
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Promotion of the collagen triple helix in a hydrophobic environment†
Vladimir
Kubyshkin
 *a and
Nediljko
Budisa
*ab
*a and
Nediljko
Budisa
*ab
aInstitute of Chemistry, Technical University of Berlin, Müller-Breslau-Str. 10, Berlin, 10623, Germany. E-mail: kubyshkin@win.tu-berlin.de; nediljko.budisa@tu-berlin.de; nediljko.budisa@umanitoba.ca
bUniversity of Manitoba, Faculty of Science, Dysart Rd. 144, 360 Parker Building, R3T 2N2 Winnipeg, Manitoba, Canada
First published on 6th February 2019
Abstract
In contrast to many other water-soluble peptide arrangements, the formation of a triple helix in collagen proceeds inside out: polar glycyl residues form the interior, whereas nonpolar prolyl side chains constitute the exterior. In our work, we decided to exploit this aspect of the peptide architecture in order to create hyperstable collagen mimicking peptides (CMPs). The key element of this study is the environment. Given that the peptide assembles in a nonpolar medium, the collapse of the polar peptide backbone into the triple helix should become more favorable. Following this idea, we prepared CMPs based on hydrophobic proline analogues. The synthesis was performed by a combination of liquid- and solid-phase approaches: first, hexapeptides were prepared in solution, and then these were launched into conventional Fmoc-based peptide synthesis on a solid support. The resulting peptides showed an excellent signal of the triple helix in the model nonpolar solvent (octanol) according to circular dichroism observations. In a study of a series of oligomers, we found that the minimal length of the peptides required for triple helical assembly is substantially lower compared to water-soluble CMPs. Our results suggest further explorations of the CMPs in hydrophobic media; in particular, we highlight the suggestion that collagen could be converted into a membrane protein.
Introduction
The assembly of peptide structures is a key feature in numerous natural phenomena, such as tertiary and quaternary folding of proteins, protein–protein interactions and more. Hydrophobic effect usually plays a major role in peptide assembly in aqueous media. For example, a tertiary fold is commonly described using a simplistic “apolar in – polar out” principle, also known as “hydrophobic collapse”.1 It is interesting to note that collagen assembly in a triple helix can violate this principle. This argument can be illustrated with collagen mimicking peptides (CMPs), repetitive sequences based on a prolyl-prolyl-glycyl triplet scaffold. When forming a triple helix, the parent peptide (Pro-Pro-Gly)n places most polar glycyl residues inward, thereby forming interchain hydrogen bonds. At the same time, relatively hydrophobic prolyl residues form the exterior of the resulting triple helix.2–4 Post-translational5 or synthetic6 hydroxylations of prolyl fragments partially restore the polarity of the collagen exterior,7 although it is believed that the role of this modification is different for collagen stability.8The fact that the collagen helix formation is driven by polar interaction between backbone amides (“polar in”) is rather peculiar since this assembly occurs in an aqueous medium, which is polar as well. It would be reasonable to assume that the collagen assembly is better suited for a nonpolar environment, where the lipophobic collapse of the chains into the triple helix would hide polar NH fragments from their exposure to the medium. Interestingly, in this case, the formation of the triple helix would resemble the assembly principle of transmembrane α-helical bundles. In the latter structures, compact glycine residues are often utilized to form interhelical interfaces, thereby causing protein assembly in the hydrophobic environment of a lipid membrane.9–11 We were intrigued by this comparison and asked ourselves whether it is possible to construct a CMP capable of assembling in a hydrophobic environment of organic solvents or biomembranes. Thereby, collagen could be converted into a membrane handle or even a fully artificial membrane protein. CMPs capable of self-assembling in hydrophobic media might also be functionalized with various cargoes to mimic the microenvironment of the different biological settings (e.g., extracellular matrix, cell adhesion or signalling) important for applications in materials science and medicine.12
The literature has hitherto described a large number of CMPs chemically modified at the prolyl residues. Nonetheless, these peptides usually contain additional ionisable, polar, metal coordinating and other functional groups that are not significantly hydrophobic.13,14 The modifications aim to manipulate, preferably enhance, the stability of the triple helix formed by CMPs,15,16 drive their high-order self-assembly,17,18 or otherwise enable the use of CMPs as a scaffold for functionalization.19,20 As an alternative approach to manipulate the stability of the collagen structure, there are modifications which impact the nucleation of the triple helix.21,22 For example, Cejas et al. modified CMPs with hydrophobic ‘sticky ends’ in order to facilitate the formation of collagen fibrils through hydrophobic interactions at the termini.23,24 None of these approaches targeted an assembly of the triple helix in media that would significantly differ from water. In this context, a notable observation was recently reported by Yao et al.: they have demonstrated that the addition of polar terminal residues to the CMP scaffold promotes their assembly into a separate phase in water, thereby forming nanospherical particles.25,26
As the conclusion from our literature survey, there is an unfortunate lack of information on whether CMPs would assemble in a hydrophobic environment, and how the triple helix is maintained under non-aqueous conditions. To this end, we decided to design CMPs with an amplified hydrophobicity of the triple helical exterior, with the idea that this would force the peptides to assemble in nonpolar environments. Our basic motive is to show that the collagen helix is a peptide assembly that is fully adaptable to nonpolar conditions, where it creates an enormously stable triple helix according to the “polar in – apolar out” principle.
