
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Poloxin-2HT+: changing the hydrophobic tag of Poloxin-2HT increases Plk1 degradation and apoptosis induction in tumor cells†
Stefan
Rubner
,
Sabine
Schubert
and
Thorsten
Berg
 *
*
Institute of Organic Chemistry, Leipzig University, Johannisallee 29, 04103 Leipzig, Germany. E-mail: tberg@uni-leipzig.de; Fax: (+49)341 9736599
First published on 8th March 2019
Abstract
We report the hydrophobically-tagged Plk1 PBD inhibitor Poloxin-2HT+, which selectively degrades the tumor target Plk1 and induces apoptosis in human tumor cells with higher potency than the hydrophobically-tagged inhibitor Poloxin-2HT. Our data provide further evidence that hydrophobically tagged inhibitors of protein–protein interactions can target and destroy disease-relevant proteins.
The serine/threonine kinase Plk1 is overexpressed in many human tumors and has been established as an adverse prognostic marker for cancer patients.1 Since inhibition of Plk1 induces mitotic arrest and apoptosis in tumor cells, substantial drug development efforts have been made towards the modulation of Plk1 activity in tumor cells. Most Plk1 inhibitors target the enzyme's ATP binding site, and some compounds have even progressed to clinical trials.2 However, the conserved nature of the ATP binding site in protein kinases and other ATP hydrolyzing enzymes often results in ATP-competitive inhibitors experiencing selectivity problems.
In addition to its enzymatic domain, Plk1 has a protein–protein interaction domain, the polo-box domain (PBD), by which the enzyme binds to some of its targets and its intracellular anchorage sites.3,4 Because the PBD is essential for Plk1 functions and the viability of tumor cells, its inhibition has been proposed as an approach by which to induce apoptosis in tumor cells.5 Since the PBD is unique to the polo-like kinase family,1 the Plk1 PBD can be postulated to have a higher intrinsic potential for selective targeting by small-molecule inhibitors than the ATP binding site. This has inspired the development of small-molecule inhibitors6–12 and peptide-based inhibitors13–18 of the Plk1 PBD.19
Hydrophobic tagging of small molecules has been demonstrated to be a powerful approach by which to degrade, rather than inhibit, the target proteins of small bioactive molecules.20–24 The hydrophobic tag attached to the protein-bound small molecule is displayed on the surface of the protein, and is mistaken for a partially unfolded protein by the cell's protein repair machinery. After a failed refolding attempt by chaperones, the protein is targeted for proteasomal degradation.25
