
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical benzylic bromination in continuous flow using BrCCl3 and its application to telescoped p-methoxybenzyl protection†
Yuma
Otake‡
 a,
Jason D.
Williams
ab,
Juan A.
Rincón
c,
Oscar
de Frutos
c,
Carlos
Mateos
c and
C. Oliver
Kappe
*ab
a,
Jason D.
Williams
ab,
Juan A.
Rincón
c,
Oscar
de Frutos
c,
Carlos
Mateos
c and
C. Oliver
Kappe
*ab
aInstitute of Chemistry, University of Graz, NAWI Graz, Heinrichstrasse 28, 8010 Graz, Austria. E-mail: oliver.kappe@uni-graz.at; Web: http://goflow.at
bCenter for Continuous Flow Synthesis and Processing (CC FLOW), Research Center Pharmaceutical Engineering GmbH (RCPE), Inffeldgasse 13, 8010 Graz, Austria
cCentro de Investigación Lilly S.A., Avda. de la Industria 30, 28108 Alcobendas-Madrid, Spain
First published on 22nd January 2019
Abstract
BrCCl3 represents a rarely used benzylic brominating reagent with complementary reactivity to other reagents. Its reactivity has been revisited in continuous flow, revealing compatibility with electron-rich aromatic substrates. This has brought about the development of a p-methoxybenzyl bromide generator for PMB protection, which was successfully demonstrated on a pharmaceutically relevant intermediate on 11 g scale, giving 91% yield and a PMB-Br space–time-yield of 1.27 kg L−1 h−1.
Photochemical benzylic bromination (the Wohl–Ziegler reaction) represents one of the most well used methods within synthetic organic photochemistry.1,2 This simple procedure is initiated by the photolysis of the Br–Br bond in Br2, present in low levels in the synthetic reagent N-bromosuccinimide (NBS).3–6 However, despite the simplicity and wide implementation of this methodology, it suffers from a number of drawbacks; most notably incompatibility with electron-rich aromatic substrates, due to competing electrophilic (ring) bromination.7 Although multiple alternative bromine sources for benzylic bromination have been described, these are almost exclusively comprised of other N-Br compounds,8 which can also function as electrophilic Br sources, or in situ Br2 generation methods.9–12
In recent years continuous flow and photochemistry, which previously lay in the realm of specialist users, have been increasingly applied to challenges faced by synthetic organic chemists. The combination of these two methods has proved to be fruitful in many areas, particularly in scaling up, due to improved and evenly distributed light penetration throughout the reaction medium.13–18 Indeed, many examples of Wohl–Ziegler reactions under continuous flow conditions have been reported.19–22 Furthermore, the advent of continuous flow photochemistry has encouraged re-evaluation of many previously examined reaction methodologies which, upon their initial discovery, were often limited in their application. A greater potential for these procedures may now be realized, owing to significant advances in synthesis technology over the past two decades.
Photochemical benzylic bromination using BrCCl3 as a source of bromine was initially reported in 1960![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23,24 and expanded upon in 1976,25 where a small number of substrates were examined using simple irradiation by a “sun lamp”, in traditional batch apparatus. Outside of these three short reports, no further use of this approach has been disseminated in the following four decades.26 As a reagent, BrCCl3 is readily available,27 does not have any significant identified health concerns28 and is not listed as an ozone depleting substance.29 It has seen numerous applications in atom-transfer radical addition (ATRA) reactions, both photochemically30,31 or by classical radical methods32–35 and also as an oxidizing agent; used directly,36–38 or within a photoredox catalytic cycle.39–42 It was anticipated that by utilizing advancements in light source and reactor technology, an improved methodology could be developed. Accordingly, the advantages of simple separation of a single by-product (CHCl3), highly concentrated reactions without precipitation of solid reagents and the possibility of accessing electron-rich benzyl bromides may be realized (Scheme 1).
23,24 and expanded upon in 1976,25 where a small number of substrates were examined using simple irradiation by a “sun lamp”, in traditional batch apparatus. Outside of these three short reports, no further use of this approach has been disseminated in the following four decades.26 As a reagent, BrCCl3 is readily available,27 does not have any significant identified health concerns28 and is not listed as an ozone depleting substance.29 It has seen numerous applications in atom-transfer radical addition (ATRA) reactions, both photochemically30,31 or by classical radical methods32–35 and also as an oxidizing agent; used directly,36–38 or within a photoredox catalytic cycle.39–42 It was anticipated that by utilizing advancements in light source and reactor technology, an improved methodology could be developed. Accordingly, the advantages of simple separation of a single by-product (CHCl3), highly concentrated reactions without precipitation of solid reagents and the possibility of accessing electron-rich benzyl bromides may be realized (Scheme 1).
