Substrate selectivity and its mechanistic insight of the photo-responsive non-nucleoside triphosphate for myosin and kinesin†
Received
1st November 2018
, Accepted 3rd December 2018
First published on 4th December 2018
Abstract
Linear motor proteins including kinesin and myosin are promising biomaterials for developing nano-devices. Photoswitchable substrates of these biomotors can be used to optically regulate the motility of their associated cytoskeletal filaments in in vitro systems. Here, we describe the discovery of the myosin selective azobenzene-tethered triphosphate. It enables the specific photocontrol over myosin in a reversible mode with the composite motility assay composed of both kinesin and myosin. The mechanistic insight into this myosin selectivity is also explained with the docking simulation study.
Introduction
Molecular motor proteins including the linear and rotary motors are mechanical enzymes that can move by catalytically converting chemical energy of adenosine triphosphate (ATP) into mechanical work.1–3 In particular, cytoskeletal motor proteins including conventional kinesin-14,5 and myosin II6,7 that can move unidirectionally along their associated cytoskeletal filaments are pivotal for maintaining the cellular functions. Kinesin-1 mainly conducts the intracellular active transport of organelles and macromolecular assemblies along microtubules from the cell center towards the cell periphery (i.e. anterograde transport). Myosin II moving along actin filaments toward the barbed end is a leading contributor to muscle contraction and cell migration. In spite of the different associated filaments, these two biomolecular motors possess similar engines using ATP to generate the mechanical force. Due to this immense potential of producing force with nano-sized motors, numerous attempts have been made to develop functional nano-devices.8,9
Gliding motility assay is a well-established model system to study and engineer bio-molecular motors in vitro.4,10 In this versatile approach, the gliding motility of fluorescently labeled microtubules or actin filaments driven by bio-motors (kinesin or myosin) which are bound to the glass surface can be visualized by fluorescence microscopy. To develop controllable nano-devices using bio-molecular motors, various external stimuli including electricity,11–13 heat,14–21 light22–27 and so on have been applied. In particular, photo-controllable approaches should be drawing the center of attention due to their high spatiotemporal operability and high switchability. For instance, Higuchi et al.22 and Hess et al.23 demonstrated caged ATP (photolabile protected ATP) as the photocontrolled one-way switch that was from motility OFF to ON using ultraviolet light. In addition to these excellent methods, our group recently reported the reversibly photocontrollable ATP analogues, i.e. non-nucleoside triphosphate molecules based on azobenzene (AzoTPs), for kinesin-128 and myosin II.29 AzoTPs in the trans state of their azobenzene moiety can be recognized as the enzyme substrate for both kinesin and myosin as well as natural energy molecule ATP for powering the motor proteins, whereas they in the cis state cannot be recognized (Fig. 1). These AzoTPs are rapidly photoconverted without any side products due to the photoisomerization reaction of azobenzene with the picosecond time scale.30,31 In contrast, caged ATP including the 2-nitrobenzyl type of the photoreactive group is photoactivated with a millisecond time scale32 to produce ATP with a highly reactive nitroso compound with proteins as the byproduct. Using these highlighted photochemical properties of AzoTPs, we demonstrated the repetitive switching of kinesin or myosin-based cytoskeletal filament translocation with external light stimuli without any significant fatigue of cytoskeletal filament motility.
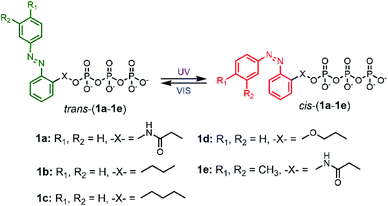 |
| | Fig. 1 Schematic representation of cis–trans photo isomerization of 1a–1e with UV and VIS light irradiation. | |
Due to the well-conserved engine mechanism in kinesin and myosin,33 ATP can efficiently power each kinesin and myosin. In sharp contrast, other nucleotides including synthetic ATP analogues exhibited unique specificity towards kinesin and/or myosin.34–43 The specific substrate should be a useful tool for analyzing which proteins are responsible for the motor-based motility in a crude system. Herein, we describe the successful discovery of the myosin-selective photoswitchable energy molecule. Through the structure–activity relationship study in both microtubule–kinesin and actin–myosin systems, we found the myosin-selective AzoTP derivative (1e) with hydrophobic bulky substitution on the azobenzene moiety retaining high photoswitchability, which was also demonstrated in the kinesin–myosin composite system.44 Compared with ATP as a standard substrate, 1e exhibited 9.4-fold higher selectivity toward the actin–myosin system than the microtubule–kinesin system. This selectivity mechanism was rationally explained through the docking simulation study.
Results and discussion
Design and synthesis of AzoTP derivatives and their photoresponsibility
To explore the powering properties towards kinesin and myosin, four AzoTP derivatives (1a–1e) except for 1c with different types of linkages and azobenzene substituents were prepared according to our previous reports.28,29 AzoTP derivative 1c was newly synthesized as depicted in Scheme 1 (Experimental section) with the details of synthetic procedures. The characterization (1H NMR, 13C NMR, 31P NMR and HR-MS) and the purity of 1c are also shown in Fig. S1 and S9–S17 (ESI†). These AzoTPs were designed to compare the length and flexibility of linkers in ethyl (1b), propyl (1c) and ethyl ether linkages (1d) with the original acetamide linker in 1a and the steric and hydrophobic effects of 3′- and 4′-methyl groups on an azobenzene ring in 1e.
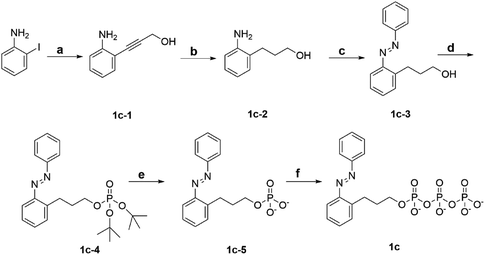 |
| | Scheme 1 Synthetic scheme of 1c. Reagent and condition: (a) CuI, bis(triphenylphosphine)palladium(II) dichloride, 2-propyn-1-ol, triethylamine, RT, 18 h. (b) 10% Pd/C, H2, MeOH, RT, 48 h. (c) Nitrosobenzene, AcOH, toluene, N2 atmosphere, 60 °C, 21 h. (d) Di-tert-butyl N,N-diisopropylphosphoramidite, 1H-tetrazol, dry THF, Ar atmosphere, RT, 6 h, then mCPBA, 0 °C, 1 h. (e) TFA, dry DCM, Ar atmosphere, RT, 6 h. (f) Tributylamine, carbonyldiimidazole, tributylammonium pyrophosphate, dry DMF, Ar atmosphere, RT, overnight. | |
Photochromic azobenzene triphosphates can undergo reversible cis–trans isomerization by absorbing ultraviolet (UV) and visible (VIS) light, respectively (Fig. 1). UV-VIS absorption spectra of 1a–1e in BRB-80 buffer before irradiation (BI) and after UV (365 nm) or VIS (436 nm) irradiation are shown in Fig. 2 and Fig. S2 (ESI†). By the illumination of UV light to form cis rich states of AzoTPs (UV photostationary state, UV PSS), the π–π* band at around 327 nm in absorbance spectra decreased, while the n–π* band at around 430 nm increased. An opposite trend (namely, the increment of the π–π* band and the decrement of the n–π* band) was observed by applying VIS light to reach the trans rich states (VIS PSS). The cis–trans isomer ratio of 1a–1e at UV PSS and VIS PSS was calculated by 1H NMR measurements as shown in Table S1 and Fig. S3 (ESI†). The reversible photoisomerization of 1a–1e could be recycled several times by alternating UV and VIS light irradiation without any fatigue. cis isomers of all AzoTPs shown here were stable in BRB-80 buffer at 25 °C (t1/2 > 3 days) enough to show an insignificant thermal back reaction during the motility experiments (Fig. S4, ESI†).
