
Cascade cyclization and intramolecular nitrone dipolar cycloaddition and formal synthesis of 19-hydroxyibogamine†
Ziad T. I.
Alkayar‡
 and
Iain
Coldham
*
and
Iain
Coldham
*
Department of Chemistry, University of Sheffield, Sheffield, S3 7HF, UK. E-mail: i.coldham@sheffield.ac.uk
First published on 5th December 2018
Abstract
A cascade or domino sequence of condensation of hydroxylamine and an aldehyde to give an oxime, cyclization to a nitrone, and intramolecular 1,3-dipolar cycloaddition has been successfully employed where there is branching at C-4 as a route to the iboga alkaloids. Cyclization occurs with displacement of chloride as a leaving group and intramolecular cycloaddition occurs with an alkene as a dipolarophile. The reaction gives an azabicyclo[2.2.2]octane product containing a fused isoxazolidine as a single stereoisomer and this was converted to an isoquinuclidine that completed a formal synthesis of the alkaloid (±)-19-hydroxyibogamine.
Introduction
Iboga alkaloids have been isolated from plants of the Tabernanthe genus, found particularly in West Africa.1 The iboga alkaloids are related by a common pentacyclic structure found in ibogamine 1, that consists of an isoquinuclidine core with a fused indoloazepine ring and representative examples are shown in Fig. 1. Their pharmacological properties have been studied widely and they have attracted attention due to their ability to treat drug addiction.2 Ibogaine 2 is the most abundant of the alkaloids in Tabernanthe iboga and has been used clinically. The alkaloid catharanthine 4 has potent antagonist activity against transient receptor potential melastatin 8 (TRPM8), which is expressed in sensory neurons and involved in thermoregulation and pain.3 This compound is an important intermediate in the synthesis of vinblastine and analogues.4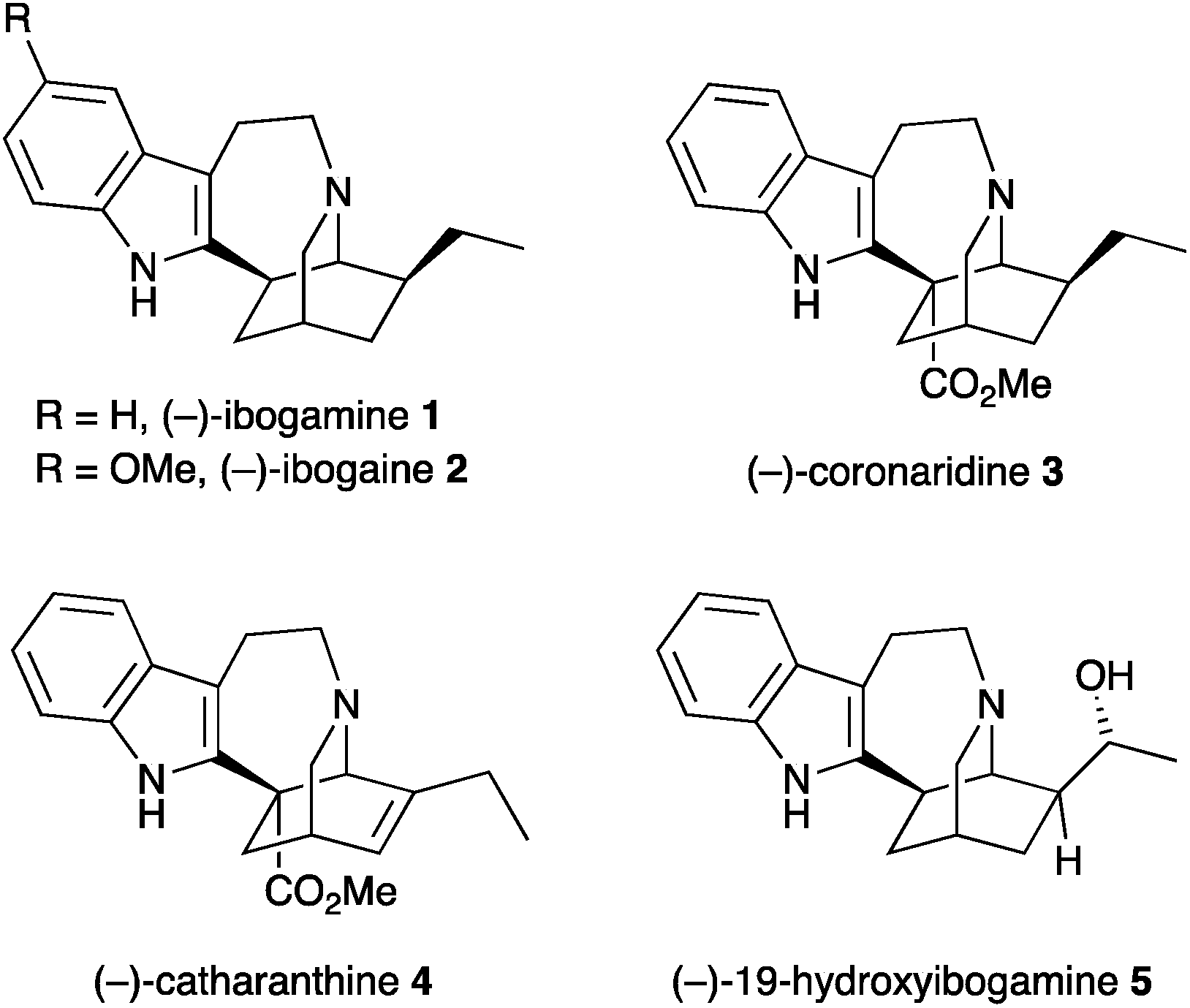 | ||
| Fig. 1 Representative iboga alkaloids, 1–5. | ||
Many syntheses of the alkaloid ibogamine have been reported.5,6 In contrast, there is only one report of the preparation of the iboga alkaloid (–)-19-hydroxyibogamine 5,7 which shows marked antibiotic activity.8 There is a growing number of iboga alkaloids with a hydroxyethyl group or other oxygenated side chain at C-20 of the core ring system.9 Here we report an efficient synthesis of the isoquinuclidine core with a hydroxyethyl side chain that makes use of a nitrone cycloaddition as part of a cascade process involving simple condensation of hydroxylamine with an aldehyde followed by cyclization on to an alkyl halide and cycloaddition. We have applied this to a formal synthesis of 19-hydroxyibogamine 5.
We have reported a number of examples of the formation of polycyclic amines by use of a cascade strategy that incorporates a condensation reaction of an amine and an aldehyde (or ketone) followed by in situ cyclization of the imine (or oxime or hydrazone) on to an alkyl halide, followed by in situ dipolar cycloaddition.10–15 Our efforts have been centred mostly on fused ring systems (Scheme 1a) and this chemistry leads to the synthesis of several natural products (aspidosperma alkaloids, myrioxazine A).10,11 An intermolecular cycloaddition alternatively leads to bicyclic products and has been used to prepare crispine A and macronecine.12 We have extended the methodology to bridged polycyclic compounds (Scheme 1b),13 and recently to spirocyclic compounds (Scheme 1c).14 The research on the bridged compounds located the branch point (between the alkene dipolarophile and the alkyl halide) β- to the aldehyde. This led to compounds with a one-carbon bridge. We were interested to see if the chemistry could be extended to compounds that would give a two-carbon bridge and hence access the isoquinuclidine ring system. Here we report the results of this study in the context of the synthesis of the iboga alkaloids.