We recently presented the first application of hydrophobic tagging to an existing inhibitor of a protein–protein interaction domain.26 By adding a hydrophobic adamantyl tag to Poloxin-2,27 an established selective inhibitor of the Plk1 PBD, we created the fusion molecule Poloxin-2HT (1, Fig. 1A).26 The design of 1 was based on the previously reported structure–activity profile of Poloxin and derivatives.27 We had previously demonstrated that Poloxin-type molecules are irreversible inhibitors of the Plk1 PBD,27,28 and had identified two possible modes of action: either a nucleophilic attack of an amino acid within the PBD to the activated ester functionality, leading to protein acylation (Fig. 1B, upper panel), or nucleophilic addition to the imino quinone system (Fig. 1B, lower panel). Protein acylation (Fig. 1B, upper panel) appears to be more likely based on the structure–activity relationships of Poloxin and derivatives,27 mass spectrometric analysis,29 and attempts to soak Poloxin into the Plk1 PBD.30Via either mechanism, fusion of the hydrophobic tag (HT) to the o-toluoyl moiety would display the tag on the protein surface of Plk1 after binding of the fusion molecule to Plk1 within the cell (Fig. 1B).26 The validity of the design principle was confirmed by cell-based data obtained after treatment with 1, which led to selective degradation of Plk1 in a human tumor cell line, and exerted a stronger effect on cell viability and the induction of apoptosis than the parent compound Poloxin-2.26
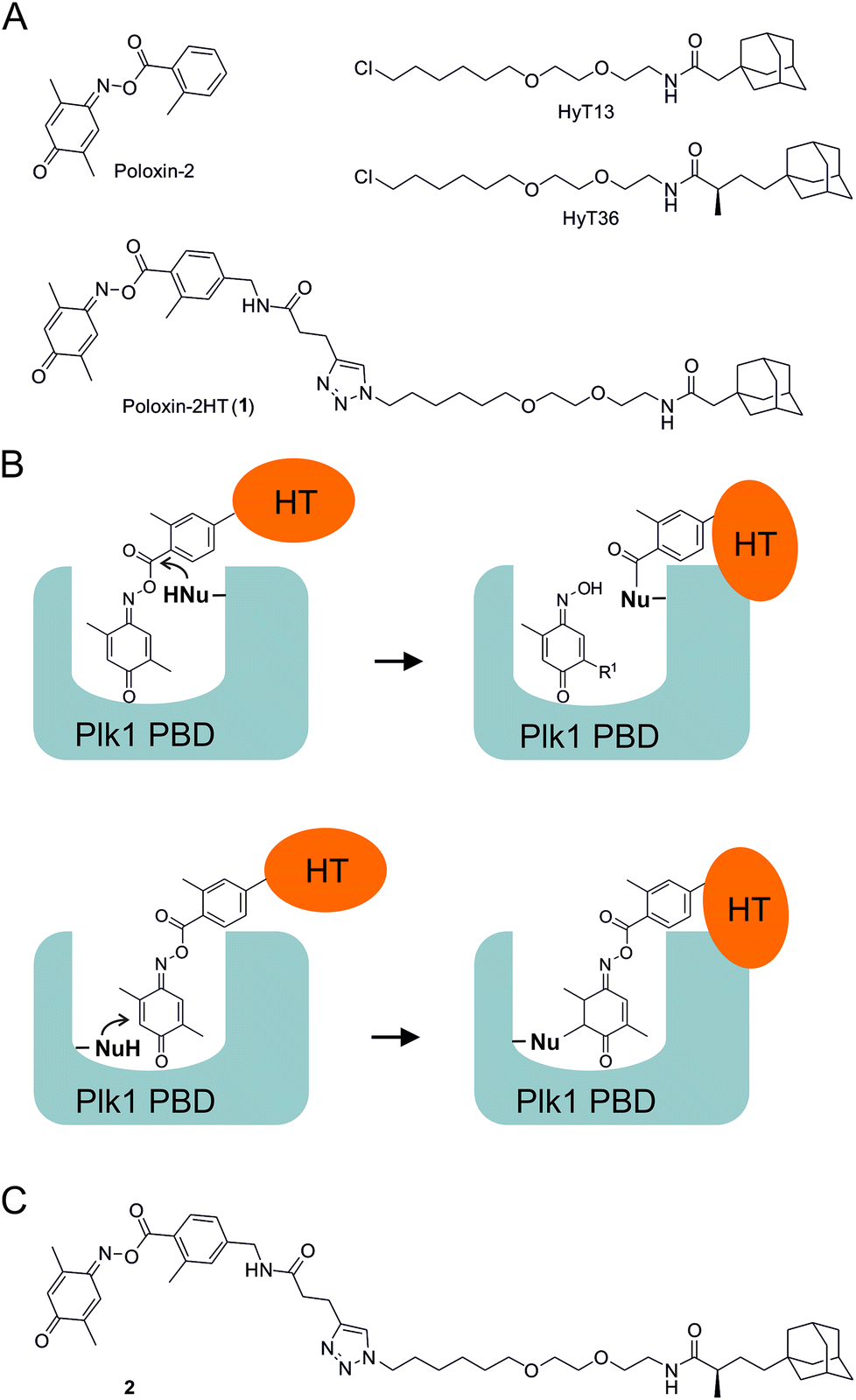 | ||
| Fig. 1 (A) Structures of Poloxin-2, the hydrophobic tags HyT13 and HyT36, and Poloxin-2HT (1). (B) Rationale for placement of the hydrophobic tag (HT) on the acyl component of Poloxin-2. (C) Structure of 2. | ||
The hydrophobic tag used in the design of 1 (HyT13, Fig. 1A) was shown by Crews and coworkers20 to degrade fusions between various proteins and HaloTag2, a modified bacterial dehalogenase that covalently reacts with halogenated alkanes.31 In a subsequent study, the Crews group demonstrated that an adamantyl tag with a methyl group in the α-position to the peptide carbonyl group (HyT36, Fig. 1A) was superior to HyT13 in degrading HaloTag7 fusion proteins, which are more resistant to degradation than HaloTag2 fusion proteins.21 Similarly, HyT36 was more effective than HyT13 in degrading the pseudokinase Her3 when fused to a covalently binding ligand of Her3.32
In this study, we aimed to explore whether the superior degradation-targeting properties of HyT36 over HyT13 also apply to chimeras between the hydrophobic tags and Poloxin-2. To this end, we synthesized a chimera consisting of Poloxin-2 and HyT36 (compound 2, Fig. 1C), in order to compare its ability to degrade Plk1 with that of 1, the chimera between Poloxin-2 and HyT13 (Fig. 1A).26
For synthesis of 2, HyT3620 was converted to the corresponding azide 3 (Fig. 2). This was coupled to the alkyne 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33via Cu(I)-catalyzed [3 + 2]-cycloaddition. Esterification of the triazole 5 with the oxime 6 produced the target compound 2. In addition, we reacted the azide 3 with the methyl ester 7 to furnish the negative control compound 8 lacking the reactive parts of Poloxin-2.
33via Cu(I)-catalyzed [3 + 2]-cycloaddition. Esterification of the triazole 5 with the oxime 6 produced the target compound 2. In addition, we reacted the azide 3 with the methyl ester 7 to furnish the negative control compound 8 lacking the reactive parts of Poloxin-2.