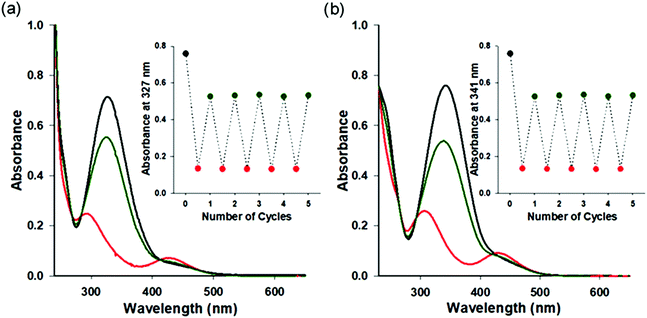 |
| | Fig. 2 UV-VIS absorption spectra of 1a and 1e before irradiation and at UV photo-stationary state (PSS) and VIS PSS. UV-VIS absorption spectra of (a) 1a (4.4 × 10−4 M) and (b) 1e (5.2 × 10−4 M) in BRB-80 buffer at 25 °C. Before irradiation (black line), UV PSS (red line), and VIS PSS (dark green line). Insets: Absorbance changes after the alternate irradiation with UV (20 s) and VIS (150 s) light for 5 cycles. | |
AzoTP derivatives in actin–myosin in vitro motility assay
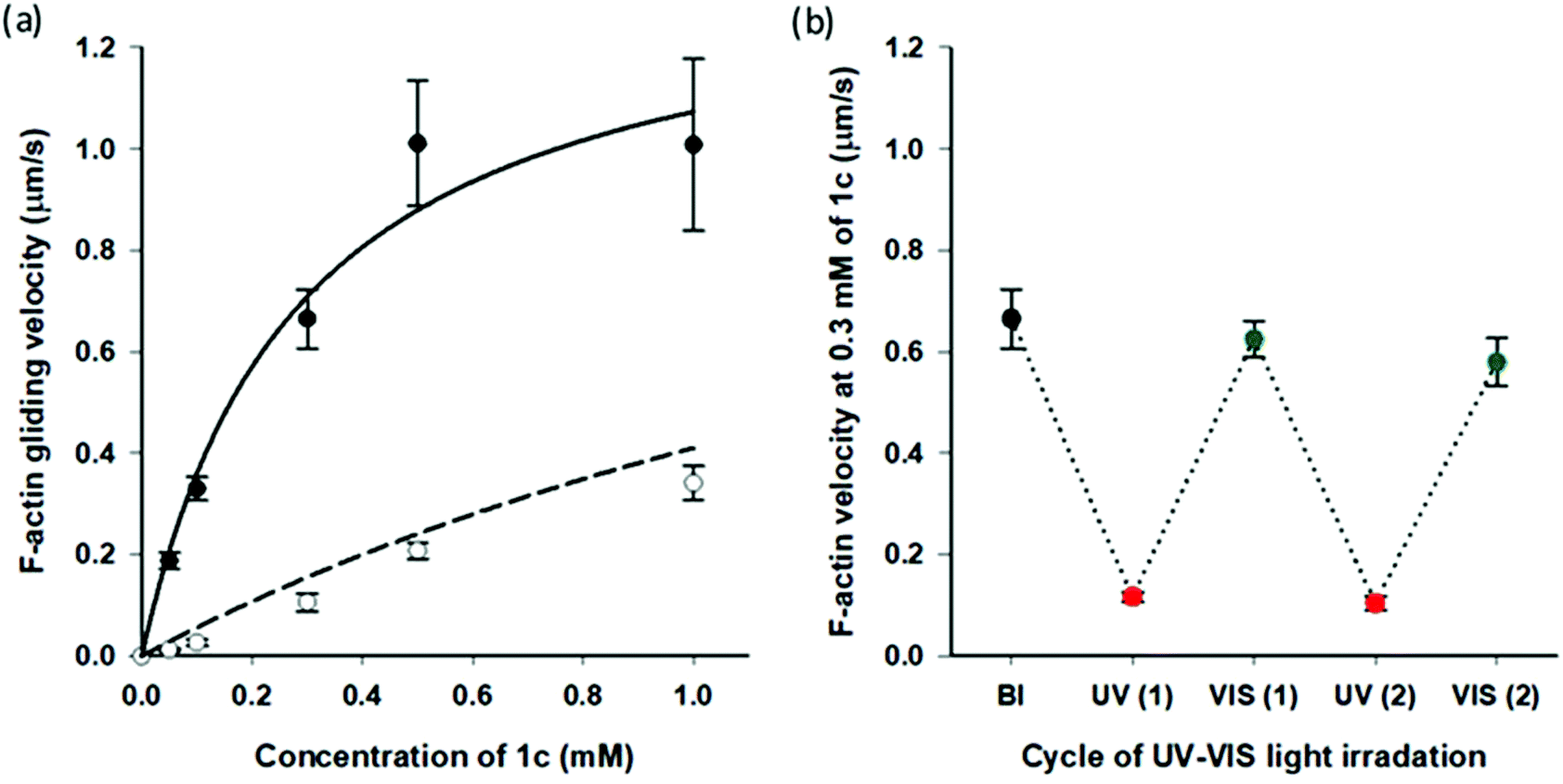
Very recently, we reported that AzoTPs 1a, 1b, 1d and 1e worked as photo-responsive myosin substrates in a photoreversible mode in actin–myosin gliding motility assay (Table S2†).29 The newly synthesized 1c was studied in the actin–myosin gliding motility assay (Fig. 3). The maximum gliding velocity (Vmax) of actin filaments with 1c was 1.4 μm s−1, which was 27% of Vmax (5.1 μm s−1) compared with ATP (Fig. S5†). The gliding velocity of actin filaments was reversibly controlled for several cycles with repeated UV and VIS light illumination (Fig. 3b). Similarly to other AzoTPs in the actin–myosin system,29 the cis isomer of 1c had no contribution to powering the myosin. The motility at UV PSS originated from the remaining trans isomer of 1c (12%) as plotted in the theoretical curves (dashed black line in Fig. 3a). Therefore, we concluded that in the actin–myosin system, a wide range of AzoTP derivatives with different linkers and azobenzene moieties exhibited the myosin-powering activities in a photoswitchable mode.
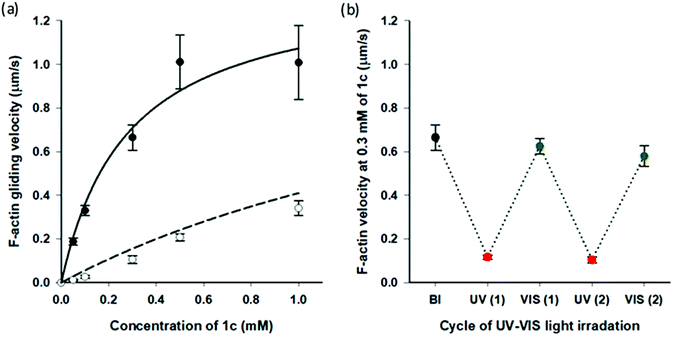 |
| | Fig. 3 Photoresponsive actin gliding motility with 1c (a) Gliding velocity depending on the concentration of 1c. (Filled circle (●): BI condition and open circle (○): after the UV irradiation condition). Solid black line: curve fitting using the Michaelis–Menten equation (Km = 0.28 ± 0.09 mM, Vmax = 1.4 ± 0.2 μm s−1 for 1c) and dashed black line: theoretical curve derived from the solid black line for the remaining trans isomer (12%) in the cis rich state of 1c. (b) Reversible photoregulation of the actin gliding velocity using 1c (0.30 mM) with repeated UV and VIS light irradiation. Error bars represent the standard deviation of 10 actin filaments in a single flow cell. | |
AzoTP derivatives in microtubule–kinesin in vitro motility assay
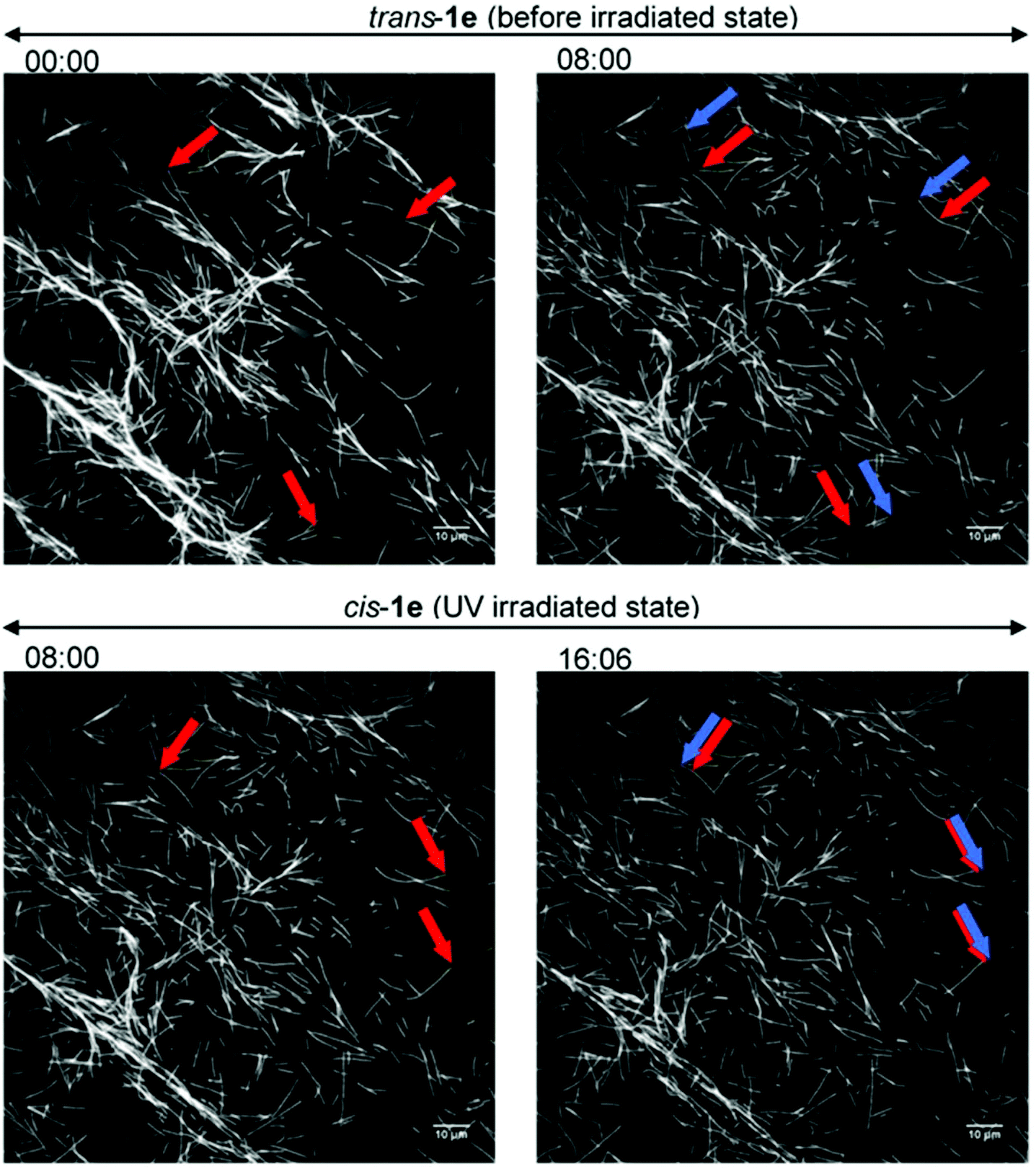
So far, only 1a28 was exploited in microtubule–kinesin in vitro motility assay in our previous study. Using 1a, the microtubule gliding velocity was drastically photo-switched in concert with cis–trans photoisomerization of 1a by illumination with two different wavelengths of light (BI: 0.51 μm s−1, UV: 0.12 μm s−1 at 3.0 mM). Encouraged by successful application of our AzoTPs to the actin–myosin system, we investigated AzoTP derivatives, 1b–1e, as the substrates of kinesin-1 to drive microtubule gliding in the microtubule–kinesin system (Fig. 4, S6 (ESI†) and Table 1). Photoswitchability of gliding velocity of microtubules by alternating illumination with UV and VIS light was also studied. AzoTP derivatives 1b (0.51 μm s−1 at 3.0 mM) and 1c (0.45 μm s−1 at 3.0 mM) exhibited comparable velocity with 1a under the BI condition. Upon the subsequent UV irradiation, the gliding velocity decreased to 0.31 μm s−1 at 3.0 mM with 1b and 0.34 μm s−1 at 3.0 mM with 1c (Fig. 4a, b and Fig. S6, ESI†), but their changes between BI and UV conditions were not drastic compared with the actin–myosin system (Table S2† and Fig. 3). The reversible photoswitching of microtubule gliding velocity using 1b and 1c was also achieved with alternating irradiation of UV and VIS light (Fig. S7, ESI†). These results indicated that the flexibility and length of linker chains between the azobenzene moiety and triphosphate group (acetamide linker for 1a, ethyl linker for 1b and propyl linker for 1c) had a trivial effect on powering the microtubule–kinesin system. AzoTP 1d with the ethyl ether linker showed less activity (BI: 0.07 μm s−1 at 3.0 mM) than the original AzoTP 1a. There was no significant change in the microtubule velocity between BI and UV conditions (UV: 0.10 μm s−1 at 3.0 mM) although the clear photo-isomerization from the trans isomer to cis isomer was confirmed by the UV-VIS spectra (Fig. S2c, ESI†). The ethyl ether linker in 1d had almost the same length as the propyl linker in 1c, but it was found that the oxygen atom in the ether group of 1d negatively affected the