
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubstituted polyfluoroaryl interactions with an arginine side chain in galectin-3 are governed by steric-, desolvation and electronic conjugation effects†
Rohit
Kumar‡
a,
Kristoffer
Peterson‡
b,
Majda
Misini Ignjatović‡
c,
Hakon
Leffler
 d,
Ulf
Ryde
c,
Ulf J.
Nilsson
b and
Derek T.
Logan
*a
d,
Ulf
Ryde
c,
Ulf J.
Nilsson
b and
Derek T.
Logan
*a
aBiochemistry and Structural Biology, Centre for Molecular Protein Science, Department of Chemistry, Lund University, Box 124, SE-221 00 Lund, Sweden. E-mail: derek.logan@biochemistry.lu.se
bCentre for Analysis and Synthesis, Department of Chemistry, Lund University, Box 124, SE-221 00 Lund, Sweden
cTheoretical Chemistry, Department of Chemistry, Lund University, Box 124, SE-221 00 Lund, Sweden
dDepartment of Laboratory Medicine, Section MIG, Lund University BMC-C1228b, Klinikgatan 28, 221 84 Lund, Sweden
First published on 3rd January 2019
Abstract
In the β-D-galactopyranoside-binding protein galectin-3, synthetic inhibitors substituted at the 3-position of a thiodigalactoside core cause the formation of an aglycone binding pocket through the displacement of an arginine residue (Arg144) from its position in the apoprotein. To examine in detail the role of different molecular interactions in this pocket, we have synthesized a series of nine 3-(4-(2,3,5,6-tetrafluorophenyl)-1,2,3-triazol-1-yl)-thiogalactosides with different para substituents and measured their affinities to galectin-3 using a fluorescence polarization assay. High-resolution crystal structures (<1.3 Å) have been determined for five of the ligands in complex with the C-terminal domain of galectin-3. The binding affinities are rationalised with the help of the three-dimensional structures and quantum-mechanical calculations. Three effects seem to be involved: Firstly, the binding pocket is too small for the largest ligands with ethyl and methyl. Secondly, for the other ligands, the affinity seems to be determined mainly by desolvation effects, disfavouring the polar substituents, but this is partly counteracted by the cation–π interaction with Arg144, which stacks on top of the substituted tetrafluorophenyl group in all complexes. The results provide detailed insight into interactions of fluorinated phenyl moieties with arginine-containing protein binding sites and the complex interplay of different energetic components in defining the binding affinity.
1. Introduction
Structure-based drug design relies on careful analysis of protein–ligand interactions and the structure and dynamics of ligand and binding sites. Improving binding affinity involves modulating the specific interactions that the ligand makes with the binding site by modifying or substituting chemical moieties in the ligand.1 Investigating such specific interactions requires information about the protein–ligand complex that is often obtained from crystal structures and affinity data.1 Structural analysis of protein–ligand complexes identifies potential binding interactions and steric restrictions, providing insight into design of new ligands with enhanced binding affinity. However, the energetic components contributing to the binding affinity are not always self-evident from an inspection of the crystal structure.The drug target of interest here, galectin-3, belongs to the galectin super-family that has 14 members in humans. All galectins have a conserved carbohydrate recognition domain (CRD) that binds β-D-galactopyranosides, and the binding site is a shallow, hydrophilic pocket formed by β-sheets and loops.2 Galectins are found everywhere in the cell. They are involved in cell growth, differentiation, cell-cycle regulation.3 Their role in cancer, immunity and inflammatory conditions is well-documented, making them attractive therapeutic targets.4–9 Galectins bind galactosides with affinities in the millimolar range. Suitable modifications of galactose at the C3 position to introduce specific groups improves the binding affinity drastically to micromolar and even nanomolar affinity. A wealth of structural data is available for the galectin-3 CRD in complex with different compounds10–15 and we have recently reported the structures of high-affinity phenyltriazole thiogalactosides in complex with galectin-3.16 This ample availability of structural and affinity data makes galectin-3 an excellent model protein for studying protein–ligand interactions. The high galectin-3 affinity of thiodigalactosides with mono- to trifluorinated 3-(4-aryl-1,2,3-triazol-1-yl) moieties at C3 has been explained using X-ray crystallography by orthogonal multipolar fluorine–amide interactions with backbone amides and a cation–π interaction with Arg144.15 Arg144 is raised from its normal position in a water-mediated salt bridge on the surface of galectin-3 by the influence of fluorinated phenyl moieties on synthetic ligands, which creates a small pocket beneath Arg144 that could accommodate a larger substituent than fluorine in the para position on the phenyl ring.
Herein we report on a systematic probing of the binding interactions near Arg144 in galectin-3 by varying the para substituent on 2,3,5,6-tetrafluorophenyltriazoles through affinity measurements using fluorescence polarisation combined with structural analysis and quantum-mechanical calculations.
