. Ito, T. Katayama, M. Hattie, H. Sakurama, J. Wada, R. Suzuki, H. Ashida, T. Wakagi, K. Yamamoto, K. A. Stubbs and S. Fushinobu, J. Biol. Chem., 2013, 288, 11795 CrossRef CAS PubMed; (b) J. Wada, T. Ando, M. Kiyohara, H. Ashida, M. Kitaoka, M. Yamaguchi, H. Kumagai, T. Katayama and K. Yamamoto, Appl. Environ. Microbiol., 2008, 74, 3996 CrossRef CAS PubMed; (c) C. Val-Cid, X. Biarnés, M. Faijes and A. Planas, PLoS One, 2015, 10, e0128075 CrossRef PubMed.
- (a) T. Murata, T. Inukai, M. Suzuki, M. Yamagishi and T. Usui, Glycoconjugate J., 1999, 16, 189 CrossRef CAS; (b) F. Baumgärtner, J. Conrad, G. A. Sprenger and C. Albermann, ChemBioChem, 2014, 15, 1896 CrossRef PubMed; (c) M. Hederos, G. Dekany, S. Demkó, I. Kovács and I. Bajza, U. S. Patent, 20140323705A1, 2014 Search PubMed; (d) M. R. E. Aly, E.-S. I. Ibrahim, E.-S. H. El Ashry and R. R. Schmidt, Carbohydr. Res., 1999, 316, 121 CrossRef CAS PubMed; (e) T. Takamura, T. Chiba, H. Ishihara and S. Tejima, Chem. Pharm. Bull., 1980, 28, 1804 CrossRef CAS; (f) F. Baumgärtner, G. A. Sprenger and C. Albermann, Enzyme Microb. Technol., 2015, 75–76, 37 CrossRef PubMed.
- (a) J. P. Giddens, J. V. Lomino, M. N. Amin and L.-X. Wang, J. Biol. Chem., 2016, 291, 9356 CrossRef CAS PubMed; (b) C. Li and L.-X. Wang, Chem. Rev., 2018, 118, 8359 CrossRef CAS PubMed; (c) T. B. Parsons, W. B. Struwe, J. Gault, K. Yamamoto, T. A. Taylor, R. Raj, K. Wals, S. Mohammed, C. V. Robinson, J. L. P. Benesch and B. G. Davis, Angew. Chem., Int. Ed., 2016, 55, 2361 CrossRef CAS PubMed.
- (a) A. G. Santana, G. Vadlamani, B. L. Mark and S. G. Withers, Chem. Commun., 2016, 52, 7943 RSC; (b) K. Slámová, J. Krejzová, P. Marhol, L. Kalachova, N. Kulik, H. Pelantová, J. Cvačka and V. Křen, Adv. Synth. Catal., 2015, 357, 1941 CrossRef; (c) D. Laaf, P. Bojarová, B. Mikulová, H. Pelantová, V. Křen and L. Elling, Adv. Synth. Catal., 2017, 359, 2101 CrossRef CAS.
- M. Umekawa, W. Huang, B. Li, K. Fujita, H. Ashida, L.-X. Wang and K. Yamamoto, J. Biol. Chem., 2008, 283, 4469 CrossRef CAS PubMed.
- M. Nishimoto and M. Kitaoka, Biosci., Biotechnol., Biochem., 2007, 71, 2101 CrossRef CAS PubMed.
- M. Noguchi, T. Fujieda, W. C. Huang, M. Ishihara, A. Kobayashi and S.-I. Shoda, Helv. Chim. Acta, 2012, 95, 1928 CrossRef CAS.
- S. Kobayashi, T. Kiyosada and S.-i. Shoda, J. Am. Chem. Soc., 1996, 118, 13113 CrossRef CAS.
- (a) J. Muschiol and A. S. Meyer, Z. Naturforsch., 2019, 74c, 85 Search PubMed; (b) C. Nyffenegger, R. T. Nordvang, B. Zeuner, M. Lezyk, E. Difilippo, M. J. Logtenberg, H. A. Schols, A. S. Meyer and J. D. Mikkelsen, Appl. Microbiol. Biotechnol., 2015, 99, 7997 CrossRef CAS PubMed; (c) S. B. Jamek, J. Muschiol, J. Holck, B. Zeuner, P. K. Busk, J. D. Mikkelsen and A. S. Meyer, ChemBioChem, 2018, 19, 1858 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and supporting figures. See DOI: 10.1039/c9ob00424f |
| This journal is © The Royal Society of Chemistry 2019 |