
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Silent catalytic promiscuity in the high-fidelity terpene cyclase δ-cadinene synthase†
Marianna
Loizzi
,
David J.
Miller
 and
Rudolf K.
Allemann
*
and
Rudolf K.
Allemann
*
School of Chemistry, Cardiff University Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: allemannrk@cardiff.ac.uk
First published on 17th January 2019
Abstract
δ-Cadinene synthase (DCS) is a high-fidelity sesquiterpene synthase that generates δ-cadinene as the sole detectable organic product from its natural substrate (E,E)-FDP. Previous work with this enzyme using substrate analogues revealed the ability of DCS to catalyse both 1,10- and 1,6-cyclisations of substrate analogues. To test whether this apparent promiscuity was an artefact of alternate substrate use or an inherent property of the enzyme, aza analogues of the proposed α-bisabolyl cation intermediate were prepared since this cation would be formed after an initial 1,6-cyclisation of FDP. In the presence of 250 μM inorganic disphosphate both (R)- and (S)-aza-bisaboyl cations were potent competitive inhibitors of DCS (Ki = 2.5 ± 0.5 mM and 3.44 ± 1.43 μM, respectively). These compounds were also shown to be potent inhibitors of the 1,6-cyclase amorpha-4,11-diene synthase but not of the 1,10-cyclase aristolochene synthase from Penicillium roquefortii, demonstrating that the 1,6-cyclase activity of DCS is most likely an inherent property of the enzyme even when the natural substrate is used and not an artefact of the use of substrate analogues.
Introduction
Terpene synthases catalyse some of the most complex reactions in the natural world. From a small pool of isoprenyl diphosphates they generate a myriad of hydrocarbons and alcohols that are often processed into thousands of terpenoids with diverse biological activities with many potential applications for instance as agrochemicals or therapeutic agents.1The details of terpene synthase chemistry2 have been investigated by site directed mutagenesis and with non-natural amino acids,3 analogues of substrates,4 and putative reaction intermediates,5 X-ray crystallography,2b,c,6 and computational modelling.2d,7 Together these investigations revealed a fascinating, yet still incomplete picture. A series of X-ray crystal structure of aristolochene synthase from Aspergillus terreus in both closed and open conformations along with complexes containing the complete substrate (or analogue), diphosphate anion and/or Mg2+ co-factors8 revealed the physical steps of the catalytic cycle. Binding of a Mg2+-ion is followed by coordination of the prenyl diphosphate substrate and a second Mg2+ ion; coordination of a third Mg2+ ion triggers active site closure to form the Michaelis complex.8 Diphosphate cleavage is then triggered to form an initial carbocation and the hydrophobic active site shelters this high energy intermediate from bulk solvent.2b,c The active site, lined with hydrophobic and aromatic amino acid residues then steers the initial carbocation through a series of ring closures and rearrangements prior to quench of the final carbocation either by proton loss or nucleophilic attack by water.2b,c,6e Usually this is tightly controlled by the enzyme, with a single enantiomer dominating the product pool whereby several rings and stereocentres are often generated in a single chemical step from an achiral precursor. Control of this process is thought to arise from a product-like active site contour in combination with direction of carbocation location in the intermediates through the negative charge on the diphosphate anion and aromatic amino acid side chains that can stabilise carbocations at certain locations through cation–π interaction.2b,c,4a A small subset of terpene synthases, on the other hand, exhibit significant promiscuity, presumably through having a less structured and/or flexible active site that allows the intermediates to sample a large number of reactive conformations prior to final carbocation quench. For example, δ-selinine synthase and γ-humulene synthases from Abies grandis generate 34 and 52 products from farnesyl diphosphate (1), respectively.9 Terpene synthases have been postulated to evolve through such promiscuous intermediates prior to further evolution into high-fidelity synthases.3e The modern δ-cadinene synthase (DCS) from Gossypium arboreum is a high-fidelity sesquiterpene synthase that catalyses the formation of the bicyclic hydrocarbon (+)-δ-cadinene (7),10 the first committed step in the biosynthesis of the phytoalexin gossypol.11 The catalytic domain is situated in the C-terminal domain and adopts the α-helical fold domain, typical of class 1 terpene synthases.2b,c,12 It contains the conserved aspartate rich motif D307DTYD311 on helix D, but instead of the usual characteristic NSE/DTE Mg2+ binding motif, DCS has a second aspartate rich motif D451DVAE455 on helix H.6e Despite only generating a single detectable hydrocarbon product, extensive mechanistic analysis of the DCS-catalysed reaction pathway has not unambiguously defined the chemical steps of its catalytic cycle. Moreover, conversion of fluorinated and stereochemically altered FDP analogues with DCS revealed an underlying mechanistic promiscuity with products arising from an initial 1,10-, 1,6- or 1,11-ring closure depending