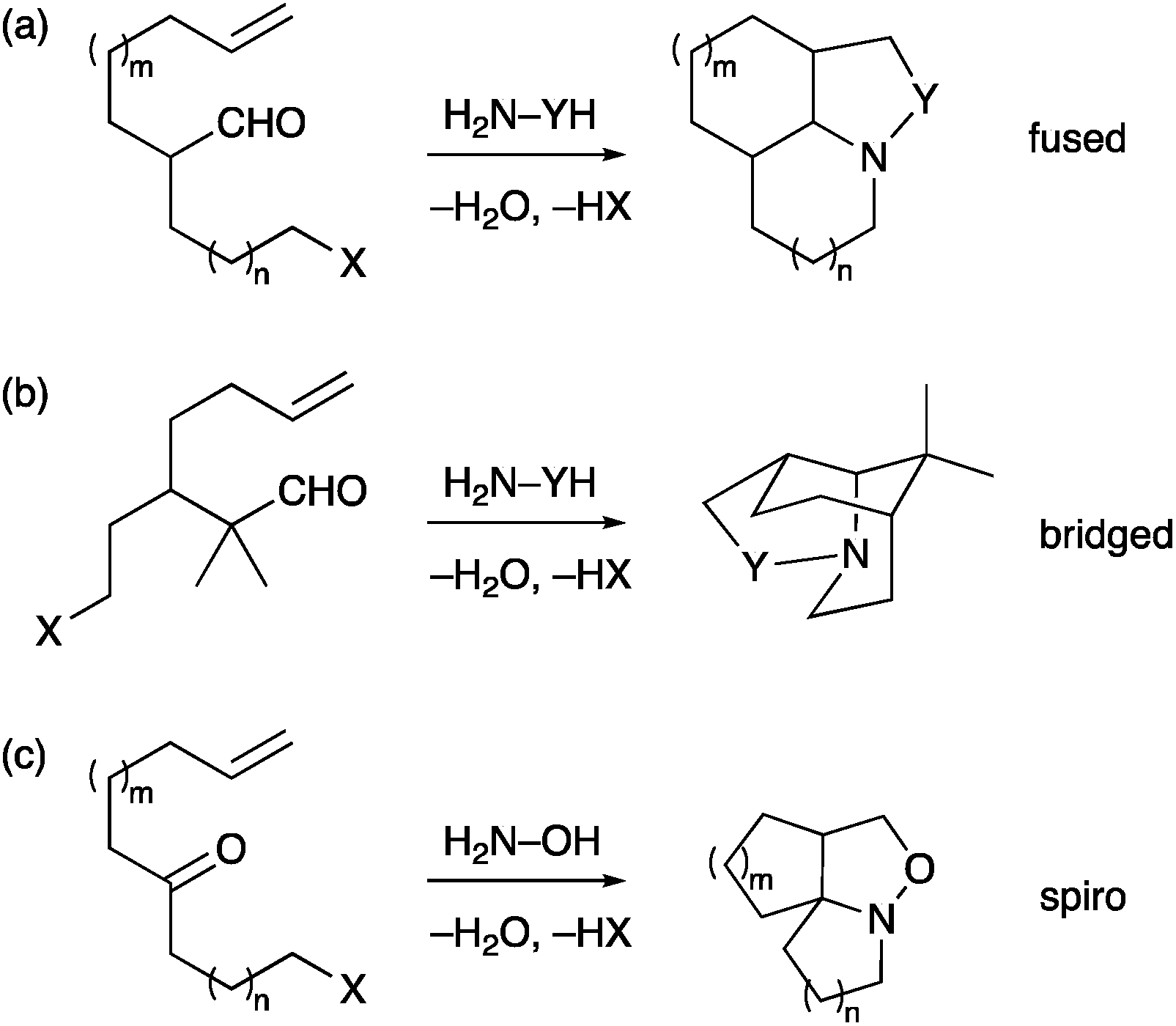 | ||
| Scheme 1 Cascade chemistry to polycyclic amines (X = Br, Cl; Y = O, NR, CHR).10–14 | ||
Results and discussion
To explore the ability to conduct the cascade chemistry towards a two-carbon bridged compound that could lead to the iboga alkaloids, we needed to prepare an aldehyde such as compound 7 with branching between the alkene and the alkyl halide γ- to the aldehyde (Scheme 2). We anticipated that condensation of hydroxylamine with this aldehyde would provide an intermediate oxime that would undergo cyclization with displacement of the chloride and subsequent 1,3-dipolar cycloaddition of the resulting nitrone with the tethered alkene to give the isoquinuclidine core 6. | ||
| Scheme 2 Retrosynthesis for 19-hydroxyibogamine 5. | ||
To test the chemistry and to access the required aldehyde rapidly, we prepared the racemic iodide 11 by reduction of the known diester 816 followed by two separate mono-halogenations (Scheme 3). The diester 8 was formed by allylation with crotyl bromide as a mixture of stereoisomers E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z 5.5:1 and could be reduced without purification. Treatment of iodide 11 with the anion formed from (EtO)2CHCN and LDA or NaH failed to give any alkylated product.17 We therefore turned to a dithiane anion. Deprotonation of dithiane 12 with n-BuLi followed by addition of the iodide 11 gave the desired alkylated product 13. As expected, this occurred by displacement of the iodide not the chloride and best conditions involved addition of the iodide 11 at −40 °C followed by warming to room temperature.
:Z 5.5:1 and could be reduced without purification. Treatment of iodide 11 with the anion formed from (EtO)2CHCN and LDA or NaH failed to give any alkylated product.17 We therefore turned to a dithiane anion. Deprotonation of dithiane 12 with n-BuLi followed by addition of the iodide 11 gave the desired alkylated product 13. As expected, this occurred by displacement of the iodide not the chloride and best conditions involved addition of the iodide 11 at −40 °C followed by warming to room temperature.
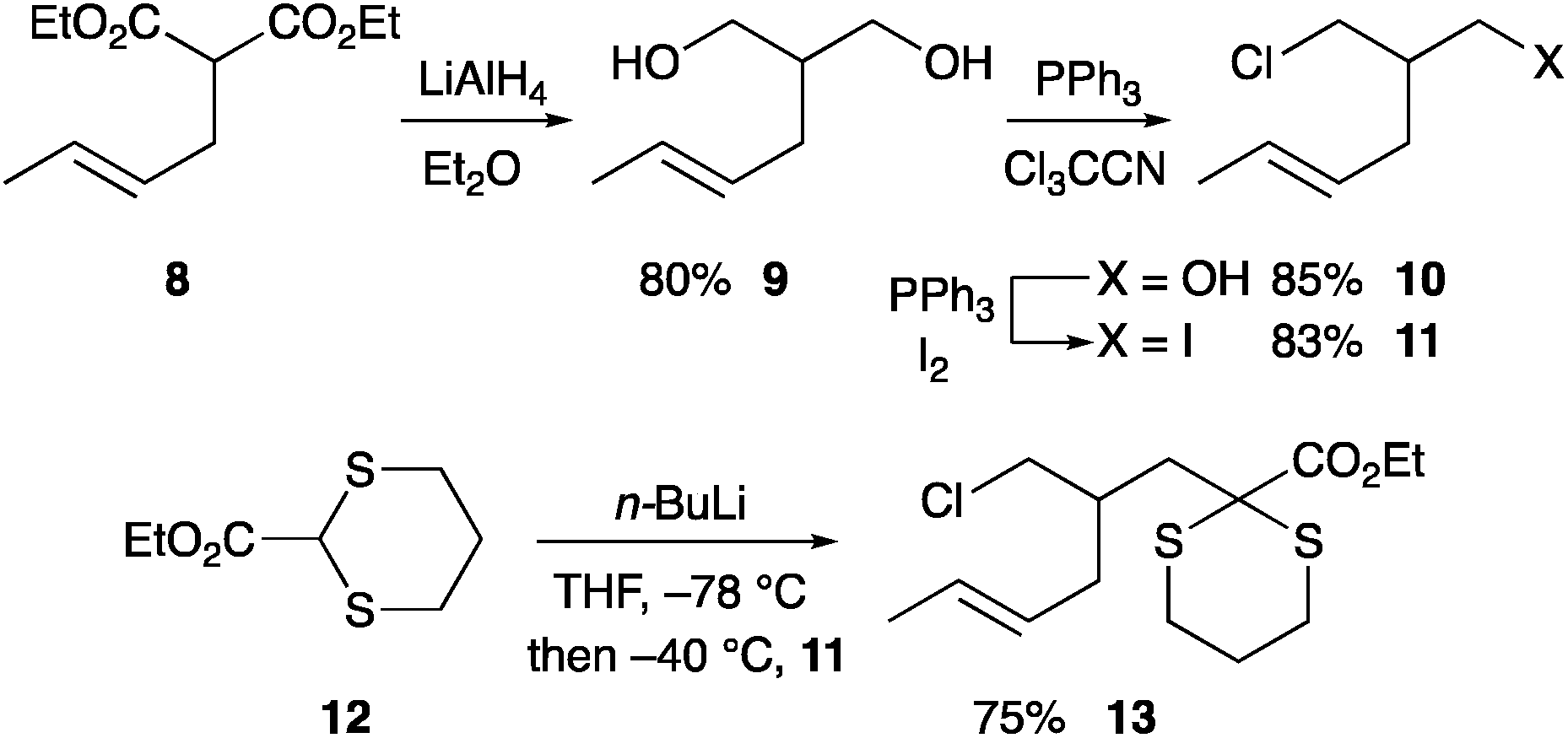 | ||
| Scheme 3 Synthesis of the dithiane 13. Ratio E:Z 5.5:1 for 8–11, 13. | ||
The ester 13 was reduced with LiAlH4 to give the alcohol 14 (Scheme 4). The presence of a dimethyl acetal (rather than a dithiane) adjacent to the required aldehyde has previously been found to be beneficial in an intramolecular dipolar cycloaddition reaction.18 Therefore, initially, alcohol 14 was treated with methanol, silver nitrate and N-chlorosuccinimde to promote transacetalation from the dithiane to the corresponding dimethyl acetal, although this new acetal proved to be unstable to subsequent Swern oxidation. Therefore, alcohol 14 was oxidised to give aldehyde 15. The aldehyde 15 could be converted to the dimethyl acetal 16.
 | ||
| Scheme 4 Synthesis of the aldehydes 15 and 16. Ratio E:Z 5.5:1 for 13–16. | ||
We now had in-hand two aldehydes to test the cascade chemistry. Taking the aldehyde 16 (which was still an inseparable mixture of E:Z stereoisomers in a ratio of 5.5:1) and heating with hydroxylamine at 60 °C gave the expected oxime 17 (Scheme 5). This oxime could be isolated or, alternatively, directly raising the temperature to heat under reflux gave a low yield (15%) of a single stereoisomer of the cycloadduct 18. No other isolable compounds could be obtained and it appeared that decomposition was taking place predominantly. Heating the purified oxime 17 in toluene in the presence of iPr2NEt gave the same result, with cycloadduct 18 being isolated in only 15% yield. Carrying out this cascade reaction in the presence of Bu4NI or by using xylene instead of toluene failed to provide any cycloadduct 18. The stereochemistry of the cycloadduct 18 was assigned on the basis of similar coupling constants in its 1H NMR spectrum in comparison with the cycloadduct 20 (and on the basis of the single crystal X-ray analysis of ketone 21, see below).
 | ||
| Scheme 5 Cycloaddition from the aldehyde 16 to give (±)-18. | ||
With the disappointing result in the formation of the cycloadduct 18, our attention turned to the corresponding dithiane 15. Heating aldehyde 15 with the hydrochloride salt of hydroxylamine in toluene and iPr2NEt resulted in a low yield (18%) of the cycloadduct 20 (Scheme 6). Addition of Bu4NI or heating in xylene did not change the outcome significantly (22% of 20 isolated). However, addition of MgSO4 did improve the yield, particularly if the oxime was allowed to form at 60 °C prior to heating at 110 °C. In this case the cycloadduct 20 was isolated in 36% yield. The difficulty appeared to be the poor formation of the oxime intermediate (19). Therefore an alternative strategy was adopted, whereby the oxime 19 was formed (in quantitative yield) under modified conditions (with sodium acetate as the base in methanol). The oxime was then heated in toluene with iPr2NEt and MgSO4 to give the desired cycloadduct 20 in more acceptable yield (57%). In the absence of either iPr2NEt or MgSO4, the yields were lower. A single stereoisomer of the cycloadduct 20 was formed; the stereochemistry was confirmed as discussed below and this fits with a concerted cycloaddition across the E-alkene.
 | ||
| Scheme 6 Cycloaddition from the aldehyde 15 to give (±)-20. | ||
With a successful synthesis of the cycloadduct 20, we sought to carry out further transformations towards the iboga alkaloid structures. Attempts to reduce the N–O bond in the cycloadduct 20 with zinc in aqueous acetic acid gave only recovered starting material. Alternatively, hydrolysis of the dithiane was carried out and we found that [bis(trifluoroacetoxy)iodo]benzene in aqueous acetonitrile resulted in the desired ketone 21 (Scheme 7). The relative stereochemistry of the ketone 21 was confirmed by single crystal X-ray analysis (see ESI†). At this point it was possible to reduce the N–O bond with activated zinc in acetic acid and methanol to give the isoquinuclidine 22. The crude material was coupled with 3-indoleacetic acid to give the isoquinuclidine 23 in quantitative yield from the isoquinuclidine 21. Finally, we were able to convert in high yield the ketone functional group to the dimethyl acetal 24 and the alcohol to the acetate so as to prepare racemic isoquinuclidine 25. The NMR spectroscopic data of the isoquinuclidine 25 matched those reported by Höck and Borschberg.7b Therefore this chemistry provides a formal synthesis of the alkaloid 19-hydroxyibogamine 5.
 | ||
| Scheme 7 Completion of the formal synthesis of 19-hydroxyibogamine 5 by preparation of (±)-25. | ||
Experimental
Reactions were carried out under nitrogen or argon using oven-dried glassware. Petrol refers to petroleum ether (b.p. 40–60 °C). Thin layer chromatography was carried out using silica plates, visualising by UV irradiation at 254 nm or by staining with an alkaline aqueous KMnO4 dip. The silica gel used for column chromatography was 40–63 micron mesh. The chemical shifts in the 1H NMR spectra are reported in ppm with respect to residual solvent peaks, with multiplicities s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, and br = broad. Coupling constants, J, are quoted to the nearest 0.5 Hz. High resolution (accurate mass) mass spectra (HRMS) used electrospray ionisation (ESI) with a time-of-flight (TOF) mass analyzer.2-[But-2-en-1-yl]propane-1,3-diol 9
Diethyl malonate (24.0 g, 100 mmol) was added to sodium hydride (1.44 g, 60 mmol) in THF (40 mL) at room temperature. After 15 min, the suspension was added dropwise to crotyl bromide (6.69 g, 50 mmol, E:Z 5.5:1) in THF (100 mL). After 3 h, saturated aqueous ammonium chloride (100 mL) and H2O (100 mL) were added. The aqueous layer was extracted with Et2O (3 × 75 mL) and the combined organic extracts were dried (MgSO4) and the solvent was evaporated. The crude product was added dropwise to a suspension of Et2O (50 mL) and LiAlH4 (3.7 g, 100 mmol) at 0 °C. After 10 min, the mixture was warmed to room temperature. After 3 h, aqueous sodium hydroxide (25 mL, 1 M) was added until a white precipitate was observed. The mixture was filtered through Celite®, washed with CH2Cl2–MeOH (9:1) (200 mL) and the solvent was evaporated. Purification by column chromatography on silica gel, eluting with petrol–EtOAc (3:2), gave the diol 9 (5.2 g, 80%) as an oil as a 5.5:1 E:Z mixture; Rf 0.2 [petrol–EtOAc (1:1)]; IR νmax(film)/cm−1 3315, 2915, 2885; 1H NMR (400 MHz, CDCl3, peaks for the major E isomer) δ = 5.57–5.37 (2H, m), 3.80 (2H, dd, J 10.5, 4 Hz), 3.66 (2H, dd, J 10.5, 7.5 Hz), 2.55 (2H, br s), 1.98 (2H, t, J 7 Hz), 1.87–1.79 (1H, m, CH), 1.67 (3H, dd, J 6, 1 Hz); 13C NMR (101 MHz, CDCl3, peaks for the major E isomer) δ = 128.5, 127.2, 65.8, 42.2, 31.2, 17.9; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C7H15O2 131.1070; found: 131.1072.
2-(Chloromethyl)hex-4-en-1-ol 10
Triphenylphosphine (7.25 g, 27.6 mmol) was added to the diol 9 (3.0 g, 23 mmol) in CH2Cl2 (230 mL) at 0 °C. After 10 min, trichloroacetonitrile (2.31 mL, 23.0 mmol) was added dropwise and the mixture was allowed to warm to room temperature. After 4 h, the solvent was evaporated. Purification by column chromatography on silica gel, eluting with petrol–EtOAc (4:1), gave the chloroalcohol 10 (2.89 g, 85%) as an oil as a 5.5:1 E:Z mixture; Rf 0.27 [petrol–EtOAc (4:1)]; IR νmax(film)/cm−1 3330, 2935, 2915, 1435, 725; 1H NMR (400 MHz, CDCl3, peaks for the major E isomer) δ = 5.57–5.50 (1H, m), 5.43–5.35 (1H, m), 3.72–3.68 (2H, m), 3.67–3.60 (2H, m), 2.13–2.08 (2H, m), 1.97–1.90 (1H, m), 1.68 (3H, dd, J 6, 1 Hz); 13C NMR (101 MHz, CDCl3, peaks for the major E isomer) δ = 128.0, 127.7, 62.8, 45.6, 42.8, 31.8, 18.0; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C7H1435ClO 148.0655; found: 148.0655; LRMS m/z (ES) 150 (60%), 148 (100).
6-Chloro-5-(iodomethyl)hex-2-ene 11
Imidazole (1.64 g, 25.9 mmol) and triphenylphosphine (6.80 g, 25.9 mmol) were added to 2-(chloromethyl)hex-4-en-1-ol 10 (3.20 g, 21.6 mmol) in THF (75 mL) at room temperature. After 15 min, ground iodine (6.00 g, 23.8 mmol) was added portionwise. After 3 h, CH2Cl2 (50 mL) was added, the suspension was filtered through Celite® and the solvent was evaporated. Purification by column chromatography on silica gel, eluting with petrol, gave the iodide 11 (4.62 g, 83%) as an oil as a 5.5:1 E:Z mixture; Rf 0.5 (petrol); IR νmax(film)/cm−1 2960, 1435; 1H NMR (400 MHz, CDCl3, peaks for the major E isomer) δ = 5.62–5.54 (1H, m), 5.38–5.29 (1H, m), 3.66 (1H, dd, J 11, 4.5 Hz), 3.52 (1H, dd, J 11, 6.5 Hz), 3.40 (1H, dd, J 10, 4.5 Hz), 3.30 (1H, dd, J 10, 6 Hz), 2.16–2.06 (2H, m), 1.80–1.72 (1H, m), 1.69 (3H, dd, J 6.5, 1.5 Hz); 13C NMR (101 MHz, CDCl3, peaks for the major E isomer) δ = 128.9, 126.7, 47.9, 41.8, 35.1, 18.0, 10.5; HRMS (ESI-TOF) m/z: [M]+ calcd for C7H1235ClI 257.9672; found: 257.9669; LRMS m/z (EI) 260 (2%), 258 (6), 55 (100).
Ethyl 2-[2-(chloromethyl)hex-4-en-1-yl]-1,3-dithiane-2-carboxylate 13
n-BuLi (6.2 mL, 15.6 mmol, 2.5 M in hexanes) was added to ethyl 1,3-dithiane-2-carboxylate 12 (2.0 mL, 13 mmol) in THF (35 mL) at −78 °C. After 15 min, the iodide 11 (4.0 g, 15.6 mmol) in THF (5 mL) was added at −40 °C and the mixture was allowed to warm to room temperature. After 16 h, H2O (40 mL) was added and the aqueous layer was extracted with Et2O (3 × 50 mL), dried (MgSO4), and the solvent was evaporated. Purification by column chromatography on silica gel, eluting with petrol–EtOAc (98:2), gave the ester 13 (3.2 g, 75%) as an oil as a 5.5:1 E:Z mixture; Rf 0.3 [petrol–EtOAc (98:2)]; IR νmax(film)/cm−1 2980, 2920, 1720; 1H NMR (400 MHz, CDCl3, peaks for the major E isomer) δ = 5.60–5.51 (1H, m), 5.40–5.32 (1H, m), 4.31–4.24 (2H, m), 3.68–3.61 (2H, m), 3.33–3.25 (2H, m), 2.73–2.67 (2H, m), 2.27–2.11 (5H, m), 2.05 (1H, dd, J 14, 4 Hz), 1.93–1.82 (1H, m), 1.68 (3H, dd, J 6, 1 Hz), 1.36 (3H, t, J 7 Hz); 13C NMR (101 MHz, CDCl3, peaks for the major E isomer) δ = 171.0, 128.4, 127.5, 62.1, 52.9, 49.1, 39.8, 36.8, 35.9, 27.95, 27.9, 24.4, 18.0, 14.2; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H24O2S235Cl 323.0906; found: 323.0909; LRMS m/z (ES) 325 (45%), 323 (100).
2-[2-(Chloromethyl)hex-4-en-1-yl]-1,3-dithian-2-ylmethanol 14
The ester 13 (3.17 g, 9.8 mmol) in Et2O (5 mL) was added to a suspension of LiAlH4 (0.56 g, 14.7 mmol) in Et2O (25 mL) at room temperature. After 2.5 h, aqueous NaOH (30 mL, 1 M) was added until a white precipitate was observed. The mixture was filtered through Celite® washing with CH2Cl2–MeOH (9:1) (100 mL) and the solvent was evaporated. Purification by column chromatography on silica gel, eluting with petrol–EtOAc (4:1), gave the alcohol 14 (2.2 g, 80%) as an oil as a 5.5:1 E:Z mixture; Rf 0.5 [petrol–EtOAc (4:1)]; IR νmax(film)/cm−1 3275, 2935, 2910; 1H NMR (400 MHz, CDCl3, peaks for the major E isomer) δ = 5.59–5.50 (1H, m), 5.42–5.34 (1H, m), 3.77–3.74 (2H, m), 3.72–3.66 (2H, m), 2.99–2.90 (2H, m), 2.64–2.59 (2H, m), 2.28–2.19 (3H, m), 2.15–2.06 (2H, m), 1.93–1.78 (2H, m), 1.73 (1H, dd, 14, 3 Hz), 1.68 (3H, dd, J 6, 1 Hz); 13C NMR (101 MHz, CDCl3, peaks for the major E isomer) δ = 128.3, 127.7, 64.0, 54.8, 49.6, 39.0, 36.4, 35.9, 25.9, 25.8, 24.6, 18.0; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C12H22OS235Cl 281.0801; found: 281.0796; LRMS m/z (ES) 283 (3%), 281 (8), 265 (45), 263 (100).
2-[2-(Chloromethyl)hex-4-en-1-yl]-1,3-dithiane-2-carbaldehyde 15
DMSO (0.80 mL, 11 mmol) in CH2Cl2 (1 mL) was added dropwise to oxalyl chloride (0.50 mL, 5.5 mmol) in CH2Cl2 (15 mL) at −60 °C. After 5 min, alcohol 14 (1.4 g, 5.0 mmol) in CH2Cl2 (1 mL) was added dropwise. After 10 min, N,N-diisopropylethylamine (4.5 mL, 25 mmol) was added. After 15 min at −60 °C the mixture was allowed to warm to room temperature, then water (20 mL) was added. The mixture was extracted with CH2Cl2 (3 × 30 mL), the combined organic layers were washed successively with aqueous HCl (2 × 15 mL, 1 M), H2O (15 mL), aqueous Na2CO3 (15 mL, 5%), and H2O (20 mL), then dried (MgSO4) and the solvent was evaporated. Purification by column chromatography on silica gel, eluting with petrol–EtOAc (9:1) gave the aldehyde 15 (1.15 g, 83%) as an oil as a 5.5:1 E:Z mixture; Rf 0.5 [petrol–EtOAc (9:1)]; IR νmax(film)/cm−1 2930, 2855, 1705; 1H NMR (400 MHz, CDCl3, peaks for the major E isomer) δ = 9.03 (1H, s), 5.57–5.50 (1H, m), 5.36–5.28 (1H, m), 3.61 (1H, dd, J 11.5, 4 Hz), 3.56 (1H, dd, J 11.5, 4 Hz), 3.17–3.10 (1H, m), 3.01–2.93 (1H, m), 2.65–2.58 (2H, m), 2.24–2.17 (1H, m), 2.13–2.02 (4H, m), 1.87–1.75 (2H, m), 1.68 (3H, dd, J 6, 1 Hz); 13C NMR (101 MHz, CDCl3, peaks for the major E isomer) δ = 188.7, 128.9, 127.1, 57.7, 48.2, 37.0, 35.9, 30.1, 26.8, 26.7, 24.3, 18.0; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C12H20OS235Cl 279.0644; found: 279.0646; LRMS m/z (ES) 281 (45%), 279 (100).
4-(Chloromethyl)-2,2-dimethoxyoct-6-enal 16
Bis(trifluoroacetoxy)iodobenzene (3.00 g, 6.85 mmol) was added to aldehyde 15 (1.12 g, 4.04 mmol) in anhydrous methanol (5 mL) at room temperature. After 15 min, saturated aqueous sodium bicarbonate (5 mL) was added. The mixture was extracted with Et2O (3 × 10 mL), dried (MgSO4) and the solvent was evaporated. Purification by column chromatography on silica gel, eluting with petrol–Et2O (9:1), gave the aldehyde 16 (580 mg, 62%) as an oil as a 5.5:1 E:Z mixture; Rf 0.3 [petrol–Et2O (9:1)]; IR νmax(film)/cm−1 2945, 2840, 1750, 1440; 1H NMR (400 MHz, CDCl3, peaks for the major E isomer) δ = 9.46 (1H, s), 5.56–5.47 (1H, m), 5.34–5.25 (1H, m), 3.60–3.55 (2H, m), 3.31 (3H, s), 3.30 (3H, s), 2.17–2.05 (3H, m), 1.87–1.80 (1H, m), 1.75 (1H, dd, J 15, 5 Hz), 1.67 (3H, dd, J 6, 1 Hz); 13C NMR (101 MHz, CDCl3, peaks for the major E isomer) δ = 199.5, 128.7, 127.3, 102.1, 49.9, 49.7, 48.4, 35.6, 34.9, 32.6, 18.0; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C11H20O335Cl 235.1101; found: 235.1097; LRMS m/z (ES) 237 (35%), 235 (100).
4-(Chloromethyl)-2,2-dimethoxyoct-6-enal oxime 17
Aldehyde 16 (100 mg, 0.43 mmol), hydroxylamine hydrochloride (0.04 g, 0.64 mmol), N,N-diisopropylethylamine (0.18 mL, 1.06 mmol) and MgSO4 in toluene (4 mL) were heated at 60 °C. After 30 min, the mixture was cooled to room temperature and the solvent was evaporated. Purification by column chromatography on silica gel, eluting with CH2Cl2–MeOH (96:4), gave the oxime 17 (96 mg, 90%) as an oil as a mixture of E and Z alkene and oxime isomers; Rf 0.3 [CH2Cl2–MeOH (96:4)]; IR νmax(film)/cm−1 3340, 2935, 1440; 1H NMR (400 MHz, CDCl3) δ = 7.45 (1H, s), 7.31 (1H, s), 5.62–5.49 (1H, m), 5.37–5.29 (1H, m), 3.63–3.60 (2H, m), 3.28 (3H, s), 3.26 (3H, s), 2.19–2.10 (2H, m), 2.08 (1H, dd, J 15, 7.5 Hz), 1.95–1.88 (1H, m), 1.75 (1H, dd, J 15, 5 Hz), 1.68 (3H, dd, J 6, 1.5 Hz); 13C NMR (101 MHz, CDCl3, peaks for the major isomer) δ = 151.0, 128.4, 127.6, 100.7, 49.3, 49.2, 48.6, 35.6, 35.5, 35.4, 18.0. HRMS and LRMS could not be obtained.
8,8-Dimethoxy-5-methyl-4-oxa-3-azatricyclo[4.3.1.03,7]decane 18
The oxime 17 (E:Z 5.5:1) (100 mg, 0.43 mmol), N,N-diisopropylethylamine (0.18 mL, 1.07 mmol), hydroxylamine hydrochloride (40 mg, 0.64 mmol) and MgSO4 (50 mg) in toluene (4 mL) were heated under reflux. After 16 h, the mixture was cooled to room temperature and the solvent was evaporated. Purification by column chromatography on silica gel, eluting with CH2Cl2–MeOH (97:3), gave the cycloadduct 18 (14 mg, 15%) as an oil as a single stereoisomer; Rf 0.36 [CH2Cl2–MeOH (9.7:0.3)]; IR νmax(film)/cm−1 2950, 2900, 1460; 1H NMR (400 MHz, CDCl3) δ = 4.10 (1H, q, J 6 Hz), 3.49 (1H, d, J 3.5 Hz), 3.37–3.01 (4H, m), 3.26 (3H, s), 2.97 (1H, dt, J 14.5, 3 Hz), 2.18 (1H, dd, J 9.5, 3.5 Hz), 1.96–1.89 (1H, m), 1.82–1.80 (1H, m), 1.68–1.65 (2H, m), 1.52 (1H, dd, J 13, 4 Hz), 1.21 (3H, d, J 6 Hz); 13C NMR (101 MHz, CDCl3) δ = 100.0, 85.6, 60.6, 60.3, 48.7, 47.8, 40.6, 39.0, 34.1, 22.8, 20.9; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C11H20NO3 214.1438; found: 214.1433; LRMS m/z (ES) 214 (100%), 182 (50).
2-(2-(Chloromethyl)hex-4-en-1-yl)-1,3-dithiane-2-carbaldehyde oxime 19
Hydroxylamine hydrochloride (135 mg, 2.0 mmol) and sodium acetate (365 mg, 4.5 mmol) were added to aldehyde 15 (500 mg, 1.8 mmol) in dry methanol (25 mL) at room temperature. After 5 h, the solvent was evaporated. The residue was washed with CH2Cl2 and then concentrated to give the oxime 19 (527 mg, 100%) which was used without further purification; 1H NMR (400 MHz, CDCl3) δ = 7.44 (1H, s), 5.62–5.50 (1H, m), 5.38–5.30 (1H, m), 3.67–3.60 (2H, m), 3.16–3.04 (2H, m), 2.73–2.66 (2H, m), 2.27–2.07 (5H, m), 1.94–1.83 (2H, m), 1.69 (3H, dd, J 6, 1 Hz); 13C NMR (101 MHz, CDCl3, peaks for the major isomer) δ = 153.2, 128.7, 127.4, 51.5, 48.9, 41.0, 36.2, 35.9, 27.3, 27.2, 24.8, 18.0; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C12H21NOS235Cl 294.0753; found: 294.0746; LRMS m/z (ES) 296 (48%), 294 (100).5′-Methyl-4′-oxa-3′-azaspiro-1,3-dithiane-2,8′-tricyclo[4.3.1.03,7]decane 20
Oxime 19 (527 mg, 1.8 mmol), N,N-diisopropylethylamine (0.32 mL, 1.8 mmol) and MgSO4 (100 mg) in toluene (15 mL) were heated under reflux. After 16 h, the mixture was cooled to room temperature and the solvent was evaporated. Purification by column chromatography on silica gel, eluting with CH2Cl2–MeOH (97:3), gave the cycloadduct 20 (263 mg, 57%) as an oil as a single stereoisomer; Rf 0.3 [CH2Cl2–MeOH (97:3)]; IR νmax(film)/cm−1 2925, 2900, 1440; 1H NMR (400 MHz, CDCl3) δ = 4.19 (1H, q, J 6 Hz), 3.87 (1H, d, J 3.5 Hz), 3.46 (1H, dd, J 14.5, 2 Hz), 3.20–3.13 (1H, m), 3.09–3.00 (2H, m), 2.86–2.76 (2H, m), 2.42 (1H, dd, J 9.5, 3.5 Hz), 2.13–2.00 (3H, m), 1.91–1.88 (2H, m), 1.79–1.77 (1H, m), 1.55 (1H, dd, J 13.5, 4.5 Hz), 1.26 (3H, d, J 6 Hz); 13C NMR (101 MHz, CDCl3) δ = 86.6, 61.8, 59.4, 44.7, 44.5, 41.9, 33.2, 27.5, 27.2, 24.7, 23.0, 21.1; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C12H20NOS2 258.0986; found: 258.0994; LRMS m/z (ES) 258 (100%).
5-Methyl-4-oxa-3-azatricyclo[4.3.1.03,7]decan-8-one 21
Bis(trifluoroacetoxy)iodobenzene (500 mg, 1.16 mmol), and trifluoroacetic acid (10 equiv.) was added to dithiane 20 (300 mg, 1.16 mmol) in acetonitrile/water (1:1) (44 mL) at room temperature. Additional bis(trifluoroacetoxy)iodobenzene (1.9 g, 4.4 mmol) was added over a 1 h period, and the mixture was stirred for 2.5 h, before being neutralised with saturated aqueous sodium bicarbonate (30 mL). The mixture was extracted with CH2Cl2 (3 × 10 mL), dried (MgSO4) and the solvent was evaporated. Purification by column chromatography on silica gel, eluting with CH2Cl2–MeOH (97:3), gave the ketone 21 (155 mg, 80%) as needles, m.p. 62–63.5 °C; Rf 0.32 [CH2Cl2–MeOH (97:3)]; IR νmax(film)/cm−1 2965, 1730, 1450; 1H NMR (400 MHz, CDCl3) δ = 4.26 (1H, q, J 6 Hz), 3.62 (1H, d, J 3.5 Hz), 3.51 (1H, dd, J 14.5, 2.5 Hz), 3.30–3.20 (1H, m), 2.45 (1H, dd, J 9, 3.5 Hz), 2.25–2.23 (2H, m), 2.20–2.18 (1H, m), 2.03–1.96 (1H, m), 1.79 (1H, dd, J 13.5, 4.5 Hz), 1.21 (3H, d, J 6 Hz); 13C NMR (101 MHz, CDCl3) δ = 209.1, 85.6, 68.9, 61.5, 44.8, 44.5, 34.1, 23.3, 20.5; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C9H14NO2 168.1019; found: 168.1020; LRMS m/z (ES) 168 (100%).
7-[(1RS)-1-Hydroxyethyl]-2-[2-(1H-indol-3-yl)acetyl]-2-azabicyclo[2.2.2]octan-6-one 23
Activated Zn powder (168 mg, 2.6 mmol) [washed with freshly prepared aqueous HCl (1.0 M), EtOH, then Et2O] was added to the cycloadduct 21 (85 mg, 0.51 mmol) in AcOH–MeOH (5 mL, 4:1) at room temperature. After 4 h, the mixture was filtered through Celite® and the solvent was evaporated. The residue was dissolved in MeOH (5 mL) and poured onto aqueous sodium hydroxide (5 mL, 2 M). The aqueous layer was extracted with CH2Cl2 (3 × 5 mL) and the organic layers were dried (MgSO4) and evaporated to give the crude aminoalcohol 22 (86 mg, 100%). Indole acetic acid (89 mg, 0.51 mmol) and EDCI (97 mg, 0.51 mmol) were added to the crude amino alcohol (86 mg, 0.51 mmol) in CH2Cl2 (6 mL) at room temperature. After 1.5 h, the mixture was extracted with CH2Cl2 (3 × 15 mL). The organic layers were combined and were washed with aqueous HCl (1.5 mL, 0.01 M) and with saturated aqueous K2CO3 (1.5 mL), dried (MgSO4) and the solvent was evaporated. Purification by column chromatography on silica gel, eluting with CH2Cl2–MeOH (9:1), gave the amide 23 (165 mg, 100%) as a foam; Rf 0.39 [CH2Cl2–MeOH (9:1)]; IR νmax(film)/cm−1 3320, 2970, 1730, 1625, 1460; 1H NMR (400 MHz, CDCl3, as a mixture of rotamers 64:36, data for major rotamer) δ = 8.50 (1H, br s), 7.59 (1H, d, J 7.5 Hz), 7.38 (1H, d, J 7.5 Hz), 7.24–7.11 (3H, m), 5.01 (1H, d, J 1.5 Hz), 3.96–3.93 (2H, m), 3.59–3.52 (2H, m), 3.32–3.28 (1H, m), 2.49–2.33 (3H, m), 1.89–1.81 (2H, m), 1.16 (3H, d, J 6 Hz), 1.07–1.03 (1H, m); 13C NMR (101 MHz, CDCl3, as a mixture of rotamers, data for major rotamer) δ = 206.6, 173.3, 136.1, 127.1, 122.9, 122.3, 119.8, 118.3, 111.5, 107.7, 68.7, 54.7, 50.3, 43.7, 42.5, 30.7, 28.4, 27.7, 19.6; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C19H22N2O3 327.1703; found: 327.1703; LRMS m/z (ES) 327 (100%).
1-[7-(1-Hydroxyethyl-6,6-dimethoxy-2-azabicyclo[2.2.2]octan-2-yl)]-2-(1H-indol-3-yl)ethanone 24
p-Toluenesulfonic acid (7.5 mg, 0.04 mmol) was added to crude 23 (100 mg, 0.31 mmol), trimethylorthoformate (1.21 mL, 11 mmol) and MeOH (1.2 mL) at room temperature. After 7 h, saturated aqueous NaHCO3 (1.5 mL) was added and the mixture was extracted with CH2Cl2 (3 × 10 mL), dried (MgSO4) and the solvent was evaporated. Purification by column chromatography on silica gel, eluting with CH2Cl2–MeOH (97:3), gave amide 24 (98 mg, 85%) as a foam; Rf 0.35 [CH2Cl2–MeOH (95:5)]; IR νmax(film)/cm−1 3320, 2970, 1625, 1460; 1H NMR (400 MHz, CDCl3, as a mixture of rotamers ∼9:1, data for major rotamer) δ = 8.33 (1H, br s), 7.61 (1H, d, J 8 Hz), 7.38 (1H, d, J 8 Hz), 7.23–7.15 (3H, m, ArH), 4.78 (1H, d, J 1 Hz), 4.17–4.10 (1H, m), 3.83–3.80 (2H, m), 3.53–3.49 (1H, m), 3.36 (1H, dd, J 10, 3 Hz), 3.23 (3H, s), 3.14 (3H, s), 2.09–2.05 (1H, m), 1.96–1.88 (1H, m), 1.76–1.69 (3H, m), 1.15 (3H, d, J 6 Hz), 0.84–0.79 (1H, m); 13C NMR (101 MHz, CDCl3) δ = 172.7, 136.1, 127.2, 122.7, 122.2, 119.6, 118.5, 111.3, 108.6, 101.4, 69.6, 49.2, 48.6, 48.1, 47.1, 40.9, 37.2, 30.4, 28.7, 27.6, 19.6; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H28N2O4 373.2122; found: 373.2122; LRMS m/z (ES) 373 (100%).
1-{2-[2-(1H-Indol-3-yl)acetyl]-7,7-dimethoxy-2-azabicyclo[2.2.2]octan-6-yl}ethyl acetate 257b
Acetyl chloride (0.02 mL, 0.3 mmol) and pyridine (0.024 mL, 0.3 mmol) were added to the alcohol 24 (100 mg, 0.28 mmol) in CH2Cl2 (6 mL) at room temperature. After 30 min, the solvent was evaporated. Purification by column chromatography on silica gel, eluting with CH2Cl2–MeOH (97.5:2.5), gave the ester 25 (98 mg, 87%) as a foam; Rf 0.26 [CH2Cl2–MeOH (97.5:2.5)]; IR νmax(film)/cm−1 3280, 2940, 1730, 1640, 1450; 1H NMR (400 MHz, CDCl3, as a mixture of rotamers ∼8:1, data for major rotamer) δ = 8.30 (1H, br s), 7.57 (1H, dd, J 8, 1 Hz), 7.36 (1H, dd, J 8, 1 Hz), 7.23–7.22 (1H, m), 7.19 (1H, ddd, J 8, 7, 1 Hz), 7.13 (1H, ddd, J 8, 7, 1 Hz), 4.91 (1H, d, J 1.5 Hz), 4.52 (1H, dq, J 10.5, 6 Hz), 3.77–3.74 (2H, m), 3.39 (1H, dt, J 10, 2.5 Hz), 3.29 (1H, dt, J 10, 2.5 Hz), 3.26 (3H, s), 3.20 (3H, s), 2.29–2.20 (1H, m), 2.13 (3H, s), 2.05–2.02 (1H, m), 1.78 (1H, dt, J 13.5, 2.5 Hz), 1.74–1.69 (1H, m), 1.66 (1H, dt, J 13.5, 2.5 Hz), 1.22 (3H, d, J 6 Hz), 0.92 (1H, ddt, 13.5, 5.5, 2.5 Hz); 13C NMR (101 MHz, CDCl3, as a mixture of rotamers, data for major rotamer) δ = 171.2, 171.1, 136.1, 127.4, 122.9, 121.9, 119.3, 118.4, 111.2, 108.9, 101.7, 71.4, 49.0, 48.9, 48.1, 45.8, 38.1, 36.9, 30.7, 28.2, 27.5, 21.5, 17.8; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H31N2O5 415.2227; found: 415.2227; LRMS m/z (ES) 415 (100%). Data match the literature.7b
Conclusions
We have demonstrated that cascade chemistry involving condensation of hydroxylamine and an aldehyde to give an oxime, cyclization on to an alkyl halide to give a nitrone, and intramolecular dipolar cycloaddition is successful in which the branch point for the alkyl halide and alkene tethers is at C-4. This leads to bridged compounds with the azabicyclo[2.2.2]octane framework and thereby extends the methodology described previously (Scheme 1) to a new class of ring system. In addition, the chemistry has been applied to a formal synthesis of the alkaloid 19-hydroxyibogamine. The cascade sequence has potential for the formation of a variety of iboga alkaloid frameworks.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the Ministry of Higher Education and Scientific Research of Iraq and the University of Sheffield for support of this work. We are grateful to Harry Adams for the single crystal X-ray data and the Faculty of Science Mass Spectrometry Centre, University of Sheffield. We would like to thank Hélène Guerrand and Rachel Furnival for discussions and related studies.Notes and references
- K. R. Alper, Alkaloids: Chem. Biol., 2001, 56, 1 CAS.
- P. Popik, R. T. Layer and P. Skolnick, Pharmacol. Rev., 1995, 47, 235 CAS.
- Y. Terada, M. Kitajima, F. Taguchi, H. Takayama, S. Horie and T. Watanabe, J. Nat. Prod., 2014, 77, 1831 CrossRef CAS PubMed.
- (a) J. E. Sears and D. L. Boger, Acc. Chem. Res., 2015, 48, 653 CrossRef CAS PubMed; (b) Y. Zhang, Y. Xue, G. Li, H. Yuan and T. Luo, Chem. Sci., 2016, 7, 5530 RSC.
- For reviews, see: (a) M. Faisal, D. Shahzad, A. Saeed, B. Lal, S. Saeed, F. A. Larik, P. A. Channar, P. A. Mahesar and J. Mahar, Tetrahedron: Asymmetry, 2017, 28, 1445 CrossRef CAS; (b) C. Lavaud and G. Massiot, in Progress in the Chemistry of Organic Natural Products, ed. A. D. Kinghorn, H. Falk, S. Gibbons and J. Kobayashi, Springer, Switzerland, 2017, vol. 105, p. 89 Search PubMed; (c) G. K. Jana, S. Paul and S. Sinha, Org. Prep. Proced. Int., 2011, 43, 541 CrossRef CAS.
- For some recent references, see: ref. 4b and (a) G. K. Jana and S. Sinha, Tetrahedron, 2012, 68, 7155 CrossRef CAS; (b) A. C. Kruegel, S. Rakshit, X. Li and D. Sames, J. Org. Chem., 2015, 80, 2062 CrossRef CAS PubMed; (c) G. Zhao, X. Xie, H. Sun, Z. Yuan, Z. Zhong, S. Tang and X. She, Org. Lett., 2016, 18, 2447 CrossRef CAS PubMed.
- (a) S. Höck, F. Koch and H. J. Borschberg, Tetrahedron: Asymmetry, 2004, 15, 1801 CrossRef; (b) S. Höck and H. J. Borschberg, Helv. Chim. Acta, 2006, 89, 542 CrossRef.
- H. Achenbach, B. Raffelsberger and G.-U. Brillinger, Phytochemistry, 1980, 19, 2185 CrossRef CAS.
- (a) D.-B. Zhang, D.-G. Yu, M. Sun, X.-X. Zhu, X.-J. Yao, S.-Y. Zhou, J.-J. Chen and K. Gao, J. Nat. Prod., 2015, 78, 1253 CrossRef CAS PubMed; (b) C.-E. Nge, K.-W. Chong, N. F. Thomas, S.-H. Lim, Y.-Y. Low and T.-S. Kam, J. Nat. Prod., 2016, 79, 1388 CrossRef CAS PubMed; (c) C.-E. Nge, K.-S. Sim, S.-H. Lim, N. F. Thomas, Y.-Y. Low and T.-S. Kam, J. Nat. Prod., 2016, 79, 2709 CrossRef CAS PubMed.
- (a) I. Coldham, A. J. M. Burrell, L. E. White, H. Adams and N. Oram, Angew. Chem., Int. Ed., 2007, 46, 6159 CrossRef CAS PubMed; (b) A. J. M. Burrell, I. Coldham, L. Watson, N. Oram, C. D. Pilgram and N. G. Martin, J. Org. Chem., 2009, 74, 2290 CrossRef CAS PubMed.
- (a) A. J. M. Burrell, I. Coldham and N. Oram, Org. Lett., 2009, 11, 1515 CrossRef CAS PubMed; (b) A. J. M. Burrell, L. Watson, N. G. Martin, N. Oram and I. Coldham, Org. Biomol. Chem., 2010, 8, 4530 RSC; (c) I. Coldham, L. Watson, H. Adams and N. G. Martin, J. Org. Chem., 2011, 76, 2360 CrossRef CAS PubMed; (d) I. Coldham, A. J. M. Burrell, L. Watson, N. Oram and N. G. Martin, Heterocycles, 2012, 84, 597 CrossRef CAS; (e) I. Coldham, A. J. M. Burrell, H. D. S. Guerrand, L. Watson, N. G. Martin and N. Oram, Beilstein J. Org. Chem., 2012, 8, 107 CrossRef CAS PubMed.
- (a) I. Coldham, S. Jana, L. Watson and N. G. Martin, Org. Biomol. Chem., 2009, 7, 1674 RSC; (b) H. D. S. Guerrand, H. Adams and I. Coldham, Org. Biomol. Chem., 2011, 9, 7921 RSC; (c) R. C. Furnival, R. Saruengkhanphasit, H. E. Holberry, J. R. Shewring, H. D. S. Guerrand, H. Adams and I. Coldham, Org. Biomol. Chem., 2016, 14, 10953 RSC.
- I. Coldham, A. J. M. Burrell, H. D. S. Guerrand and N. Oram, Org. Lett., 2011, 13, 1267 CrossRef CAS PubMed.
- R. Saruengkhanphasit, D. Collier and I. Coldham, J. Org. Chem., 2017, 82, 6489 CrossRef CAS PubMed.
- For selected reports of other cascade cyclization then dipolar cycloaddition reactions, see: (a) H. A. Dondas, R. Grigg, M. Hadjisoteriou, J. Markandu, P. Kennewell and M. Thornton-Pett, Tetrahedron, 2001, 57, 1119 CrossRef; (b) M. S. Karatholuvhu, A. Sinclair, A. F. Newton, M.-L. Alcaraz, R. A. Stockman and P. L. Fuchs, J. Am. Chem. Soc., 2006, 128, 12656 CrossRef CAS PubMed; (c) H. Yuan, J. Gong and Z. Yang, Org. Lett., 2016, 18, 5500 CrossRef CAS PubMed; (d) R. Yamada, Y. Adachi, S. Yokoshima and T. Fukuyama, Angew. Chem., Int. Ed., 2016, 55, 6067 CrossRef CAS PubMed; (e) H.-J. Zhang, L. Hu, Z. Ma, R. Li, Z. Zhang, C. Tao, B. Cheng, Y. Li, H. Wang and H. Zhai, Angew. Chem., Int. Ed., 2016, 55, 11638 CrossRef CAS PubMed; (f) J. Boudreault, F. Lévesque and G. Bélanger, J. Org. Chem., 2016, 81, 9247 CrossRef CAS PubMed; (g) C. Hauduc and G. Bélanger, J. Org. Chem., 2017, 82, 4703 CrossRef CAS PubMed; (h) P. Boissarie and G. Bélanger, Org. Lett., 2017, 19, 3739 CrossRef CAS PubMed; (i) S. Sugita, N. Takeda, N. Tohnai, M. Miyata, O. Miyata and M. Ueda, Angew. Chem., Int. Ed., 2017, 56, 2469 CrossRef CAS PubMed; (j) J. M. E. Hughes and J. L. Gleason, Angew. Chem., Int. Ed., 2017, 56, 10830 CrossRef CAS PubMed; (k) J. M. E. Hughes and J. L. Gleason, Tetrahedron, 2018, 74, 759 CrossRef CAS.
- R. Berthold, Chem. Ber., 1957, 90, 793 CrossRef CAS.
- For alkylation of lithiated 2,2-dimethoxyacetonitrile, see: K. Utimoto, Y. Wakabayashi, Y. Shishiyama, M. Inoue and H. Nozaki, Tetrahedron Lett., 1981, 22, 4279 CrossRef CAS.
- (a) I. Coldham, K. M. Crapnell, J.-C. Fernàndez, J. D. Moseley and R. Rabot, J. Org. Chem., 2002, 67, 6181 CrossRef CAS PubMed; (b) R. Pathak, B. C. Dobson, N. Ghosh, K. A. Ageel, M. R. Alshawish, R. Saruengkhanphasit and I. Coldham, Org. Biomol. Chem., 2015, 13, 3331 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Copies of NMR spectra and X-ray data. CCDC 1858480. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ob02839g |
| ‡ New address: Department of Chemistry, University of Diyala, Diyala, Iraq. |
| This journal is © The Royal Society of Chemistry 2019 |