Results and discussion
Design of the peptides
We started with the design of the peptide sequence based on the typical CMP peptide scaffold composed of repeating amino acid triplets (ProX-ProY-Gly)n (Fig. 1A). It is well documented in the literature that the stability of the triple helix depends on the side-chain conformation of the prolyl residues.8 Stabilization of the triple helix occurs when ProX adopts a C4-endo envelope conformation of the pyrrolidine ring, while ProY adopts the C4-exo conformation (Fig. 1B). In the first step, we decided to replace the ProY residue with (2S,3aS,7aS)-octahydroindole-2-carboxylic acid (Oic, Fig. 1C). Previously we have demonstrated that this proline analogue combines a strong bias towards the C4-exo conformation with the stabilized trans-amide bond,27 and exhibits high hydrophobicity.28 For example, oligomeric Oic sequences form a stable polyproline-II helix, and these peptides can be dissolved in a variety of organic solvents including alkanes.28 Therefore, we designed the sequence acetyl-(Pro-Oic-Gly)10-OH (AcPOG10) as the target sequence for the peptide synthesis (Fig. 1D).‡ In addition to this peptide, we have thought of improving the peptide hydrophobicity by replacing ProX with a more hydrophobic analogue, 5-azaspiro[2.4]heptane-6-carboxylic acid (Ash, Fig. 1C). According to our design, the more hydrophobic target sequence was pivaloyl-(Ash-Oic-Gly)10-OH (PivσOG10). Both Oic and Ash are considerably cheap proline analogues, since Oic is a component of an antihypertensive drug perindopril, while Ash is a precursor to ledipasvir, an anti-HCV drug launched on the market in 2014. We believe that accessibility of the starting amino acids can become an important prerequisite for further research exploration of the designed peptide sequences. | ||
| Fig. 1 Design of the study. | ||
Lipophilicity of the peptide triplets
Next, to illustrate the hydrophobicity of the peptide triplets, we prepared a series of model peptides with C-terminal 2,2-difluoroethyl esters, which were installed according to a recently described method.29 The resulting peptides were subjected to octan-1-ol/water partitioning, and equilibrium (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) values were read-out by 19F NMR analysis (Fig. 2). As can be seen from the resulting experimentally determined logP values, the original Pro-Pro-Gly triplet is rather hydrophilic (logP < 0), while the hydroxylation at the ProY position notably enhances this hydrophilicity. At the same time, the triplets containing Oic at the ProY position are hydrophobic (logP > 0), as desired for the final target sequences. Therefore, we expected sufficient hydrophobicity of the designed hydrophobic CMPs that should enable their solubility in nonpolar solvents.
P) values were read-out by 19F NMR analysis (Fig. 2). As can be seen from the resulting experimentally determined logP values, the original Pro-Pro-Gly triplet is rather hydrophilic (logP < 0), while the hydroxylation at the ProY position notably enhances this hydrophilicity. At the same time, the triplets containing Oic at the ProY position are hydrophobic (logP > 0), as desired for the final target sequences. Therefore, we expected sufficient hydrophobicity of the designed hydrophobic CMPs that should enable their solubility in nonpolar solvents.
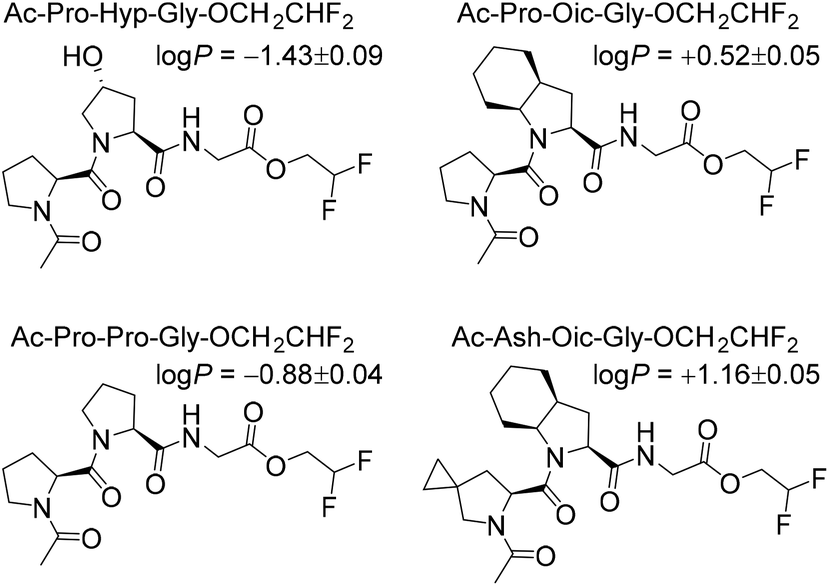 | ||
| Fig. 2 Octan-1-ol/water partitioning constants (logP) for model peptides (at 22 °C). | ||
Peptide synthesis
We then performed the optimization of the synthesis for the target peptides. It is common for CMPs to be synthesized by a combination of liquid- and solid-phase peptide syntheses. This strategy allows the minimization of coupling steps and helps to ensure better purity of the resulting peptides. Thus, we first explored the potential of synthesis steps in the solution. All peptide couplings were performed using HATU as the coupling reagent in a 1:1 dichloromethane–dimethylformamide mixture as a solvent. Using these conditions, we prepared the triplet peptide Boc-Pro-Oic-Gly-OBn in an excellent 90% yield on a gram scale (Scheme 1A). Then, we tried to couple two triplets together to form Boc-(Pro-Oic-Gly)2-OBn, which resulted in a moderate yield of about 53% (Scheme 1B). In addition, the separation from the residual unreacted starting triplet Boc-Pro-Oic-Gly-OH was quite tedious when using this approach.
 | ||
| Scheme 1 Optimization of the peptide synthesis. | ||
We thus decided to synthesize the hexapeptide by adding amino acids one-by-one instead of coupling two triplets together. Although the coupling of the next three amino acids proceeded smoothly, and the yield in each step was again reduced to approximately 80%, which did not improve the overall yield of the peptide Boc-(Pro-Oic-Gly)2-OBn (55%, Scheme 1C). Nonetheless, the advantage of this approach is that it allows easy and complete separation of the peptide from excessive N-Boc amino acids at each step of the synthesis. The peptide Boc-(Ahx-Oic-Gly)2-OBn was prepared analogously using this approach on a gram scale (Scheme 1D). We then converted the peptides into the N-terminally Fmoc-protected hexapeptides (Scheme 1E), to allow further construction of the peptides on a solid support.
The solid phase peptide synthesis was then performed on 2-chlorotrityl resin under low loading conditions (Scheme 1F). The peptides were assembled from the N-Fmoc hexapeptides using the same peptide coupling conditions as before. The final peptide cleavage was performed with a hexafluoroisopropanol–dichloromethane cocktail,30 thereby affording the peptide species.
Peptide studies
The two resulting peptides AcPOG10 and PivσOG10 turned out to be insoluble in water and poorly soluble in most of the pure NMR solvents, such as methanol or dichloromethane, but were soluble in DMSO to a concentration of a few mg ml−1. The addition of sodium dodecyl sulphate (SDS) micelles enabled the solubilisation of the peptide in an aqueous medium. Diffusion-ordered spectroscopic studies of the resulting solutions demonstrated that the peptides were bound to micelles, confirming their hydrophobicity (see the ESI†).Subsequently, we analysed the triple helical assembly by circular dichroism (CD) spectroscopy, which is a conventional method for CMP studies. In the CD spectra, the triple helical arrangement is usually seen by the presence of a positive CD at 227 nm (Δε227), while it becomes negative if the assembly conditions are not met.
We first examined the CD spectra of the peptides in trifluoroethanol, a moderately hydrophobic solvent that also stabilizes hydrogen bonding in dissolved peptides (Fig. 3A). For the peptide AcPOG10, we observed a weak positive Δε227 in this solvent. This is a clear indication that the triple helix formation has indeed occurred. In contrast, more hydrophobic PivσOG10 failed to produce a triple helical signal, and we assume that this occurs due to the low solubility of the peptide in trifluoroethanol. The spectra in SDS micelles showed the absence of the triple helix. We then examined the CD spectra in octanol. In this case, both peptides showed excellent solubility and the CD spectra revealed the formation of the triple helical structure. We thus found conditions to observe the triple helical stability in a hydrophobic environment.
 | ||
| Fig. 3 Examination of the conditions for triple helical assembly by circular dichroism spectroscopy (A). The peptides were mixed with the solvents to achieve 0.2 mM final concentration of peptides. The spectra in detergent micelles were at 25 mM concentration of sodium dodecyl sulphate (SDS). The spectra were acquired at 25 °C. Temperature dependence of the characteristic circular dichroism signal at 227 nm in octan-1-ol (B). The machine units (mdeg) were converted into Δε using the total amide concentration (peptide concentration multiplied by the number of amide bonds). | ||
It is interesting to note that in the case of a hydrophobic polyproline-II helix formed by oligomeric Oic sequences, the structure is preserved in both octanol and SDS micelles.28 In the case of CMP, however, the assembly was seen in the former solvent and was fully absent in the latter. We assume that this difference occurs because the triple helix is not a secondary structure but an assembly, whereby a detergent micelle exerts a disrupting effect on it.
The assembly of the peptides in octanol proved to be very stable as there were no signs of melting when heated up to 95 °C (Fig. 3B). Moreover, the signal of the triple helix was still observed even when the peptide was diluted to 16 μM concentration (see the ESI†).
The next step in our investigations was to determine at which peptide length the triple helical assembly becomes stable. For example, for water-soluble CMPs, a minimal length of 7 (for repetitive Pro-Hyp-Gly), 10 (for repetitive Pro-Pro-Gly) or more triplets is usually required for stability.8 We thus synthesised peptides with the sequences acetyl-(Ash-Oic-Gly)n-OH, where n = 2, 4, 6, 8 and 10 (AcσOG2, AcσOG4, AcσOG6, AcσOG8, and AcσOG10), in order to test the minimal length.
We examined their CD spectra (Fig. 4A) as well as the temperature dependence of the CD signal at 227 nm (Fig. 4B). The results show that the formation of the triple helix occurred already with four triplets and persisted in higher oligomers. The melting of the peptide AcσOG4 occurred between 42 and 76 °C with a melting point at 59 °C. For the longest peptides with 8 and 10 triplets (AcσOG8 and AcσOG10), the melting process did not start even at 95 °C!