upon the substrate analogue used (vide infra).12 Two chemical mechanisms remain plausible for the formation of δ-cadinene from FDP (Scheme 1). Both pathways involve initial formation of (3R)-nerolidyl diphosphate ((3R)-NDP, (2)) as an enzyme-bound intermediate. In pathway (a), a 1,10-macrocyclisation occurs to generate cis-germacradienyl cation (4). A subsequent [1,3]-hydride shift is followed by a 1,6-electrophilic ring closure to cadinenyl cation (6), from which δ-cadinene (7) is formed after proton loss from C6. In pathway (b), a 1,6 ring-closure of 2 is followed by a [1,3]-hydride shift from C1 to C7; subsequently a second ring closure and a [1,5]-hydride shift lead to cadinenyl cation, an intermediate common to both pathways. In previous work, using substrate analogues we were unable to definitively rule out pathway (b) and indeed when 6-fluorofarnesyl diphosphate (6F-FDP) was used as a substrate analogue it proved to be a potent inhibitor (Ki = 2.4 μM), giving no detectable pentane-extractable products when incubated with DCS. This result is consistent with an initial 1,6-cyclisation pathway since it would be expected to undergo 1,10-ring closure and give an abortive product rather than inhibit the enzyme in the latter scenario. On the other hand, 2-fluorofarnesyl diphosphates (2F-FDP) and 10-fluorofarnesyl diphosphate (10F-FDP) gave products arising from 1,10- and 1,11 ring-closures, respectively, consistent with an initial 1,10-ring closure mechanism.12
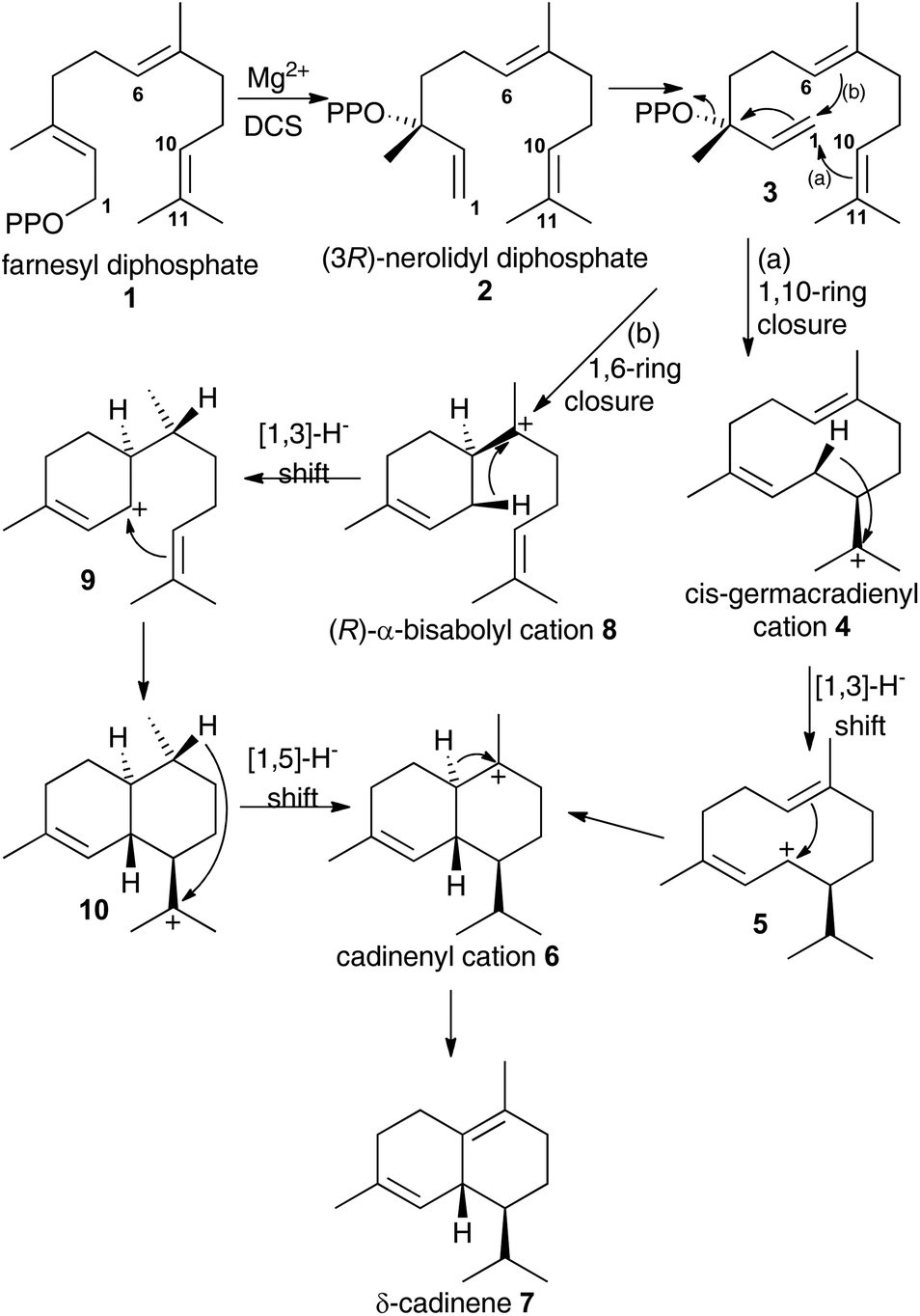 | ||
| Scheme 1 Possible chemical steps for DCS catalysed production of δ-cadinene from FDP (1). | ||
Hence examination of the catalytic mechanism of DCS using FDP analogues has led to inconclusive, yet intriguing results, showing that this enzyme has the potential to use alternative reaction pathways. Yet the question arises, is this simply an artefact of the substrate used or is this an inherent property of the enzyme? The work described here provides alternative mechanistic data for the DCS-catalysed transformation of FDP to δ-cadinene using aza-analogues of putative carbocation intermediates. Although the highly unstable carbocationic intermediates formed during terpene synthase catalysis, cannot be isolated, it is possible to replace the sp2 hybridised carbocationic carbon of a given intermediate with an sp3 hybridised nitrogen in an amine analogue or with a sp2 hybridised nitrogen in an iminium ion. Although the tetrahedral tertiary ammonium ions inherently are imperfect geometric analogues of the planar carbocations, these aza-terpenoids are thought to mimic the topological and electrostatic properties of carbocations generated by these enzymes.5 However, since they cannot be processed by the enzyme, they often act as tightly bound competitive inhibitors of terpene synthases.5c,13
Hence, the use of strategically designed aza-analogues may enable the disentanglement of the possible reaction mechanisms catalysed by DCS. Here we report the stereoselective synthesis of the two enantiomers of aza-bisabolyl cation and their kinetic evaluation as inhibitors of DCS. Comparison of their effect upon catalysis by AS and amporpha-4,11-diene synthase (ADS), enzymes that follow 1,10- and 1,6-ring-closure mechanisms, validate the result that DCS has inherent 1,6- as well as 1,10 ring closure activity.
Results and discussion
If α-bisabolyl cation 8 is a reaction intermediate on the pathway to δ-cadinene (7), one or both of enantiomeric aza-analogues of 11 (Fig. 1) should act as competitive inhibitors of DCS.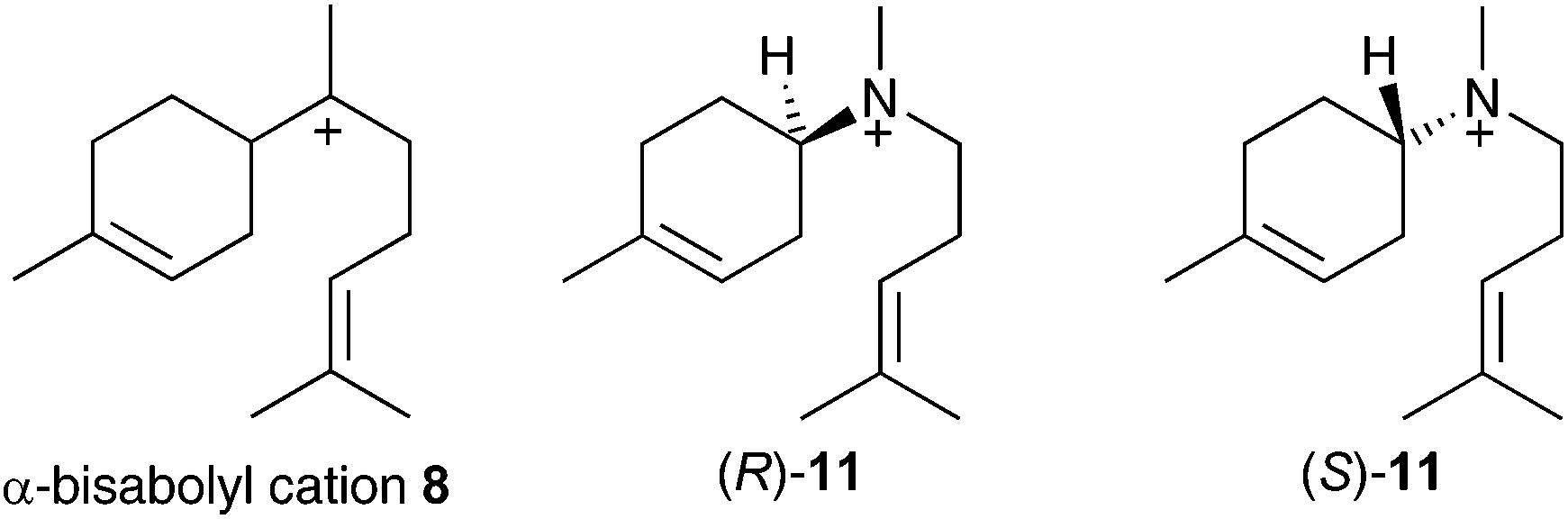 | ||
| Fig. 1 Chemical structures of the α-bisabolyl cation (8) and corresponding aza analogues (R)-11 and (S)-11. | ||
Both enantiomers of 11 have previously been prepared.14a Here we report an alternative synthesis that is more concise and avoids the use of harsh reaction conditions. Key to the synthesis of both enantiomers is an enantioselective synthesis of the two enantiomers of carboxylic acid 18 (Scheme 2). This was achieved through asymmetric Diels–Alder reaction of an acrylate derivatised with a chiral auxiliary with a butadiene.15 Oxazolidin2-one 12 was alkylated with acryloyl chloride after deprotonation with n-butyl lithium with 35% yield. The resulting ester 13 was then subjected to an asymmetric Diels–Alder reaction with 2-methylbutadiene.15 The enantioselectivity and yield were optimal at −100 °C in CH2Cl2 (52%, ee >95%, de >95% (see ESI† for details). This produced the key compound to generate the S enantiomer of the aza-analogue 11. The equivalent R configured ester was generated using D-pantolactone (15) as a chiral auxiliary.15 After alkylation with acryloyl chloride, using NEt3 as the base in CH2Cl2, diester 16 was isolated in 60% yield. Again, an asymmetric Diels–Alder reaction with 2-methylbutadiene was carried out, this time at −10 °C in CH2Cl2 using TiCl4 as a Lewis acid catalyst yielding the R ester in 84% yield (ee = 92% and de = 97%).16 The latter procedure was in fact optimal for both enantiomers but due to the high cost of L-pantolactone not used for bulk preparation for the R-enantiomer of 18. Optical purity of all subsequent compounds was checked using chiral GC, HPLC and/or polarimetry and in all cases no loss of optical purity was detected in later synthetic steps.