2. Experimental section
2.1 General
All reagents and solvents were dried prior to use according to standard methods. Commercial reagents were used without further purification. 2,3,5,6-tetrafluoro-4-hydroxyphenylacetylene was synthesized following a published procedure17 for the alkene analogue and it used without purification. Analytical TLC was performed using on silica gel 60 F254 (Merck) with detection by UV absorption and/or by charring following immersion in a 7% ethanolic solution of sulfuric acid. Purification of compounds was carried out by column chromatography on silica gel (40–60 μm, 60 Å) and/or preparative HPLC (Agilent 1260 infinity system, column SymmetryPrep-C18, 17 ml min−1 H2O–MeCN gradient 10–100% 15 min with 0.1% formic acid). Specific rotations were measured on a PerkinElmer model 341 polarimeter. NMR spectra 1H, 13C, 19F, 2D COSY, HMQC and HMBC were recorded with a Bruker Avance II 400 MHz spectrometer (400 Hz for 1H, 100 Hz for 13C and 376 Hz for 19F) at ambient temperature. Chemical shifts are reported in δ parts per million (ppm). In the 13C NMR spectra no signals were observed for the carbons in the fluorinated phenyl or the C4 triazole carbon, due to signal splitting caused by short- and long-range fluorine couplings. However, in the HMBC spectra the cross peak of the triazole C4 and H5 was observed (exemplified in the ESI† for compound 3). HRMS was determined by direct infusion on a Waters XEVO-G2 QTOF mass spectrometer using electrospray ionization (ESI). Compounds 2–10 were of >95% purity according to HPLC-analysis (Agilent series 1100 system, column Eclipse XDB-C18, 0.8 ml min−1 H2O–MeCN gradient 5–95% 13 min with 0.1% trifluoroacetic acid).2.2 Synthesis of compounds (2–5)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH 14:1–>5:1) to give 2 (14 mg, 48%) as an amorphous white solid. [α]20D 56.6 (c 0.93, CH3OH). 1H NMR (CD3OD, 400 MHz): δ 8.32 (s, 1H, Ph), 7.50 (d, J = 8.1 Hz, 2H, Ph), 7.15 (d, J = 8.1 Hz, 2H, Ph), 4.93 (obscured by water H-3), 4.78 (d, J = 9.5 Hz, 1H, H-1), 4.26 (t, J = 10.0 Hz, 1H, H-2), 4.16 (d, J = 2.8 Hz, 1H, H-4), 3.84–3.70 (m, 3H, H-5 and H-6), 2.33 (s, 3H, CH3). 13C NMR (CD3OD, 100 MHz): δ 138.8, 133.1, 131.6, 130.7, 125.5, 91.8, 80.9, 69.5, 69.2, 68.0, 62.3, 22. 19F NMR (CD3OD, 376 MHz): δ −145.6 (d, J = 16.0 Hz, 2F), −165.0 (d, J = 15.7 Hz, 2F). HRMS calculated for [C21H19F4N3O5SNa]+, 524.0879; found: 524.0880.
:EtOAc 1:1–>1:2) to give 3 (295 mg, 99%) as an amorphous white solid. [α]20D 57.6 (c 0.59, CH3OH). 1H NMR (CD3OD, 400 MHz): δ 8.44 (s, 1H, Ph), 7.50 (d, J = 8.1 Hz, 2H, Ph), 7.15 (d, J = 8.1 Hz, 2H, Ph), 4.95 (obscured by water H-3), 4.78 (d, J = 9.5 Hz, 1H, H-1), 4.26 (t, J = 10.0 Hz, 1H, H-2), 4.16 (d, J = 2.8 Hz, 1H, H-4), 3.84–3.70 (m, 3H, H-5 and H-6), 2.33 (s, 3H, CH3). 13C NMR (CD3OD, 100 MHz): δ 138.8, 133.2, 131.6, 130.7, 126.3, 91.7, 80.9, 69.5, 69.3, 68.0, 62.3, 21.1. 19F NMR (CD3OD, 376 MHz): δ −142.3 (dd, J = 13.7, 7.0 Hz, 2F), −157.8 (t, J = 20.0 Hz, 1F), −165.0 (m, 2F). HRMS calculated for [C21H19F5N3O4S]+, 504.1016; found: 504.1019.
:EtOAc 2:3–>1:2) to give 4 (23 mg, 87%) as an amorphous white solid. [α]20D 53.8 (c 0.89, CH3OH). 1H NMR (CD3OD, 400 MHz): δ 8.42 (s, 1H, Ph), 7.50 (d, J = 8.1 Hz, 2H, Ph), 7.15 (d, J = 8.1 Hz, 2H, Ph), 4.95 (dd, J = 10.5, 3.0 Hz, 1H, H-3), 4.78 (d, J = 9.5 Hz, 1H, H-1), 4.26 (t, J = 10.0 Hz, 1H, H-2), 4.16 (d, J = 2.8 Hz, 1H, H-4), 3.84–3.70 (m, 3H, H-5 and H-6), 2.33 (s, 3H, CH3). 13C NMR (CD3OD, 100 MHz): δ 138.8, 133.2, 131.6, 130.7, 126.2, 91.7, 80.9, 69.5, 69.3, 68.0, 62.3, 21.1. 19F NMR (CD3OD, 376 MHz): δ −143.2 (dd, J = 20.0, 9.0 Hz, 2F), −154.6 (dd, J = 20.0, 9.0 Hz, 2F). HRMS calculated for [C21H19F4N6O4S]+, 527.1125; found: 527.1124.
:EtOAc 1:1–>1:2) to give 5 (11 mg, 96%) as an amorphous white solid. [α]20D 58.9 (c 0.79, CH3OH). 1H NMR (CD3OD, 400 MHz): δ 8.25 (s, 1H, Ph), 7.50 (d, J = 8.1 Hz, 2H, Ph), 7.15 (d, J = 8.1 Hz, 2H, Ph), 4.91 (obscured by water H-3), 4.78 (d, J = 9.5 Hz, 1H, H-1), 4.25 (t, J = 10.0 Hz, 1H, H-2), 4.16 (d, J = 2.8 Hz, 1H, H-4), 3.83–3.70 (m, 3H, H-5 and H-6), 2.33 (s, 3H, CH3). 13C NMR (CD3OD, 100 MHz): δ 138.8, 133.1, 131.7, 130.7, 125.0, 91.8, 80.9, 69.5, 69.2, 68.0, 62.3, 21.1. 19F NMR (CD3OD, 376 MHz): δ −146.5 (m, 2F), −164.9 (m, 2F). HRMS calculated for [C21H20F4N4O4SNa]+, 523.1039; found: 523.1034.