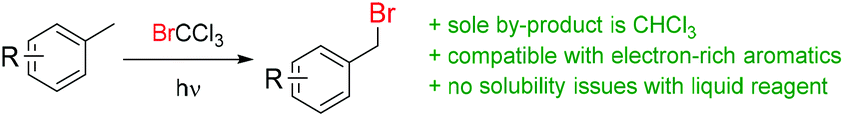 | ||
| Scheme 1 Proposed photochemical bromination in continuous flow using BrCCl3. | ||
Initial screening of the benzylic bromination of toluene (1a) in continuous flow using different light sources demonstrated only a minor extent of reaction, aside from under irradiation by a 254 nm lamp (light source A, Table 1 entry 1) or medium pressure Hg lamp (light source B, entry 2).43 This can be simply explained, since BrCCl3 shows significant absorption at wavelengths shorter than 300 nm.44 Accordingly, only short wavelength light-sources are capable of initiating this reaction to any significant extent, by directly homolyzing the C–Br bond. The computationally examined triplet energy of BrCCl3 reveals that a very low energy (46.4 kcal mol−1) is required to form the diradical species.45 Capitalizing on this observation, addition of benzophenone (4) as a triplet sensitizer resulted in an increased extent of reaction when using the 365 nm lamp (light source C, entry 3 vs. entry 4). However, no improvement was observed using the 254 nm lamp (light source A entry 5). Furthermore, an unselective reaction was observed using the medium pressure Hg lamp (light source B entry 6), due to the multiple discrete emission bands of this light source which excite both BrCCl3 and benzophenone, leading to a high transient radical concentration and reduced selectivity. This effect was exacerbated upon increasing the reaction temperature to 70 °C (entry 7), which prompted far higher selectivity towards dibrominated product (3a). Although the dibrominated product is undesired in this case, it should be noted that its derivitization to yield the corresponding aldehyde is facile, so in many cases it may be the preferred reaction product.46 Surprisingly, it was found that an array of high power LEDs at a narrow emission of 365 nm gave a far lesser extent of reaction than the 365 nm 8 W fluorescent lamp (see ESI Table S1†). This is likely due to the shorter wavelengths reached by the broad emission of the 365 nm fluorescent lamp (light source C).43
|
|
|||||
|---|---|---|---|---|---|
| Entry | Light source | PhCOPh 4 loading [mol%] | Remaining 1aa [%] |
2aa [%] |
3aa [%] |
| Light sources are as follows: A = 254 nm UV-C lamp (8 W), 7.7 min residence time; B = medium pressure Hg lamp (150 W), 10 min residence time; C = 365 nm UV-A lamp (8 W), 7.7 min residence time. See ESI for full details of each setup.a Product ratios were determined by NMR.b Reaction was run at 70 °C using a 5 bar backpressure regulator.c Reaction was run at 0.5 M in DCM.d Reaction was run using 2 equiv. BrCCl3.e Reaction was run using 6 equiv. BrCCl3.f Reaction was run using 15 min residence time.g Reaction was run using 30 min residence time.h Reaction was run at 80 °C, with no light source. | |||||
| 1 | A | — | 75 | 23 | 2 |
| 2 | B | — | 71 | 26 | 3 |
| 3 | C | — | 96 | 4 | 0 |
| 4 | C | 2 | 58 | 38 | 4 |
| 5 | A | 2 | 75 | 24 | 1 |
| 6 | B | 2 | 17 | 40 | 43 |
| 7b | B | 2 | 1 | 26 | 73 |
| 8 | C | 1 | 63 | 35 | 2 |
| 9 | C | 5 | 46 | 44 | 10 |
| 10c | C | 2 | 78 | 22 | 0 |
| 11d | C | 2 | 63 | 34 | 3 |
| 12e | C | 2 | 37 | 49 | 14 |
| 13 | C | 2 | 29 | 52 | 19 |
| 14g | C | 2 | 17 | 50 | 33 |
| 15h | — | 2 | 99 | 1 | 0 |
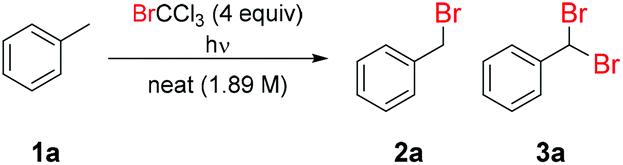
Use of a sensitizer bestows additional reaction control and allows the use of lower energy wavelengths which, in turn, lowers the likelihood of observing reactor fouling by degradation and polymerization pathways.47,48 For these reasons, optimization using the 365 nm lamp (light source C) was pursued further. Both higher and lower benzophenone loadings were examined, whereby it was observed that a lower loading (1 mol%) provided lower conversion (entry 8 vs. entry 4), yet a higher loading (5 mol%) began to significantly reduce selectivity (entry 9 vs. entry 4). Several alternative triplet sensitizers and photoredox catalysts were examined (see ESI, Table S2†), but none were observed to provide a combination of product formation and selectivity comparable to that of benzophenone. Diluting the reaction mixture with a range of different solvents was observed to have a detrimental effect on the progress of this reaction. DCM (entry 10) performed far better than other solvents, yet still decreased reaction rate substantially, hence neat reaction conditions were retained.43
Subsequently, the impact of brominating agent loading was evaluated, finding that a decrease from 4 equiv. (1.89 M toluene solution) to 2 equiv. (3.31 M toluene solution) resulted in a slightly lower rate of reaction (entry 4 vs. entry 11). However, increasing this loading to 6 equiv. (1.44 M toluene solution) was observed to reduce reaction selectivity (entry 4 vs. entry 12). Accordingly, we opted to remain with 4 equiv. (1.89 M toluene solution) and examine longer residence times in attempt to achieve complete reaction. A 15 min residence time (entry 13) gave an improvement in yield of monobrominated product (2a) to 52%, yet further increasing this to 30 min (entry 14) actually resulted in a drop in yield of the desired product, due to preferential formation of the dibrominated product (3a). Unfortunately, as described in previous reports, there is a low energy barrier to H-atom abstraction from both starting material (1a) and monobrominated product (2a), implying that differentiation between the two can be difficult to achieve in many cases.20 Finally, a control reaction run at high temperature without light demonstrated that this process does not proceed to any appreciable extent when performed thermally (entry 15). Although conversion to the desired product (2a) was relatively low, reaction selectivity was maintained, throughput is high due to neat conditions and CHCl3 is the only reaction by-product, so its removal is straightforward.
In order to assess the generality of this procedure, reactions were carried out with various substrates (Fig. 1). For ease of isolation (separation of mono and dibrominated products),49 and to avoid handling the hazardous benzyl bromide itself, the continuous flow reactor output was collected and reacted directly with morpholine (2 equiv.). The toluene-derived product (5a) was isolated in a 47% yield, corroborating well with NMR yields from previous optimization experiments (Table 1 entry 13). Ethylbenzene proved to be less susceptible to dibromination, furnishing its tertiary amine (5b) in 81% yield, when processed using a longer residence time of 30 min. To varying degrees, para-substitution was also tolerated (products 5c to 5g), but a more significantly electron-deficient aromatic (product 5e) required higher sensitizer loading and residence time to achieve a synthetically useful level of conversion.