powering activity and photoswitchability in the microtubule–kinesin system. This might be because the ether group worked as the acceptor of hydrogen bonding, which could interrupt the substrate recognition of 1d by kinesin. To quantitatively assess the powering ability of cis-isomers of AzoTPs in the microtubule–kinesin system, we plotted theoretical curves (dashed black line in Fig. 4a–c and S6a†) corresponding to the remaining trans isomer (Table S1, ESI†) of AzoTPs under UV irradiation conditions. Inconsistency in 1b, 1c and 1d between the theoretical curves and the actual velocity under UV conditions implied that the cis isomers had significant activity on gliding motility. In contrast to 1a–1d (0.47 μm s−1 for 1a, 0.48 μm s−1 for 1b, 0.41 μm s−1 for 1c and 0.09 μm s−1 for 1d at 2.0 mM concentration), to our surprise, 1e with two methyl groups in the azobenzene moiety showed almost no powering activity (0.027 μm s−1 at 2.0 mM) and photoswitchability towards kinesin-1 (Fig. 4d and 5).45 Representative fluorescence images of the gliding motility of microtubules with 1e at 8 min interval for both in the BI state and after UV illumination state are shown in Fig. 5. Vmax (0.058 μm s−1) with 1e was only 6.3% of Vmax (0.92 μm s−1) with ATP (Fig. S6, ESI†) in the microtubule–kinesin motility assay. Therefore, we concluded that the methyl substitution critically impeded the substrate recognition of AzoTP in the kinesin motor.
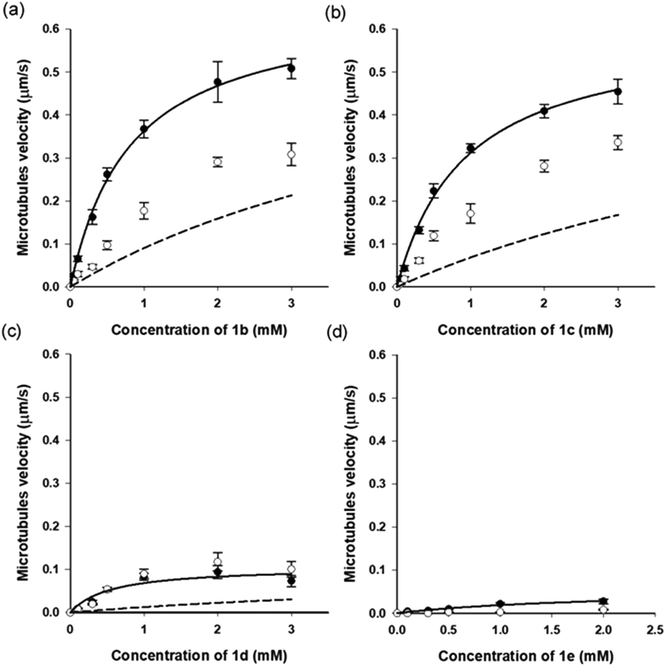 |
| | Fig. 4 Microtubule gliding velocity (filled circle (●): BI condition and open circle (○): UV condition) with AzoTP derivatives (a) 1b, (b) 1c, (c) 1d and (d) 1e.45 Solid black line: curve fitting using the Michaelis–Menten equation (Km = 0.82 ± 0.07 mM, Vmax = 0.66 ± 0.02 μm s−1 for 1b; Km = 0.93 ± 0.09 mM, Vmax = 0.60 ± 0.02 μm s−1 for 1c, and Km = 1.0 ± 0.4 mM, Vmax = 0.15 ± 0.03 μm s−1 for 1d; Km = 2.1 ± 1 mM, Vmax = 0.058 ± 0.02 μm s−1 for 1e). Dashed black line: theoretical curve calculated from the remaining trans isomer in the cis rich state of the respective AzoTP derivatives. Error bars represent the standard deviation of 10 microtubules in a single flow cell. | |
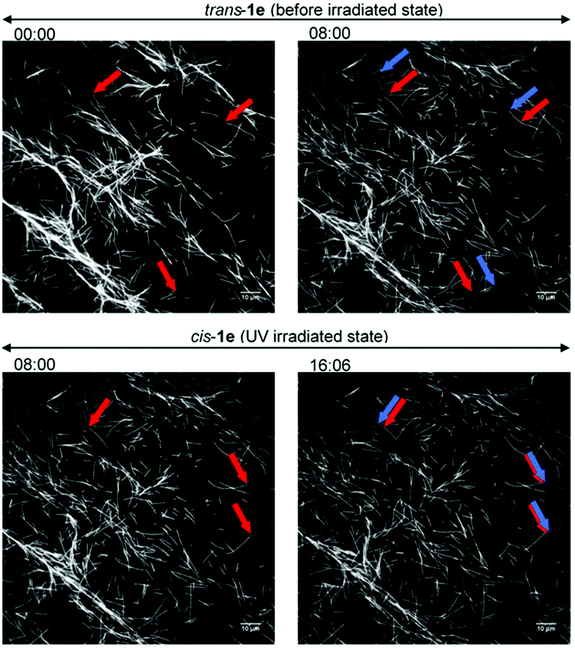 |
| | Fig. 5 Fluorescence images of the gliding microtubules driven by 1e (2.0 mM) on microtubule–kinesin gliding motility assay. (Top) Before the irradiation state; (bottom) after the UV irradiation state; (left side) starting frame; (right side) frame after 8 min; red arrows: leading points of the microtubules at the starting frame; blue arrows: leading points of the same microtubules after 8 min. | |
Table 1 Summary of AzoTP derivatives in the microtubule–kinesin system
| AzoTP derivatives |
V
max (μm s−1) |
K
m (mM) |
Gliding velocity switchinga (%) |
|
Gliding velocity switching between trans and cis-rich states of AzoTP derivatives at saturated concentration, 3.0 mM for 1a, 1b, 1c and 1d; 2.0 mM for 1e.45 N.D.: not determined.
|
|
1a
23
|
0.83 ± 0.06 |
1.7 ± 0.2 |
76 |
|
1b
|
0.66 ± 0.02 |
0.82 ± 0.07 |
39 |
|
1c
|
0.60 ± 0.02 |
0.93 ± 0.09 |
26 |
|
1d
|
0.15 ± 0.03 |
1.0 ± 0.4 |
27 |
|
1e
|
0.058 ± 0.02 |
2.1 ± 1 |
N.D. |
Comparison of AzoTPs between myosin and kinesin systems
Guanosine triphosphate (GTP) has been reported as a kinesin-selective natural nucleoside triphosphate (NTP).35,39 We also confirmed the kinesin selectivity of GTP in our motility system. GTP could power kinesin to drive microtubules efficiently (0.21 μm s−1 at 1.0 mM), whereas it could not work efficiently (0.049 μm s−1 at 1.0 mM) in the actin–myosin motility system (Fig. S5 and S6c, ESI†). This special trend encouraged us to compare AzoTPs in both myosin and kinesin systems in a quantitative way. To directly demonstrate the powering activities and efficiencies of our AzoTPs for myosin and kinesin, ATP was used as the standard substrate for both the kinesin and myosin motor protein systems (Table 2). AzoTPs (1a–1d) exhibited moderate activity towards both myosin (>27%Vmax of ATP) and kinesin (>16%Vmax of ATP). However, AzoTP 1e showed different trends toward myosin and kinesin. In the myosin, 1e showed 59% of Vmax and 16% of 1/Km (3.7 mM−1) compared with ATP. In the kinesin, 1e showed 6.3% of Vmax and 3.8% of 1/Km (0.48 mM−1) compared with ATP. This high activity specificity of 1e (Myo/Kin = 9.4) with high photoswitchability (Table 2) indicated that 1e was recognized as a substrate of not kinesin-1 but myosin II, although other AzoTPs (1a–1d) worked as substrates for both motor proteins.