:MeOH 14:1–>5:1) to give 2 (14 mg, 48%) as an amorphous white solid. [α]20D 56.6 (c 0.93, CH3OH). 1H NMR (CD3OD, 400 MHz): δ 8.32 (s, 1H, Ph), 7.50 (d, J = 8.1 Hz, 2H, Ph), 7.15 (d, J = 8.1 Hz, 2H, Ph), 4.93 (obscured by water H-3), 4.78 (d, J = 9.5 Hz, 1H, H-1), 4.26 (t, J = 10.0 Hz, 1H, H-2), 4.16 (d, J = 2.8 Hz, 1H, H-4), 3.84–3.70 (m, 3H, H-5 and H-6), 2.33 (s, 3H, CH3). 13C NMR (CD3OD, 100 MHz): δ 138.8, 133.1, 131.6, 130.7, 125.5, 91.8, 80.9, 69.5, 69.2, 68.0, 62.3, 22. 19F NMR (CD3OD, 376 MHz): δ −145.6 (d, J = 16.0 Hz, 2F), −165.0 (d, J = 15.7 Hz, 2F). HRMS calculated for [C21H19F4N3O5SNa]+, 524.0879; found: 524.0880.
:EtOAc 1:1–>1:2) to give 3 (295 mg, 99%) as an amorphous white solid. [α]20D 57.6 (c 0.59, CH3OH). 1H NMR (CD3OD, 400 MHz): δ 8.44 (s, 1H, Ph), 7.50 (d, J = 8.1 Hz, 2H, Ph), 7.15 (d, J = 8.1 Hz, 2H, Ph), 4.95 (obscured by water H-3), 4.78 (d, J = 9.5 Hz, 1H, H-1), 4.26 (t, J = 10.0 Hz, 1H, H-2), 4.16 (d, J = 2.8 Hz, 1H, H-4), 3.84–3.70 (m, 3H, H-5 and H-6), 2.33 (s, 3H, CH3). 13C NMR (CD3OD, 100 MHz): δ 138.8, 133.2, 131.6, 130.7, 126.3, 91.7, 80.9, 69.5, 69.3, 68.0, 62.3, 21.1. 19F NMR (CD3OD, 376 MHz): δ −142.3 (dd, J = 13.7, 7.0 Hz, 2F), −157.8 (t, J = 20.0 Hz, 1F), −165.0 (m, 2F). HRMS calculated for [C21H19F5N3O4S]+, 504.1016; found: 504.1019.
:EtOAc 2:3–>1:2) to give 4 (23 mg, 87%) as an amorphous white solid. [α]20D 53.8 (c 0.89, CH3OH). 1H NMR (CD3OD, 400 MHz): δ 8.42 (s, 1H, Ph), 7.50 (d, J = 8.1 Hz, 2H, Ph), 7.15 (d, J = 8.1 Hz, 2H, Ph), 4.95 (dd, J = 10.5, 3.0 Hz, 1H, H-3), 4.78 (d, J = 9.5 Hz, 1H, H-1), 4.26 (t, J = 10.0 Hz, 1H, H-2), 4.16 (d, J = 2.8 Hz, 1H, H-4), 3.84–3.70 (m, 3H, H-5 and H-6), 2.33 (s, 3H, CH3). 13C NMR (CD3OD, 100 MHz): δ 138.8, 133.2, 131.6, 130.7, 126.2, 91.7, 80.9, 69.5, 69.3, 68.0, 62.3, 21.1. 19F NMR (CD3OD, 376 MHz): δ −143.2 (dd, J = 20.0, 9.0 Hz, 2F), −154.6 (dd, J = 20.0, 9.0 Hz, 2F). HRMS calculated for [C21H19F4N6O4S]+, 527.1125; found: 527.1124.
:EtOAc 1:1–>1:2) to give 5 (11 mg, 96%) as an amorphous white solid. [α]20D 58.9 (c 0.79, CH3OH). 1H NMR (CD3OD, 400 MHz): δ 8.25 (s, 1H, Ph), 7.50 (d, J = 8.1 Hz, 2H, Ph), 7.15 (d, J = 8.1 Hz, 2H, Ph), 4.91 (obscured by water H-3), 4.78 (d, J = 9.5 Hz, 1H, H-1), 4.25 (t, J = 10.0 Hz, 1H, H-2), 4.16 (d, J = 2.8 Hz, 1H, H-4), 3.83–3.70 (m, 3H, H-5 and H-6), 2.33 (s, 3H, CH3). 13C NMR (CD3OD, 100 MHz): δ 138.8, 133.1, 131.7, 130.7, 125.0, 91.8, 80.9, 69.5, 69.2, 68.0, 62.3, 21.1. 19F NMR (CD3OD, 376 MHz): δ −146.5 (m, 2F), −164.9 (m, 2F). HRMS calculated for [C21H20F4N4O4SNa]+, 523.1039; found: 523.1034.
2.3 General procedure for the preparation of compounds (6–10)
Method A for compounds 6–7: Compound 3 (25 mg, 0.050 mmol) was dissolved in ROH (3 mL) and NaOR (1 M, 1 mL) and stirred for 2 days at rt before quenching with dowex. The mixture was filtered and following evaporation of the filtrate the residue was purified with column chromatography (heptane:EtOAc 1:1–>1:2).
Method B for compounds 8–10: A mixture of compound 3 (20 mg, 0.040 mmol) and K2CO3 (16.5 mg, 0.12 mmol), amine (x, 3 equiv.) and DMF (3 mL) was stirred for (t) time at 50 °C. After evaporation of the solvent the residue was purified with column chromatography (heptane:EtOAc 1:1–>1:2).