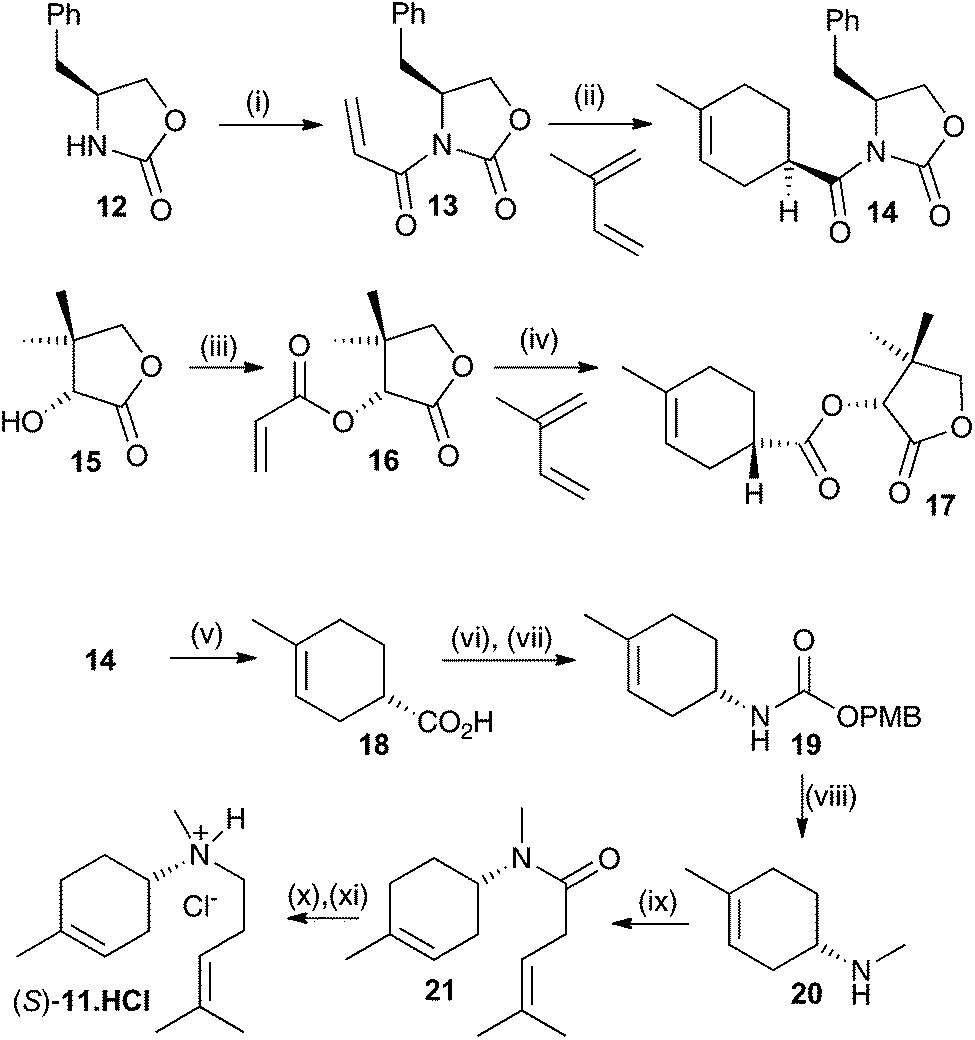 | ||
| Scheme 2 Synthesis of aza-analogues (R)- and (S)-11. Reagents and conditions: (i) acryloyl chloride, BuLi, THF, 35%. (ii) Et2AlCl, CH2Cl2, −100 °C, 54%. (iii) Acryloyl chloride, NEt3, CH2Cl2, 60%. (iv) TiCl4, CH2Cl2, −10 °C, 84%. (v) LiOH, THF, H2O, MeOH, 50 °C, 99%. (vi) DPPA, NEt3. (vii) p-Methoxybenzyl alcohol, toluene, 60% over two steps. (viii) LiAlH4, Et2O, 50%, (ix) 4-methylpent-3-enoic acid, EtNPri2, HBTU, DMF, 70%. (x) LiAlH4, Et2O. (xi) HCl in Et2O, 55% two steps. | ||
Both syntheses now proceeded in identical manner and Scheme 2 only illustrates the synthesis of the S-enantiomer of 11. Hydrolysis of 14 using LiOH in an equivolume mixture of THF, water and methanol for 1 h at 50 °C gave carboxylic acid 18 in near quantitative yield. 18 was converted to p-methoxybenzyl urethane derivative 19 by treatment with diphenylphosphorylazide (DPPA) followed by a Curtius rearrangement in the presence of p-methoxybenzyl alcohol, which proceeded with strict retention of stereochemistry.15b The overall yield of the urethane product 19 was 60% over the two steps. Final conversion to (S)-11 was achieved first through reduction with LiAlH4 in anhydrous Et2O (50%) then HBTU mediated coupling to 4-methypent-3-enoic acid (70%) followed by a second reduction with LiAlH4 in Et2O. To prevent air oxidation upon storage the product was converted to its hydrochloride salt with HCl in ether, yielding (S)-11·HCl in 55% yield over the final two steps. The optical purity of (S)-11 was estimated to be ≥98% by comparison with previously reported data.14a Similar results were obtained for the synthesis of (R)-11.
To validate any results obtained for these compounds as inhibitors of DCS, they were tested as inhibitors of aristolochene synthase from Penicillium roqueforti (AS) and amorpha-4,11-diene synthase (ADS). These two enzymes are known to proceed via 1,10- and 1,6-cyclisations of the initial carbocation during their catalytic cycle (Scheme 3).1g Hence aza-bisabolyl cations 11 should act as poor inhibitors of AS and potent inhibitors of ADS, as they closely resemble a reaction intermediate in the latter case only.
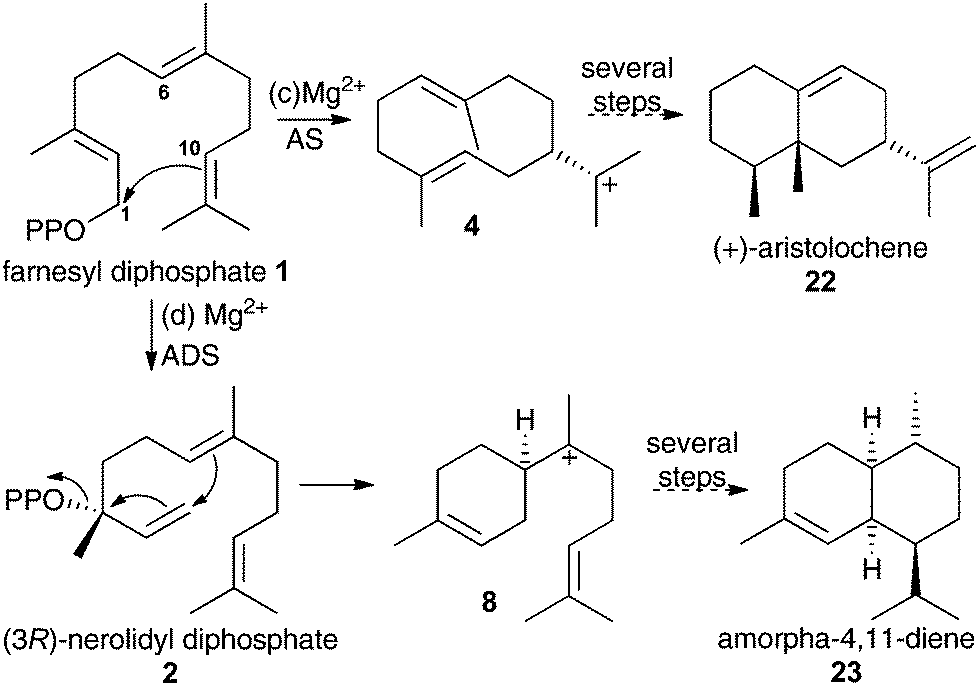 | ||
| Scheme 3 Initial catalytic chemical steps leading to (c) (+)-aristolochene (22) and (d) amorpha-4,11-diene (23) follow 1,10- and 1,6-cyclisation of FDP, respectively. | ||
Recombinant AS and ADS were prepared and purified according to previously published procedures17,18 and both (R)-11 and (S)-11 were tested as inhibitors using a standard radiolabelled assay involving conversion of tritium labelled FDP by each enzyme and scintillation counting of the pentane extractable products.5b Terpene synthases are known to efficiently bind cation-PPi pairs and inhibition was assessed both in the presence and absence of 250 μM diphosphate (Table 1). Synergistic inhibition of aza-analogues 11 with diphosphate has been observed previously for a variety of other terpene synthases.5d,13c,14
| Enzyme | Aza-analogue | K i (μM) (+250 μM PPi) | K i (μM) |
|---|---|---|---|
| ADS | (S)-11 | 1.5 ± 0.5 | 25 ± 5 |
| (R)-11 | 3.7 ± 1.9 | 50 ± 17 | |
| AS | (S)-11 | 255 ± 23 | 295 ± 23 |
| (R)-11 | 489 ± 62 | 472 ± 48 | |
| DCS | (S)-11 | 3.44 ± 1.43 | 273 ± 77 |
| (R)-11 | 2.5 ± 0.5 | 1700 ± 300 |
Kinetic data were fitted by non-linear regression to the Michaelis–Menten equation (v0 = kcat[E][S]/(KM + [S]). The mode of inhibition was determined by inspection of double reciprocal plots and observed to be competitive in all cases where inhibition was significant at low concentrations of 11. KI was determined from a plot of inhibitor concentration versus K′M/(kcat[E]) where K′M = KM(1 + [I]/KI).