Table 2 Summary of efficacy of AzoTP derivatives as the photo-responsive energy molecule in microtubule–kinesin and actin–myosin systems
| NTPs/AzoTPs |
Microtubule–kinesin system |
Actin–myosin system |
Activity specificity (Myo/Kin) under BI condition |
| MT velocitya (μm s−1) |
F-actin velocityb (μm s−1) |
| BI |
UVc |
BI |
UVc |
|
Microtubule (MT) velocity at Vmax.
F-actin filament (F-actin) velocity at Vmax.
Under UV conditions, the saturated concentration of AzoTPs in the corresponding motility assays was considered.
Velocity at 1.0 mM of GTP was considered because Vmax is not determinable due to too low activity.
The different Vmax (5.1 ± 0.2 μm s−1) of ATP was used for the calculation, because we used the different batch of myosin and actin from our previous report in this new experiment. Activity specificity, Myo/Kin is the ratio of myosin driven actin filament standardized velocity and kinesin driven microtubule standardized velocity at Vmax. The standardized velocity is shown in the parentheses with the actual velocity where the Vmax of ATP is considered as the standard velocity (100%) for each of the motor protein systems. N.D.: not determined.
|
| ATP |
0.92 ± 0.01 (100%) |
0.92 ± 0.01 (100%) |
2.9 ± 0.124 (100%) |
2.9 ± 0.124 (100%) |
1.0 |
| GTP |
0.55 ± 0.03 (60%) |
0.55 ± 0.03 (60%) |
0.049 ± 0.01d (1.0%) |
0.049 ± 0.01d (1.0%) |
0.02 |
|
1a
|
0.83 ± 0.06 (90%) |
0.12 ± 0.01 (13%) |
1.5 ± 0.0424 (52%) |
0.84 ± 0.124 (29%) |
0.6 |
|
1b
|
0.66 ± 0.02 (72%) |
0.31 ± 0.03 (34%) |
1.0 ± 0.124 (35%) |
0.13 ± 0.0224 (4.5%) |
0.5 |
|
1c
|
0.60 ± 0.02 (65%) |
0.34 ± 0.02 (37%) |
1.4 ± 0.2e (27%) |
0.34 ± 0.03e (6.7%) |
0.4 |
|
1d
|
0.15 ± 0.03 (16%) |
0.10 ± 0.02 (11%) |
1.9 ± 0.124 (66%) |
0.38 ± 0.0424 (13%) |
4.1 |
|
1e
|
0.058 ± 0.02 (6.3%) |
N.D. |
1.7 ± 0.224 (59%) |
0.39 ± 0.0524 (13%) |
9.4 |
Computational docking simulation of AzoTP derivatives with the motor domains of kinesin-1 and myosin II
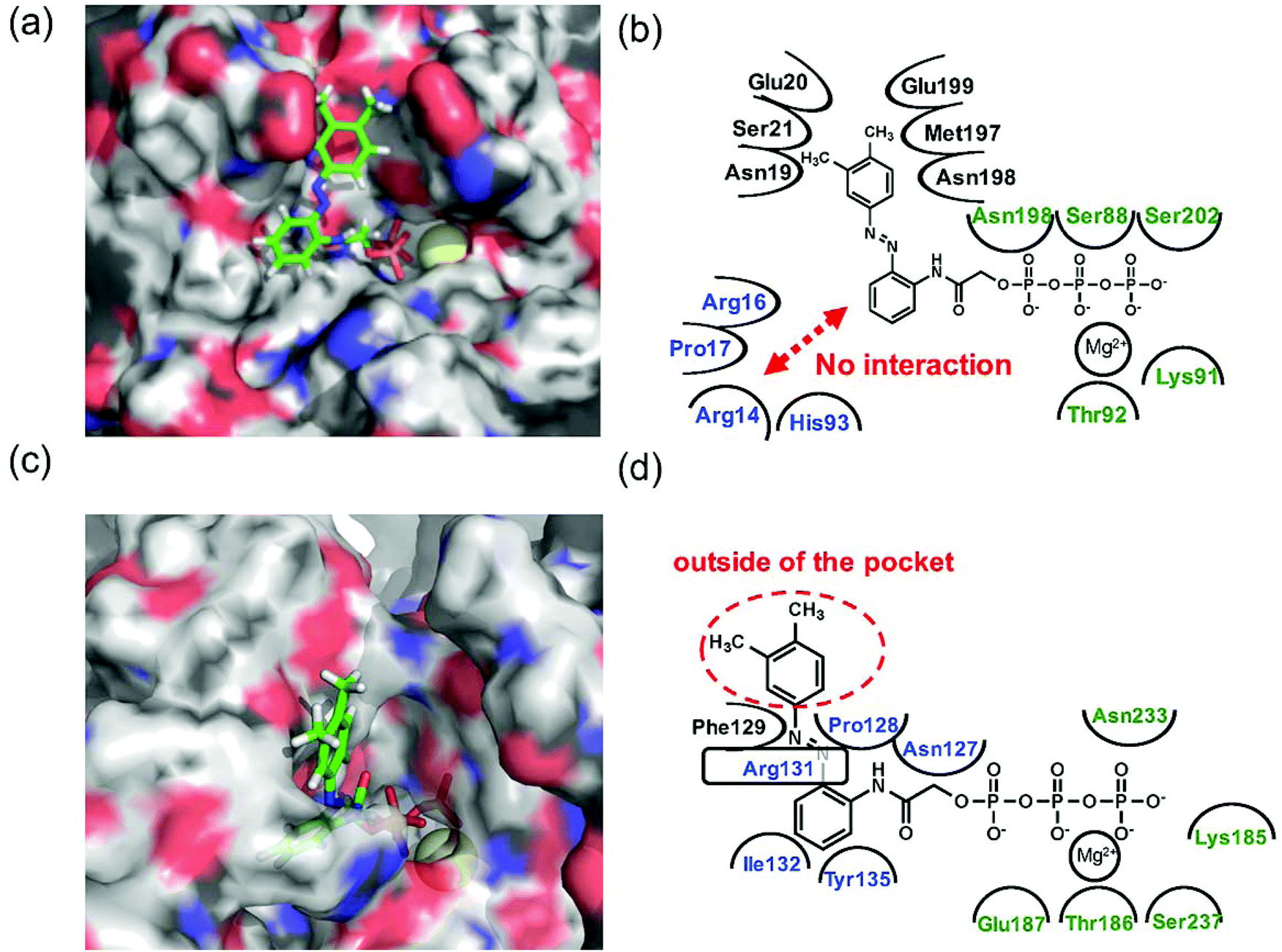
Since 1e displayed high selectivity towards myosin II compared with kinesin-1, the putative binding modes of AzoTPs 1a (the good substrate for both kinesin and myosin) and 1e with kinesin-1 and myosin II were investigated through the docking simulations (Fig. 6–8). In the simulation results of kinesin-1 and myosin II with the trans isomer of 1a, triphosphate groups were chelated with magnesium ions at the same binding site of the triphosphate group in ATP (Fig. 6a and b). In the azobenzene moiety, the benzene ring connected with the triphosphate group through an acetamide linker was well-overlapped with the adenine ring of ADP in the original X-ray structures of both kinesin and myosin (Fig. S8, ESI†). In myosin, the azo group (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) can interact via a hydrogen bond with Arg131 of covering the adenine ring binding site (ARBS), which could fix the angle of the azobenzene moiety. Another benzene ring in the azobenzene moiety was located outside of the ATP binding site to face the solvent direction. Therefore, it was likely that there was no interaction of that benzene ring with the surrounding amino acids. In kinesin, His93 was the key residue for exhibiting ATPase activity.33 From the simulation, the benzene unit in 1a was estimated to show the π–π interaction with His93 as well as ATP. Due to no amino acids covering ARBS unlike myosin, the azo group (–NN–) was relatively flexible. This flexibility possibly contributed to arranging the outside benzene ring of the azobenzene moiety not to facing the solvent direction but to being buried in other space near the ATP binding site (Fig. 6b). Despite slightly different binding modes of the trans isomer of 1a with kinesin and myosin, these well-fittings were robustly consistent with the results on the motility assays that 1a exhibited relatively high activities for powering kinesin (4.7% of 1/Km and 90% of Vmax compared with ATP) and myosin (43% of 1/Km and 52% of Vmax compared with ATP) under the BI condition. Conversely, the cis isomer of 1a has almost no contribution to the gliding velocity of cytoskeletal filaments in both the motor protein systems.28,29 We also docked the cis isomer of 1a with kinesin and myosin for explaining the low affinity of the cis isomer of 1a in kinesin and myosin. As shown in Fig. 7a and b, the total incompatibility of the cis isomer of 1a in ARBS was observed, although the triphosphate group was well-bound. This suggested that the bent azobenzene unit prevented the approach of 1a into ARBS. Almost similar binding modes of AzoTPs were applied except for 1e, which exhibited high selectivity to myosin over kinesin. In the docking results of the trans isomer of 1e with myosin, the benzene ring in 1e was well-occupied with ARBS as well as other AzoTPs and the dimethyl-substituted benzene ring was located facing to solvents (Fig. 8). In contrast, the docking of 1e with kinesin showed the incompatibility of the benzene ring of the azobenzene moiety with ARBS, because the steric hindrance of two methyl groups on the azobenzene moiety interrupted the access of 1e to ARBS. Thus, these computational docking studies could explain our experimental results of myosin selectivity in 1e over kinesin.