2.4 Competitive fluorescence polarization experiments determining galectin-3 affinities
Human galectin-3 was expressed and purified as earlier described.18 Fluorescence polarization experiments were performed on a PheraStarFS plate reader with software PHERAstar Mars version 2.10 R3 (BMG, Offenburg, Germany) and fluorescence anisotropy of fluorescein tagged probes measured with excitation at 485 nm and emission at 520 nm. Experiments were performed at 20 °C with galectin-3 at 0.20 μM and the fluorescent probe 3,3′-dideoxy-3-[4-(fluorescein-5-yl-carbonylaminomethyl)-1H-1,2,3-triazol-1-yl]-3′-(3,5-di-methoxybenzamido)-1,1′-sulfanediyl-di-β-D-galactopyranoside19 (Kd 80 nM) at 0.02 μM as previously described.10,15,19 Compounds were dissolved in neat DMSO at 20 mM and diluted in PBS to 3–6 different concentrations to be tested in duplicate. Kd averages and SEM were calculated from 4 to 25 single-point measurements from at least two independent experiments showing between 20–80% inhibition.2.5 Crystallization of galectin-3 C-terminal domain with compounds (2–5) and (8)
Solutions of the C-terminal CRD of galectin-3C2 (19.2 mg ml−1 in 10 mM phosphate pH 7.4, 100 mM NaCl, 10 mM β-mercaptoethanol and 2 mM EDTA) were mixed with crystallization solution (20% PEG 4000, 0.1 M Tris/HCl pH 7.5, 0.4 M NaSCN, 7.9 mM β-mercaptoethanol). Crystallization drops of 2 + 2 μL were set up over 0.5 mL reservoir solution. The crystals obtained were soaked with compounds. Compounds 2–5 and 8 were dissolved in DMSO to obtain highly concentrated stocks. These stocks were then diluted with PEG400 (final concentration 30%), as the compounds were highly insoluble in water, then a ligand cocktail was prepared using crystallization reservoir and the ligand stock to obtain a final compound concentration of 10 mM. Crystals were placed in 4 μl of these cocktails and left for 15–20 hours. These soaked crystals were flash-cooled in cryoprotectant solution (15% PEG400, 25.5 w/v % PEG 4000, 250 mM NaSCN, 85 mM Tris/HCl pH 7.5, 2.5 mM ligand concentration).2.6 Data collection and structure solution of galectin-3C in complex with compounds (2–5) and (8)
Data for compounds 3–5 and 8 were collected at 100 K at station I911-3 of the MAX-II synchrotron, Lund, Sweden (λ = 1.0000 Å), equipped with a marMosaic 225 mm CCD detector. 300–360 images with 0.5° rotation and 1–3 seconds exposure times were collected for 3–5 and 8. Data for 2 were collected at ID23-2, ESRF, France on a DECTRIS PILATUS3 2M detector. 600 images were collected with 0.5° rotation and 0.2 seconds exposure time. Data for all structures were integrated using XDS and scaled using XSCALE.21 The structures were refined using phenix.refine22 and PDB entry 3ZSL (stripped of water molecules and alternate conformations) as starting model, first by rigid-body refinement. Five percent of the total reflections chosen at random were set aside for cross validation. The models were then subjected to model building and maximum likelihood refinement, gradually increasing the resolution to the highest resolutions with anisotropic B factors. After initial refinement of the protein coordinates in phenix.refine,22 the coordinates of 2–5 and 8 were fitted to the electron density using Coot.23 Further model building and manipulations were done in Coot. Restraints were generated using eLBOW24 from Phenix for 3–7. The structures were refined until convergence and individual anisotropic atomic displacement parameters for each atom were refined. Water molecules were added to positive difference density peaks more than 4.5 or 5 σ above the mean and also present in the 2m|Fo| − D|Fc| map at the 1 σ level. Riding hydrogen atoms were added in the final stages of refinement. Refinement statistics are listed in Table S1.† Molecular images were generated using PyMOL (Schrodinger LLC). Model validation and analysis were performed using MolProbity25 and PDB_REDO.26 Coordinates have been deposited in the Protein Data Bank with accession numbers 6I75 for compound 2, 6I74 for compound 3, 6I76 for compound 4, 6I77 for compound 5 and 6I78 for compound 8. For detailed structure refinement statistics, please refer to Table 1 in the ESI.†2.7 Quantum mechanical calculations
Four sets of quantum-mechanical (QM) calculations at different levels of theory were employed to obtain energies that can help to explain the differences in binding affinity of compounds 2–9 to galectin-3. All QM calculations were performed with the Turbomole 7.2 software.27,28 In all systems, ligands were modelled as the isolated fluorine-substituted benzene moiety by replacing the remaining part of the ligand with a hydrogen atom.In the first set, we calculated the energy of rotating the variable group of compounds 2, 4, 5, 6 and 8 out of the plane of the tetrafluorophenyl group by changing the value of a C–C–X–Y dihedral angle from 0° to 90° in increments of 10°, where the first and the second carbon atom belong to the ring, whereas the X and Y atoms belong to the varying group (in case of compound 8, Y atom is carbon). For each dihedral angle value, optimization of all the other degrees of freedom was performed at the B3LYP-D3/def2-SV(P) level of theory.29–33
In the next two sets of calculations, we calculated the interaction energy between ligands and three nearby residues, Ser237–Gly238 and Arg144. We performed separate calculations for each of the two residues. The amide group of Ser237–Gly238 was modelled as CH3–CO–NH–CH3, and the side chain of Arg144 was modelled as [CH3–NH–C(NH2)2]+. The coordinates were taken from the crystal structures. Arg144 has two conformations in complex with compounds 3 and 5, and for these, we performed separate calculations on both conformations. The calculations were performed at the TPSS-D3/def2-TZVP level of theory.32,34,35 The interaction energy for each compound–residue system was calculated from three single-point calculations as ΔE = Ecomplex − Eresidue − Eligand.