The inhibition data for AS and ADS validate both of these compounds as valuable mechanistic probes for the present investigation since they are poor inhibitors of AS and potent inhibitors of ADS. PPi had little effect on the ability to inhibit AS (KI > 200 μM in both the presence and absence of PPi for AS). Both enantiomers of 11 acted as competitive inhibitor of ADS, showing that they are able to compete effectively with the natural substrate FDP at the active site. As these aza-compounds cannot be turned over by ADS, these result support the intermediacy of an α-bisabolyl cation in the biosynthesis of amorpha-4,11-diene, in agreement with the findings of Picaud et al.18b who used deuterated farnesyl diphosphate and deuterium exchange experiments to suggest that the R-enantiomer of the α-bisabolyl cation is the sole intermediate formed in the biosynthesis of amorpha-4,11-diene. Therefore, only the R enantiomer of 11 would be expected inhibit ADS; however, if the S-enantiomer was a slightly more potent inhibitor (Ki = 50 μM for (R)-11versus 25 μM for (S)-11) Table 1. This is consistent with a flexible model for sesquiterpene active sites, according to which an active site can accommodate a variety of intermediates of different shape and charge distribution without being rigidly complementary to a single intermediate or transition state species. For example, work by Cane et al. showed that both enantiomers of the aza-analogue 11 were equally effective inhibitors of trichodiene synthase.14a It is also notable that the presence of PPi enhanced inhibition of ADS by both enantiomers, improving the Ki approximately 20-fold (Ki = 1.5 and 3.7 μM for the S and R enantiomers respectively) demonstrating that the active site of ADS prefers a cation–anion pair in its active site.5d,13
Recombinant DCS was generated with a C-terminal hexahistidine tag (DCS-His6) as previously described.19 Inhibition assays were carried out using the same protocol used for AS and ADS. Both aza analogues were found to be competitive inhibitors of DCS-His6 in the presence of PPi but only poor inhibitors in its absence (Table 1). DCS clearly requires a cation–anion pair in its active site for effective inhibition by aza-analogues. Our results provide strong evidence for 1,6-cyclase activity for DCS.
Conclusions
The aza-bisabolyl cations 11 were potent competitive inhibitors of ADS, a 1,6-cyclase yet were much poorer inhibitors of PR-AS, a known 1,10-cyclase. When DCS was challenged with these aza-analogues in the presence of diphosphate anion they were potent inhibitors of the conversion of FDP to (+)-δ-cadinene (7), which would only be expected if DCS had a 1,6-cyclase activity. The use of a variety of substrate analogues possessing different stereochemistry and heteroatoms did not lead to clear results regarding whether DCS follow a 1,6 or 1,10 pathway.12 If the proposed initial isomerism of the substrate to nerolidyl diphosphate (2) was suppressed using a fluorine atom at C2 then a 1,10 cyclisation was observed (Fig. 2). 2-Fluorogemacrene A (25) was the DCS catalysed product from the transoid (2Z,6E)-2-fluorofarnesyl diphosphate (24) while the cisoid substrate analogue 26 gave the cisoid product 2F-helminthogermacrene A (27).12 However, in nearly every other case involving the use of substrate analogues with DCS, 1,6-cyclisation was observed at least in-part (Fig. 2).12 These results may simply reflect the use of different substrates rather than an inherent ability of DCS to catalyse the conversion of FDP to 7 along two distinct reaction paths.20 The observation that 11 acts as a competitive inhibitor of the DCS catalysed conversion of FDP provides strong evidence that DCS can efficiently use a 1,6-cyclisation pathway.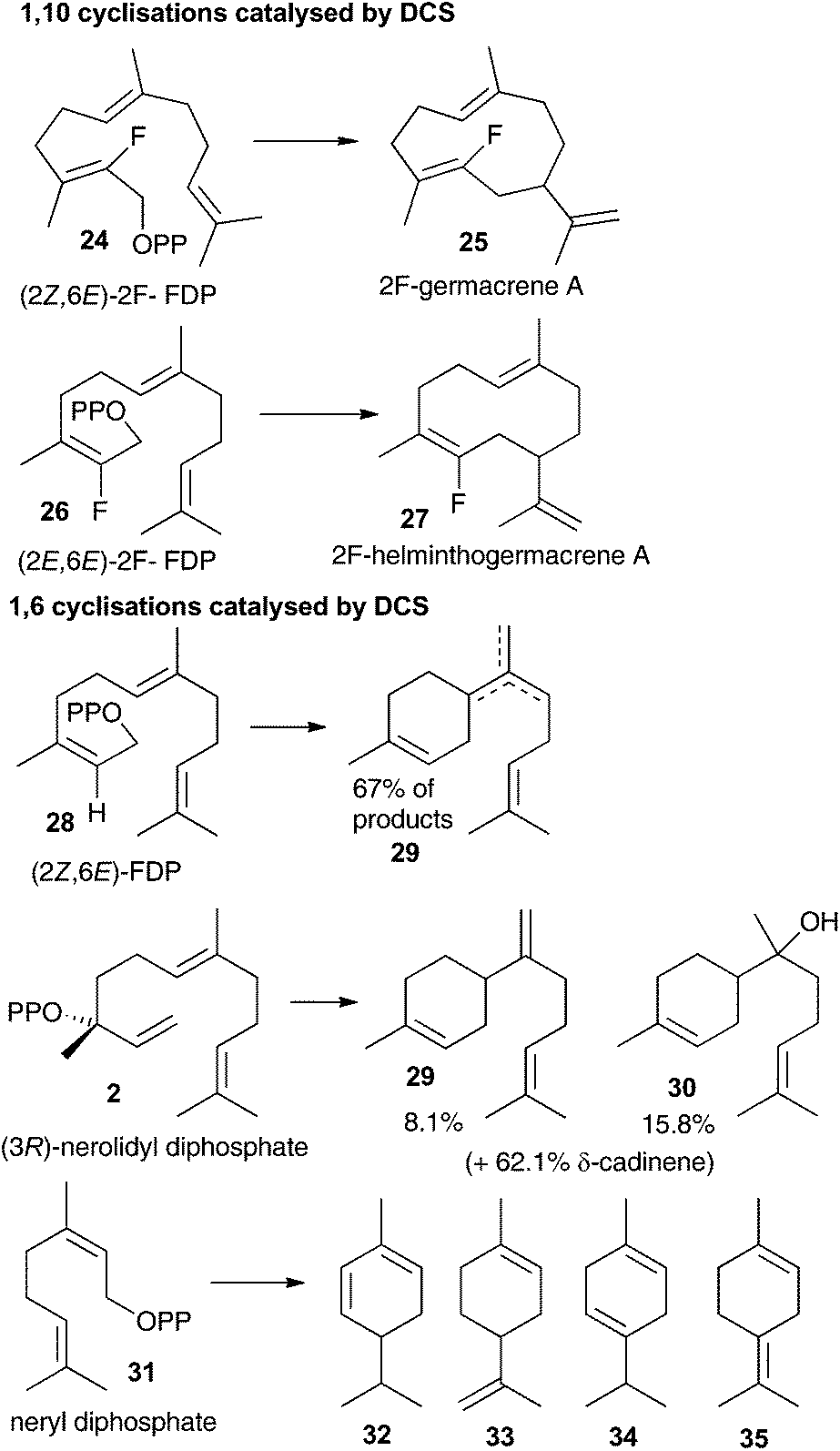 | ||
| Fig. 2 Summary of 1,10 and 1,6 cyclisation products generated through DCS catalysis. | ||
The fact that inorganic diphosphate led to a more tightly bound active site carbocation/diphosphate ion pair is consistent with previous work where often the active site recognises a cation-PPi pair more effectively than the cation alone.5b,c–7a,14 The fact that (R)-11 acts as a weak inhibitor in the absence of PPi is more difficult to explain. It was previously suggested that, in the DCS active site pocket, the alkenyl chain of (3R)-nerolidyl diphosphate (2) is ideally positioned to ensure the formation of the α-bisabolyl cation with an R configuration at C6 (Scheme 1).12 The C1–C7 hydride shift from 8 to 9 then occurs to the same Si face of C7 in cation 8, therefore a (7R)-9 formation is expected (Scheme 1). Hence the (R)-11 should mimic better the α-bisabolyl cation generated by this enzyme, and therefore act as competitive inhibitor with higher binding affinity when compared with the S-enantiomer. The evidence that both enantiomers of 11 are equally as effective in the presence of PPi is consistent with a permissive model of the active site structure, according to which an active site should accommodate a variety of rearranged intermediates of different shape and charge distribution without being rigidly complementary to a single intermediate. On the other hand, their lack of inhibitory effects on the 1,10-cyclase PR-AS shows that a major difference in the connectivity of the aza-analogue compared to the carbocation intermediate (i.e. bisabolyl cation rather than the 10-membered ring containing germacrenyl cation) renders them ineffective as inhibitors; hence the 1,6-cyclase activity of DCS postulated previously12 is intrinsic to the enzyme.