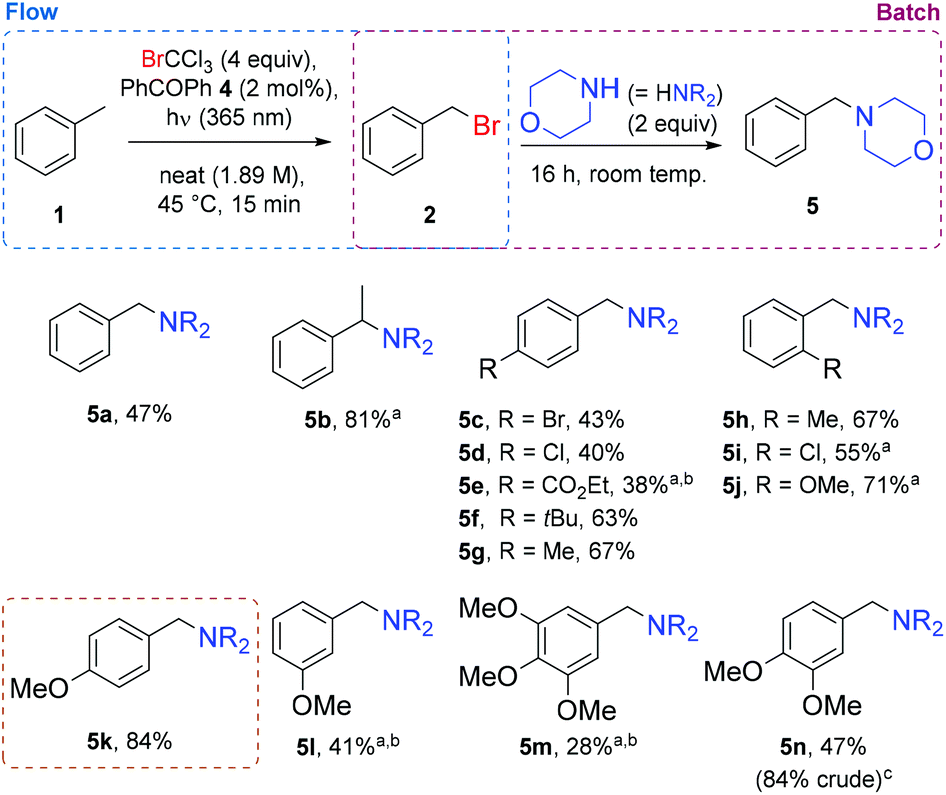 | ||
| Fig. 1 Substrate scope of benzylic bromination in continuous flow. Yields shown are those of isolated morpholine adducts. aContinuous flow reaction was carried out with a 30 min residence time. bAn increased quantity (5 mol%) of benzophenone 4 was used. cCrude yield by NMR is displayed, since product isolated in poor yield, despite effective bromination. | ||
Methylbenzenes bearing ortho-substitutents were reasonably well tolerated (products 5h to 5i), and a good level of selectivity was observed when an o-methyl group was present (product 5h). As previously observed,24p-methoxytoluene undergoes reaction quickly using this reagent, and the corresponding amine (5k) was isolated in 84% yield. The influence of methoxy substituents was probed further, and was found to follow the expected reactivity trend, where a meta-methoxylated substrate (more electron-deficient) required more forcing conditions to achieve 41% yield of its product (5l), similarly to trimethoxy product (5m), where the negative reactivity contribution from two m-methoxy groups appears to outweigh the positive contribution from a single p-methoxy group. When the p-methoxy group is counteracted by only one m-methoxy group, reactivity returns to a good level (product 5n).
Of these substrates, the successful reaction of p-methoxytoluene (1k) is of particular interest, since p-methoxybenzyl (PMB) is a commonly used protecting group in organic synthesis.50 This protection is generally performed by alkylation, but since p-methoxybenzyl bromide (2k) is unstable under long-term storage conditions,51 the corresponding chloride (which also suffers from stability issues)52 is often used in its place. This less reactive benzyl halide can require more forcing conditions such as strong bases,53 or addition of an (undesirable) iodide salt for reaction to proceed via the benzyl iodide.54 For these reasons, an in situ method of generating the alkylating agent in a mild manner would be of clear advantage. The concept of generating and consuming hazardous or unstable reagents such as these in situ has received significant attention in recent years, for the resulting improvement in safety and reaction reproducibility.55 The electron-rich nature of p-methoxytoluene means that its benzylic bromination by NBS under photochemical conditions is challenging due to competing electrophilic bromination processes.7 Consequently, the procedure reported herein is particularly fitting for the novel application of this brominating agent.56
We sought to apply the developed p-methoxybenzyl bromide (2k) generation to an intermediate in API (active pharmaceutical ingredient) synthesis. Cipargamin is a potent anti-malarial candidate currently in phase II clinical trials and, as a spiroindole, represents the first new family of antimalarial drugs in two decades.57 An asymmetric route to this compound begins with PMB protection of 5-chloroisatin (6); a more challenging protection which requires addition of potassium iodide, due to the poor nucleophilicity of the aromatic amide nitrogen.52 To achieve this transformation using in situ generated alkylating agent, p-methoxytoluene (1k) was continuously flowed through a tubing-based photoreactor, under the developed bromination conditions, before entering a batch vessel containing substrate (6) and potassium carbonate in acetonitrile (Fig. 2). An elevated temperature was used to ensure the rapid consumption of benzyl bromine (2k) as it entered the reaction flask. Under these conditions, the PMB-protecting isatin derivative (7) was isolated in 91% yield on a 0.6 mmol scale then scaled up, with no decrease in yield, to isolate 11 grams following 4 hours of continual p-methoxybenzyl bromide (2k) dosing.
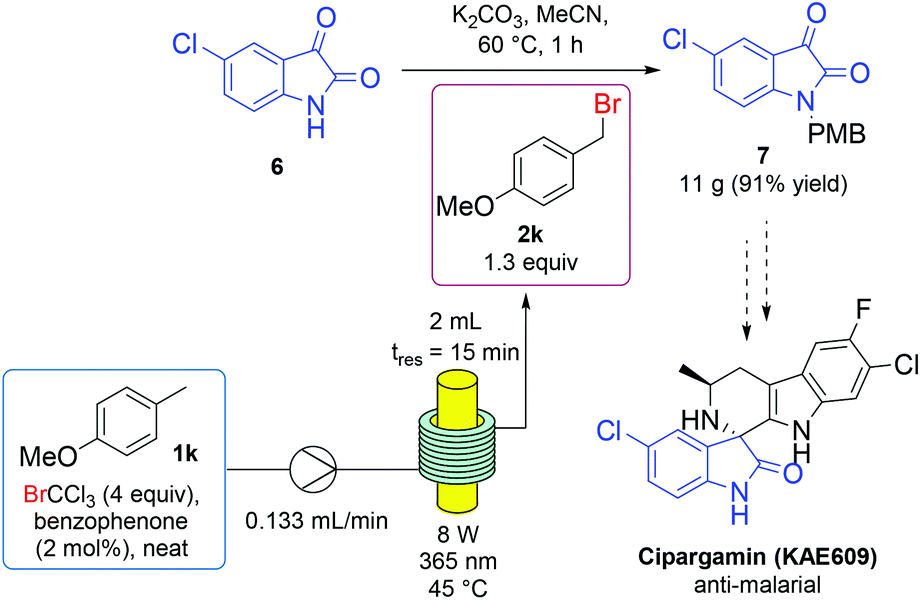 | ||
| Fig. 2 Continuous flow photochemical generation of p-methoxybenzyl bromide 2k, fed into a batch protection of API precursor 6 on a multigram scale. | ||
Conclusions
Within this study a previously reported, but seldom used, photochemical bromination method has been examined in detail under continuous flow photochemical conditions. BrCCl3 represents an underutilized bromine source for benzylic halogenation, since it is readily available and possesses no significant safety or environmental issues. Optimization was performed by considering multiple distinct light sources, which also highlighted the benefits of introducing a photosensitizer to the reaction mixture. The optimized conditions were examined against a number of substrates with reasonable yields attained across different substitution patterns. Most significantly, p-methoxytoluene, an electron-rich substrate which is challenging for NBS-based benzylic bromination procedures, reacted smoothly. This appeared suitable in the application of a p-methoxybenzyl (PMB) protection, where the alkylating agent is generated from cheap and safe starting materials in situ, with high space–time-yield of 1.27 kg L−1 h−1, then consumed in the reaction. The protection method was exemplified on an 11 g scale, achieving facile reaction and an excellent 91% yield, which demonstrates the compatibility of the bromination reaction mixture with these general PMB protection conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The CC Flow Project (Austrian Research Promotion Agency FFG No. 862766) is funded through the Austrian COMET Program by the Austrian Federal Ministry of Transport, Innovation and Technology (BMVIT), the Austrian Federal Ministry of Science, Research and Economy (BMWFW), and by the State of Styria (Styrian Funding Agency SFG).Notes and references
- C. Djerassi, Chem. Rev., 1948, 43, 271–317 CrossRef CAS
.
- I. Saikia, A. J. Borah and P. Phukan, Chem. Rev., 2016, 116, 6837–7042 CrossRef CAS PubMed
- J. H. Incremona and J. C. Martin, J. Am. Chem. Soc., 1970, 92, 627–634 CrossRef CAS
- J. C. Day, M. J. Lindstrom and P. S. Skell, J. Am. Chem. Soc., 1974, 96, 5616–5617 CrossRef CAS
- G. A. Russell, C. DeBoer and K. M. Desmond, J. Am. Chem. Soc., 1963, 85, 365–366 CrossRef CAS
- R. E. Pearson and J. C. Martin, J. Am. Chem. Soc., 1963, 85, 354–355 CrossRef CAS
- G.-J. M. Gruter, O. S. Akkerman and F. Bickelhaupt, J. Org. Chem., 1994, 59, 4473–4481 CrossRef CAS
- L. S. De Almeida, P. M. Esteves and M. C. S. De Mattos, Tetrahedron Lett., 2015, 56, 6843–6845 CrossRef CAS
- T. Raju, G. Kalpana Devi and K. Kulangiappar, Electrochim. Acta, 2006, 51, 4596–4600 CrossRef CAS
- P. Galloni, M. Mancini, B. Floris and V. Conte, Dalton Trans., 2013, 42, 11963–11970 RSC
- A. Podgoršek, S. Stavber, M. Zupan and J. Iskra, Green Chem., 2007, 9, 1212–1218 RSC
- R. Mestres and J. Palenzuela, Green Chem., 2002, 4, 314–316 RSC
- D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed
- Y. Su, N. J. W. Straathof, V. Hessel and T. Noël, Chem. – Eur. J., 2014, 20, 10562–10589 CrossRef CAS
- T. Noël, J. Flow Chem., 2017, 7, 87–93 CrossRef
- F. Politano and G. Oksdath-Mansilla, Org. Process Res. Dev., 2018, 22, 1045–1062 CrossRef CAS
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed
- J. P. Knowles, L. D. Elliott and K. I. Booker-Milburn, Beilstein J. Org. Chem., 2012, 8, 2025–2052 CrossRef CAS PubMed
- D. Cantillo, O. De Frutos, J. A. Rincon, C. Mateos and C. Oliver Kappe, J. Org. Chem., 2014, 79, 223–229 CrossRef CAS
- H. E. Bonfield, J. D. Williams, W.-X. Ooi, S. G. Leach, W. J. Kerr and L. J. Edwards, ChemPhotoChem, 2018, 2, 938–944 CrossRef CAS
- Y. Chen, O. de Frutos, C. Mateos, J. A. Rincón, D. Cantillo and C. O. Kappe, ChemPhotoChem, 2018, 2, 906–912 CrossRef CAS
- D. Šterk, M. Jukič and Z. Časar, Org. Process Res. Dev., 2013, 17, 145–151 CrossRef
- E. S. Huyser, J. Am. Chem. Soc., 1960, 82, 391–393 CrossRef CAS
- E. S. Huyser, J. Am. Chem. Soc., 1960, 82, 394–396 CrossRef CAS
- S. W. Baldwin and T. H. O'Neill, Synth. Commun., 1976, 6, 109–112 CrossRef CAS
- Aside from a handful of reactions using this reagent in a photoredox catalytic cycle: T. Hou, P. Lu and P. Li, Tetrahedron Lett., 2016, 57, 2273–2276 CrossRef CAS
- Price of EUR134 per 500 g and MSDS: Sigma Aldrich Catalogue, https://www.sigmaaldrich.com/catalog/product/aldrich/b82822 (accessed December 2018).
-
H. Shinokubo, K. Oshima, Z. Gu and A. Zakarian, Bromotrichloromethane, in e-EROS Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd., Chichester, UK, 2012, pp. 1–7 Search PubMed
- Not listed as an depleting substance: United States Environmental Protection Agency, https://www.epa.gov/ozone-layer-protection/ozone-depleting-substances (accessed December 2018).
- C. J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875–8884 CrossRef CAS PubMed
- T. Kimura, M. Fujita, H. Sohmiya and T. Ando, J. Org. Chem., 1998, 63, 6719–6720 CrossRef CAS
- K. O. Proinsias, A. Jackowska, K. Radzewicz, M. Giedyk and D. Gryko, Org. Lett., 2018, 20, 296–299 CrossRef CAS PubMed
- T. Nakamura, H. Yorimitsu, H. Shinokubo and K. Oshima, Synlett, 1998, 1351–1352 CrossRef CAS
- C. Wang and G. A. Russell, J. Org. Chem., 1999, 64, 2346–2352 CrossRef CAS
- N. J. Cornwall, S. Linehan and R. T. Weavers, Tetrahedron Lett., 1997, 38, 2919–2922 CrossRef CAS
- J. F. Franz, W. B. Kraus and K. Zeitler, Chem. Commun., 2015, 51, 8280–8283 RSC
- A. M. Nauth, J. C. Orejarena Pacheco, S. Pusch and T. Opatz, Eur. J. Org. Chem., 2017, 6966–6974 CrossRef CAS
- A. M. Nauth, A. Lipp, B. Lipp and T. Opatz, Eur. J. Org. Chem., 2017, 2099–2103 CrossRef CAS
- G. Pandey, S. Koley, R. Talukdar and P. K. Sahani, Org. Lett., 2018, 20, 5861–5865 CrossRef CAS PubMed
- S. S. Shah, A. Paul, M. Bera, Y. Venkatesh and N. D. P. Singh, Org. Lett., 2018, 20, 5533–5536 CrossRef CAS PubMed
- G. Pandey, R. Laha and D. Singh, J. Org. Chem., 2016, 81, 7161–7171 CrossRef CAS PubMed
- F. Liu, Z. Zhang, Z. Bao, H. Xing, Y. Yang and Q. Ren, Tetrahedron Lett., 2017, 58, 2707–2710 CrossRef CAS
- See ESI section 1.2† for experimental setup and lamp emission spectra.