Finally, we calculated the solvation free energies for compounds 2–9, using the conductor-like screening model for real solvents (COSMO-RS),36,37 with the dielectric constant for water εr = 80 and optimized radii for all atoms.38 These calculations were based on two single point BP86 calculations29,35,39 with the TZVP basis set,40 as is requested by the method, one in vacuum and one in a continuum solvent with an infinite dielectric constant.36,37
3. Results and discussion
3.1 Synthesis and galectin-3 affinities of 3-(4-aryl-1,2,3-triazol-1-yl)-thiogalactosides
A 1,3-dipolar cycloadditions with alkynes and azide 116 produced penta- and tetrafluoroaryltriazoles 2–3 (Scheme 1). Nucleophilic aromatic substitution of the p-fluorine in 3 with alcohols, amines and NaN3 gave tetrafluoroaryltriazoles 4 and 6–10, while reduction of azide 4 resulted in amine 5.
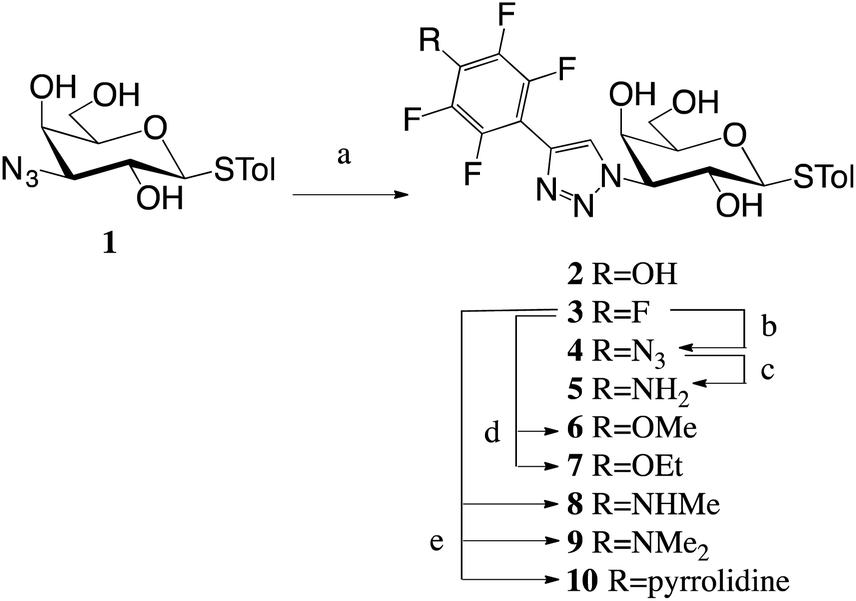 | ||
| Scheme 1 Synthesis of triazoles 2–10. Reagents and conditions: (a) Alkyne, CuI, DIPEA, MeCN, 50 °C; (b) NaN3, DMF, 60 °C; (c) 1,3-propanedithiol, Et3N, MeOH, rt; (d) NaR, HR, rt; (e) amine, K2CO3, DMF, 50 °C. Tol = p-methylphenyl. | ||
The inhibition potencies of thiogalactosides 2–10 were evaluated towards galectin-3 using a previously described competitive fluorescence polarization assay10,20 and the results are presented in Table 1. The pentafluorophenyl 3 had an affinity of 3.4 μM to galectin-3. Any replacement of the fluorine in the para position led to a drop in affinity. Replacing the fluorine with an amine (5) or azide (4) resulted in a 2–3-fold decrease, while replacement with a hydroxyl (2) resulted in a 7-fold decrease. Adding methyl groups (8–9) to amine 5 further decreased the affinity 2-fold per methyl group, while adding a methyl group (6) to the hydroxide 2 did not affect the affinity. Fluorine replacement with a bulkier ethoxy group (7) resulted in an almost 5-fold decrease in affinity compared to methoxide 6, which is indicative of steric restrictions in the binding pocket. This is further demonstrated by the even bulkier pyrrolidine (10) that does not bind galectin-3 at all at the concentrations tested.
3.2 Structural analysis of thiogalactosides 2–5 and 8 in complex with the galectin-3 CRD (galectin-3C)
In order to further investigate the binding interactions in the pocket below Arg144, high-resolution X-ray structures (all <1.3 Å resolution; see Table S1 in the ESI†) of thiogalactosides 2–5 and 8 in complex with galectin-3C were determined. X-ray structures of thiogalactosides 6–7 and 9–10 could not be obtained owing to solubility issues. The structures revealed a virtually identical binding mode for the galactose unit as earlier reported in many publications, and the triazole extends the para-substituted tetrafluorophenyl group into the pocket below Arg144.15The superimposition of all crystal structures (Fig. 1) shows that the ligands reside in the binding pocket in a similar manner to that reported previously. The anomeric S-tolyl group of the ligands is disordered, and in this work, focus is on the phenyl group below Arg144 and its para-substituents. Fig. 1 also shows that Arg144 adopts two principal conformations, either directly above the phenyl ring or above the para substituent, depending on the nature of the phenyl substitutions. Arg144 has split occupancy in the crystal structures of 3 and 5. This is likely due to a weakened cation–π interaction as a result of the electron-withdrawing fluorines. The N-methyl group in 8 is oriented above the phenyl ring plane towards Arg144. As a result, Arg144 resides only above the phenyl ring in this complex, while for both 2 and 4, Arg144 shows a single conformation directly above the para substituent.