Terpene synthases can generate great structural and stereochemical complexity in one synthetic step and have therefore potential as powerful synthetic biocatalysts for the generation of many bioactive compounds.4f,18a,21,22 A clear understanding of the catalytic strategies employed by these enzymes can aid their redesign to produce nature-like compounds that are not found in the biosphere.23,24
Experimental
General experimental procedures, enzyme preparation and purification are described in ESI† along with kinetics data, gas chromatograms, mass spectra and NMR spectra.(R)-4,4-Dimethyl-2-oxotetrahydrofuran-3-yl acrylate (16)
Freshly distilled propenoyl chloride (0.41 mL, 5 mmol) was added over 1 h to a stirred solution of (R)-pantolactone (500 mg, 3.84 mmol) and Et3N (583 mg, 5.76 mmol) in anhydrous CH2Cl2 (10 mL) at −24 °C. The resulting mixture was stirred for 5 h at −24 °C, and subsequently washed with aqueous 1 M HCl (10 mL). The aqueous phase was then extracted with CH2Cl2 (3 × 20 mL). The combined organic phases were washed with saturated NaHCO3 solution (3 × 20 mL), water (3 × 20 mL) and brine (3 × 20 mL). The organic phase was dried over MgSO4, concentrated under reduced pressure and the residue was purified by flash chromatography on silica (EtOAc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane 4:6) to yield the pure compound as a yellow oil (375 mg, 53%). δH (300 MHz, CDCl3) 6.48 (1 H, d, J = 17.5, CHH
:hexane 4:6) to yield the pure compound as a yellow oil (375 mg, 53%). δH (300 MHz, CDCl3) 6.48 (1 H, d, J = 17.5, CHH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 6.18 (1H, dd, J = 17.5, 10.5 Hz, CHCH2), 5.93 (1H, d, J = 10.5 Hz, CHHC), 5.40 (1H, s, COCH–O), 4.03 (2 H, s, CH2–OCO), 1.17 (3 H, s, CH3), 1.08 (s, 3 H, CH3); δC (63 MHz, CDCl3) 174.7 (OCOCHO), 172.5 (OCOCH2), 134.0 (H2CCHCO), 118.7 (H2CCHCO), 76.1 (OCOCHO), 74.6 (OCH2CH), 40.2 (C–(CH3)2), 23.4 (CH3), 23.0 (CH3). αD +10° (CH2Cl2, c = 17). Data are in agreement with previous work.15a
C), 6.18 (1H, dd, J = 17.5, 10.5 Hz, CHCH2), 5.93 (1H, d, J = 10.5 Hz, CHHC), 5.40 (1H, s, COCH–O), 4.03 (2 H, s, CH2–OCO), 1.17 (3 H, s, CH3), 1.08 (s, 3 H, CH3); δC (63 MHz, CDCl3) 174.7 (OCOCHO), 172.5 (OCOCH2), 134.0 (H2CCHCO), 118.7 (H2CCHCO), 76.1 (OCOCHO), 74.6 (OCH2CH), 40.2 (C–(CH3)2), 23.4 (CH3), 23.0 (CH3). αD +10° (CH2Cl2, c = 17). Data are in agreement with previous work.15a
(R)-4.4-Dimethyl-2-oxotetrahydrofuran-3-yl-(S)-4-methylcyclohex-3-ene-1-carboxylate (17)
To a solution of 16 (302 mg, 1.67 mmol) in anhydrous CH2Cl2 (5 mL) at −10 °C, TiCl4 (0.82 mL, 0.82 mmol, 1.0 M solution in CH2Cl2) was added, and the resulting solution was stirred under argon at −10 °C for 1 h. 2-Methylbutadiene (0.23 mL, 2.3 mmol) was then added over 5 min and the mixture was left stirring for 3 h at −10 °C. The reaction was quenched by addition of 10% Na2CO3 in water (5 mL). The aqueous phase was then extracted with CH2Cl2 (3 × 20 mL). The organic layers were combined, washed with H2O (3 × 10 mL), brine (3 × 10 mL), dried over anhydrous MgSO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography on silica (EtOAc:hexane 2:8) to yield pure 17 as a colourless oil (353 mg, 85% yield). δH (300 MHz, CDCl3) 5.32 (1 H, s, OCHCO,) and (1 H, br CH), 3.98 (2 H, s, CHCH2O), 2.69–2.51 (1 H, m, CH–CO), 2.19 (2 H, m broad, CHO–CH2C), 1.98 (3 H, m, CHH–CH2), 1.73 (1H, m, –CHH–CH2), 1.58 (s, 3H, CH3C), 1.13 (3H, s, CH3), 1.04 (3 H, s, CH3). δC (75 MHz, CDCl3) 174.7 (OCOCHO), 172.5 (OCOCH2), 134.0 (HCCCH3), 118.7 (HCCCH3), 76.1 (OCOCHO), 74.6 (OCH2C), 40.2 (C–(CH3)2), 38.9 (CH2CH–CO), 28.9 (HCCCH2), 27.7 (CCHCH2), 25.2 (HCCCH2CH2), 23.4 (CCH3), 23.0 (CCH3), 19.8 (CH3CCH). LRMS (EI+) m/z: 252.13 (50%), 122.07 (20), 94.08 (100), 79.05 (30), 67.05 (10). αD −51.3 (CHCl3, c = 1). Data are in agreement with previous work.16
(S)-3-Acryloyl-4-benzyloxazolidin-2-one (13)
To a solution of (S)-4-benzyloxazolidin-2-one (1.00 g, 5.60 mmol) in anhydrous THF (12 mL) at −78 °C, n-BuLi (2.1 M in THF, 3.14 mL, 6.59 mmol) was added dropwise over 30 minutes and the mixture stirred for a further 3 h at −78 °C. Freshly distilled acryloyl chloride (557 mg, 6.16 mmol) was added dropwise over 20 minutes and the reaction stirred for 2 h at −78 °C. The reaction was then allowed to warm to room temperature overnight. The reaction was quenched with sat. NH4Cl (20 mL) and extracted with diethyl ether (3 × 30 mL). The organic layer was washed with water (3 × 40 mL), saturated aqueous NaHCO3 (3 × 40 mL), dried (MgSO4), filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (hexane:ethyl acetate 6:4) afforded 13 as a colorless solid (452 mg, 35%). δH (CDCl3, 300 MHz) 7.45 (dd, 1H, J = 6.0, 18.0, CHCH2), 7.23 (m, 5H, aromatic Hs), 6.54 (dd, 1H, JH, H = 18.0, 18.0, CHHCH2), 5.87 (dd, 1H, J = 9.0, 9.0, CHHC), 4.68 (m, 1H, CHN), 4.14 (m, 2H, CH2O), 3.29 (dd, 1H, J = 9.0, 9.0, CCHHPh), 2.74 (dd, 1H, J = 12.0, 12.0, CHHPh) αD −86° (CH2Cl2, c = 0.65). Data are in agreement with previous work.14a
(R)-4-Benzyl-3-((S)-4-methylcyclohex-3-enecarbonyl)oxazolidin-2-one (14)
To a stirred solution of 13 (200 mg, 0.86 mmol) at −100 °C, were added 2-methylbutadiene (1.72 mL, 17.2 mmol) in anhydrous CH2Cl2 (5.0 mL) and Et2AlCl (1.2 mL, 1.5 eq.). The reaction was stirred at −100 °C for 30 min then the mixture was poured into ice cold aqueous hydrochloric acid (1 M, 20 mL). The mixture was extracted with CH2Cl2 (2 × 10 mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The product was purified by flash chromatography on silica (EtOAc:hexane:Et3N 92:7:1) to yield the Diels Alder adduct (14) as a white crystalline solid (139 mg, 54%). δH (300 MHz, CDCl3) 7.34–7.10 (5 H, m, ArCH), 5.36 (1 H, br., CHC), 4.63 (1 H, dt, J = 16.6, 6.9, CHN), 4.23–3.95 (2 H, m, CH2O), 3.60 (1 H, t, J = 8.8, CHCO), 3.20 (1 H, dd, J = 13.2, 3.3, CHHPh), 2.70 (1 H, dd, J = 13.3, 9.5, CHHPh), 2.20–1.6 (6 H, m, CH2CH2CHCH2CC), 1.64 (3H, s, CH3) αD +79 (CH2Cl2, c = 1.4). LR-MS (EI+) m/z: 299.15 (100% M+), 300.16 (15), 269.06 (18), 267.07 (50), 232.10 (20), 178.08 (100), 146.07 (30), 140.03 (55), 122.07 (20), 91.00 (65), 63.00 (30). Data are in agreement with previous work.14a