N–) can interact via a hydrogen bond with Arg131 of covering the adenine ring binding site (ARBS), which could fix the angle of the azobenzene moiety. Another benzene ring in the azobenzene moiety was located outside of the ATP binding site to face the solvent direction. Therefore, it was likely that there was no interaction of that benzene ring with the surrounding amino acids. In kinesin, His93 was the key residue for exhibiting ATPase activity.33 From the simulation, the benzene unit in 1a was estimated to show the π–π interaction with His93 as well as ATP. Due to no amino acids covering ARBS unlike myosin, the azo group (–NN–) was relatively flexible. This flexibility possibly contributed to arranging the outside benzene ring of the azobenzene moiety not to facing the solvent direction but to being buried in other space near the ATP binding site (Fig. 6b). Despite slightly different binding modes of the trans isomer of 1a with kinesin and myosin, these well-fittings were robustly consistent with the results on the motility assays that 1a exhibited relatively high activities for powering kinesin (4.7% of 1/Km and 90% of Vmax compared with ATP) and myosin (43% of 1/Km and 52% of Vmax compared with ATP) under the BI condition. Conversely, the cis isomer of 1a has almost no contribution to the gliding velocity of cytoskeletal filaments in both the motor protein systems.28,29 We also docked the cis isomer of 1a with kinesin and myosin for explaining the low affinity of the cis isomer of 1a in kinesin and myosin. As shown in Fig. 7a and b, the total incompatibility of the cis isomer of 1a in ARBS was observed, although the triphosphate group was well-bound. This suggested that the bent azobenzene unit prevented the approach of 1a into ARBS. Almost similar binding modes of AzoTPs were applied except for 1e, which exhibited high selectivity to myosin over kinesin. In the docking results of the trans isomer of 1e with myosin, the benzene ring in 1e was well-occupied with ARBS as well as other AzoTPs and the dimethyl-substituted benzene ring was located facing to solvents (Fig. 8). In contrast, the docking of 1e with kinesin showed the incompatibility of the benzene ring of the azobenzene moiety with ARBS, because the steric hindrance of two methyl groups on the azobenzene moiety interrupted the access of 1e to ARBS. Thus, these computational docking studies could explain our experimental results of myosin selectivity in 1e over kinesin.
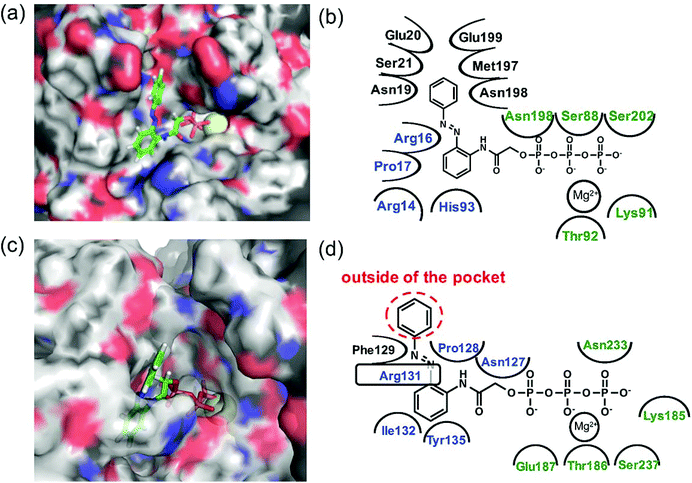 |
| | Fig. 6 Computational docking simulation of trans isomers of 1a with kinesin-1 and myosin II motor domains. Docking simulation of AzoTPs was conducted with CLC Drug Discovery Workbench (QIAGEN). X-ray crystal structures obtained from RCSB Protein Data Bank (PDB entry code: 4HNA for kinesin-1 and 1MMD for myosin II) were used. All figures of simulated results were created with pymol (DeLano Scientific). (a) Simulated result and (b) binding modes of the trans isomer of 1a in kinesin-1. (c) Simulated result and (d) binding modes of the trans isomer of 1a in myosin II. In simulated results, AzoTPs were represented by a stick mode. Magnesium ions were denoted by a yellow ball. In the figures of binding modes, the amino acid resides composing ARBS were colored in blue and the residues composing the triphosphate binding site were colored in green. | |
 |
| | Fig. 7 Computational docking simulation of cis isomers of 1a with (a) kinesin-1 and (b) myosin II motor domains. | |
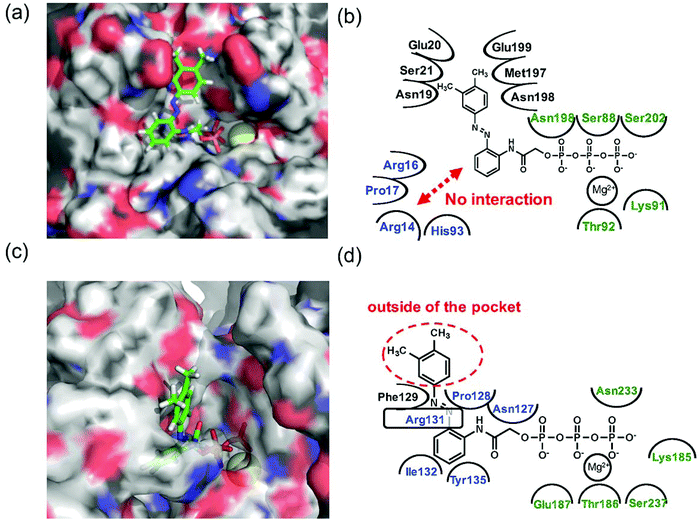 |
| | Fig. 8 Computational docking simulation of trans isomers of 1e with kinesin-1 and myosin II motor domain. Docking simulation of AzoTPs was conducted with CLC Drug Discovery Workbench (QIAGEN). X-ray crystal structures obtained from RCSB Protein Data Bank (PDB entry code: 4HNA for kinesin-1 and 1MMD for myosin II) were used. All figures of simulated results were created with pymol (DeLano Scientific). (a) Simulated result and (b) binding modes of the trans isomer of 1e in kinesin-1. (c) Simulated result and (d) binding modes of the trans isomer of 1e in myosin II. In simulated results, AzoTPs were represented by a stick mode. Magnesium ions were denoted by a yellow ball. In the figures of binding modes, the amino acid resides composing ARBS were colored in blue and the residues composing the triphosphate binding site were colored in green. | |
Selective photoregulation of myosin motility using 1e in kinesin–myosin composite motility assay
Since 1e exhibited high selectivity to myosin over kinesin, the selective photoregulation of myosin was explored in kinesin–myosin composite motility assay,44 where both kinesin and myosin were attached on the same glass surface and microtubules and actin filaments were glided over the motor proteins. The gliding motility of fluorescence labeled microtubules and actin filaments driven by respective motor proteins was observed with a fluorescence microscope.
The gliding motility of microtubules and actin filaments in the kinesin–myosin composite assay with 1e (1.0 mM) was examined under different light irradiation conditions (Fig. 9, Movie S1 in ESI†). AzoTP 1e could power myosin to drive actin filaments with photoreversibility (BI: 1.1 ± 0.04 μm s−1, UV: 0.27 ± 0.01 μm s−1 and VIS: 0.88 ± 0.03 μm s−1 at 1.0 mM) in the composite motility assay. In contrast, it could not power kinesin efficiently to drive microtubules (BI: 0.054 ± 0.01 μm s−1 at 1.0 mM). As the control experiment, ATP (0.20 mM) was applied to our kinesin–myosin composite system. It was shown that both kinesin and myosin motors were driven efficiently to afford the velocity of microtubules (0.71 ± 0.02 μm s−1) and actin filaments (3.3 ± 0.3 μm s−1) (Movie S2, ESI†). These findings suggested that AzoTP 1e also worked as the photoswitchable substrate with myosin specificity in kinesin–myosin composite motility assay.
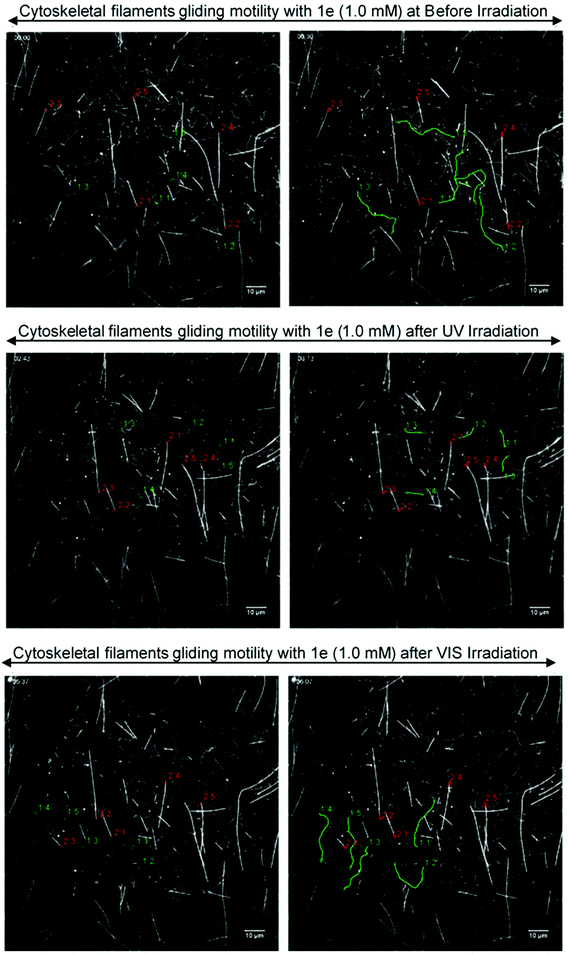 |
| | Fig. 9 Fluorescence images of the gliding actin filaments and microtubules driven by 1e (1.0 mM) on kinesin–myosin composite motility assay. (Top) Before irradiation, trans state; (middle) cis-rich state after UV irradiation; (bottom) trans-rich state after VIS light irradiation; (left side) starting frame; (right side) frame after 30 s. The distance traveled by five actin filaments (green lines) and five microtubules (red lines) were shown using tracking the path of each actin filament or microtubule tails. | |
Conclusion
The myosin-specific AzoTP molecule 1e with high photoswitchability was discovered through studying the powering activity of several AzoTPs in the kinesin and myosin motility assays, respectively. Even in the kinesin–myosin composite system, 1e could drive the myosin selectively. This myosin selectivity in 1e was explained by its binding modes with kinesin or myosin motors through the docking simulation. This rational analysis of binding modes of AzoTPs using both the experimental assays and the computational studies might contribute to the other motor-specific substrate molecules. We envision that our motor-specific AzoTP can be a chemical tool for unveiling the motor-based biological processes and constructing nanotechnological systems using the microtubule and actin composite system.