 | ||
| Fig. 1 Superimposed view of the five crystal structures showing the ligand and neighbouring protein residues. The galactose moiety forms a hydrophobic stacking interaction with Trp181, the triazole linker extends the tetrafluorophenyl group in to the pocket near Arg144, which makes a cation–π interaction with the tetrafluorophenyl group. Ligands 2, 3, 4, 5, and 8 are shown in yellow, green, purple, magenta and light blue colours, respectively (also for Arg144). | ||
Compound 2 has the lowest affinity among the successfully crystallized ligands. The phenolic proton in 2 is, based on the pKa value of 5.7 for pentafluorophenol,42 likely to be deprotonated, and the resulting anion could interact favourably with the cation of Arg144. However, as will be discussed below, it will be more disfavoured by desolvation effects than the other ligands. Besides the interaction with Arg144, the binding site is unable to stabilise the negative charge on the ligand. For example, Ser237 shows two conformations as for the other ligands, and only one of these forms a rather weak hydrogen bond to the hydroxyl group of the ligand (O–O distance of 3.4 Å).
The azide in 4 is located in-plane with the phenyl ring and pointing outwards to solution, because the connecting nitrogen is sp2 hybridized and orienting it inwards to the protein would result in a steric clash with Ile145. The single occupancy of Arg144 in the complex with 4 may be due to a more favourable interaction with the π-system of the azide than with the phenyl ring of 3. The azide group is within hydrogen bonding distance of three water molecules (Fig. 2c), which may stabilize the ligand, resulting in better affinity than other compounds except 3. Most of these water molecules are present at very similar positions in all complexes, but they make more interactions with 4 than with the other ligands.
 | ||
| Fig. 2 Close-up view of the binding pocket in the crystal structures of galectin-3C in complex with phenyltriazoles (a) 2, (b) 3, (c) 4, (d) 5 and (e) 8. (f) Superimposed view of 2 and 3 showing the important hydrogen bonded water and fluorine–amide interactions. Water in 3 is coloured red and water in 2 is coloured cyan. | ||
Compound 3 has the highest affinity, showing that fluorine is the best candidate at the para position. This fluorine atom forms multipolar orthogonal interactions with a nearby peptide bond (Gly238; Fig. 2f) which increases the affinity. The fluorine atom is at a distance of 3.0 Å from the N atom of the backbone and 3.6 Å from the carbonyl C atom.
3.3 Quantum mechanical calculations
To rationalize the affinities and the structural observations, we have made a number of quantum mechanical (QM) calculations with compounds 2–9. First, we calculated the potential-energy surface for rotation of the varying para substituent of 2, 4–6, and 8 out of the plane of the tetrafluorophenyl group (Fig. 3). It can be seen that ligands with OH (2) and N3 (4) attain their energy minima with the substituent in the plane of the phenyl group. This is in accordance with the crystal structure of the azide 4 in complex with galectin-3. The other three ligands (5, 6, and 8) attain a shallow minimum around ∼20°, which probably reflects a competition between conjugation (favouring an angle of 0°) and the bulk of the methyl groups of 6 and 8, which prefer a larger dihedral. The figure indicates that ligand 8 with NHMe, for which the angle is 72° in the crystal structure, is strained by ∼8 kJ mol−1, which might be compensated by polar interactions with the NH groups, although no such interactions are obvious from the crystal structure.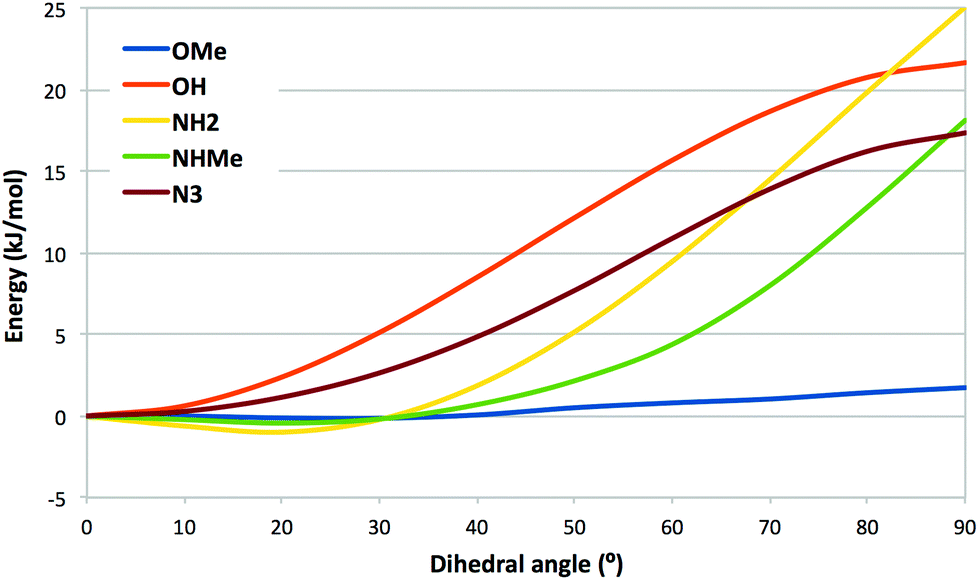 | ||
| Fig. 3 Potential-energy surface for the C–C–X–Y dihedral angle of the varying group in compounds 2 (OH), 4 (N3), 5 (NH2), 6 (OMe) and 8 (NHMe). | ||
Next, we calculated the interaction energy between the pentafluorophenyl group of 3 and the backbone amide group of Ser237–Gly238 (modelled by CH3–CO–NH–CH3 with coordinates taken from the crystal structure). It was 6 kJ mol−1, which is of the expected size for an F–amide interaction.43 On the other hand, ligand 5 gave the same interaction energy (6 kJ mol−1) with an amide group.