(R)- and (S)-4-Methylcyclohex-3-ene-1-carboxylic acid (18)
To a solution of 14 or 17 (0.32 mmol) in THF:MeOH:H2O (1:1:1, 1.5 mL), LiOH (67 mg, 1.6 mmol) was added, and the resulting mixture was vigorously stirred for 1 h at 50 °C. The reaction was then cooled to room temperature and concentrated under reduced pressure. The resulting slurry was dissolved in H2O (10 mL) and extracted with CH2Cl2 (3 × 5 mL). The resulting aqueous phase was acidified to pH = 2 at 0 °C with 15% HCl, extracted with a mixture of n-pentane:CH2Cl2 (98:2 3 × 10 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure to give 18 as a white powder (35 mg, 80%). δH (CDCl3, 300 MHz) δ 5.32 (1H, s, CH), 2.54–2.39 (1 H, m, CH–COOH), 2.17 (2H, m, –CH2CHC), 1.93 (3 H, m, CHH–CH2), 1.69 (1H, m, CHH–CH2), 1.59 (3H, s, CH3). δC (100 MHz, CDCl3) δ 182.3 (HOCO), 133.8 (HCCCH3), 119.0 (CH2–HCCCH3), 39.0 (HC–COOH), 29.13 (CH2CCH3), 27.3 (CH2CHCCH3), 25.5 (CH2CH2CCH), 23.5 (CH3). (S)-18: αD −80.6 (CHCl3, c = 0.5); −106.4 (95% EtOH, c = 4). (R)-18: αD +93 (CHCl3, c = 0.5); +105.5 (95% EtOH, c = 4). M.p. 82–92 °C. LRMS (EI+) m/z 140.06 (100%, M+), 136.06 (15), 125.05 (40), 122.06 (100), 95.87 (100), 94.06 (100), 93.07 (80), 79.04 (100), 77.03 (100), 68.06 (90), 67.04 (100) Data are in agreement with previous work.15,16
(R)- and (S)-4-Methoxybenzyl [(4-methylcyclohex-3-en-1-yl) methyl]-carbamate (19)
To a solution of 18 (1.3 g, 9.3 mmol) in anhydrous toluene (20 mL) at 0 °C, diphenylphosphoryl azide (2.2 mL, 10.2 mmol) and Et3N (3.9 mL, 27.8 mmol) were added. The resulting mixture was left stirring for 3 h at 100 °C before 4-methoxybenzyl alcohol (1.27 mL, 10.2 mmol) was added, and the reaction was left to stir for 16 h at 100 °C. The reaction was then allowed to cool to room temperature and the solution was concentrated under reduced pressure. The residue was the purified by flash chromatography on silica gel (EtOAc:n-hexane 1:9) to yield 19 as a yellow crystalline solid (1.28 g, 80%). δH (300 MHz, CDCl3) δ 7.23 (2 H, dt, J = 2.9 and 5.3 CH ArCH), 6.81 (2 H, dt, J = 2.9 and 5.3, ArCH), 5.21 (1 H, br, HCC), 4.95 (2 H, s, –OCH2Ph), 4.67–460 (1 H, m, CHN), 3.74 (3 H, s, –OCH3), 2.29–2.20 (2 H, m, CH2–CH2CHN), 1.93 (2 H, m, CH2–CH2CHN), 2.01–1.86 (2 H, m, CH–CH2CHN), 1.55 (3 H, s, CH3CCH). δC (75 MHz, CDCl3) δ 159.5 (CO–CH3), 155.8 (NHCO), 134.1 (CCCH3), 130.0 (CC aromatic), 128.7 (CCH2O), 118.3 (CCCH3), 113.9 (CC aromatic), 66.3 (CCH2O), 62.8 (CHN), 55.3 (OCH3), 31.9 (CH2CHN), 28.4 (CH2CC), 28.0 (CH2CC), 23.4 (CCH3). νmax (thin film, cm−1) 3300 (N–H stretch), 2900–2700 (C–H stretch), 1650 (CO ester stretch), 1250 (C–N stretch), 830 (aromatic CH bending); (S)-19: αD −9.3, (c = 0.6, CHCl3) (R)-19: αD +12 (c = 0.6, CHCl3) m.p. 69–71 °C LRMS (EI+) m/z: 275.15 (100% M+), 276.15 (20), 259.12 (18), 258.12 (60), 231.12 (25), 228.1128(12), 214.16 (100). HRMS (EI+) 275.1522; C16H21NO3 requires 275.1521.
(R)- & (S)-N-Methyl-1-(4-methylcyclohex-3-en-1-yl)methanamine (20)
To a stirred solution of carbamate 19 (100 mg, 0.4 mmol) in anhydrous diethyl ether (7 mL) at 0 °C, was added LiAlH4 (50 mg, 1.28 mmol). The mixture was then heated to reflux for 5 h. The reaction was cooled to 0 °C before it was quenched by the addition of water (6 mL) and an excess of 15% NaOH solution (6 mL). The resulting mixture was left to stir at 0 °C for 1 h and the precipitate was removed by filtration through a Celite pad. The organic phase was extracted with water (2 × 10 mL) and the pooled organic layers were then washed with 10% HCl (2 × 10 mL) and the organic fraction was discarded. The combined aqueous layers were adjusted to pH 12 by dropwise addition of 10% NaOH (15 mL). The product was extracted with diethyl ether (4 × 15 mL), then dried over anhydrous MgSO4, and filtered. The product was then concentrated carefully under reduced pressure to give 20 as a volatile colorless oil (20 mg, 40%). δH (300 MHz, CDCl3) 5.24 (1 H, m br, CHC), 2.55 (1 H, dt, J = 16.6 and 8.0, CHNH2), 2.37 (3 H, s, HNCH3), 2.25–2.10 (1 H, m, NH), 1.99–1.87 (2 H, m, CH2CHC), 1.87–1.66 (2 H, m, CH2CH2CCH), 1.61 (1 H, broad m, CH2CH2CCH), 1.60 (3 H, s, H2CCCH3), 1.45–1.26 (1 H, m, CH2CH2CCH). δC (75 MHz, CDCl3) 134.0 (CCCH3), 119.1 (CCCH3), 54.8 (CHNH), 32.7 (CH2CHNH), 32.1 (CCHCH2CH), 29.7 (CH3N), 29.0 (CH2C), 23.4 (CH3C) (S)-20: αD −79 (c 1.00, CHCl3) (R)-20: αD +84 (c 1.00, CHCl3) Data in agreement with previous work.14a
(R)- and (S)-N,4-Dimethyl-N-(4-methylcyclohex-3-en-1-yl)pent-3-enamide (21)
To a stirred solution of 20 (172 mg, 1.5 mmol) and DIPEA (775 mg, 6.0 mmol) in anhydrous DMF (6 mL), HBTU (1.15 g, 3.0 mmol) was added and the resulting mixture was stirred at room temperature for 20 min before 20 (358 mg, 1.5 mmol) was added. The reaction was then stirred for 24 h at room temperature. The mixture was concentrated under reduced pressure and the residue was dissolved in diethyl ether (20 mL). The solution was washed with water (2 × 25 mL), 10% NaHCO3 (2 × 25 mL), 10% HCl (2 × 10 mL), and brine (25 mL) before it was dried over anhydrous MgSO4 and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (EtOAc:hexane 4:6) yielding 21 as a colorless oil (166 mg, 50%). δH (400 MHz, CDCl3) 5.24 (2 H, dd, J = 14.6 and 8.0, NCHCH2CH and CHCH2CO), 4.70–4.54 (0.5 H, m, CHN), 3.75 (0.5 H, dd, J = 12.6 and 9.7, CHN), 3.01 (2 H, dd, J = 12.6, 6.8 Hz, CH2CO), 2.75 (1.5 H, s, CH3N), 2.72 (1.5 H, s, CH3N), 2.20–1.86 (5 H, m, CH2CH2CHCH2), 1.71–1.88 (1 H, m, CH2CH2CHCH2), 1.75 (s, 3 H, CNCH2CHCH3), 1.68 (3 H, s, CCH3), 1.66 (3 H, s, CCH3). (S)-21: αD −9.5, (CHCl3, c = 0.9) (R)-21: αD +10, (CHCl3, c = 0.9) HRMS (EI+) 221.1779; C14H23NO requires 221.1780. Data are in agreement with previous work.14a
(R)- and (S)-N,4-Dimethyl-N-(4-methylpent-3-en-3-yl)cyclohex-3-en-1 ammonium chloride (11)
To a stirred solution of 21 (41 mg, 0.19 mmol) in anhydrous diethyl ether at 0 °C, was added LiAlH4 (33 mg, 0.87 mmol). The mixture was heated to reflux for 6 h then allowed to cool to room temperature and stirred for a further 12 h. The reaction was quenched by the addition of water (6 mL) and 15% NaOH (6 mL) at 0 °C and stirred for 1 h at 0 °C. The white precipitate was removed by filtration on Celite and the filtrate was extracted with diethyl ether (2 × 25 mL). The combined organic layers were dried over anhydrous Na2SO4, concentrated under reduced pressure, and the residue was purified by flash chromatography on silica (Et2O:MeOH 1:9) to yield the amine as a yellow oil (32 mg, 65% yield). δH (300 MHz, CDCl3) 5.27 (1 H, d, J = 2.4, H3CCCH), 5.03 (1H, t, J = 5.6, (CH3)2CCH), 2.64–2.44 (1 H, m, CHN), 2.38 (2 H, ddd, J = 7.6, 5.8 and 2.6, CH2N), 2.23 (3 H, s, CH3N), 2.08 (2 H, dd, J = 15.5 and 7.2, CH2CH2N), 2.04–1.85 (4 H, m, CH2CH2CHN), 1.85–1.67 (2 H, m, CCHCH2CHN), 1.62 (3 H, s, CH3CCH), 1.55 and 1.57 (2 × 3 H, 2 × s, (CH3)2CCH). δC (75 MHz, CDCl3) δ 133.9 (HCCCH3), 132.6 (CHCCH3), 122.1 (CHCCH3), 120.0 (NCH2CH2CHCCH3), 58.9 (CHN), 53.5 (CH2N), 37.9 (CH2CH2CHN), 30.8 (CHCH2N), 27.2 (CH2CCH3), 26.5 (CH3N), 25.7(CH2CH2N), 25.6 (CH3CCH3), 23.2 (CH3CCH3), 17.8 (CH3C). HRMS (EI+) 207.1990; C14H25N requires 207.1987. (S)-11: αD −61 (CHCl3, c = 1) (R)-11αD +63 (CHCl3, c = 1). Data are in agreement with previous work.14a The amine was then dissolved in Et2O (1 mL) and HCl (1 M in anhydrous Et2O) was added slowly. A light yellow precipitate was formed. The ether was concentrated under reduced pressure and the salt stored in 1.2 mL of deionised water. δH (300 MHz, MeOD) 5.38 (1 H, br, H3CCCH), 5.14 (1 H, t, J = 5.0, (CH3)2CCH), 3.58–3.42 (1 H, m, CHN), 3.32 (1 H, dd, J = 4.9, 1.6, CHHCH2N), 3.25–2.97 (1 H, m, CHHCH2N), 2.85 (3 H, s, CH3N), 2.59–2.39 (2 H, m, CH2CH2CHN), 2.39–2.26 (2 H, m, CH2CH2CHN), 2.26–2.05 (2 H, m, CCHCH2CH2N), 1.84 (2 H, dd, J = 12.2, 10.4 Hz, CH3CCHCH2), 1.76 (3 H, s, CH3CCH), 1.72 (6 H, 2 × s, (CH3)2CCH). δC (75 MHz, MeOD) δ 136.1 (HCCCH3), 134.3 (CHC(CH3)2) 117.6 (CHCCH3) 116.5 (NCH2CH2CHCCH3), 61.8, 61.5 (CHN), 53.0, 52.5 (CH2N), 35.9, 35.2 (CH3N) 29.1, 29.0 (CH2CH2CHN), 25.7 (CHCH2N), 24.5 (CH2CCH3), 24.2, 24.0 (NCH2CH2), 23.25, 22.64 (CH3C), 21.64 (CH3CCH3), 16.65 (CH3CCH3). νmax (neat, cm−1) 2972 (broad, N–H stretch), 1379 (C–N stretch), 1161, 1051 and 1022 (C–N stretch), 950, 879, 815; HRMS (APCI+) 208.2057, C14H26N requires 208.2065; m.p. 131–133 °C, (S)-11αD −62.2 (MeOH, c = 0.09), (R)-11αD +57.1 (MeOH, c = 0.09). Data are in agreement with previous work.14a
Conflicts of interest
There are on conflicts of interest to declare.Acknowledgements
We thank Dr Robert Jenkins and Simon Waller for assistance with GC-MS experiments and Dr Juan A. Faraldos (all Cardiff University) for valuable scientific discussions during this work. This work was supported by the UK's Biotechnology and Biological Sciences Research Council (BBSRC) through grants (BB/H01683X/1, BB/M022463/1, BB/N012526/1). We thank Cardiff University for the provision of a postgraduate studentship to M. L.Notes and references
- (a) J. D. Connolly and R. A. Hill, Dictionary of Terpenoids, Chapman and Hall, London, 1991 CrossRef; (b) J. S. Glasby, Encyclopedia of Terpenoids, Wiley, Chichester, 1982 Search PubMed; (c) E. M. Davis and R. Croteau, in Biosynthesis: Aromatic Polyketides, Isoprenoids, Alkaloids, Springer, Berlin Heidelberg, 2000, vol. 209, p. 53 Search PubMed; (d) E. M. Davis, in Comprehensive Natural Products II, ed. L. Mander and H.-W. Liu, Elsevier, 2010, vol. 1, p. 585 Search PubMed; (e) T. Toyomasu and T. Sassa, in Comprehensive Natural Products II, ed. L. Mander and H.-W. Liu, Elsevier, 2010, vol. 1, p. 643 Search PubMed; (f) T. Kushiro and Y. Ebizuka, in Comprehensive Natural Products II, ed. L. Nander and H.-W. Liu, Elsevier, 2010, vol. 1, p. 673 Search PubMed; (g) D. J. Miller and R. K. Allemann, Nat. Prod. Rep., 2012, 29, 60 RSC; (h) Y. Gao, R. B. Honzatko and R. J. Peters, Nat. Prod. Rep., 2012, 29, 1153 RSC.
- (a) D. Cane, Chem. Rev., 1990, 90, 1089 CrossRef CAS; (b) D. W. Christianson, Chem. Rev., 2006, 106, 3412 CrossRef CAS; (c) D. W. Christianson, Chem. Rev., 2017, 117, 11570 CrossRef CAS PubMed; (d) D. J. Tantillo, Angew. Chem., Int. Ed., 2017, 56, 10040 CrossRef CAS PubMed.
- (a) B. Felicetti and D. E. Cane, J. Am. Chem. Soc., 2004, 126, 7212 CrossRef CAS PubMed; (b) M. Seemann, G. Z. Zhai, J. W. de Kraker, C. M. Paschall, D. W. Christianson and D. E. Cane, J. Am. Chem. Soc., 2002, 124, 7681 CrossRef CAS; (c) J. A. Aaron, X. Lin, D. E. Cane and D. W. Christianson, Biochemistry, 2010, 49, 1787 CrossRef CAS PubMed; (d) B. T. Greenhagen, P. E. O'Maille, J. P. Noel and J. Chappell, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9826 CrossRef CAS PubMed; (e) Y. Yoshikuni, T. E. Ferrin and J. D. Keasling, Nature, 2006, 440, 1078 CrossRef CAS PubMed; (f) V. González, S. Touchet, D. J. Grundy, J. A. Faraldos and R. K. Allemann, J. Am. Chem. Soc., 2014, 136, 14505 CrossRef PubMed; (g) J. A. Faraldos, A. K. Antonczak, V. González, R. Fullerton, E. M. Tippmann and R. K. Allemann, J. Am. Chem. Soc., 2011, 133, 13906 CrossRef CAS PubMed.
- (a) J. Degenhardt, T. G. Kollner and J. Gershenzon, Phytochemistry, 2009, 70, 1621 CrossRef CAS PubMed; (b) D. E. Cane, P. C. Prabhakaran, J. S. Oliver and D. B. McIlwaine, J. Am. Chem. Soc., 1990, 112, 3209 CrossRef CAS; (c) S. H. Kim, K. Heo, Y. J. Chang, S. H. Park, S. K. Rhee and S. U. Kim, J. Nat. Prod., 2006, 69, 758 CrossRef CAS PubMed; (d) C. D. Poulter, J. C. Argyle and E. A. Mash, J. Biol. Chem., 1978, 253, 7227 CAS; (e) R. Croteau, Arch. Biochem. Biophys., 1986, 251, 777 CrossRef CAS PubMed; (f) D. J. Miller, F. L. Yu, D. W. Knight and R. K. Allemann, Org. Biomol. Chem., 2009, 7, 962 RSC; (g) J. A. Faraldos, Y. X. Zhao, P. E. O'Maille, J. P. Noel and R. M. Coates, ChemBioChem, 2007, 8, 1826 CrossRef CAS PubMed.
- (a) J. Y. Jiang, X. F. He and D. E. Cane, Nat. Chem. Biol., 2007, 3, 711 CrossRef CAS PubMed; (b) J. A. Faraldos, B. Kariuki and R. K. Allemann, J. Org. Chem., 2010, 75, 1119 CrossRef CAS PubMed; (c) J. A. Faraldos and R. K. Allemann, Org. Lett., 2011, 13, 1202 CrossRef CAS PubMed; (d) A. S. Narula, A. Rahier, P. Beneviste and F. Shuber, J. Am. Chem. Soc., 1981, 103, 2408 CrossRef CAS; (e) C. D. Poulter, Acc. Chem. Res., 1990, 23, 70 CrossRef CAS.
- (a) J.-X. Li, X. Fang, Q. Zhao, J.-X. Ruan, C.-Q. Yang, L. J. Wang, D. J. Miller, J. A. Faraldos, R. K. Allemann, X. Y. Chen and P. Zhang, Biochem. J., 2013, 451, 417 CrossRef CAS PubMed; (b) D. J. Grundy, M. Chen, V. González, S. Leoni, D. J. Miller, D. W. Christianson and R. K. Allemann, Biochemistry, 2016, 55, 2112 CrossRef CAS PubMed; (c) K. Y. Shishova, F. Yu, D. J. Miller, J. A. Faraldos, Y. Zhao, R. M. Coates, R. K. Allemann, D. E. Cane and D. W. Christianson, J. Biol. Chem., 2008, 283, 15431 CrossRef PubMed; (d) M. Chen, N. Al-lami, M. Janvier, E. L. D'Antonio, J. A. Faraldos, D. E. Cane, R. K. Allemann and D. W. Christianson, Biochemistry, 2013, 52, 5441 CrossRef CAS PubMed; (e) H. A. Gennadios, V. González, L. Di Costanzo, A. Li, F. L. Yu, D. J. Miller, R. K. Allemann and D. W. Christianson, Biochemistry, 2009, 48, 6175 CrossRef CAS PubMed.
- (a) J. A. Faraldos, D. J. Grundy, O. Cascon, S. Leoni, M. W. van der Kamp and R. K. Allemann, Chem. Commun., 2017, 52, 14027 RSC; (b) M. W. van der Kamp, J. Sirirak, J. Zurek, R. K. Allemann and A. J. Mulholland, Biochemistry, 2013, 52, 8094 CrossRef CAS PubMed; (c) R. K. Allemann, N. J. Young, S. Ma, D. G. Truhlar and J. Gao, J. Am. Chem. Soc., 2007, 129, 13008 CrossRef CAS PubMed; (d) S. R. Hare and D. J. Tantillo, Beilstein J. Org. Chem., 2017, 13, 1669 CrossRef PubMed; (e) D. J. Tantillo, Nat. Prod. Rep., 2011, 28, 1035 RSC.
- C. L. Steele, J. Crock, J. Bohlmann and R. Croteau, J. Biol. Chem., 1998, 273, 2078 CrossRef CAS.
- G. D. Davis and M. Essenberg, Phytochemistry, 1995, 39, 553 CrossRef CAS.
- (a) C. R. Benedict, J. L. Lu, D. W. Pettigrew, J. Liu, R. D. Stipanovic and H. J. Williams, Plant Physiol., 2001, 125, 1754 CrossRef CAS; (b) S. Opitz, G. Kunert and J. Gershenzon, J. Chem. Ecol., 2008, 34, 508 CrossRef CAS PubMed.
- (a) G. M. Bianchini, R. D. Stipanovic and A. A. Bell, J. Agric. Food Chem., 1999, 47, 4403 CrossRef CAS; (b) F. Chen, D. Tholl, J. Bohlmann and E. Pichersky, Plant J., 2011, 66, 212 CrossRef CAS PubMed.
- J. A. Faraldos, D. J. Miller, V. González, Z. Yoosuf-Aly, O. Cascón, A. Li and R. K. Allemann, J. Am. Chem. Soc., 2012, 134, 5900 CrossRef CAS PubMed.
- (a) J. A. Faraldos, B. M. Kariuki and R. M. Coates, Org. Lett., 2011, 13, 836 CrossRef CAS PubMed; (b) S. C. Karcher and S. A. Laufer, J. Med. Chem., 2009, 52, 1778 CrossRef CAS PubMed; (c) A. N. van Nhien, C. Tomassi, C. Len, J. L. Marco-Contelles, J. Balzarini, C. Pannecouque, E. de Clercq and D. Postel, J. Med. Chem., 2005, 48, 4276 CrossRef CAS PubMed; (d) M. Taton, M. Ceruti, L. Cattel and A. Rahier, Phytochemistry, 1996, 43, 75 CrossRef CAS; (e) A. Steiger, H.-J. Pyun and R. M. Coates, J. Org. Chem., 1992, 57, 3444 CrossRef CAS; (f) D. A. Evans, K. T. Chapman and J. Bisaha, J. Am. Chem. Soc., 1988, 110, 1238 CrossRef CAS; (g) D. A. Evans, K. T. Chapman and J. Bisaha, J. Am. Chem. Soc., 1984, 106, 4261 CrossRef CAS.
- (a) D. E. Cane, G. Yang and R. M. Coates, J. Org. Chem., 1992, 57, 3454 CrossRef CAS; (b) M. M. Ravn, R. J. Peters, R. M. Coates and R. Croteau, J. Am. Chem. Soc., 2002, 124, 6998 CrossRef CAS PubMed; (c) K. Zhou, R. J. Peters and R. M. Coates, J. Am. Chem. Soc., 2007, 124, 12453 Search PubMed.
- (a) T. Poll, A. Sobczak, H. Hartmann and G. Helmchen, Tetrahedron Lett., 1985, 26, 3095 CrossRef CAS; (b) R. M. Sandifer, M. D. Thompson, R. G. Gaughan and C. D. Poulter, J. Am. Chem. Soc., 1982, 4, 7376 CrossRef.
- (a) L. Argenti, F. Bellina, A. Carpita and R. Rossi, Synth. Commun., 1995, 25, 1909 CrossRef; (b) W. J. Watkins, Preparation of thiophencarboxylic acid derivatives for use as flaviviridae viruses inhibitors, US Patent20130052161, 2013 Search PubMed.
- M. Calvert, S. E. Taylor and R. K. Allemann, Chem. Commun., 2002, 2384 RSC.
- (a) M. Demiray, X. Tang, J. A. Faraldos, T. Wirth and R. K. Allemann, Angew. Chem., Int. Ed., 2017, 56, 4347 CrossRef CAS PubMed; (b) S. Picaud, P. Mercke, X. He, O. Sterner, M. Brodelius, D. E. Cane and P. E. Brodelius, Arch. Biochem. Biophys., 2006, 448, 150 CrossRef CAS PubMed.
- M. Loizzi, V. González, D. J. Miller and R. K. Allemann, ChemBioChem, 2018, 19, 100–105 CrossRef CAS PubMed.
- D. J. Tantillo, Angew. Chem., Int. Ed., 2017, 56, 10040 CrossRef CAS PubMed.
- X. Tang, M. Demiray, T. Wirth and R. K. Allemann, Bioorg. Med. Chem., 2017, 19, 100 Search PubMed.
- D. J. Miller, F. Yu and R. K. Allemann, ChemBioChem, 2007, 15, 1819 CrossRef PubMed.
- S. Touchet, K. Chamberlain, C. M. Woodcock, D. J. Miller, M. A. Birkett, J. A. Pickett and R. K. Allemann, J. Am. Chem. Soc., 2015, 136, 7550 Search PubMed.
- F. Huynh, D. J. Miller and R. K. Allemann, ChemBioChem, 2018, 19, 1834 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental procedures, enzyme preparation and purification, kinetics data, gas chromatograms, mass spectra and NMR spectra. See DOI: 10.1039/c8ob02821d |
| This journal is © The Royal Society of Chemistry 2019 |