Experimental
Chemicals
All chemical and biochemical reagents were purchased from commercial sources (Wako Pure Chemical, Kanto Chemical, TCI Chemical, Sigma-Aldrich, Biotium and Dojindo Molecular Technologies) and used without further purification. Thin-layer chromatography (TLC) was performed on silica gel 60 F254-precoated aluminum sheets (Merck) and visualized by fluorescence microscopy. Chromatographic purification was performed using flash column chromatography on silica gel 60 N (neutral, 63–210 μm, Kanto Chemical).
Synthesis of AzoTP 1c
3-(2-Aminophenyl)prop-2-yn-1-ol (1c-1).
A reaction mixture of copper(I) iodide (32 mg, 0.2 mmol), bis(triphenylphosphine)palladium(II) dichloride (140 mg, 0.2 mmol), and 2-iodoaniline (4.4 g, 20 mmol) in 120 mL of triethylamine was stirred under an Ar atmosphere. After 30 min of stirring, 2-propyn-1-ol (1.2 mL, 20 mmol) was added to the reaction mixture and it was kept stirring for 18 h at room temperature. The reaction mixture was evaporated and partitioned between water and DCM. The DCM part was washed several times with water to remove triethylamine completely, then dried over MgSO4 and concentrated. The residue was purified by silica gel column chromatography (MeOH/DCM, 3/97) to afford 1.95 g (65%) of a maroon solid 3-(2-aminophenyl)prop-2-yn-1-ol (1c-1). 1H NMR (400 MHz, CHLOROFORM-D) δ 7.28 (d, J = 1.3 Hz, 1H), 7.13 (t, J = 7.9 Hz, 1H), 6.70–6.66 (m, 2H), 4.55 (d, J = 4.0 Hz, 2H), 4.21 (br, 2H), 1.70 (br, 1H).
3-(2-Aminophenyl)propan-1-ol (1c-2).
A solution of 1c-1 (1.95 g, 13.2 mmol) in dry MeOH (60 mL) was added to 10% Pd/C (0.5 g) under a N2 atmosphere at room temperature. Then N2 gas was changed to H2 gas with a constant pressure from a rubber bladder. The reaction mixture was stirred for 48 h and filtered through a Celite pad. The solvent was evaporated and the residue was purified by silica gel column chromatography (hexane/EtOAc, 1/1) to afford 1.03 g (51%) of 3-(2-aminophenyl)propan-1-ol (1c-2) as a dark brown solid. 1H NMR (400 MHz, CHLOROFORM-D) δ 7.06–7.02 (m, 2H), 6.76 (td, J = 7.4, 1.2 Hz, 1H), 6.69 (d, J = 7.4 Hz, 1H), 3.64 (t, J = 6.0 Hz, 2H), 2.64 (t, J = 7.3 Hz, 2H), 1.87 (quin, J = 6.5 Hz, 2H), 1.67 (br, 1H).
(E)-3-(2-(Phenyldiazenyl)phenyl)propan-1-ol (1c-3).
A solution of 1c-2 (0.276 g, 1.8 mmol) in toluene (13 mL) was degassed under a stream of N2 gas for 15 min. To this solution, nitrosobenzene (0.193 g, 1.8 mmol) and acetic acid (0.5 mL) were added under an Ar atmosphere. The reaction mixture was stirred at 60 °C for 21 h. The solvent of the reaction mixture was evaporated and partitioned between water and DCM. The DCM part was washed several times with water and dried over MgSO4. The solvent was evaporated in vacuo. The residue was purified by silica gel column chromatography (hexane/EtOAc, 4/3) to afford 0.374 g (86%) of (E)-3-(2-(phenyldiazenyl)phenyl)propan-1-ol (1c-3) as a dark brown solid. 1H NMR (400 MHz, CHLOROFORM-D) δ 7.89 (dd, J = 8.3, 1.4 Hz, 2H), 7.67 (dd, J = 8.0, 1.2 Hz, 1H), 7.56–7.46 (m, 3H), 7.44–7.36 (m, 2H), 7.31 (t, J = 7.5 Hz, 1H), 3.63 (q, J = 6.1 Hz, 2H), 3.24 (t, J = 7.2 Hz, 2H), 2.05 (t, J = 6.1 Hz, 1H), 1.97 (quin, J = 6.6 Hz, 2H).
(E)-Di-tert-butyl (3-(2-(phenyldiazenyl)phenyl)propyl) phosphate (1c-4).
To a solution of 1c-3 (1.22 g, 5.1 mmol) and di-tert-butyl N,N-diisopropylphosphoramidite (2.1 mL, 6.5 mmol) in dry THF (20 mL), 1H-tetrazole (1.04 g, 15.0 mmol) was added. The reaction mixture was stirred for 7 h at room temperature. A solution of m-chloroperoxybenzoic acid (65%, 2.3 g, 8.6 mmol) in dry CH2Cl2 (10 mL) was added. The mixture was stirred for 1 h in an ice bath and then for 30 min at room temperature. Saturated aqueous NaHCO3 (60 mL) was added and the mixture was stirred for further 30 min. The reaction mixture was partitioned between EtOAc and water. The organic phase was washed with brine (once), dried over MgSO4 and concentrated. The residue was purified through silica gel column chromatography (hexane/EtOAc, 3/2) to afford 1.18 g (38%) of (E)-di-tert-butyl (3-(2-(phenyldiazenyl)phenyl)propyl) phosphate (1c-4) as a red oil. 1H NMR (400 MHz, CHLOROFORM-D) δ 7.92 (dd, J = 8.3, 1.4 Hz, 2H), 7.67 (d, J = 7.8 Hz, 1H), 7.55–7.34 (m, 5H), 7.33–7.28 (m, 1H), 4.03 (q, J = 6.4 Hz, 2H), 3.25 (t, J = 7.7 Hz, 2H), 2.06 (quin, J = 7.2 Hz, 2H), 1.47 (s, 18H).
(E)-3-(2-(Phenyldiazenyl)phenyl)propyl phosphate (1c-5).
Trifluoroacetic acid (1.8 mL) was added to a solution of compound 1c-4 (0.65 g, 1.5 mmol) in dry DCM (16 mL). The reaction mixture was stirred for 6 h at room temperature. MeOH (30 mL) was added and the mixture was evaporated. This procedure was repeated several times to remove CF3COOH completely. The residue was dried in vacuo. Water (5 mL) was added to the residue and neutralized with 1 M NaOH. This crude product was purified by using a DEAE Sephadex A-25 column with 0.5 M triethylammonium hydrogencarbonate (TEAB) buffer (pH 8.0) as an eluent at 4 °C. The fractions containing the product were coevaporated with EtOH to remove water and TEAB to afford 0.60 g (96%) of (E)-3-(2-(phenyldiazenyl)phenyl)propyl phosphate (1c-5) (triethyl ammonium salt) as a red oil. 1H NMR (400 MHz, CD3OD) δ 7.91 (dd, J = 8.4, 1.3 Hz, 2H), 7.65 (d, J = 7.9 Hz, 1H), 7.57–7.41 (m, 5H), 7.29 (t, J = 8.4 Hz, 1H), 3.94 (q, J = 6.4 Hz, 2H), 3.26 (t, J = 7.8 Hz, 2H), 3.17 (q, J = 7.3 Hz, 6H), 2.00 (quin, J = 7.1 Hz, 2H), 1.29 (t, J = 7.3 Hz, 9H).
(E)-3-(2-(Phenyldiazenyl)phenyl)propyl triphosphate, azopropylTP (1c).
The triethylammonium salt of compound 1c-5 (0.58 g, 1.4 mmol) was converted into its tributylammonium salt through the addition of tributylamine (1 mL, 4 mmol) in dry MeOH (7 mL). Triethylamine and MeOH were evaporated. The tributylammonium salt was dissolved in dry DMF (12 mL). While stirring, a solution of 1,1′-carbonyldiimidazole (1.2 g, 7.5 mmol) in dry DMF (10 mL) was added under an Ar atmosphere and then the reaction mixture was left for 16 h at room temperature. Excess 1,1′-carbonyldiimidazole was decomposed through the addition of dry MeOH (0.3 mL, 6 mmol) and stirring for 1 h. This solution was added dropwise with mixing to a solution of the tributylammonium salt of pyrophosphate, (prepared from tetrasodium pyrophosphate) (2.0 g, 7.5 mmol) in dry DMF (10 mL). After reacting overnight at room temperature, the mixture was cooled to 0 °C in an ice bath. Cold water (15 mL, 4 °C) was added with mixing and then the pH was adjusted to 7 using 1 M NaOH. The reaction mixture was partitioned between diethyl ether and H2O; the aqueous phase was evaporated with EtOH at 30 °C and dried. The crude product was purified by using the DEAE Sephadex A-25 column with the eluent of a linear gradient of 0.2–1.0 M TEAB solution (over 400 min) at 4 °C. The fractions were checked with ESI MS and the desired fractions were mixed with EtOH and evaporated. The resulting residue was dried in vacuo. The obtained product was dissolved in dry MeOH (1 mL); upon addition of NaI into acetone (1 M, 10 mL) sodium salt of the product was precipitated. The precipitate was washed several times with acetone and lyophilized to afford 0.40 g (50%) sodium salt of compound 1c as a red powder. 1H NMR (400 MHz, D2O) δ 8.00 (dd, J = 7.9, 1.7 Hz, 2H), 7.71–7.54 (m, 6H), 7.46 (t, J = 8.4 Hz, 1H), 4.08 (q, J = 6.4 Hz, 2H), 3.27 (t, J = 7.8 Hz, 2H), 2.03 (quin, J = 7.1 Hz, 2H). 13C NMR (100 MHz, D2O with some drops of CD3OD as a standard) δC 153.25, 151.32, 141.91, 132.66, 132.62, 132.00, 130.54, 128.15, 123.57, 116.66, 67.13, 33.18, 28.16. 31P NMR (160 MHz, D2O (H3PO4 as the external standard)) δP −9.72 (d, J = 19.5 Hz), −10.71 (d, J = 19.7 Hz), −22.84 (t, J = 19.4 Hz). HR-MS (ESI, m/z) calculated for C15H19N2O10P3 [M − H]−: 479.01798; found 479.01855.
Physical measurements
NMR spectroscopy (1H, 13C and 31P) was carried out on a 400 MHz spectrometer (ECX-400, JEOL). Purity of AzoTP derivatives was analyzed using a reversed phase HPLC system (Prominence, Shimadzu). High-resolution mass spectroscopy (HR-MS) experiments were performed by using a Thermo Scientific Exactive mass spectrometer with electrospray ionization. A FDU-2200 (EYELA) lyophilization system was used for freeze-drying. UV-Visible spectrophotometry was carried out on a UV-1800 (Shimadzu) UV-Visible spectrophotometer, in stoppered 1 mm path length quartz cells. cis–trans photoisomerization of AzoTP derivatives was performed by using a 365 nm UV LED (C11924-101, Hamamatsu) and a mercury lamp (Ushio) with an appropriate bandpass filter for 436 nm. Fluorescence imaging of in vitro gliding motility assay was performed in an EMCCD digital camera (DL-604M-0EM-H1, Andor Technology) equipped with an epifluorescence microscope (IX71, Olympus) with a 60×/1.45 NA oil-immersion objective lens (PlanApo, Olympus) and an appropriate excitation filter set for 640 nm light from a mercury lamp.
Preparation of proteins
The kinesin used in this study was a recombinant kinesin consisting of 573 amino acid residues from the N-terminus of a conventional human kinesin. This recombinant kinesin, fused with His-tag at the N-terminus (plasmid; pET 30b), was expressed in E. coli Rossetta (DE3) pLysS and purified through the general method using Ni-NTA-agarose. Purified kinesin was distributed in small aliquots and quickly frozen in liquid N2 and stored at −80 °C until use. Tubulin was purified from porcine brains through two successive cycles of polymerization and depolymerization in the presence of a high-molarity PIPES buffer.46 To prepare fluorescently labelled microtubules, tubulin was labelled with CF®633 Succinimidyl Ester (92133, Biotium). CF®633-microtubules were polymerized by copolymerizing labelled and unlabeled tubulin at a ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 for 60 min at 37 °C and stabilized with paclitaxel.
:4 for 60 min at 37 °C and stabilized with paclitaxel.
Skeletal muscle myosin was purified from chicken pectoralis muscles and heavy meromyosin (HMM) was produced by digestion of myosin using α-chymotrypsin followed by dialysis.47 The HMM was distributed in small aliquots and quickly frozen in liquid N2 and stored at −80 °C until use. F-actin was extracted from acetone powder of rabbit skeletal muscle and prepared by partially modifying the previously reported method48 and labelled with a phalloidin conjugated CF®633 dye (00046T, Biotium) at a 1/1.2 (actin/phalloidin) ratio. The fluorescently labelled actin was stored on ice for up to 4 weeks. Concentrations of HMM and actin were determined from the absorbance at 280 nm using the extinction coefficients of 0.63 and 1.1 for 1 mg ml−1 solutions, respectively.
In vitro microtubule–kinesin gliding motility assay
Microtubule gliding on surface bound kinesin motors in a flow cell was observed using fluorescence microcopy at 25 °C, as described previously.28 A flow chamber was first filled with 3.5 μL solution of kinesin (300 nM) with casein (0.5 mg mL−1) in BRB-80 buffer (pH 6.9; PIPES, 80 mM; MgCl2, 2 mM; EGTA, 1 mM) and incubated at room temperature for 3 min. Then paclitaxel stabilized microtubules (0.25 μM as tubulin dimer) in BRB-80 buffer containing paclitaxel (10 μM) and casein (0.1 mg mL−1) were perfused and incubated at room temperature for 3 min. After washing with 3.5 μL of BRB80 buffer containing paclitaxel (10 μM) and casein (0.1 mg mL−1) they were incubated for 1 min. Then BRB80 buffer containing paclitaxel (10 μM), β-mercaptoethanol (0.14 M), glucose oxidase (0.1 mg mL−1), catalase (20 μg mL−1), glucose (20 mM), casein (0.5 mg mL−1) and energy molecule (AzoTP derivatives, 0.05–2.0 mM) was perfused into the flow chamber. Then the flow cell was subjected to microtubule gliding motility observation under a fluorescence microscope. Microtubule filament gliding was imaged using an epifluorescence microscope based on an IX 70 (Olympus) equipped with a 60×/NA 1.45 oil immersion objective lens (PlanApo, Olympus). Images obtained under the fluorescence microscope were projected onto an EMCCD camera (DL-604M-0EM-H1, Andor Technology). The camera was controlled by the andor solis software platform ver. 4.23.30008.0. The typical frame rate was 1 s and the observation duration was 40–120 s depending on the faster/slower movement of the microtubules. The position of the microtubules was determined manually by mouse clicking on the tip of the filament in the Fiji image processing software package based on ImageJ and tracked typically first and last frames of the observation. The gliding velocity of at least 10 microtubules in randomly chosen microscopic fields was measured in each experiment. The velocity of each filament was determined by a linear least-squares fit, assuming that the filaments move in a straight line. To study whether cis–trans rich state AzoTP derivatives induced microtubule gliding velocity, the flow cell was irradiated with UV 365 nm (0.68 W cm−2) light for 8 s and visible 436 nm (21.7 mW cm−2) light for 25 s, respectively.
In vitro actin–myosin gliding motility assay
Actin filament gliding on nitrocellulose coated surface bound HMM motors in a flow cell was observed using fluorescence microcopy at 23.5 °C, as described previously.29 In brief, 3.5 μL HMM (300 μg ml−1) solution in assay buffer (pH 7.4; HEPES, 100 mM; MgCl2, 4 mM; EGTA, 1 mM; NaCl, 25 mM; DTT, 10 mM) was added to the flow chamber and incubated for 60 seconds. After 60 seconds, unbound HMM molecules were washed by using 3.5 μL assay buffer with BSA (0.5 mg ml−1) and incubated for 3 minutes. Then 3.5 μL of CF®633 phalloidin labelled actin (1.2 μg ml−1) was perfused to the chamber and incubated for 60 seconds. After the incubation 3.5 μL solution of a scavenger cocktail [glucose oxidase (0.1 mg mL−1), catalase (20 μg mL−1), glucose (20 mM)], BSA (0.5 mg ml−1) and AzoTPs/NTPs (0.05–2.0 mM) in assay buffer was perfused to the flow cell. Then the flow cell was subjected to actin filament gliding motility observation under an epifluorescence microscope (IX 70, Olympus) equipped with an EMCCD camera (DL-604M-0EM-H1, Andor Technology). The typical frame rate was 0.5 s and the observation duration was 10–120 s depending on the faster/slower movement of the actin filaments. The gliding velocity of at least 10 actin filaments in randomly chosen microscopic fields was measured in each experiment; however, stationary filaments that did not move at all were excluded from velocity measurements. The gliding velocities were measured using MTrackJ plugin in the Fiji image processing software package based on ImageJ; typically each actin filament was tracked every 0.5 s or 10 s depending on the faster/slower movement of actin filaments.
Computational docking simulation
The computational docking simulation was carried out with the program of CLC Drug Discovery Workbench (QIAGEN, Denmark) using the parameters and scoring function of PLANTS(PLP) score.49 We manually inspected the docking modes with high-scored results.