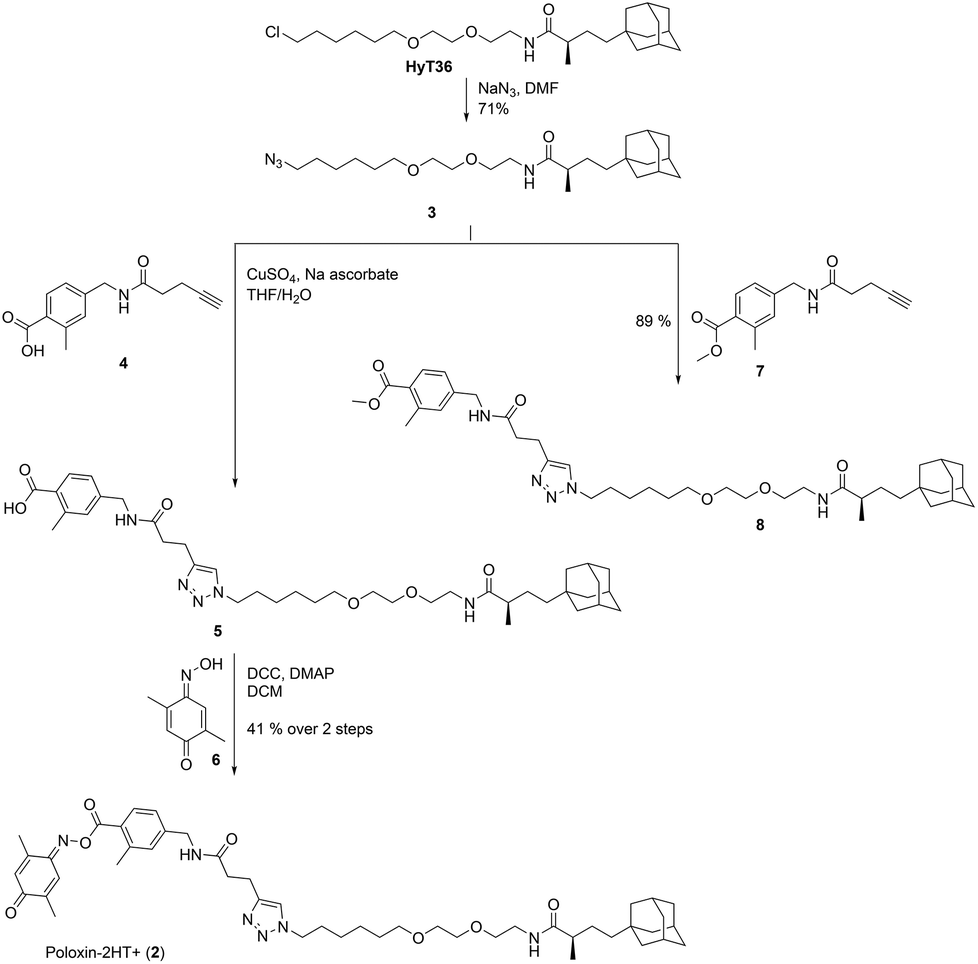 | ||
| Fig. 2 Synthesis of 2 and the negative control compound 8. | ||
2 was tested in competitive binding assays based on fluorescence polarization (FP) against the PBDs of Plk1, Plk2, and Plk3.34,352 showed slight selectivity for the Plk1 PBD (apparent IC50 = 48.2 ± 4.9 μM) over the Plk2 PBD (apparent IC50 = 54.8 ± 11.0 μM), and substantially higher selectivity over the Plk3 PBD (34 ± 4% inhibition at 100 μM, the highest concentration tested, Fig. 3A). The decrease in activity against Plk1 of 2 as compared to Poloxin-2 (apparent IC50 = 1.4 μM)27 and 1 (app. IC50 = 10.5 μM)26 demonstrates that the presence of a hydrophobic tag, as well as the tag's precise chemical structure, can influence the binding characteristics of the fusion molecules (Table S1†). Reduction of in vitro binding affinity of a bioactive molecule by addition of hydrophobic tags has previously been reported.23 The negative control compound 8 showed little activity against the Plk1 PBD (14 ± 4% inhibition at 100 μM, the highest concentration tested, Fig. 3B), demonstrating the importance of the protein-reactive parts of Poloxin-2 for inhibition of the Plk1 PBD.