Third, we calculated the COSMO-RS solvation free energies of the nine para-substituted tetrafluorophenyl groups 2–9 (not pyrrolidine 10). The results are shown in Fig. 4 as a function of the measured binding affinities. It can be seen that for the OEt (7), NMe2 (6) and OMe (9), the estimated solvation free energies are small, −1 to −3 kJ mol−1, and there is no correlation with the binding free energies. However, for the F (3), N3 (4), NH2 (5) and OH (2) substituents, there is a good negative correlation to the binding affinity (R = −0.95). The ligand with the NHMe (8) substituent also falls close to the correlation line, albeit reducing the correlation to −0.83 if included. Taken together, these results indicate that the observed affinities can be explained by two effects. The alkylated substituents, especially OEt (7), are too large and steric effects give a low affinity, decreasing further with the number of methyl groups on the substituent. For the other ligands, the affinity is determined by desolvation effects: in the binding site, the ligand is partly buried by the protein and is less solvated than in water solution. This desolvation is modest for the F and N3 ligands (3 and 4), which form poor hydrogen bonds and therefore give low solvation energies (>−5 kJ mol−1). However, for the NH2 and OH ligands (5 and 2), the effect is large and pronounced. The effect would be even larger if the OH ligand 2 is deprotonated (the calculated solvation energy is −240 kJ mol−1). For the NHMe ligand 8, both steric and desolvation effects seem to be significant.
 | ||
| Fig. 4 Solvation free energies (blue squares) and interaction energies between Arg144 and the substituted tetraphenyl group (red diamonds) for compounds 2–9. | ||
Finally, we also calculated the interaction energy between Arg144 and the substituted tetrafluorophenyl groups, using the geometry from the crystal structure (and two different Arg144 conformations for the F and NH2 ligands 3 and 5). The results are also included in Fig. 4 (red symbols and line). It can be seen that all ligands give a large cation–π interaction energy of 18–27 kJ mol−1. All groups give lower interaction energies with Arg144 than an unsubstituted benzene group (−37 kJ mol−1). The interaction energies of the two Arg144 conformations for the F and NH2 ligands 3 and 5 differ by 3–6 kJ mol−1 (compared to 1 kJ mol−1 for benzene, using the two conformations for pentafluorophenyl 3). The average interaction energies show a good anti-correlation with the ligand-binding affinities (R = −0.87) and a correlation with the solvation energies (R = 0.78), illustrating that all three depend on the polarity of the ligand. Thus, the interaction with Arg144 partly counteracts the desolvation penalty and the difference of these two energies give an excellent anti-correlation to the binding free energies of −0.91, although the slope is rather large at 1.7 (the same as that of the interaction with Arg144, but half as large as that of the solvation free energy).
4. Conclusions
A series of 2,3,5,6-tetrafluorophenyl derivatives 2–10 with different para substituents were synthesized to analyse in detail the binding interactions within a small pocket beneath Arg144 in galectin-3. The most potent para substituent was the fluorine (3) that forms a fluorine–amide interaction with the backbone amide of Ser237–Gly238. However, the QM interaction energy between the backbone of Ser237–Gly238 and ligand 3 is not larger than for some of the other ligands, e.g.5. Instead, the relative affinities seem to be determined by three effects: First, the pocket beneath Arg144 is not large enough for bulkier groups, e.g. –NMe2 (9) and –OEt (7). Second, the solvation energy decreases strongly in the series 2–5–4–3 (OH–NH2–N3–F), implying that the desolvation penalty also decreases in this series, closely following the affinities of these ligands. However, this effect is partly counteracted by the interaction energy of the substituted tetrafluorophenyl group with Arg144, which becomes less favourable in this series (cf.Fig. 4). Taken together, given the frequency of employing fluorinated phenyl moieties and substituted derivatives thereof in drug design and drug discovery, the results presented here provide further in-depth insight into the sometimes conflicting driving forces behind such the interactions of such moieties in arginine-containing protein binding sites.Conflicts of interest
UJN and HL are shareholders in Galecto Biotech AB, a company developing galectin inhibitors.Acknowledgements
This work was supported by the Swedish Research Council (Grant No. 621-2009-5326, 621-2012-2978 and 2014-5540), the Royal Physiographic Society, Lund, the European Community's Seventh Framework Program (FP7-2007–2013) under grant agreement no. HEALTH-F2-2011-256986 – project acronym PANACREAS, and a project grant awarded by the Knut and Alice Wallenberg Foundation (KAW 2013.0022). We thank staff at the I911-3 beamline of the MAX IV Laboratory and the ID23-2 beamline of the ESRF, for beam time and assistance in data collection. The calculations were performed on computer resources provided by the Swedish National Infrastructure for Computing (SNIC) at Lunarc at Lund University and HPC2N at Umeå University.References
- C. Bissantz, B. Kuhn and M. Stahl, J. Med. Chem., 2010, 53, 5061–5084 CrossRef CAS PubMed.
- J. Seetharaman, A. Kanigsberg, R. Slaaby, H. Leffler, S. H. Barondes and J. M. Rini, J. Biol. Chem., 1998, 273, 13047–13052 CrossRef CAS PubMed.
- H. Leffler, S. Carlsson, M. Hedlund, Y. Qian and F. Poirier, Glycoconjugate J., 2002, 19, 433–440 CrossRef CAS PubMed.
- V. L. Thijssen, R. Heusschen, J. Caers and A. W. Griffioen, Biochim. Biophys. Acta, Rev. Cancer, 2015, 1855, 235–247 CrossRef CAS PubMed.
- A. U. Newlaczyl and L. G. Yu, Cancer Lett., 2011, 313, 123–128 CrossRef CAS PubMed.