Kinesin–myosin composite motility assay
The flow cell chamber for microscopic observation was prepared by taping a nitrocellulose coated cover slip (18 × 18 mm) and a glass slide (76 × 26 mm) together with the double-sided tape to make a flow path having a volume of 3–3.5 μL with a height of ca. 100 μm. The photocontrollable study was performed by direct irradiation of UV (365 nm, 0.68 W cm−2) and visible (436 nm, 21.7 mW cm−2) light to the flow chamber. To achieve the kinesin–myosin composite motility assay, the perfusion protocol described below was followed, 3.5 μL HMM (300 μg mL−1) solution in assay buffer (pH 7.4; HEPES, 100 mM; MgCl2, 4 mM; EGTA, 1 mM; NaCl, 25 mM; DTT, 1 mM) was added to the flow chamber and it was incubated for 60 seconds. After 60 seconds, 3.5 μL of kinesin solution containing BSA (kinesin: ca. 300 nM; BSA: 5.0 mg mL−1) in the assay buffer was perfused into the chamber and it was also incubated for 3 min. Next, 3.5 μL solution of fluorescent dye labelled microtubules (0.25 μM as tubulin dimer) containing paclitaxel (10 μM) and BSA (0.5 mg mL−1) in the assay buffer was perfused to the chamber and it was incubated for 60 seconds. Then, 3.5 μL of fluorescent dye labelled actin (1.2 μg ml−1) containing paclitaxel (10 μM) and BSA (0.5 mg mL−1) in assay buffer was perfused to the chamber and it was incubated for 60 seconds. After the incubation, 3.5 μL solution of a scavenger mixture [paclitaxel (10 μM), glucose oxidase (0.1 mg mL−1), catalase (20 μg mL−1), glucose (20 mM), BSA (0.5 mg ml−1), and 1e/ATP (desired concentration)] in the assay buffer was perfused to the flow cell. The flow cell was subjected to microtubule and actin filament motility observation under an epifluorescence microscope (IX 70, Olympus) equipped with an EMCCD camera (DL-604M-0EM-H1, Andor Technology) at 25 °C.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank Ms Miho Yamada for the HR-MS analyses at the OPEN FACILITY in Hokkaido University. This work was supported by Grant-in-Aid for Scientific Research (B) 18H02042 to N. T. and a Grant-in-Aid for Young Scientists (B) KAKENHI Grant Number 16K16635 to K. M. A part of support was also provided by the Akiyama Life Science Foundation, Grants-in-Aid for Regional R&D Proposal-Based Program from Northern Advancement Center for Science & Technology of Hokkaido Japan and Takeda Science Foundation to K. M. We also acknowledge the support from Dynamic Alliance for Open Innovation Bridging Human, Environment and Materials (Five-Star Alliance) of MEXT.
Notes and references
- R. D. Vale and R. A. Milligan, Science, 2000, 288, 88–95 CrossRef CAS PubMed.
- M. Yoshida, E. Muneyuki and T. Hisabori, Nat. Rev. Mol. Cell Biol., 2001, 2, 669–677 CrossRef CAS PubMed.
-
M. Schliwa, Molecular motors, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2003 Search PubMed.
- R. D. Vale, T. S. Reese and M. P. Sheetz, Cell, 1985, 42, 39–50 CrossRef CAS PubMed.
- J. Howard, Annu. Rev. Physiol., 1996, 58, 703–729 CrossRef CAS PubMed.
- V. Mermall, P. L. Post and M. S. Mooseker, Science, 1998, 279, 527–533 CrossRef CAS PubMed.
- S. J. Kron and J. A. Spudich, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 6272–6276 CrossRef CAS.
- A. Goel and V. Vogel, Nat. Nanotechnol., 2008, 3, 465–475 CrossRef CAS PubMed.
- S. Aoyama, M. Shimoike and Y. Hiratsuka, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16408–16413 CrossRef CAS PubMed.
- S. J. Kron and J. A. Spudich, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 6272–6276 CrossRef CAS.
- M. G. L. van den Heuvel, M. P. de Graaff and C. Dekker, Science, 2006, 312, 910–914 CrossRef CAS PubMed.
- M. Uppalapati, Y. M. Huang, T. N. Jackson and W. O. Hancock, Small, 2008, 4, 1371–1381 CrossRef CAS PubMed.
- E. Kim, K. E. Byun, D. S. Choi, D. J. Lee, D. H. Cho, B. Y. Lee, H. Yang, J. Heo, H. J. Chung, S. Seo and S. Hong, Nanotechnology, 2013, 24, 195102–195107 CrossRef PubMed.
- H. Kato, E. Muto, T. Nishizaka, T. Iga, K. Kinosita JR and S. Ishiwata, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9602–9606 CrossRef CAS.
- G. Mihajlović, N. M. Brunet, J. Trbović, P. Xiong, S. von Molnár and P. B. Chase, Appl. Phys. Lett., 2004, 85, 1060–1062 CrossRef.
- L. Ionov, M. Stamm and S. Diez, Nano Lett., 2006, 6, 1982–1987 CrossRef CAS PubMed.
- F. Wang, N. M. Brunet, J. R. Grubich, E. A. Bienkiewicz, T. M. Asbury, L. A. Compton, G. Mihajlović, V. F. Miller and P. B. Chase, J. Biomed. Biotechnol., 2011, 2011, 435271 Search PubMed.
- T. Korten, W. Birnbaum, D. Kuckling and S. Diez, Nano Lett., 2012, 12, 348–353 CrossRef CAS PubMed.
- N. M. Brunet, G. Mihajlović, K. Aledealat, F. Wang, P. Xiong, S. von Molnár and P. B. Chase, J. Biomed. Biotechnol., 2012, 2012, 657523 Search PubMed.
- C. Reuther, R. Tucker, L. Ionov and S. Diez, Nano Lett., 2014, 14, 4050–4057 CrossRef CAS PubMed.
- N. M. Brunet, P. B. Chase, G. Mihajlović and B. Schoffstall, Arch. Biochem. Biophys., 2014, 552–553, 11–20 CrossRef CAS PubMed.
- H. Higuchi, E. Muto, Y. Inoue and T. Yanagida, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4395–4400 CrossRef CAS.
- H. Hess, J. Clemmens, D. Qin, J. Howard and V. Vogel, Nano Lett., 2001, 1, 235–239 CrossRef CAS.
- R. Tucker, P. Katira and H. Hess, Nano Lett., 2008, 8, 221–226 CrossRef CAS PubMed.
- K. R. S. Kumar, T. Kamei, T. Fukaminato and N. Tamaoki, ACS Nano, 2014, 8, 4157–4165 CrossRef CAS PubMed.
- K. R. S. Kumar, A. S. Amrutha and N. Tamaoki, Lab Chip, 2016, 16, 4702–4709 RSC.
- A. S. Amrutha, K. R. S. Kumar, T. Kikukawa and N. Tamaoki, ACS Nano, 2017, 11, 12292–12301 CrossRef CAS PubMed.
- N. Perur, M. Yahara, T. Kamei and N. Tamaoki, Chem. Commun., 2013, 49, 9935–9937 RSC.
- H. M. Menezes, M. J. Islam, M. Takahashi and N. Tamaoki, Org. Biomol. Chem., 2017, 15, 8894–8903 RSC.
- I. K. Lednev, T. Q. Ye, R. E. Hester and J. N. Moore, J. Phys. Chem., 1996, 100, 13338–13341 CrossRef CAS.
- T. Nägele, R. Hoche, W. Zinth and J. Wachtveitl, Chem. Phys. Lett., 1997, 272, 489–495 CrossRef.
- A. Barth, K. Hauser, W. Mäntele, J. E. T. Corrie and D. R. Trentham, J. Am. Chem. Soc., 1995, 117, 10311–10316 CrossRef CAS.
- F. Jon Kull, E. P. Sablin, R. Lau, R. J. Fletterick and R. D. Vale, Nature, 1996, 380, 550–555 CrossRef PubMed.
- A. Weber, J. Gen. Physiol., 1969, 53, 781–791 CrossRef CAS PubMed.
- S. A. Cohn, A. L. Ingold and J. M. Scholey, J. Biol. Chem., 1989, 264, 4290–4297 CAS.
- T. Shimizu, K. Furusawa, S. Ohashi, Y. Y. Toyoshima, M. Okuno, F. Malik and R. D. Vale, J. Cell Biol., 1991, 112, 1189–1197 CrossRef CAS PubMed.
- H. D. White, B. Belknap and W. Jiang, J. Biol. Chem., 1993, 268, 10039–10045 CAS.
- E. Pate, K. Franks-Skiba, H. White and R. Cooke, J. Biol. Chem., 1993, 268, 10046–10053 CAS.
- S. Higashi-Fujime and T. Hozumi, Biochem. Biophys. Res. Commun., 1996, 221, 773–778 CrossRef CAS PubMed.
- S. M. Frisbie, J. M. Chalovich, B. Brenner and L. C. Yu, Biophys. J., 1997, 72, 2255–2261 CrossRef CAS.
- M. Regnier, D. M. Lee and E. Homsher, Biophys. J., 1998, 74, 3044–3058 CrossRef CAS PubMed.
- M. Regnier and E. Homsher, Biophys. J., 1998, 74, 3059–3071 CrossRef CAS PubMed.
- I. Amitani, T. Sakamoto and T. Ando, Biophys. J., 2001, 80, 379–397 CrossRef CAS PubMed.
- L. Farhadi, C. F. Do Rosario, E. P. Debold, A. Baskaran and J. L. Ross, Front. Phys., 2018, 6, 75 CrossRef.
- Usually at higher concentration of AzoTPs (>3.5 mM), the microtubules were detached form kinesin and floating in the flow cell. In case of 1e, the detachment of microtubules was observed at >2.0 mM concentration. Therefore, the microtubule gliding velocity was shown up to 3.0 mM for all AzoTP derivatives except for 1e (up to 2.0 mM).
- M. Castoldi and A. V. Popov, Protein Expression Purif., 2003, 32, 83–88 CrossRef CAS PubMed.
- S. S. Margossian and S. Lowey, Methods Enzymol., 1982, 85, 55–71 CAS.
- J. D. Pardee and J. A. Spudich, Methods Enzymol., 1982, 85, 164–181 CAS.
- O. Knob, T. Stutzle and T. E. Exner, J. Chem. Inf. Model., 2009, 49, 84–96 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S17, Tables S1 and S2, Supplementary movies, and NMR and HR-MS spectra. See DOI: 10.1039/c8ob02714e |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |

 ab,
Kazuya
Matsuo‡
ab,
Halley M.
Menezes
ab,
Masayuki
Takahashi
c,
Hidehiko
Nakagawa
ab,
Kazuya
Matsuo‡
ab,
Halley M.
Menezes
ab,
Masayuki
Takahashi
c,
Hidehiko
Nakagawa