- See ESI Fig. S8† for an absorption spectrum of BrCCl3.
- See ESI section 4† for details of computational experiments.
- T. Noël, K. Robeyns, L. van Meervelt, E. van der Eycken and J. van der Eycken, Tetrahedron: Asymmetry, 2009, 20, 1962–1968 CrossRef
- L. D. Elliott, M. Berry, B. Harji, D. Klauber, J. Leonard and K. I. Booker-Milburn, Org. Process Res. Dev., 2016, 20, 1806–1811 CrossRef CAS
- E. B. Corcoran, F. Lévesque, J. P. McMullen and J. R. Naber, ChemPhotoChem, 2018, 2, 931–937 CrossRef CAS
- The dibrominated product was observed to be unreactive towards morpholine, by NMR comparison before and after its addition. Indeed, in the case of 5k, the aldehyde derivative was also isolated, assumedly having reacted during column chromatography. See ESI† for characterization of aldehyde product 5k′.
- R. A. Green, K. E. Jolley, A. A. M. Al-Hadedi, D. Pletcher, D. C. Harrowven, O. De Frutos, C. Mateos, D. J. Klauber, J. A. Rincón and R. C. D. Brown, Org. Lett., 2017, 19, 2050–2053 CrossRef CAS PubMed
- S. M. Ruder and R. C. Ronald, Tetrahedron Lett., 1987, 28, 135–138 CrossRef CAS
- S. E. Brewer, T. P. Vickery, D. C. Bachert, K. M. J. Brands, K. M. Emerson, A. Goodyear, K. J. Kumke, T. Lam and J. P. Scott, Org. Process Res. Dev., 2005, 9, 1009–1012 CrossRef CAS
- A. M. Sherwood, S. E. Williamson, R. S. Crowley, L. M. Abbott, V. W. Day and T. E. Prisinzano, Org. Lett., 2017, 19, 5414–5417 CrossRef CAS PubMed
- H. Takada, N. Kumagai and M. Shibasaki, Org. Lett., 2015, 17, 4762–4765 CrossRef CAS PubMed
- A. K. Singh, D. H. Ko, N. K. Vishwakarma, S. Jang, K. I. Min and D. P. Kim, Nat. Commun., 2016, 7, 1–7 Search PubMed
- BrCCl3 has previously been applied to a photoredox PMB deprotection method, but simply as a redox mediator: J. W. Tucker, J. M. R. Narayanam, P. S. Shah and C. R. J. Stephenson, Chem. Commun., 2011, 47, 5040–5042 RSC
- M. Rottmann, C. McNamara, B. K. S. Yeung, M. C. S. Lee, B. Zou, B. Russell, P. Seitz, D. M. Plouffe, N. V. Dharia, J. Tan, S. B. Cohen, K. R. Spencer, G. E. González-Páez, S. B. Lakshminarayana, A. Goh, R. Suwanarusk, T. Jegla, E. K. Schmitt, H.-P. Beck, R. Brun, F. Nosten, L. Renia, V. Dartois, T. H. Keller, D. A. Fidock, E. A. Winzeler and T. T. Diagana, Science, 2010, 329, 1175–1180 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Equipment, procedures, product characterization and NMR spectra. See DOI: 10.1039/c9ob00044e |
| ‡ Current address: Laboratory for Chemistry and Life Science, Institute of Innovative Research, Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama 226-8503, Japan. |
| This journal is © The Royal Society of Chemistry 2019 |