 | ||
| Fig. 4 CD spectra at 25 °C (A) and temperature dependence of Δε227 (B) in a series of oligomeric peptides in octan-1-ol. The spectra were acquired at 1 mM amide concentration for all peptide samples, and this concentration was used for the conversion of the machine units (mdeg) into Δε. | ||
Conclusions and outlook
Our results illustrate a truly unprecedented stability of the designed triple helical structure in the model hydrophobic solvent, octanol. It is reasonable to assume that the stability of the supramolecular assembly is associated with the “polar in – apolar out” principle, which is a notable feature of the parent CMP scaffold. Moreover, this may as well be seen as a general principle applicable to the polyproline-II helix assembly31 or structures based on an oligoproline scaffold.32 We believe that the fact that the triple helix formation occurs through a collapse of polar groups is somehow hidden from attention when studying water soluble CMPs since competition of the assembly with water solubilisation is inevitable. Therefore, our findings illustrate how the basic peptide architecture can be exploited towards enhanced triple helical stability. Effectively, it enables the reduction of the minimal length of the peptide required for the triple helical formation about two times. One should simply change the medium.We believe that our experiments provide a starting point for the future use of the collagen triple helical scaffold in other hydrophobic environments. We are particularly intrigued by the observation that the peptide length required to form the triple helical structure (four triplets ∼3.5 nm) roughly matches the hydrophobic thickness of common lipid membranes in biochemical settings. Therefore, we anticipate no fundamental limitations for the utilization of the triple helix in a membrane environment, especially considering the fact that the individual transmembrane polyproline-II helix has been reported very recently.33 We thus believe that collagen can be converted into an artificial membrane protein in the near future. We are currently working in this direction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
VK would like to acknowledge DFG-funded research group FOR 1805 for a creative environment and a joyful atmosphere of research freedom, essential for cherishing the ideas, which effectively led to the findings presented in this article.Notes and references
- M. Rubini, S. Lepthien, R. Golbik and N. Budisa, Biochim. Biophys. Acta, 2006, 1764, 1147–1158, DOI:10.1016/j.bbapap.2006.04.012.
- R. Berisio, L. Vitagliano, L. Mazzarella and A. Zagari, Protein Sci., 2002, 11, 262–270, DOI:10.1110/ps.32602.
- K. Okuyama, C. Hongo, R. Fukushima, G. Wu, H. Narita, K. Noguchi, Y. Tanaka and N. Nishino, Pept. Sci., 2004, 76, 366–367, DOI:10.1002/bip.20107.
- M. D. Shoulders, K. A. Satyshur, K. T. Forest and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 559–564, DOI:10.1073/pnas.0909592107.
- K. L. Gorres and R. T. Raines, Crit. Rev. Biochem. Mol. Biol., 2010, 45, 106–124, DOI:10.3109/10409231003627991.
- C. Priem and A. Geyer, Org. Lett., 2018, 20, 162–165, DOI:10.1021/acs.orglett.7b03525.
- V. Kubyshkin, S. Pridma and N. Budisa, New J. Chem., 2018, 42, 13461–13470, 10.1039/C8NJ02631A.
- M. D. Shoulders and R. T. Raines, Annu. Rev. Biochem., 2009, 78, 929–958, DOI:10.1146/annurev.biochem.77.032207.120833.
- M. Eilers, S. C. Shekar, T. Shien, S. O. Smith and P. J. Fleming, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5796–5801, DOI:10.1073/pnas.97.11.5796.
- W. F. DeGrado, H. Gratkowski and J. D. Lear, Protein Sci., 2003, 12, 647–665, DOI:10.1110/ps.0236503.
- S. Kim, T.-J. Jeon, A. Oberai, D. Yang, J. J. Schmidt and J. U. Bowie, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 14278–14283, DOI:10.1073/pnas.0501234102.
- C. Renner, B. Saccà and L. Moroder, Pept. Sci., 2004, 76, 34–47, DOI:10.1002/bip.10569.
- T. Luo and K. L. Kiick, Eur. Polym. J., 2013, 49, 2998–3009, DOI:10.1016/j.eurpolymj.2013.05.013.
- K. Strauss and J. Chmielewski, Curr. Opin. Biotechnol., 2017, 46, 34–41, DOI:10.1016/j.copbio.2016.10.013.
- M. Umashankara, M. V. Sonar, N. D. Bansode and K. N. Ganesh, J. Org. Chem., 2015, 80, 8552–8560, DOI:10.1021/acs.joc.5b01032.
- N. B. Hentzen, L. E. J. Smeenk, J. Witek, S. Riniker and H. Wennemers, J. Am. Chem. Soc., 2017, 139, 12815–12820, DOI:10.1021/jacs.7b07498.
- D. E. Przybyla and J. Chmielewski, Biochemistry, 2010, 49, 4411–4419, DOI:10.1021/bi902129p.
- T. Jiang, O. A. Vail, Z. Jiang, X. Zuo and V. P. Conticello, J. Am. Chem. Soc., 2015, 137, 7793–7802, DOI:10.1021/jacs.5b03326.
- R. S. Erdmann and H. Wennemers, J. Am. Chem. Soc., 2010, 132, 13957–13959, DOI:10.1021/ja103392t.
- M. R. Aronoff, J. Egli, M. Menichelli and H. Wennemers, Angew. Chem., Int. Ed., 2019 DOI:10.1002/anie.201813048 , in press.
- B. Sarkar, L. E. R. O'Leary and J. D. Hartgerink, J. Am. Chem. Soc., 2014, 136, 14417–14424, DOI:10.1021/ja504377s.
- M. Röber, S. Laroque, S. A. Lopez, T. Scheibel and H. G. Börner, Eur. Polym. J., 2019, 112, 301–305, DOI:10.1016/j.eurpolymj.2018.12.045.
- M. A. Cejas, W. A. Kinney, C. Chen, G. C. Leo, B. A. Tounge, J. G. Vinter, P. P. Joshi and B. E. Maryanoff, J. Am. Chem. Soc., 2007, 129, 2202–2203, DOI:10.1021/ja066986f.
- M. A. Cejas, W. A. Kinney, C. Chen, J. G. Vinter, H. R. Almond Jr., K. M. Balss, C. A. Maryanoff, U. Schmidt, M. Breslav, A. Mahan, E. Lacy and B. E. Maryanoff, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8513–8518, DOI:10.1073/pnas.0800291105.
- L. Yao, M. He, D. Li, J. Tian, H. Liu and J. Xiao, RSC Adv., 2018, 8, 2404–2409, 10.1039/c7ra11855d.
- L. Yao, M. He, D. Li, H. Liu, J. Wu and J. Xiao, New J. Chem., 2018, 42, 7439–7444, 10.1039/c8nj00119g.
- V. Kubyshkin and N. Budisa, Org. Biomol. Chem., 2017, 15, 619–627, 10.1039/C6OB02306A.
- V. Kubyshkin and N. Budisa, J. Pept. Sci., 2018, 24, e3076, DOI:10.1002/psc.3076.
- (a) P. K. Mykhailiuk, I. Kishko, V. Kubyshkin, N. Budisa and J. Cossy, Chem. – Eur. J., 2017, 23, 13279–13283, DOI:10.1002/chem.201703446; (b) S.-Q. Peng, X.-W. Zhang, L. Zhang and X.-G. Hu, Org. Lett., 2017, 19, 5689–5692, DOI:10.1021/acs.orglett.7b02866; (c) V. Kubyshkin and N. Budisa, Beilstein J. Org. Chem., 2017, 13, 2442–2457, DOI:10.3762/bjoc.13.241.
- R. Bollhagen, M. Schmiedberger, K. Barlos and E. Grell, J. Chem. Soc., Chem. Commun., 1994, 2559–2560, 10.1039/C39940002559.
- M. Á. Treviño, D. Pantoja-Uceda, M. Menéndez, M. V. Gomez, M. Mompeán and D. V. Laurentis, J. Am. Chem. Soc., 2018, 140, 16988–17000, DOI:10.1021/jacs.8b05261.
- N. D. Bansode, M. V. Sonar and K. N. Ganesh, Chem. Commun., 2016, 52, 4884–4887, 10.1039/C6CC00838K.
- V. Kubyshkin, S. L. Grage, J. Bürck, A. S. Ulrich and N. Budisa, J. Phys. Chem. Lett., 2018, 9, 2170–2174, DOI:10.1021/acs.jpclett.8b00829.
Footnotes |
| † Electronic supplementary information (ESI) available: Details on the synthesis and characterization of peptides. See DOI: 10.1039/c9ob00070d |
| ‡ Throughout the text one-letter code ‘O’ designates (2S,3aS,7aS)-octahydroindole-2-carboxylic acid (Oic) residues, while Greek ‘σ’ designates 5-azaspiro[2.4]heptane-6-carboxylic acid (Ash) and emphasizes on the fact that the amino acid has ‘spiro’ in its name. Three-letter code Hyp stands for (4R)-hydroxyproline. |
| This journal is © The Royal Society of Chemistry 2019 |