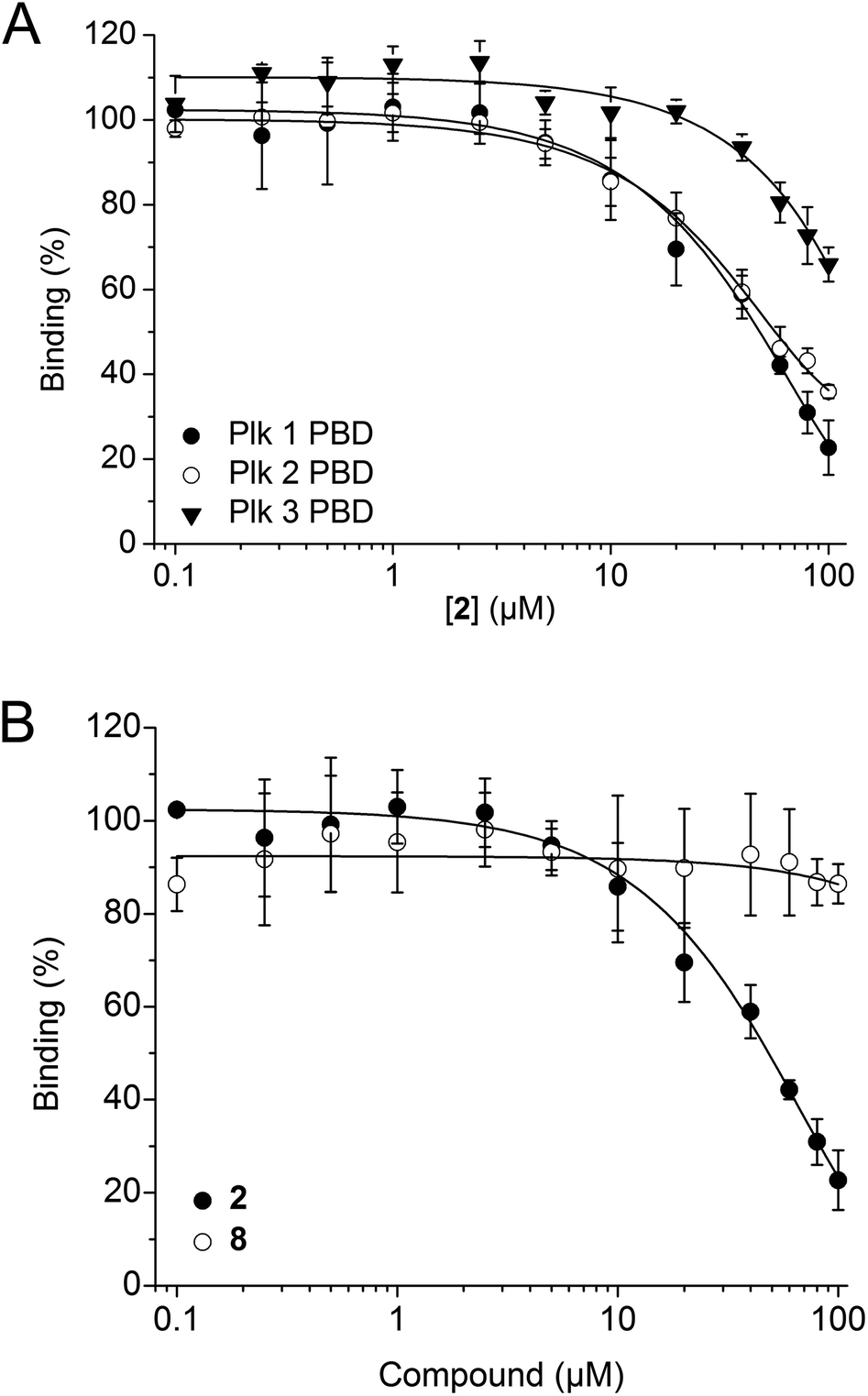 | ||
| Fig. 3 (A) Activity of 2 against the PBDs of Plk1 (n = 4), Plk2 (n = 3), and Plk3 (n = 3) in FP assays. (B) Activities of 2 (n = 4) and the negative control compound 8 (n = 3) against the Plk1 PBD. Mean values and standard deviations are shown. | ||
Functional inhibition of the Plk1 PBD by small molecules in cells induces mitotic arrest.19 Since Plk1 is upregulated in mitosis, occupancy-driven Plk1 PBD inhibitors lead to increased cellular Plk1 levels, as was reported for Poloxin-2.26 In contrast, exposure of HeLa cells to the hydrophobically tagged compound 2 for 24 h revealed a strong and dose-dependent reduction in Plk1 protein levels (apparent IC50 = 20.3 ± 3.9 μM), which exceeded the Plk1-degrading potency of 1 (apparent IC50 = 39.4 ± 2.5 μM) by twofold (Fig. 4A and B). Exposure to 50 μM 2 degraded approximately 85% of Plk1 after 24 h, while approximately 60% of Plk1 was degraded in the presence of 1. Thus, despite the lower activity of 2 compared to 1 against the Plk1 PBD in FP assays, the Plk1 degradation-targeting capabilities of HyT36 in the context of the Poloxin-2-linked chimera are superior to those of HyT13. The moderately higher activity of 2 in the cell-based assays (apparent IC50 = 20.3 ± 3.9 μM) than in the FP assays (apparent IC50 = 48.2 ± 4.9 μM) can be explained by both the longer exposure times (24 h as compared to 2 h 15 min), and the higher temperature used in the cell-based assay (37 °C) than in the FP assays (∼22 °C). Both factors, time and temperature, are expected to increase the activity of a protein-reactive compound such as 2 according to the laws of reaction kinetics. However, formal verification of this explanation is hampered by insufficient stability of the Plk1 PBD under the FP assay conditions for extended periods of time. Another possible reason for the higher activity of 2 in cells is intracellular accumulation caused by irreversible Plk1 binding.
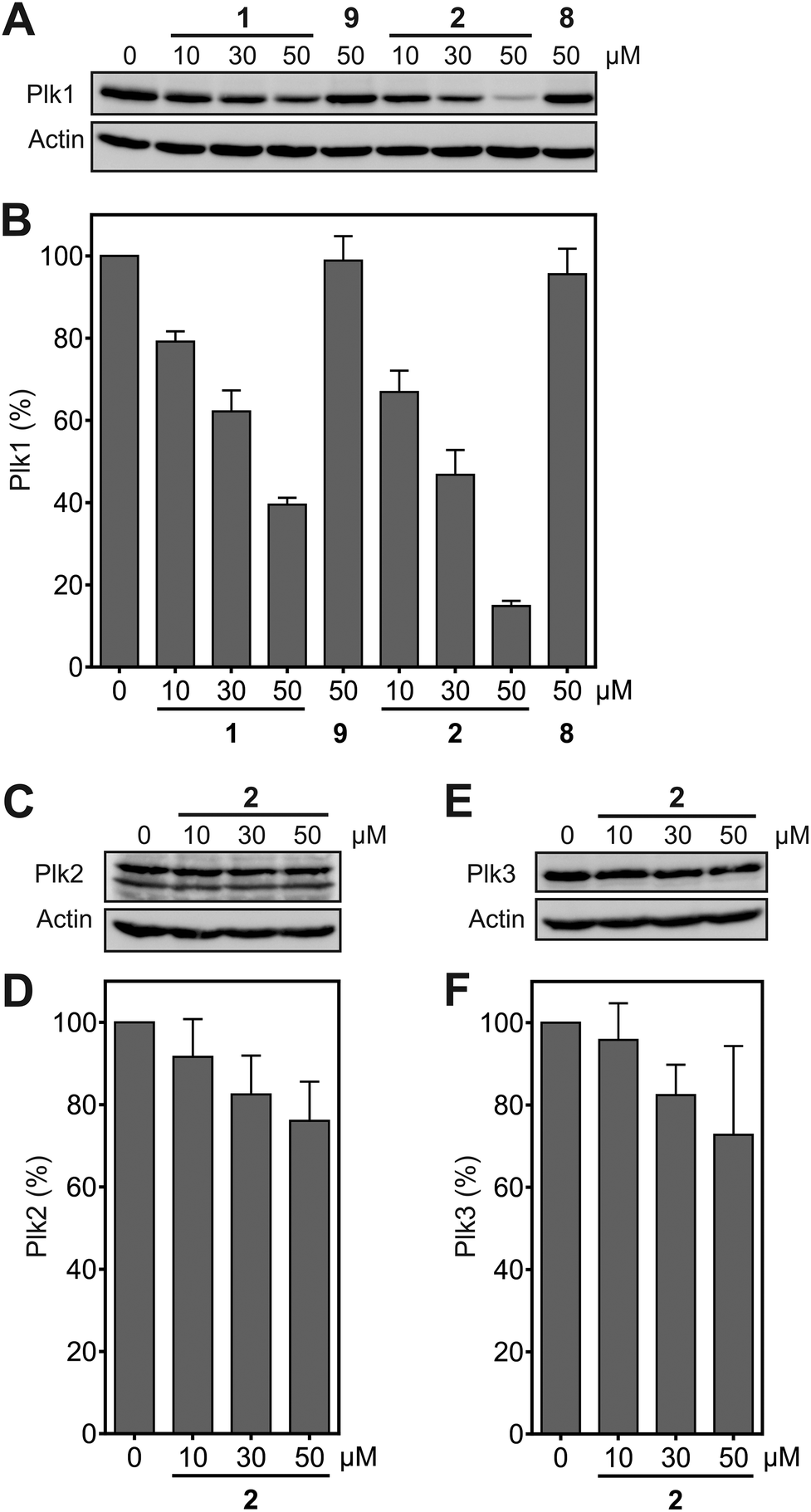 | ||
| Fig. 4 (A) Effect of 1 and 2 on protein levels of (A) Plk1 (n = 3), (B) Plk2 (n = 4), and (C) Plk3 (n = 3). (D), (E), (F) Quantification of the data shown in (A), (B), (C). Mean values and standard deviations are shown. | ||
Degradation of Plk2 (Fig. 4C and D) and Plk3 (Fig. 4E and F) under the same conditions was substantially lower than degradation of Plk1. Comparison of the selectivity of 2 for Plk1 over Plk2 in vitro (Fig. 3A) and in cell-based degradation assays (Fig. 4A–D) demonstrates a higher selectivity in cells. A similar observation has been reported for a PROTAC (proteolysis targeting chimera) between JQ1,36 a pan-selective inhibitor of the Bromo- and Extra-terminal (BET) proteins Brd2, Brd3, and Brd4, and a ligand of the E3 ubiquitin ligase von Hippel-Lindau (VHL).37 The selectivity of the PROTAC, dubbed MZ-1, for degrading Brd4 in cells was higher than the selectivity of MZ1 for Brd4 in vitro. This was rationalized by either more efficient polyubiquitination of Brd4 by MZ1 compared to Brd2 and Brd3, or alternatively, more productive formation of the ternary complex consisting of the PROTAC, its target protein Brd4, and VHL.37 The mechanism by which hydrophobic tagging induces protein degradation differs from that of PROTACS, and is still not fully understood.38,39 However, we hypothesize that, in a similar manner to that described in the PROTAC case study,37 protein degradation upon binding to 2 is more efficient for Plk1 than it is for Plk2. The negative control compounds 8, corresponding to 2, and 9 (Fig. S1†), corresponding to 1, did not cause Plk1 degradation (Fig. 4A), providing further indication for the specificity of Plk1 degradation by 2 and 1.
Since Plk1 is required for tumor cell proliferation and survival, its degradation by the hydrophobically tagged compound should reduce cell viability. Exposure of HeLa cells to 2 for 24 h induced a dose-dependent reduction of HeLa cell viability (IC50 = 9.2 ± 0.9 μM), which is a stronger effect than was observed for 1 (IC50 = 14.0 ± 0.6 μM, Fig. 5A). Similarly, flow cytometry assays revealed that 2 is a significantly more potent inducer of apoptosis in HeLa cells than 1 at concentrations up to 20 μM (Fig. 5B and Fig. S2†). Cells treated with 5 μM and 10 μM of 2 showed approximately the same percentage of apoptotic cells as cells treated with 10 μM and 20 μM 1, respectively. In order to induce apoptosis or cause cell death in virtually all tumor cells, only 20 μM of 2 was required, compared to 30 μM 1. The lower percentage of apoptotic cells upon treatment with 30 μM 2 as compared to 30 μM 1 can be ascribed to a more effective and rapid induction of apoptosis by 2, which results in a higher number of dead cells at the time of analysis. The negative control compounds 8 and 9 (Fig. S1†) did not affect cell viability or the apoptotic rate of HeLa cells, excluding the possibility of non-specific effects as the cause of the biological activities of 2 and 1 (Fig. 5A and B).
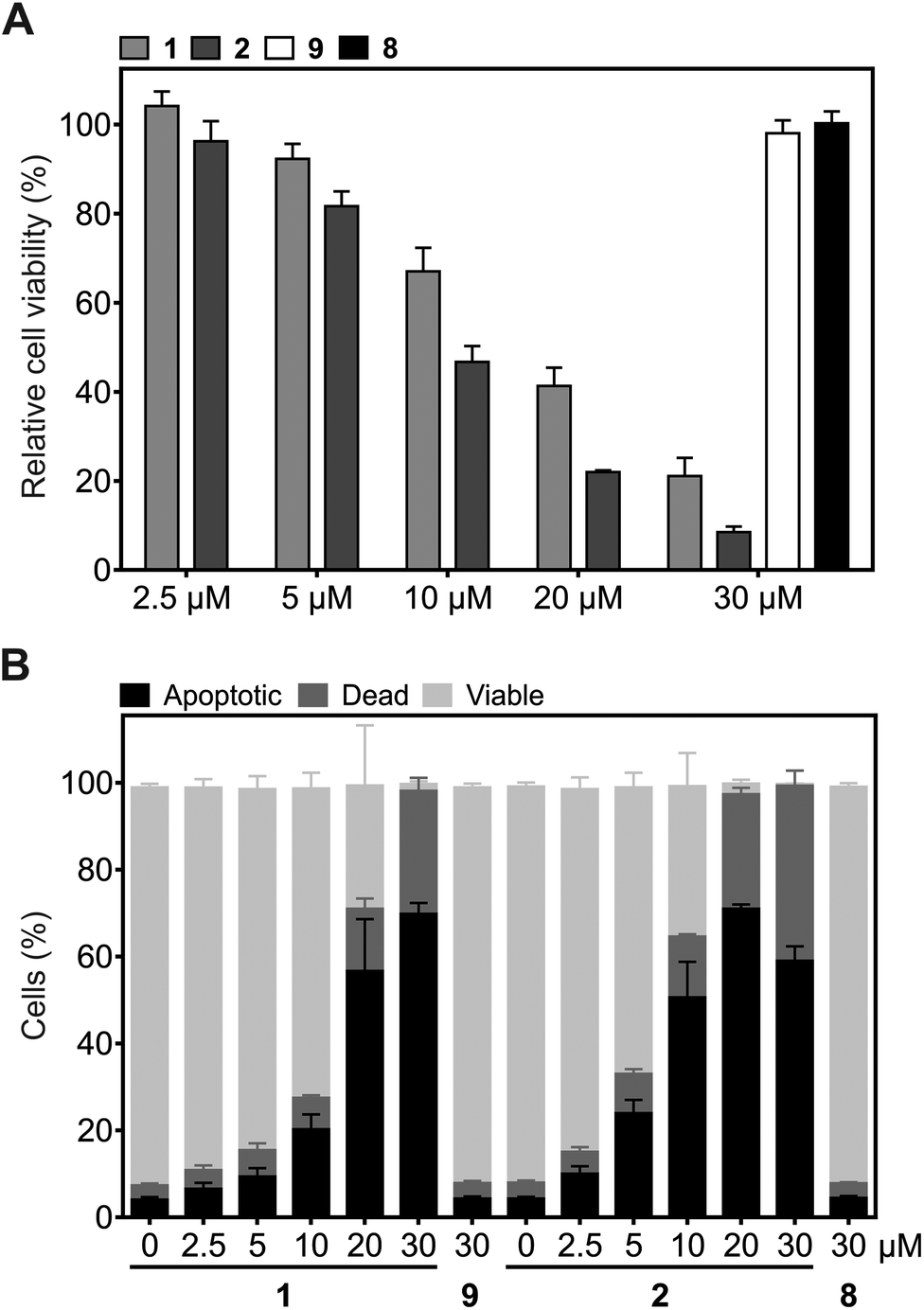 | ||
| Fig. 5 Effect of 1 and 2, and the corresponding control compounds 9 and 8, on (A) cell viability (n = 3) and (B) the induction of apoptosis in HeLa cells (n = 4). Mean values and standard deviations are shown. | ||
In conclusion, we generated compound 2 by fusing the Plk1 PBD inhibitor Poloxin-2 to the hydrophobic tag HyT36.21 Since 2 was more potent in selectively degrading Plk1 in a human tumor cell line than Poloxin-2HT (1),26 and also had a stronger effect on cell viability and the induction of apoptosis, it was dubbed Poloxin-2HT+. Our data support the notion that the hydrophobic tag HyT36 provides more potent degradation of targeted proteins than HyT13.21,32
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was generously supported by the Deutsche Forschungsgemeinschaft (BE 4572/3-1 and INST 268/281-1 FUGG), as well as the European Union and the Free State of Saxony, European Regional Development Fund. We thank the Core Unit Fluorescence Technologies of the Interdisciplinary Centre for Clinical Research (IZKF) at the Faculty of Medicine of Leipzig University (Kathrin Jäger and Andreas Lösche) for support with the flow cytometry analysis, and Andrej Scharow and Florian Nietzold for experimental support.Notes and references
- K. Strebhardt, Nat. Rev. Drug Discovery, 2010, 9, 643–660 CrossRef CAS PubMed.
- K. S. Lee, T. R. Burke Jr., J. E. Park, J. K. Bang and E. Lee, Trends Pharmacol. Sci., 2015, 36, 858–877 CrossRef CAS PubMed.
- A. E. Elia, P. Rellos, L. F. Haire, J. W. Chao, F. J. Ivins, K. Hoepker, D. Mohammad, L. C. Cantley, S. J. Smerdon and M. B. Yaffe, Cell, 2003, 115, 83–95 CrossRef CAS.
- A. E. Elia, L. C. Cantley and M. B. Yaffe, Science, 2003, 299, 1228–1231 CrossRef CAS PubMed.
- K. Strebhardt and A. Ullrich, Nat. Rev. Cancer, 2006, 6, 321–330 CrossRef CAS PubMed.
- W. Reindl, J. Yuan, A. Krämer, K. Strebhardt and T. Berg, Chem. Biol., 2008, 15, 459–466 CrossRef CAS PubMed.
- J. Yuan, M. Sanhaji, A. Kramer, W. Reindl, M. Hofmann, N. N. Kreis, B. Zimmer, T. Berg and K. Strebhardt, Am. J. Pathol., 2011, 179, 2091–2099 CrossRef CAS PubMed.
- N. Watanabe, T. Sekine, M. Takagi, J. Iwasaki, N. Imamoto, H. Kawasaki and H. Osada, J. Biol. Chem., 2009, 284, 2344–2353 CrossRef CAS PubMed.
- W. Reindl, J. Yuan, A. Krämer, K. Strebhardt and T. Berg, ChemBioChem, 2009, 10, 1145–1148 CrossRef CAS PubMed.
- Y. Chen, J. Zhang, D. Li, J. Jiang, Y. Wang and S. Si, Oncotarget, 2017, 8, 1234–1246 Search PubMed.
- A. J. Narvaez, S. Ber, A. Crooks, A. Emery, B. Hardwick, E. Guarino Almeida, D. J. Huggins, D. Perera, M. Roberts-Thomson, R. Azzarelli, F. E. Hood, I. A. Prior, D. W. Walker, R. Boyce, R. G. Boyle, S. P. Barker, C. J. Torrance, G. J. McKenzie and A. R. Venkitaraman, Cell Chem. Biol., 2017, 24, 1017–1028 CrossRef CAS PubMed.
- M. Raab, M. Sanhaji, L. Pietsch, I. Bequignon, A. K. Herbrand, E. Suss, S. L. Gande, B. Caspar, D. Kudlinzki, K. Saxena, S. Sreeramulu, H. Schwalbe, K. Strebhardt and R. M. Biondi, ACS Chem. Biol., 2018, 13, 1921–1931 CrossRef CAS PubMed.
- F. Liu, J. E. Park, W. J. Qian, D. Lim, M. Graber, T. Berg, M. B. Yaffe, K. S. Lee and T. R. Burke Jr., Nat. Chem. Biol., 2011, 7, 595–601 CrossRef CAS PubMed.
- F. Liu, J. E. Park, W. J. Qian, D. Lim, A. Scharow, T. Berg, M. B. Yaffe, K. S. Lee and T. R. Burke Jr., ChemBioChem, 2012, 13, 1291–1296 CrossRef CAS PubMed.
- F. Liu, J. E. Park, W. J. Qian, D. Lim, A. Scharow, T. Berg, M. B. Yaffe, K. S. Lee and T. R. Burke Jr., ACS Chem. Biol., 2012, 7, 805–810 CrossRef CAS PubMed.
- P. Sledz, S. Lang, C. J. Stubbs and C. Abell, Angew. Chem., Int. Ed., 2012, 51, 7680–7683 CrossRef CAS PubMed.
- P. Sledz, C. J. Stubbs, S. Lang, Y. Q. Yang, G. J. McKenzie, A. R. Venkitaraman, M. Hyvonen and C. Abell, Angew. Chem., Int. Ed., 2011, 50, 4003–4006 CrossRef CAS PubMed.
- Y. S. Tan, P. Sledz, S. Lang, C. J. Stubbs, D. R. Spring, C. Abell and R. B. Best, Angew. Chem., Int. Ed., 2012, 51, 10078–10081 CrossRef CAS PubMed.
- A. Berg and T. Berg, ChemBioChem, 2016, 17, 650–656 CrossRef CAS PubMed.
- T. K. Neklesa, H. S. Tae, A. R. Schneekloth, M. J. Stulberg, T. W. Corson, T. B. Sundberg, K. Raina, S. A. Holley and C. M. Crews, Nat. Chem. Biol., 2011, 7, 538–543 CrossRef CAS PubMed.
- H. S. Tae, T. B. Sundberg, T. K. Neklesa, D. J. Noblin, J. L. Gustafson, A. G. Roth, K. Raina and C. M. Crews, ChemBioChem, 2012, 13, 538–541 CrossRef CAS PubMed.
- T. Xie, S. M. Lim, K. D. Westover, M. E. Dodge, D. Ercan, S. B. Ficarro, D. Udayakumar, D. Gurbani, H. S. Tae, S. M. Riddle, T. Sim, J. A. Marto, P. A. Janne, C. M. Crews and N. S. Gray, Nat. Chem. Biol., 2014, 10, 1006–1012 CrossRef CAS PubMed.
- J. L. Gustafson, T. K. Neklesa, C. S. Cox, A. G. Roth, D. L. Buckley, H. S. Tae, T. B. Sundberg, D. B. Stagg, J. Hines, D. P. McDonnell, J. D. Norris and C. M. Crews, Angew. Chem., Int. Ed., 2015, 54, 9659–9662 CrossRef CAS PubMed.
- M. J. Long, D. R. Gollapalli and L. Hedstrom, Chem. Biol., 2012, 19, 629–637 CrossRef CAS PubMed.
- T. K. Neklesa and C. M. Crews, Nature, 2012, 487, 308–309 CrossRef CAS PubMed.
- S. Rubner, A. Scharow, S. Schubert and T. Berg, Angew. Chem., Int. Ed., 2018, 57, 17043–17047 CrossRef CAS PubMed.
- A. Scharow, M. Raab, K. Saxena, S. Sreeramulu, D. Kudlinzki, S. Gande, C. Dotsch, E. Kurunci-Csacsko, S. Klaeger, B. Kuster, H. Schwalbe, K. Strebhardt and T. Berg, ACS Chem. Biol., 2015, 10, 2570–2579 CrossRef CAS PubMed.
- W. Reindl, Dissertation, Technical University of Munich, 2008.
- K. Normandin, J. F. Lavallee, M. Futter, A. Beautrait, J. Duchaine, S. Guiral, A. Marinier and V. Archambault, Sci. Rep., 2016, 5, 37581 CrossRef PubMed.
- Z. Yin, Y. Song and P. H. Rehse, ACS Chem. Biol., 2013, 8, 303–308 CrossRef CAS PubMed.
- G. V. Los, L. P. Encell, M. G. McDougall, D. D. Hartzell, N. Karassina, C. Zimprich, M. G. Wood, R. Learish, R. F. Ohana, M. Urh, D. Simpson, J. Mendez, K. Zimmerman, P. Otto, G. Vidugiris, J. Zhu, A. Darzins, D. H. Klaubert, R. F. Bulleit and K. V. Wood, ACS Chem. Biol., 2008, 3, 373–382 CrossRef CAS PubMed.
- S. M. Lim, T. Xie, K. D. Westover, S. B. Ficarro, H. S. Tae, D. Gurbani, T. Sim, J. A. Marto, P. A. Janne, C. M. Crews and N. S. Gray, Bioorg. Med. Chem. Lett., 2015, 25, 3382–3389 CrossRef CAS PubMed.
- A. Scharow, D. Knappe, W. Reindl, R. Hoffmann and T. Berg, ChemBioChem, 2016, 17, 759–767 CrossRef CAS PubMed.
- W. Reindl, K. Strebhardt and T. Berg, Anal. Biochem., 2008, 383, 205–209 CrossRef CAS PubMed.
- W. Reindl, M. Gräber, K. Strebhardt and T. Berg, Anal. Biochem., 2009, 395, 189–194 CrossRef CAS PubMed.
- P. Filippakopoulos, J. Qi, S. Picaud, Y. Shen, W. B. Smith, O. Fedorov, E. M. Morse, T. Keates, T. T. Hickman, I. Felletar, M. Philpott, S. Munro, M. R. McKeown, Y. Wang, A. L. Christie, N. West, M. J. Cameron, B. Schwartz, T. D. Heightman, N. La Thangue, C. A. French, O. Wiest, A. L. Kung, S. Knapp and J. E. Bradner, Nature, 2010, 468, 1067–1073 CrossRef CAS PubMed.
- M. Zengerle, K. H. Chan and A. Ciulli, ACS Chem. Biol., 2015, 10, 1770–1777 CrossRef CAS PubMed.
- P. M. Cromm and C. M. Crews, Cell Chem. Biol., 2017, 24, 1181–1190 CrossRef CAS PubMed.
- A. C. Lai and C. M. Crews, Nat. Rev. Drug Discovery, 2017, 16, 101–114 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ob00080a |
| This journal is © The Royal Society of Chemistry 2019 |