- F. T. Liu, R. J. Patterson and J. L. Wang, Biochim. Biophys. Acta, Gen. Subj., 2002, 1572, 263–273 CrossRef CAS.
- S. Di Lella, V. Sundblad, J. P. Cerliani, C. M. Guardia, D. A. Estrin, G. R. Vasta and G. A. Rabinovich, Biochemistry, 2011, 50, 7842–7857 CrossRef CAS PubMed.
- G. A. Rabinovich and D. O. Croci, Immunity, 2012, 36, 322–335 CrossRef CAS PubMed.
- A. C. MacKinnon, M. A. Gibbons, S. L. Farnworth, H. Leffler, U. J. Nilsson, T. Delaine, A. J. Simpson, S. J. Forbes, N. Hirani, J. Gauldie and T. Sethi, Am. J. Respir. Crit. Care Med., 2012, 185, 537–546 CrossRef CAS PubMed.
- I. Cumpstey, S. Carlsson, H. Leffler and U. J. Nilsson, Org. Biomol. Chem., 2005, 3, 1922–1932 RSC.
- P. Sörme, P. Arnoux, B. Kahl-Knutsson, H. Leffler, J. M. Rini and U. J. Nilsson, J. Am. Chem. Soc., 2005, 127, 1737–1743 CrossRef PubMed.
- C. Diehl, O. Engström, T. Delaine, M. Håkansson, S. Genheden, K. Modig, H. Leffler, U. Ryde, U. J. Nilsson and M. Akke, J. Am. Chem. Soc., 2010, 132, 14577–14589 CrossRef CAS PubMed.
- P. M. Collins, C. T. Öberg, H. Leffler, U. J. Nilsson and H. Blanchard, Chem. Biol. Drug Des., 2012, 79, 339–346 CrossRef CAS PubMed.
- V. K. Rajput, A. MacKinnon, S. Mandal, P. Collins, H. Blanchard, H. Leffler, T. Sethi, H. Schambye, B. Mukhopadhyay and U. J. Nilsson, J. Med. Chem., 2016, 59, 8141–8147 CrossRef CAS PubMed.
- T. Delaine, P. Collins, A. MacKinnon, G. Sharma, J. Stegmayr, V. K. Rajput, S. Mandal, I. Cumpstey, A. Larumbe, B. A. Salameh, B. Kahl-Knutsson, H. van Hattum, M. van Scherpenzeel, R. J. Pieters, T. Sethi, H. Schambye, S. Oredsson, H. Leffler, H. Blanchard and U. J. Nilsson, ChemBioChem, 2016, 17, 1759–1770 CrossRef CAS PubMed.
- K. Peterson, R. Kumar, O. Stenström, P. Verma, P. R. Verma, M. Håkansson, B. Kahl-Knutsson, F. Zetterberg, H. Leffler, M. Akke, D. T. Logan and U. J. Nilsson, J. Med. Chem., 2018 DOI:10.1021/acs.jmedchem.7b01626.
- I. Dimitrov, K. Jankova and S. Hvilsted, J. Fluorine Chem., 2013, 149, 30–35 CrossRef CAS.
- S. M. Massa, D. N. Cooper, H. Leffler and S. H. Barondes, Biochemistry, 1993, 32, 260–267 CrossRef CAS PubMed.
- E. Salomonsson, A. Larumbe, J. Tejler, E. Tullberg, H. Rydberg, A. Sundin, A. Khabut, T. Frejd, Y. D. Lobsanov, J. M. Rini, U. J. Nilsson and H. Leffler, Biochemistry, 2010, 49, 9518–9532 CrossRef CAS PubMed.
- P. Sörme, B. Kahl-Knutsson, M. Huflejt, U. J. Nilsson and H. Leffler, Anal. Biochem., 2004, 334, 36–47 CrossRef PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- P. V. Afonine, W. Ralf, J. J. Headd and C. Thomas, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 352–367 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- N. W. Moriarty, R. W. Grosse-Kunstleve and P. D. Adams, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 1074–1080 CrossRef CAS PubMed.
- V. B. Chen, W. B. Arendall, J. J. Headd, D. A. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson and D. C. Richardson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 12–21 CrossRef CAS PubMed.
- R. P. Joosten, F. Long, G. N. Murshudov and A. Perrakis, IUCrJ, 2014, 1, 213–220 CrossRef CAS PubMed.
- F. Furche, R. Ahlrichs, C. Hättig, W. Klopper, M. Sierka and F. Weigend, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 91–100 CAS.
- TURBOMOLE version 7.1, 2016 Search PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104–115123 CrossRef PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- M. Sitarz, E. Wirth-Dzieciolowska and P. Demant, Neoplasma, 2000, 47, 148–150 CAS.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- A. Schäfer, A. Klamt, D. Sattel, J. C. W. Lohrenz and F. Eckert, Phys. Chem. Chem. Phys., 2000, 2, 2187–2193 RSC.
- A. Klamt, V. Jonas, T. Bürger and J. C. W. Lohrenz, J. Phys. Chem. A, 1998, 102, 5074–5085 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- I. Cumpstey, E. Salomonsson, A. Sundin, H. Leffler and U. J. Nilsson, Chem. – Eur. J., 2008, 14, 4233–4245 CrossRef CAS PubMed.
- D. A. Kraut, P. A. Sigala, B. Pybus, C. W. Liu, D. Ringe, G. A. Petsko and D. Herschlag, PLoS Biol., 2006, 4, e99 CrossRef PubMed.
- P. Zhou, J. Zou, F. Tian and Z. Shang, J. Chem. Inf. Model., 2009, 49, 2344–2355 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ob02888e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |