
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cascade reaction based synthetic strategies targeting biologically intriguing indole polycycles
Maria Gessica
Ciulla
a,
Stefan
Zimmermann
ab and
Kamal
Kumar
 *a
*a
aMax-Planck-Institut für Molekulare Physiologie, Abteilung Chemische Biologie, Otto-Hahn-Strasse 11, 44227 Dortmund, Germany. E-mail: kamal.kumar@mpi-dortmund.mpg.de; Tel: +49-231-133-2480
bFakultät Chemie und Chemische Biologie Technische Universität Dortmund, Otto-Hahn-Strasse 6, 44221 Dortmund, Germany
First published on 27th November 2018
Abstract
Indole polycycles are common structural frameworks of biologically intriguing small molecules of natural and synthetic origin and therefore remain interesting and challenging synthetic targets. Cascade reactions wherein a number of reactions occur in a sequential manner in the same reaction apparatus are highly efficient chemical processes which quickly build up molecular complexity. Synthetic approaches based on cascade reactions are highly useful as they tend to avoid multiple reaction work-up steps as well as purifications of all intermediary products. Therefore, in the last decade, a number of cascade reaction based approaches to build various molecular scaffolds of biological interest have been reported. However, a relatively smaller number of cascade reaction based synthetic strategies have targeted the indole polycycles. In this article, we have summarized some interesting cascade reaction based synthesis designs leading to complex indole polycycles including some biologically intriguing and natural product inspired indole frameworks.
 Maria Gessica Ciulla | M. Gessica Ciulla received her MSc degree (2013) from the University of Urbino, Italy. She obtained her Ph.D. in Chemical and Pharmaceutical Sciences in 2016 for her work on the synthesis of carbohydrate analogues and for the design and synthesis of novel cannabinoid receptor ligands. As a guest researcher in Dr Kumar's group in Max Planck Institute of Molecular Physiology (MPI), she was extensively involved in the development of asymmetric synthesis of polycyclic and biologically active indoles. Her research interests include asymmetric synthesis, natural product synthesis and drug discovery. |
 Stefan Zimmermann | Stefan Zimmermann, born in 1989, studied Chemical Biology at the Technical University of Dortmund and completed his Master's thesis under the supervision of K. Kumar. In April 2015, he joined the group of Dr Kumar at the MPI, Dortmund for his PhD studies. His research focuses on the application and development of cycloaddition and annulation reactions in the synthesis of pharmaceutically and biologically relevant core structures. |
 Kamal Kumar | Kamal Kumar was born in Amritsar, India. He studied Pharm. Sciences at Guru Nanak Dev Univ. Amritsar and later completed his Ph.D. in 2000 under the supervision of Prof. M. P. S. Ishar at the same university. He moved to Max Planck Institute of Molecular Physiology, Dortmund in 2004. Since May 2006 he has led a group in the Department of Chemical Biology at the same institute. His research interests include asymmetric cycloaddition/annulation reactions leading to natural product based libraries, cascade reactions, scaffold diversity synthesis, and probing of biological functions with small molecules. |
1. Introduction
Nature's synthetic ability to create highly complex natural products endowed with interesting bioactivities has inspired organic chemists to take up challenging de novo total synthesis studies of natural products (NPs).1–3 However, the multistep and tedious total syntheses are often marred by low efficiency and offer a limited exploration of NP chemical space for biological research. Chemists have therefore resorted to other more efficient synthetic designs and strategies leading to a wider NP-like chemical space for the discovery of bioactive small molecules.4–9 For instance, the synthesis of small molecules based on privileged scaffolds has garnered a great deal of attention from organic and medicinal chemists.10–13A large number of NPs embodying polycyclic indole scaffolds display interesting, potent and wide-ranging biological activities (Fig. 1).14–18 Consequently, in the last decade, a surge in synthesis endeavours targeting polycyclic indoles has been observed.19–27 In a multistep synthesis, even a single low yielding or stereoselectively dissatisfying reaction step can drastically reduce the overall efficiency of the whole process.28–30 A cascade or domino reaction is a chemical transformation wherein a number of reactions (at least two) happen in a sequential manner in the same reaction apparatus and the product of one reaction step is the substrate for the next sequential reaction step.31–34 Cascade reactions rapidly enhance the structural complexity of a desired scaffold. They are economically better choices for multistep synthesis as they increase manifold the efficiency of a synthetic strategy and avoid a number of purifications and reaction work-ups which may otherwise reduce the yield of the products (Fig. 2). Thus, it is not surprising that their development has taken major strides in recent years.35–37
 | ||
| Fig. 1 Biologically active natural indole polycycles. | ||
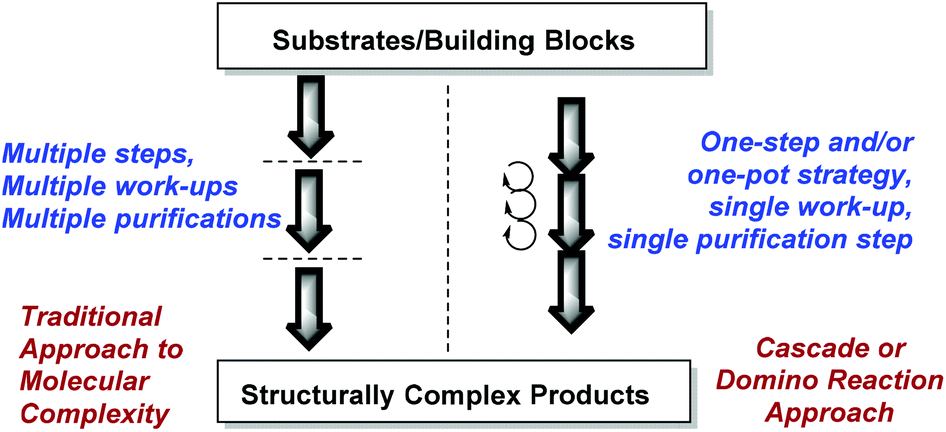 | ||
| Fig. 2 Multistep vs. cascade reaction based synthesis approach to build molecular complexity. | ||
The aim of this review is to present recent and interesting cascade reaction based synthetic strategies leading to indole polycycles including some biologically intriguing and NP-inspired indole frameworks. There are already enlightening reviews published about the concept of cascade and domino reactions38–41 and, therefore, we will not attempt to explain these concepts here. Also, we have focused only on indole polycyclic small molecules, and cascade synthesis targeting polycyclic oxindoles42,43 is not covered in this review article.
The design of a cascade reaction-driven synthesis of a polycyclic indole or for that matter any complex small molecule needs to involve some key reactions which can either generate the whole of a scaffold or an important intermediate leading to the desired molecular architecture. For the synthesis of complex indoles, a number of chemical reactions can play a key role in a cascade reaction design. Such designs may employ either a simple indole substrate or other more basic precursors that generate the indole ring during a cascade reaction sequence in addition to further molecular complexity. The key reactions may include cycloaddition and/or cyclization reactions, transition metal catalysed coupling reactions, or C–H activation routes as well as coinage metal catalysed cascade rearrangements. In order to facilitate an understanding of these key reactions in the cascade synthesis of polycyclic indoles, transition metal free i.e. either uncatalysed or organocatalysed reactions and transition metal-catalysed cascade reactions are discussed in the following in separate sections.
2. Transition metal-free cascade syntheses of indole polycycles
2.1. Cascade syntheses employing dipolar cycloaddition reaction as the key step
Cycloaddition reactions are among the most powerful tools in organic synthesis to generate complex small molecules. In particular, 1,3-dipolar cycloaddition reactions have emerged as immensely useful transformations that can be applied in cascade or domino reaction designs for rapidly building molecular complexity around a desired five-membered heterocycle.44–48 In a number of cases, dipolar cycloaddition reactions deliver products with high yields and in a regio- and stereocontrolled manner. In the following section, some cascade reaction designs making use of dipolar cycloaddition reactions for building indole polycycles are discussed.DNA interacting heterocycles are generally sp2-carbon rich and flat in their structures. However, the positioning of heteroatoms in these heterocycles plays a crucial role in ensuring DNA interaction.49 For generating similar heterocycles, Lauria et al. developed a domino reaction strategy to synthesize indolo-[2,3-e][1,2,3]-triazolo[1,5-a]-pyrimidine (6) from 3-azidoindoles (1, Scheme 1).50 Treatment of acetonitrile (2) with sodium metal generated anion 3, which entered into a dipolar cyclization with the azide moiety to form the intermediate 4. Under the same reaction conditions, the imine moiety in 4 isomerized to enamine and formed the lactam by reacting with the ester moiety on C2 of the indole ring. Thus, domino reactions of azidoindoles and acetonitriles afforded the synthetic entry to annelated indolo[2,3-e][1,2,3]triazolo[1,5-a]pyrimidines (6).
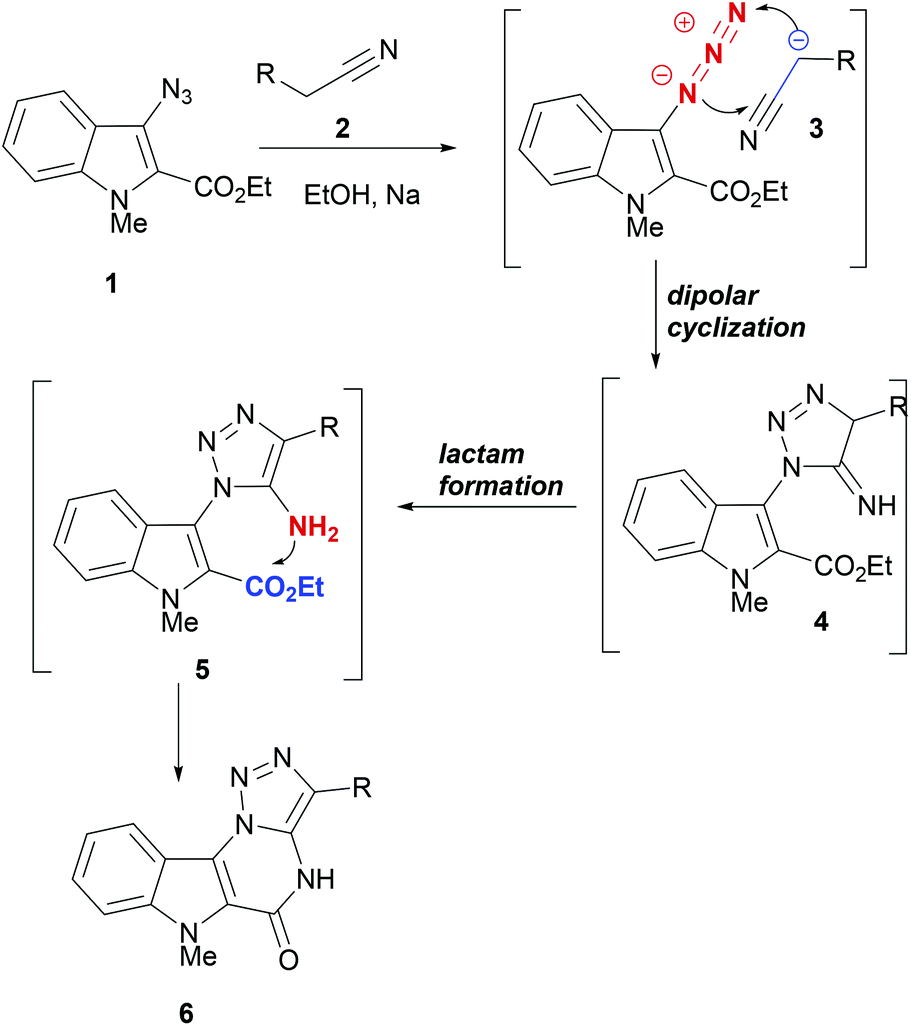 | ||
| Scheme 1 A domino synthesis of indolo[2,3-e][1,2,3]triazolo[1,5-a]pyrimidines. | ||
In docking studies to evaluate the ability of indolopyrimidines 6, with DNA, 6a (R = Ph) was found to have a greater change in free energy of binding (−13.76 kcal mol−1) than the control actinomycin D (−10.37 kcal mol−1). Thus, indolotriazolo-pyrimidine derivatives are capable of making stable complexes with DNA.
A highly useful and elegant cascade reaction strategy to build polycyclic heterocycles is through tandem and dipolar [4 + 2]–[3 + 2] cycloaddition reactions of nitroalkenes (such as oxadienes) with olefins.51 The nitronate products formed in the first cycloaddition reaction represent another 1,3-dipole that can be trapped with a number of dipolarophiles in a cascade reaction manner to deliver structurally complex indoles.52–54
Chataigner and Piettre envisaged that the chemoselectivity in cycloaddition reactions of 2-nitroindoles (7) with olefins will be directed by the LUMO of nitroindole (such as oxadiene) in the inverse electron-demand oxa-Diels–Alder reaction with electron-rich olefins and by the HOMO of nitronate intermediate (11) in the next 1,3-dipolar cycloaddition reaction with electron-poor olefins.55 The sufficiently different electron demands of the two sequential cycloadditions facilitated a selective domino process. However, establishing the right reaction conditions was not straightforward for this multicomponent [4 + 2]–[3 + 2] cascade reaction between nitroindole (7), vinyl ethers (8) and acrylates (9).
It was observed that the cascade reaction worked at room temperature albeit under high pressure in tetrahydrofuran (THF). Interestingly, the [4 + 2] inverse electron-demand cycloaddition was completely endo-selective and totally influenced the facial selectivity of the subsequent [3 + 2] cycloaddition reaction to set up the stereochemistry at the ring-junction. In two operations, a polycyclic diamine (12) featuring a quaternary stereocenter at the ring junction is efficiently generated (Scheme 2).
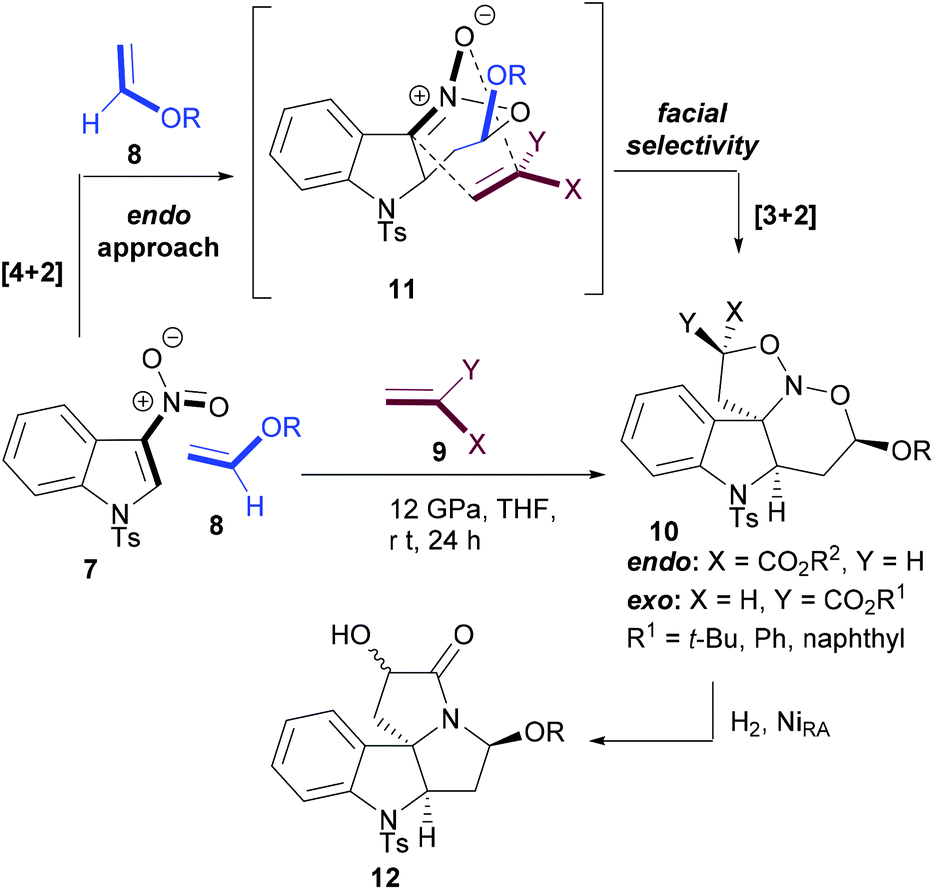 | ||
| Scheme 2 Cascade cycloaddition reactions between N-tosyl-3-nitroindole and electron-rich and -poor olefins. | ||
Boger's group has explored and exploited the tandem intramolecular Diels–Alder/1,3-dipolar cycloaddition reactions of 1,3,4-oxadiazoles in the synthesis of complex NPs and their analogues.56–58 Tryptamine derived 1,3,4-oxadiazoles (13, Scheme 3) supporting an olefin linker were employed in a cascade reaction sequence targeting the synthesis of vindoline based indole derivatives. The oxadiazoles (13) behave as electron-deficient azadienes in the intramolecular Diels–Alder reaction forming the initial [4 + 2]-cycloadducts (14), which lose N2 to generate a carbonyl ylide (15). The latter further reacts with the proximal indole moiety in a 1,3-dipolar cycloaddition reaction to deliver adduct 16 (Scheme 3).59 The reactions were most facile when initiated by an inverse electron-demand Diels–Alder reaction; however, the unactivated or electron-deficient tethered dienophiles efficiently reacted to afford polycyclic indoles in a completely stereoselective fashion to form a single diastereomer of adducts (16). The cascade of cycloadditions constructed three new rings along with the formation of four new C–C bonds. In the cascade process, all six stereocenters characteristic of vindoline NP including three contiguous among the four quaternary centers were set in a single operation. The factors that controlled the stereoselectivity involved the dienophile geometry as well as the exclusive indole endo-[3 + 2] cycloaddition reaction sterically directed to the α-face of the 1,3-dipole.
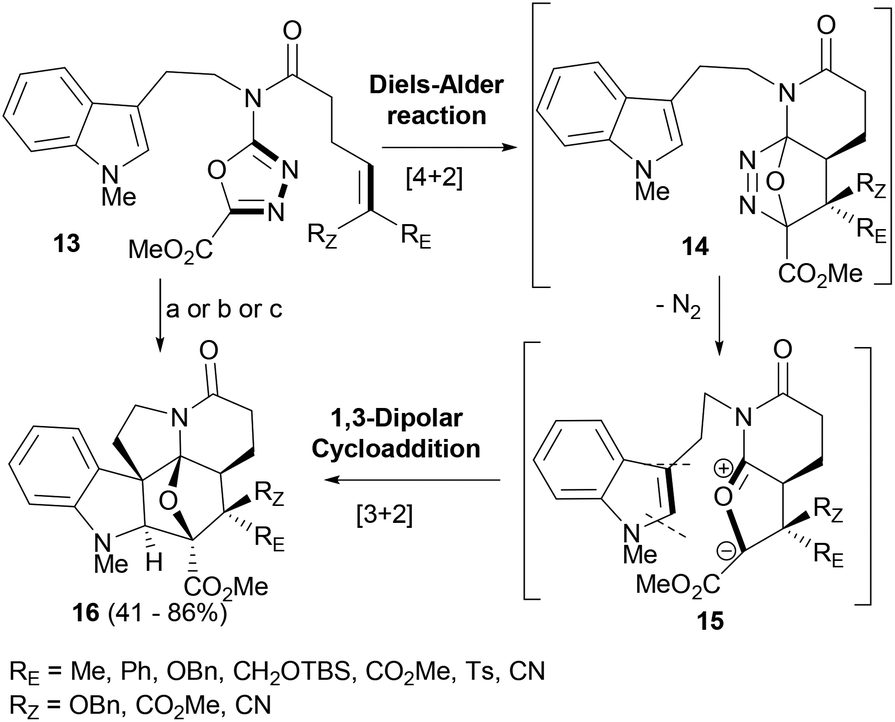 | ||
| Scheme 3 Boger's tandem Diels–Alder/1,3-dipolar tandem cycloaddition reactions to form complex polycyclic indoles. Reaction conditions: (a) o-Cl2C6H4, 180 °C, 3–144 h; (b) TIPB (triisopropylbenzene), 230 °C, 5–96 h; (c) μW, 250 °C, 30 min. | ||
2.2 Cascade syntheses of indole polycycles employing Diels–Alder type cycloaddition reactions
In a number of tandem or cascade reaction designs, Diels–Alder and related cycloaddition reactions forming six-membered rings are rationally employed to construct a desired indole polycycle. Some of these examples are discussed in the following section.Synthetic strategies targeting a combination of two privileged ring-systems are of great interest to organic as well as medicinal chemists because of the novel chemical frameworks the hybrid products offer to interact with difficult protein targets.60–62 Manian et al. developed a cascade reaction based synthesis strategy wherein an initial Knoevenagel condensation of indole-2-carbaldehyde (17) – appended with an internal dienophile (Scheme 4) – with coumarins (18) delivered an intermediate (19), supporting an oxadiene and an olefin in proximity. An intramolecular hetero Diels–Alder reaction ensued and delivered polycyclic indoles (20–21).63
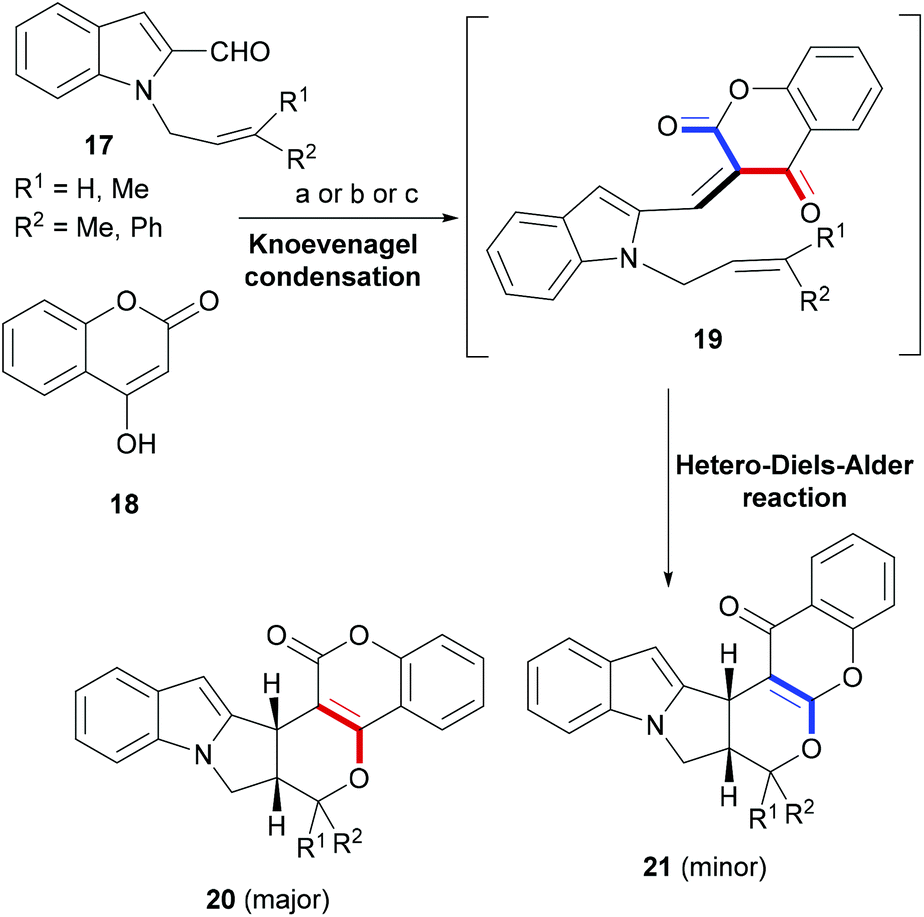 | ||
| Scheme 4 A Knoevenagel condensation–HDA reaction approach to build coumarin-indole hybrids. Reaction conditions: (a) Ethanol/reflux; (b) ethanol microwave; (c) C K-10 montmorillonite clay/microwave. | ||
The cascade reactions were performed using three different conditions. However, in all the cases, the major coumarin–indole (20) was accompanied by a chromone–indole (21) in a range of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 2:1. Nevertheless, the two regioisomeric indoles were easily separable by silica gel column-chromatography and were apparently formed via completely stereoselective hetero-Diels–Alder reactions.
:1 to 2:1. Nevertheless, the two regioisomeric indoles were easily separable by silica gel column-chromatography and were apparently formed via completely stereoselective hetero-Diels–Alder reactions.
In another similar and efficient synthesis using the domino Knoevenagel–hetero-Diels–Alder reaction, hexacyclic indole derivatives were generated (Scheme 5).64 Thus, condensation of thioindoline (22) with O-acrylated salicylaldehydes (23) led to intermediate 24 that underwent an intramolecular oxa-Diels–Alder reaction to deliver the complex indoles 25 (Scheme 5). The highlight of this reaction was that it was performed in water as a solvent and afforded the products in good to excellent yields and with high regio- and stereoselectivity.
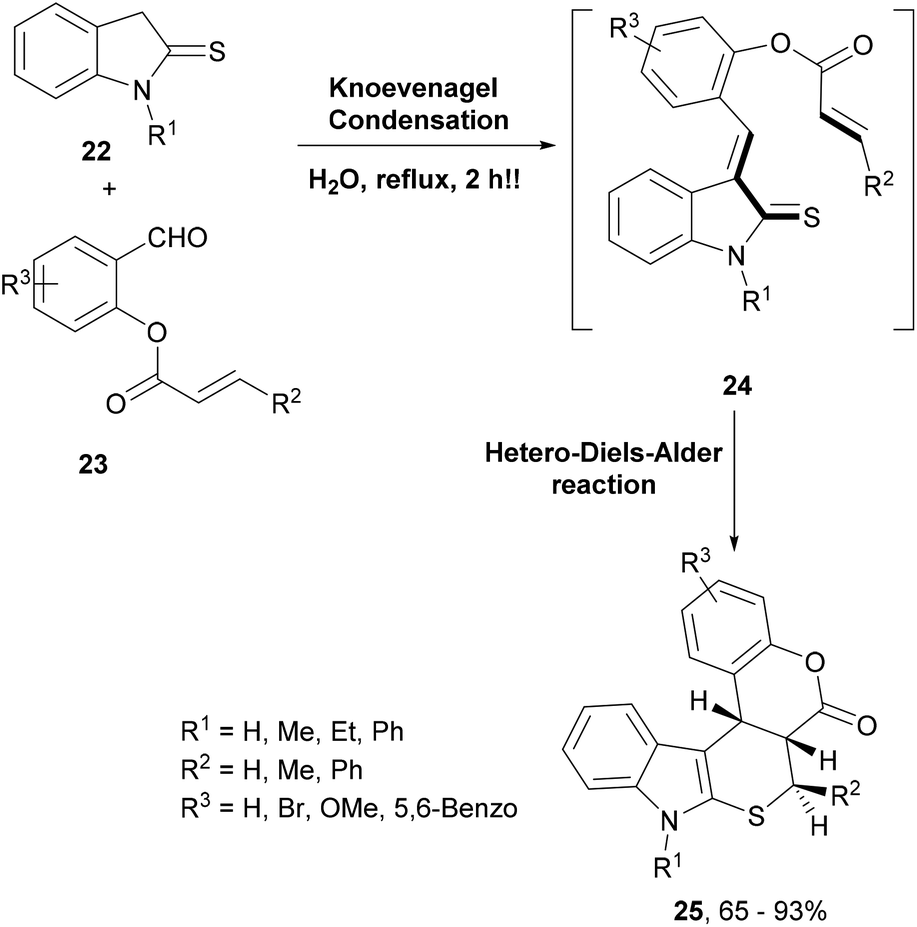 | ||
| Scheme 5 A Knoevenagel condensation–HDA reaction approach to build coumarin-indole hybrids. | ||
In an interesting domino reaction strategy developed by Palacios et al., an aza-Diels–Alder reaction of 2-azadienes (28, Scheme 6), generated in situ by a Wittig reaction, was explored in the synthesis of indole polycycles.65 Notably, as compared to 1-azadienes, only a little is known about the 2-azadiene cycloaddition reactions. Thus, reaction of conjugated phosphazene 26 with N-propargyl-2-indole carbaldehyde (27) in refluxing toluene yielded functionalized heterodiene 28 (Scheme 6). An intramolecular aza-Diels–Alder cycloaddition reaction of 28 led to tetracyclic compound 31. The latter was formed by a facile isomerization–oxidation sequence of the cycloadduct 29.
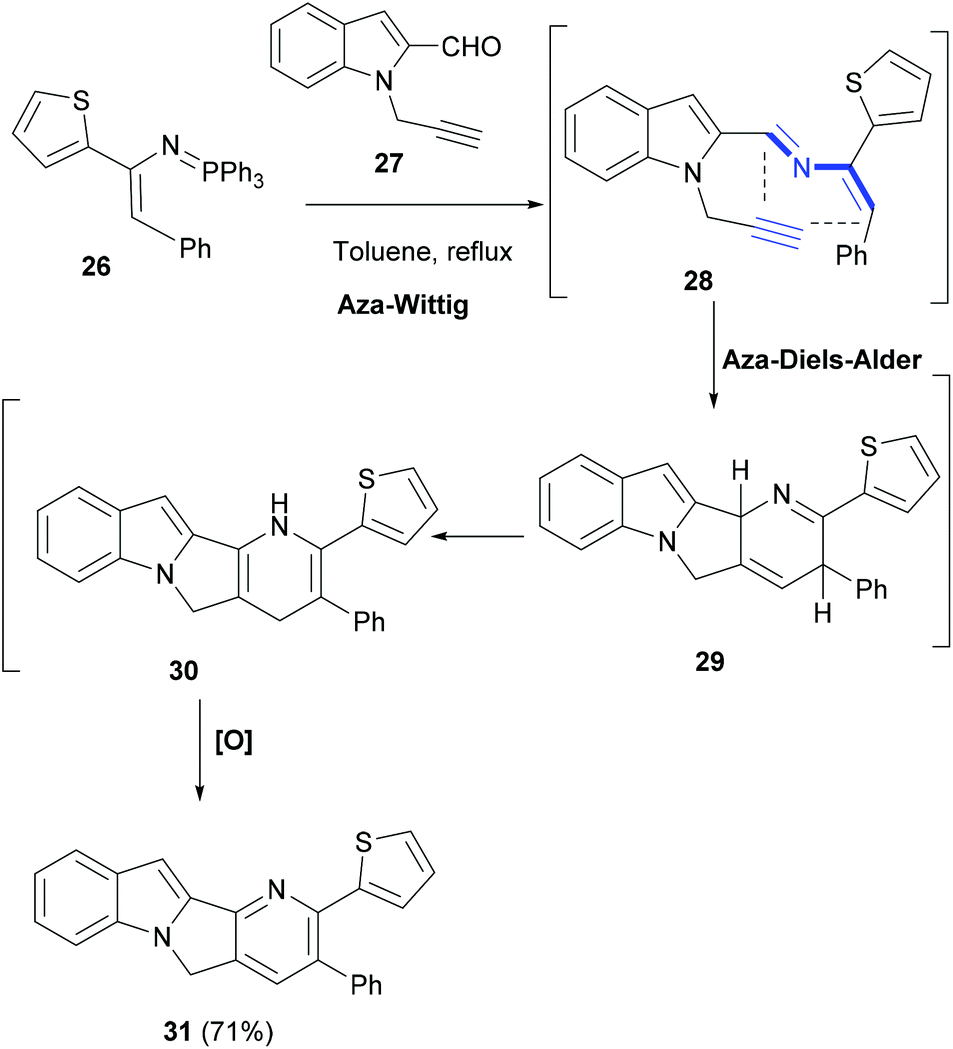 | ||
| Scheme 6 An aza-Wittig-HDA reaction approach to build tetracyclic indoles. | ||
MacMillan and co-workers recently reported an impressive intermolecular–intramolecular cycloaddition–cyclization cascade reaction sequence to construct an important common indole tetracycle (34, Scheme 7) that could afford further efficient synthesis of six structurally related alkaloid NPs.66 The cascade reaction design involved a key catalytic enantioselective iminium-mediated Diels–Alder reaction between the indole diene moiety of the substrate (32) and the activated triple bond of in situ generated catalytic iminium species (35), affording the first intermediate (36). The organocatalysed cycloaddition reaction was followed by selenide elimination (37) and terminated by an acid catalysed aza-Michael addition to deliver the desired indole tetracycle (34) in very high yield. This exquisite cascade reaction sequence in its course formed two C–C bonds, one C–N bond, two new rings and two stereocenters in a highly enantioselective fashion.
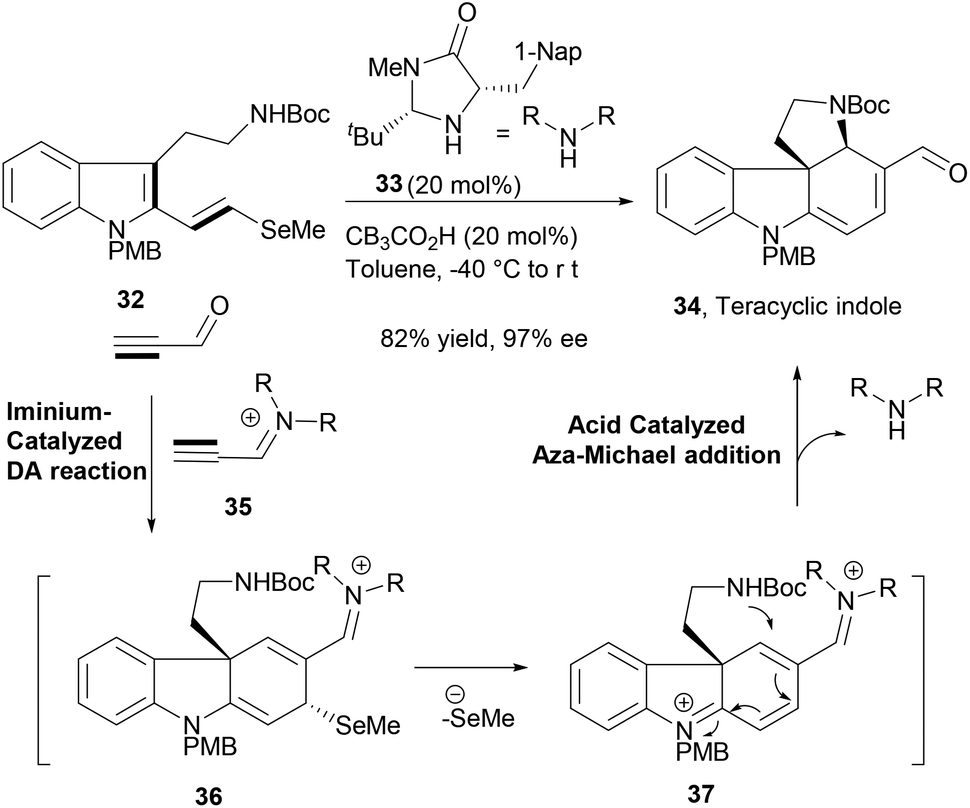 | ||
| Scheme 7 Organocatalysed-domino sequence in the synthesis of alkaloids. | ||
2.3. Cascade reactions exploiting the sequential cyclization reactions to build indole polycycles
The unique chemical space unlocked by the novel indole polycycles may offer possible interactions with difficult biological targets and therefore can be prodigiously productive in biological as well as biomedical research. A cyclization step naturally forms a cyclic structure and can be rationally introduced in a cascade reaction design to form indole polycyclic frameworks. For instance, Moghaddam et al. developed a simple yet very effective synthetic strategy of using two different alkylation steps between indolin-2-thiones (38) and N-alkylquinolinium salts (39) under basic reaction conditions to form the complex indole-annulated pentacyclic indolylhydro-quinoline derivatives 40 (Scheme 8).67 A set of more than twenty compounds (40), formed in high yields, was generated in an easy and efficient manner.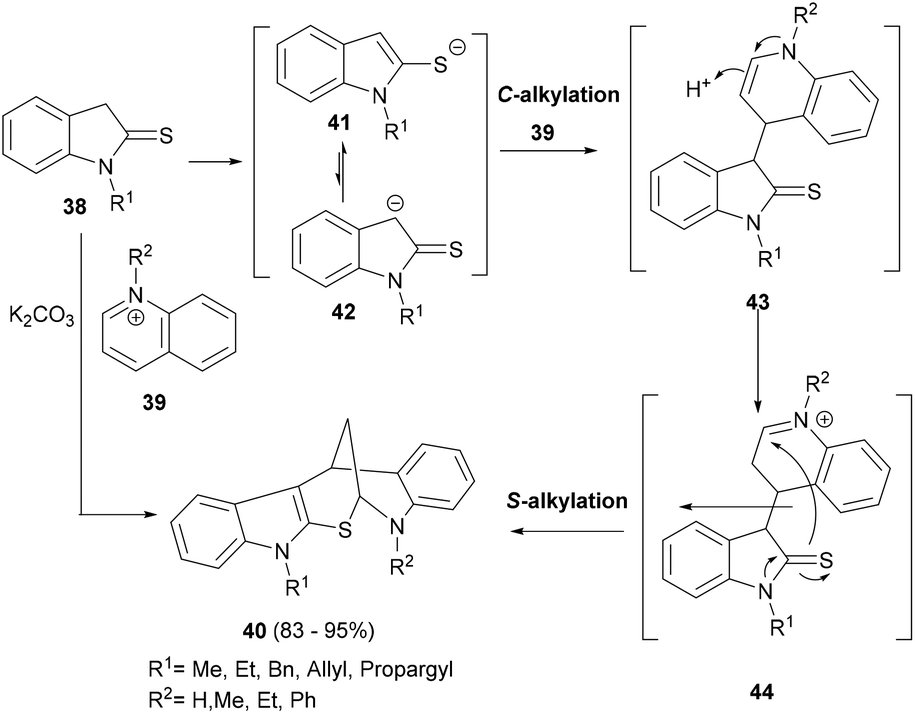 | ||
| Scheme 8 Cascade synthesis of polycyclic indolylhydroquinoline derivatives. | ||
Mechanistically, a plausible C-alkylation of anion 42 at C-4 of the quinolinium salt occurs under basic condition and forms the enamine 43. A second S-alkylation of the thiaimide moiety of thioindoline occurs after the enamine isomerization formed animinium moiety in 44, leading to the formation of pentacyclic indole 40 (Scheme 8).
Arya and co-workers explored a similar sequential cyclization approach by strategically placing internal nucleophiles and the Michael acceptor moieties in their substrates to afford different indole ring-systems.68 For instance, indoline 45 had two Michael acceptors and an aza-nucleophile for an intramolecular cyclization (Scheme 9). The cyclization required the activation of Michael acceptors and was performed by trimethylsilyl triflate (TMSOTf) affording a mixture of 46 and 47 – the tetracyclic indoles with two additional six-membered rings – in a ratio of 3:1. Plausibly, the bulky isobutyl group induced the cyclization to occur through the pathway shown in Scheme 9a. When the substrates lacking the bulky tert-butyl group (48) were exposed to TMSOTf, two isomers of tetracyclic indoles featuring additional functionalized 5- and 7-membered rings (50) were formed in a 2:1 ratio. A transition state 49 that strongly disfavoured the interaction between the side chain and an electron-deficient olefin plausibly led to the observed stereochemistry in 50 (Scheme 9b).
 | ||
| Scheme 9 Cascade synthesis of tetracyclic indoles (47 and 50). | ||
Organocatalytic transformations have proved their excellence in constructing highly complex small molecules in an enantioselective fashion. The design of the substrates is rather of greater significance in cascade reactions involving key organocatalytic steps. Cai et al. could successfully assemble the tetracyclic indoles (52) from indolyl methyl enones (51) through a novel asymmetric intramolecular Michael/Mannich cascade reaction catalysed by a quinine-derived primary amine (53, Scheme 10).69 The iminium-catalysed intramolecular conjugated addition of the indolyl enones 51 generated the spirocyclic indolenine intermediates (55–56). The tetracyclic product 52 was produced via the intramolecular Mannich reaction by enamine catalysis, thereby affording enantiopure tetracyclic indoles bearing multiple chiral centers.
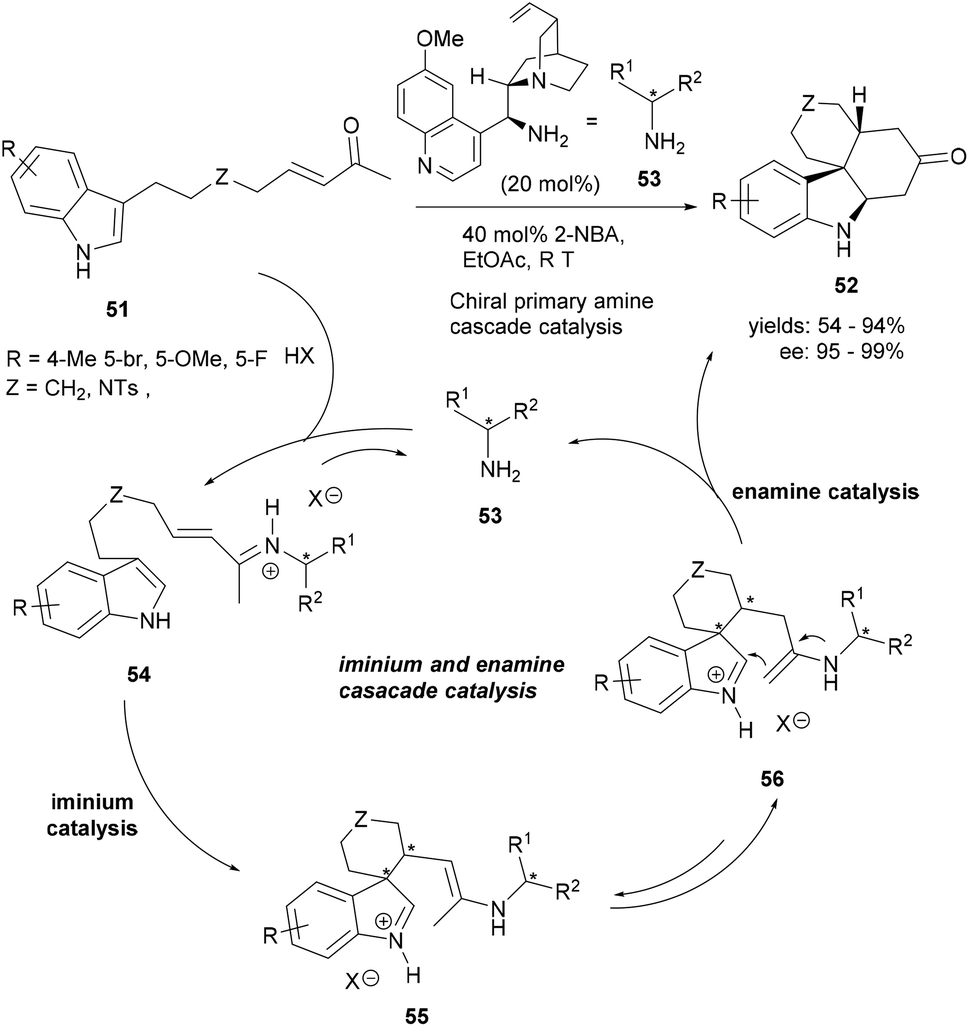 | ||
| Scheme 10 Synthesis of tetracyclic indoles (52) through enantioselective Michael/Mannich polycyclization cascade 55. | ||
In recent years, a number of reports focusing on the activation of C–H bonds and making useful applications of this chemistry have appeared.69–71 However, reactions that lead to C–H bond functionalization through redox neutral processes and wherein the C–H bond to be functionalized serves as a hydride source for a pendant acceptor moiety have only rarely been explored.72,73 Once the hydride transfer has occurred, the reduced and oxidized portions of the molecule may reassemble into a new ring system. Haibach et al. developed this redox neutral cascade approach to build polycyclic azepinoindoles (59, Scheme 11).74 In their strategy, indoles (57) reacted with aminobenzaldehydes (58) in an unprecedented cascade reaction that proceeded via a condensation (60), a 1,5-hydride shift (61) and finally ring-closure sequence, leading to complex indoles (59). Polycyclic azepinoindoles and related compounds were obtained in a single step and in good to excellent yields.
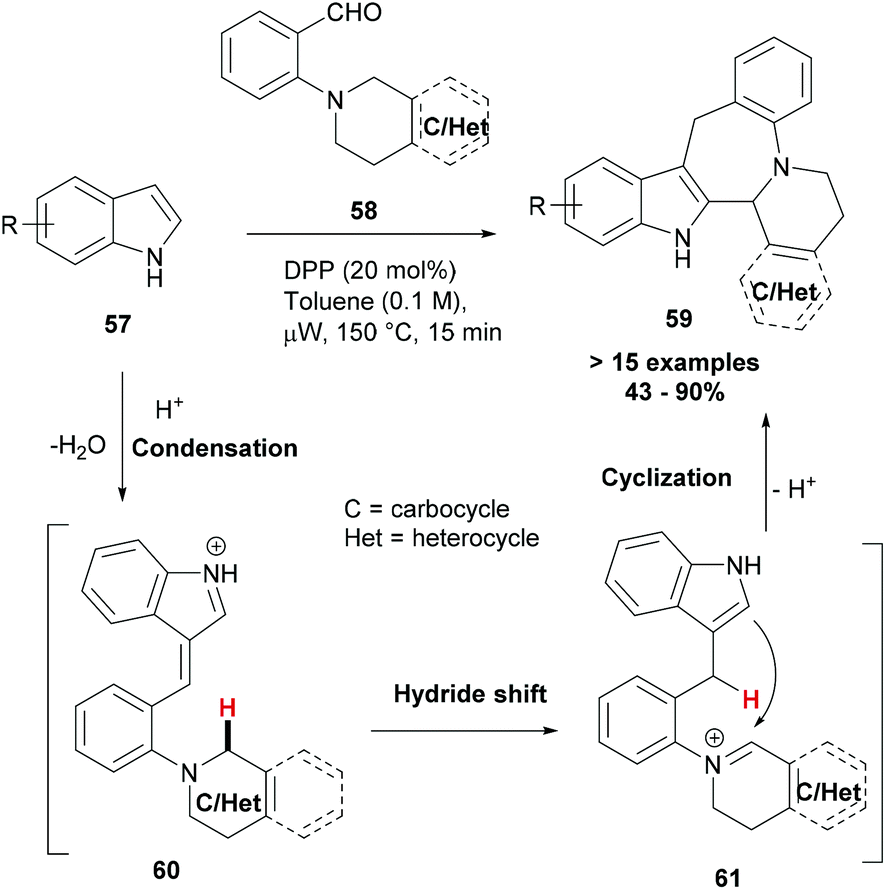 | ||
| Scheme 11 Indole polycycles via redox-neutral annulation cascades. | ||
Isocyanide-based reactions are amongst the widely used chemical tools in multi-component chemistry and the most common examples are the Passerini and the Ugi reactions.75,76 Expanding the versatility of isocyanide based cascade reactions, Wang et al. reported a 2-isocyanoethylindole (62) based chemoselective domino reaction leading to polycyclic spiroindoline derivatives.77 The reaction of 2-isocyanoethyl-indoles and gem-diactivated olefins (63) led to the polycyclic spiroindoline derivatives (64) in EtOH under reflux conditions. While the isocyanide attacked the olefin and generated a stabilized carbanion in the intermediate 65, the second nucleophilic cyclization happened by intramolecular addition of indole to form the spiro-ring intermediate (66). To the latter indolinium cation, the stabilized carbanion was added to finally deliver the product 64 in high yields where enamine isomerization had already occurred (Scheme 12).
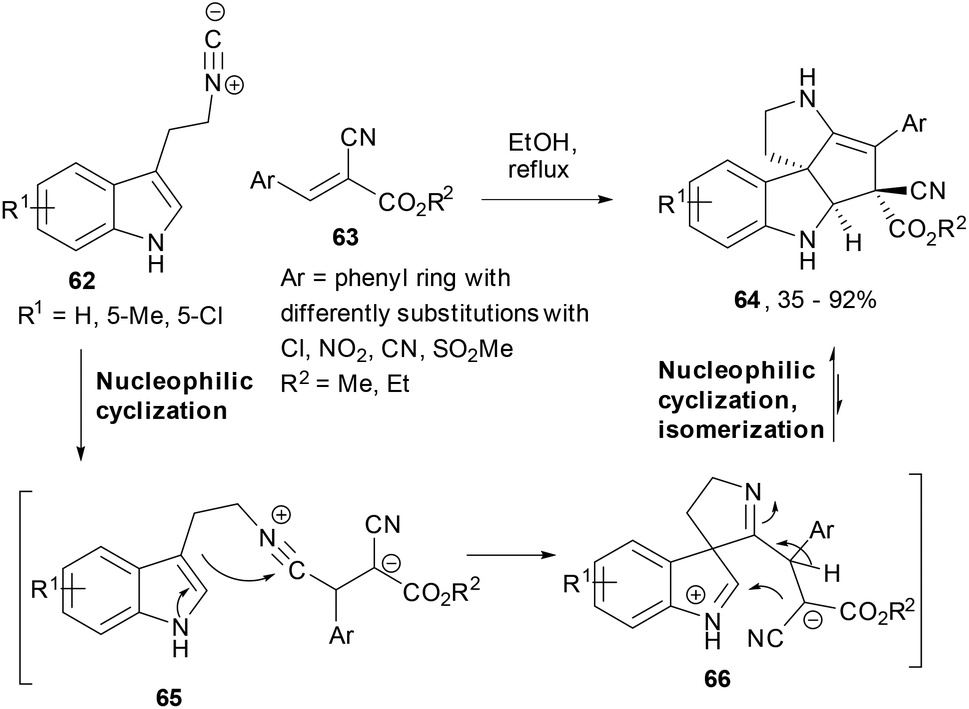 | ||
| Scheme 12 An isocyanide-driven cascade synthesis of spiroindolines. | ||
Synthesis and biological exploration of NP-inspired small molecules is one of the most significant approaches to identify functionally intriguing novel chemotypes.8 However, a cascade reaction based strategy often requires the precursors to be appropriately decorated with functional groups so that a designed reaction sequence can occur. This often requires multistep synthesis of the substrates themselves, thereby limiting the utility and reducing the efficiency of the cascade synthesis. Waldmann, Kumar and co-workers reported a long one-pot cascade reaction sequence that used all commercially available substrates and could very efficiently generate indoloquinolizine framework based small molecules.78,79 The target class of molecules was inspired by the structure of yohimbine indole alkaloids and the synthesis design by the biosynthesis of monoterpene indole alkaloids. The 12-step long cascade reaction sequence was proved by isolation and characterization of some key intermediates (in boxes, Scheme 13). Thus, to a mixture of 3-formylchromone (67), acetylene-dicarboxylates (68) and catalytic triphenylphosphine at high temperature were slowly added tryptamine (72) and camphorsulphonic acid and the mixture was further stirred for 5–30 minutes leading to the formation of tetrahydroindolo[2,3-a]quinolizines (84) in very good yields (Scheme 13).
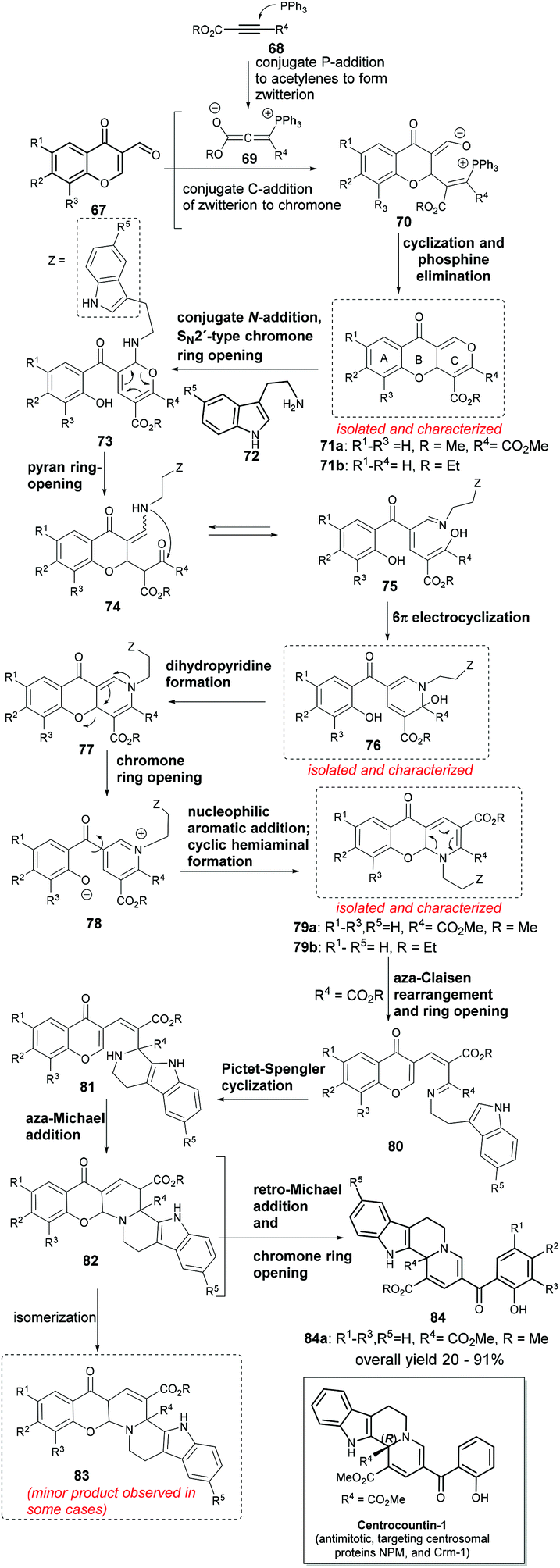 | ||
| Scheme 13 Cascade synthesis of antimitotic centrocountins. | ||
The cascade reaction sequence began with the addition of phosphine to acetylenedicarboxylates, forming phospha-zwitterion (69) that underwent conjugated addition to the C2-position of the chromone (67) and formed intermediate 70, which upon cyclization and phosphine elimination formed the first isolable intermediate – the tricyclic benzopyran (71).80 An SN2′-addition of tryptamine (72) to pyran-ring (71) led to the hemiaminal 73, which rearranged into triene (75). The latter underwent a 6π-electrocyclization and formed another cyclic hemiaminal (76). Intramolecular addition of phenol and elimination of water in 76 formed the tricyclic dihydropyridine (77). The latter is a push–pull system (cf. 71) and further rearranged to another isolable tricyclic hemiaminal 79 through pyridinium internal salt (78). The tricyclic hemiaminal (79b, R4 = H) is stable i.e. when the reaction begins with alkyl propiolate, it ends at 79b. However, with R4 = CO2R (79a), the energy barrier was lowered and this facilitated an aza-Claisen rearrangement which led to the intermediate 80 having tryptamine in proximity to the imino ester moiety. In the presence of an acid, a Pictet–Spengler cyclization occurred, forming a secondary amine (81), which was added to the highly conjugated ester leading to a hexacyclic intermediate (82). The latter underwent a retro-Michael addition and finally chromone ring-opening to yield the tetrahydroindolo[2,3-a]quinolizines (84).
Interestingly, this set of indole NP-inspired small molecules delivered potent mitotic inhibitors active against a number of cancer cell lines. Further chemical biology investigation revealed that the active molecules were targeting centrosomal proteins nucleophosmine (NPM) and Exportin 1 (Crm-1, from chromosomal maintenance 1) and therefore were named centrocountins. Centrocountin-1 is the only probe molecule known to bind NPM in a reversible non-covalent manner and can be used to as a chemical tool to unravel the functions of centrosomal proteins.
3. Transition metal-catalysed cascade synthesis of indole polycycles
Transition metal-catalysed reactions are arguably the most efficient and indispensable chemical tools for the formation of new carbon–carbon bonds that are used in academic and industrial labs. A number of diverse methods now exist to couple sp-, sp2- and sp3-carbon nucleophiles with aryl or alkenyl electrophiles.81–84 Transition metal catalysts like palladium, nickel, copper as well as rhodium metal salts offer enormous possibility to build molecular complexity and diversity.20,71,85–87 Over the years, the development of more effective metal complexes as well as novel ligands to direct chemo-, regio- and stereoselective reactions has further enhanced the power of organic chemistry to effectively build complex small molecules, including NPs and their analogues. Coinage metal complexes of gold, silver and platinum have reinvigorated the chemistry of very different sets of substrates, like enynes, and have further enriched the organic chemistry reaction toolbox. In the last decade, a number of exquisite cascade reaction routes to polycyclic indoles have been realized using transition metal catalysis.20,32,88–90 We observed that the majority of these cascade reactions exploit alkyne substrates to form indole polycycles and exploit different modes of catalytic transformations. Therefore, in the following sections, we have categorized the transition metal-catalysed cascade synthesis of indole polycycles based on non-alkyne and alkyne substrates employed in the cascade reactions.3.1 Transition metal-catalysed cascade synthesis of indole polycycles employing non-alkyne substrates
Centrocountin molecules (cf. Scheme 13) have only one stereogenic center and only R-enantiomer was active. We were curios to find if other substitutions on this stereogenic center may influence the bioactivity of the centrocountins. However, the cascade synthesis of centrocountins worked only with acetylene dicarboxylates (Scheme 13) and therefore it was not possible to replace the ester moiety. In order to replace the ester group on the stereogenic center in the indoloquinolizines, the same group developed another two-step cascade synthesis route to centrocountin analogues as well as simplified analogues where the indole ring was replaced by a phenyl ring. To this end, a novel asymmetric inverse electron-demand hetero-Diels–Alder (HDA) reaction between cyclic imines (85) and chromone dienes (86) was developed.91 In this first case of an asymmetric HDA reaction employing electron-poor carbadienes, Zn–Binol based catalyst served well to induce high enantiomeric excess in the resulting indoloquinolizines (87, Scheme 14). Thus, a two-step cascade reaction sequence of a HDA reaction (89) followed by retro-Michael and chromone ring-opening afforded the tetrahydroindolo[2,3-a]quinolizines wherein the stereogenic center had either a proton or an alkyl group (instead of ester as in centrocountin-1). A more simplified and potent analogue which was, however, less stable than centrocountin-1 was identified from this collection.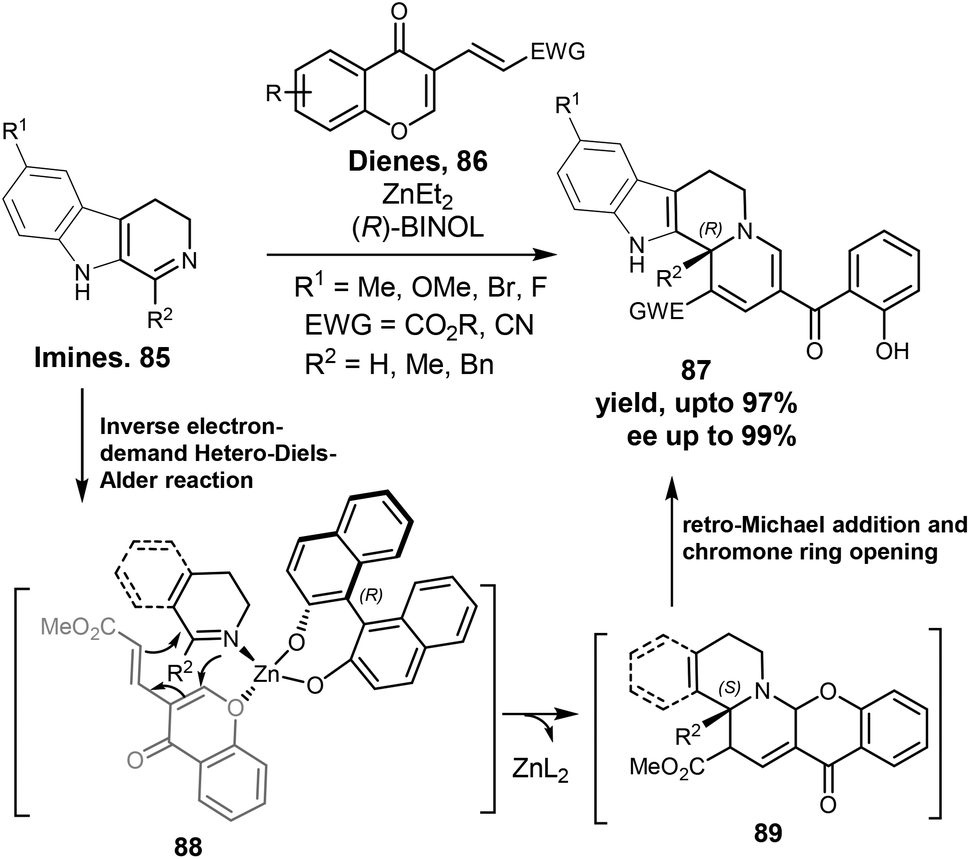 | ||
| Scheme 14 Cascade synthesis of centrocountin analogues with key inverse electron-demand hetero-Diels–Alder reactions. | ||
A number of biologically active NPs (e.g. paspaline, 90) and synthetic molecules (e.g. MK-0524 91, a prostaglandin D2 receptor antagonist) embody the cyclopenta[b]indole ring system (Scheme 15). Yokosaka et al. designed an indole based substrate – a secondary alcohol (92) that was linked to a strategically placed cinnamyl unit.92 Activation of 92 with trifluoroacetic acid or a catalytic amount of a transition metal Lewis acid as a promoter led to the elimination of water and generated a conjugated indolinium cation (93). The first cyclization of the cascade occurred by the addition of a double bond to the extended carbocation through an intramolecular ene-type addition forming another cationic intermediate (94) stabilized by the aromatic ring. Addition of nucleophilic indole to the latter cation then formed the fused-polycyclic cyclopenta[b]indoles (95, Scheme 15). Three different conditions were optimized to induce the cascade sequence efficiently. The cascade method effectively delivered structurally diverse polycyclic cyclopenta[b]indole derivatives (for instance, 96–98) including an eight-member ring-fused product (98) in moderate to excellent yield.
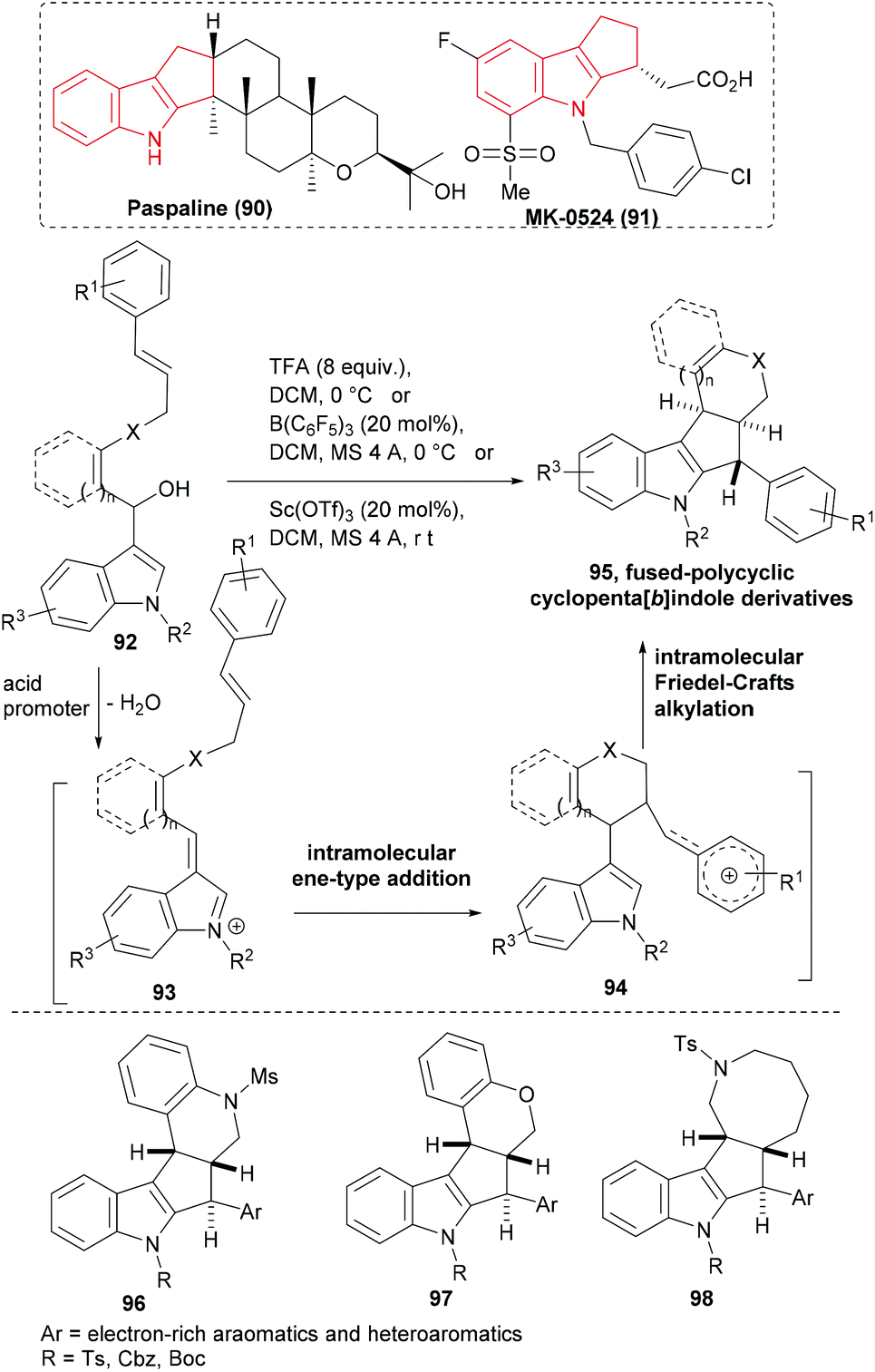 | ||
| Scheme 15 Acid-promoted cascade cyclization to build cyclopenta[b]indoles. | ||
C2–C3 unsubstituted indoles themselves can be exploited as good substrates in palladium catalysed C–H activation or cyclization strategies to synthesise more complex indoles. Wang et al.21 used N-benzyl-substituted indoles (99) in this strategy and developed a tandem fourfold C–H activation to concisely synthesize fused tetracyclic indoles (102) in moderate to good yields (Scheme 16). A combination of copper and palladium catalysts was required for the selective cyclization cascade sequence to work efficiently. Authors suggested that this one-pot process might first involve the hetero-arylation of indoles with various N-heteroarenes, such as caffeine, xanthine etc. occurring at the C3 position (101). In the next step, the nitrogen of heteroarenes (usually N3) directs C2–H bond activation of indoles to undergo an intramolecular oxidative C–H/C–H cross-coupling between the indole C2 position and the benzene ring of the benzyl group that is tethered at the N-indole moiety, affording an indole tetracycle (102, Scheme 16).
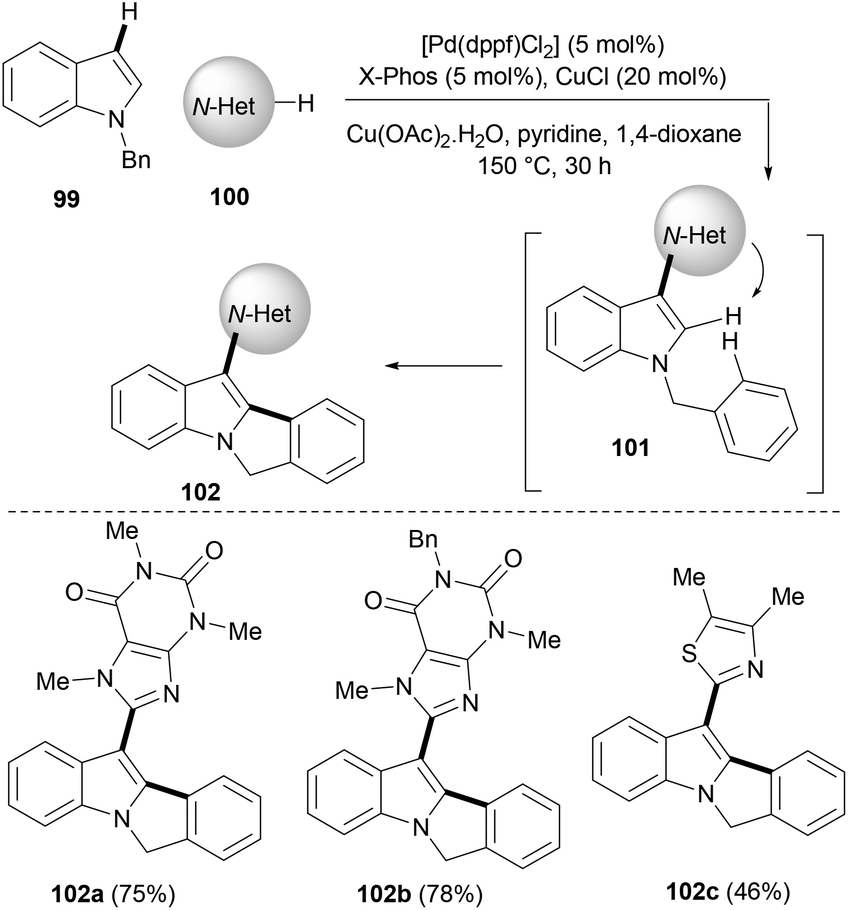 | ||
| Scheme 16 A palladium-catalysed tandem cyclization/C–H functionalization of alkynes to polycyclic indoles. | ||
Indoles often can add to Michael acceptors from their C-3 position as C-nucleophiles.93 Having a Michael acceptor attached to the indole ring via a linker can enhance the possibility of such a Michael addition happening in an intramolecular fashion. This possibility was realized by Xiao and co-workers in their elegant cascade reaction94 wherein the Lewis acid ruthenium catalyst (107) did not only mediate the cross metathesis of the substrate indole (103) with conjugated aldehydes or ketones as well as acrylates (104–106) to build the desired indole supporting an α,β-unsaturated carbonyl moiety (108) but also the hydroarylation as the second domino step to induce the cyclization and form the tricyclic indole (109, Scheme 17). In the case of acrylates, 10 mol% of BF3·Et2O had to be added to complete the intramolecular hydroarylation. Thus, a single catalyst facilitated two mechanistically distinct transformations to generate indole tricyclic small molecules.
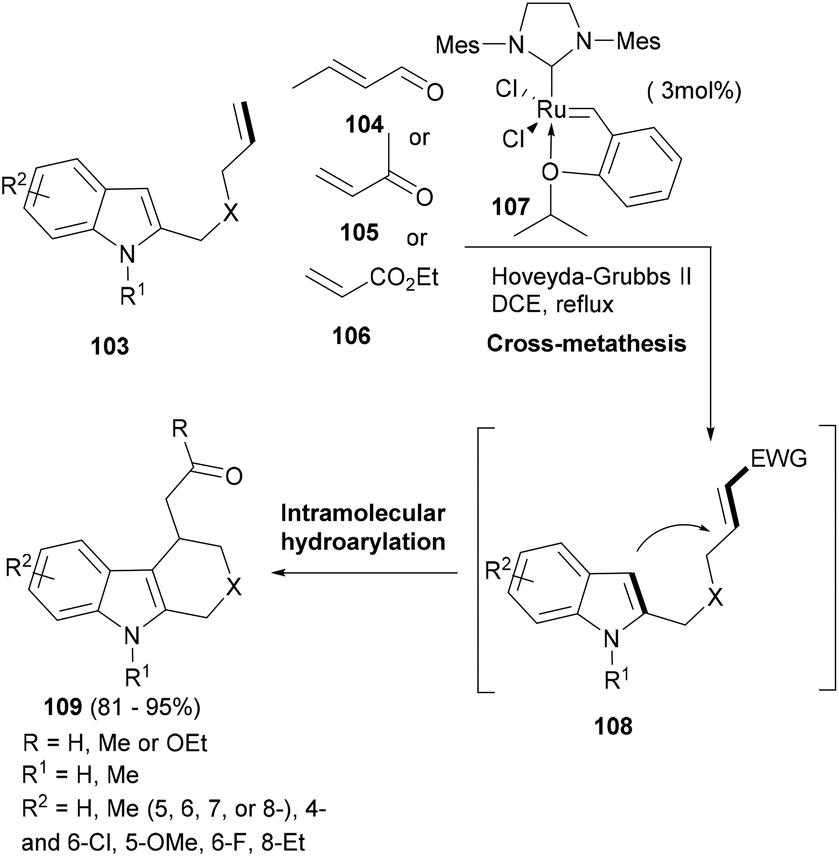 | ||
| Scheme 17 A cross-metathesis driven cascade synthesis of tricyclic indoles. | ||
Padwa and co-workers developed a synthetic strategy similar to Boger's tandem cycloaddition reactions to address the total synthesis of indole NP, aspidophytine.95 An α-diazo imide 110 was designed to generate a push–pull dipole 111. Thus, treatment of 110 with Rh2(OAc)4 generated a rhodium carbenoid species that was trapped by the imido carbonyl group to form carbonyl ylide dipole 111. An intramolecular cycloaddition of the dipole with the tethered indolyl group afforded the cycloadduct 112 in 97% yield (Scheme 18a). The complex indole polycycle 112 in a further ten-step synthesis delivered the NP aspidophytine (113, Scheme 18b).
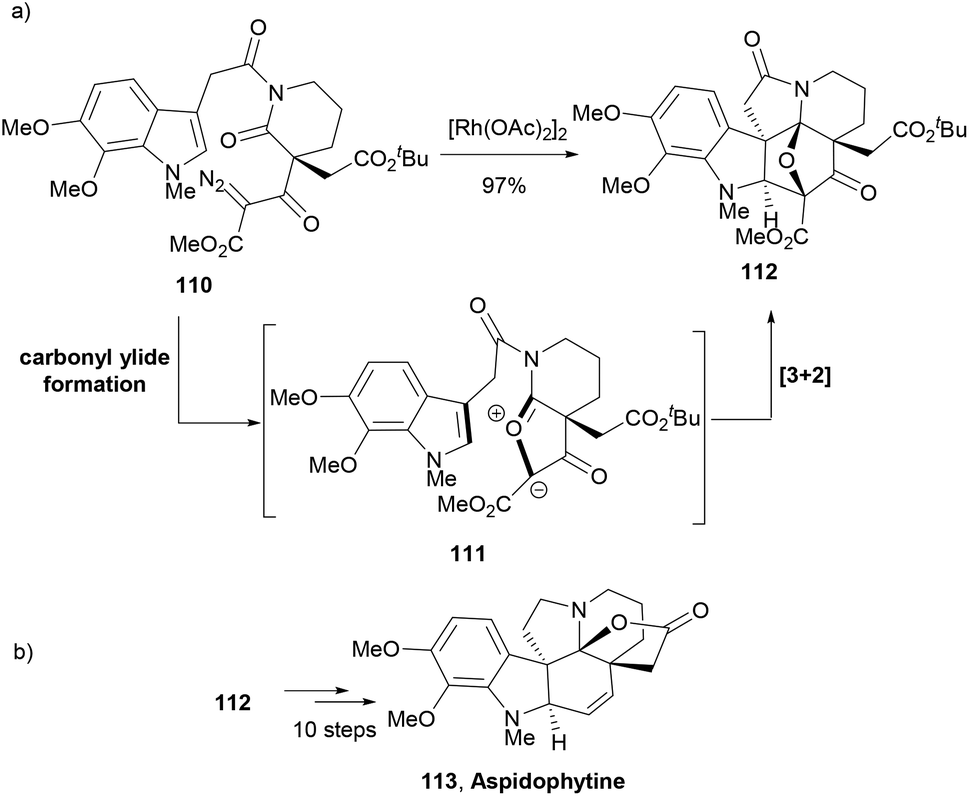 | ||
| Scheme 18 Padwa's cascade approach to the key indole 112 leading to NP aspidophytine (113). | ||
3.2 Transition metal-catalysed cascade synthesis of indole polycycles using alkyne substrates
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) failed to realise acetylene insertion and instead intramolecular arylation ensued, leading to indenone (116) through intermediates 117–118 (Scheme 19).
O) failed to realise acetylene insertion and instead intramolecular arylation ensued, leading to indenone (116) through intermediates 117–118 (Scheme 19).
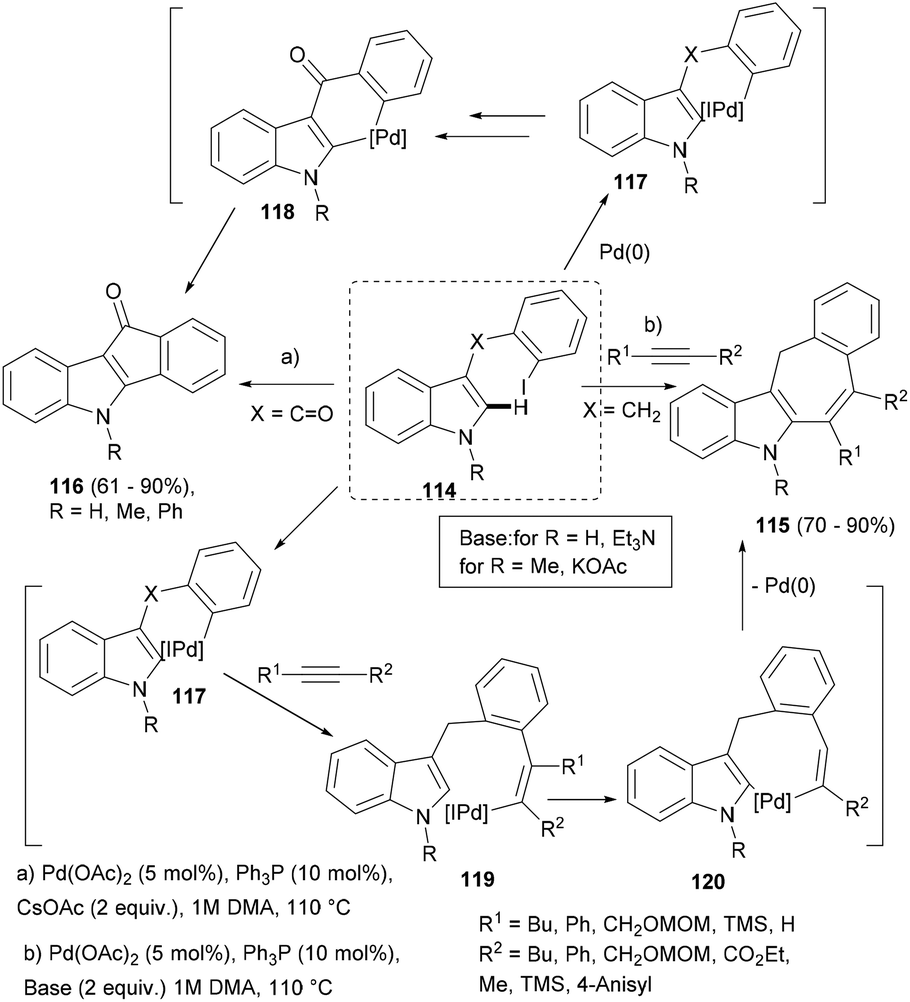 | ||
| Scheme 19 Palladium-catalysed intramolecular ortho-alkylation/direct arylation sequence to polycyclic indoles. | ||
Even the unprotected indole substrate could be successfully transformed into indenone in high yield. However, when the benzyl substrate (114, X = CH2) was used as a substrate in the presence of substituted acetylenes, the desired indoles possessing seven-membered rings (115) were obtained (Scheme 19). Interestingly, for NMe-substituted indole substrates, the cascade carbopalladation–annulation reaction worked only in the presence of KOAc as a base. Thus, under two different optimized reaction conditions, two polycyclic indole scaffolds (115–116) were constructed, supporting different substitutions.
In 2006, Liu and co-workers97 reported a one-step method to construct a variety of polycyclic indoles with a palladium-catalysed intramolecular indolization of 2-chloroanilines (121, Scheme 20) bearing tethered acetylenes. The reaction worked nicely with the 1,1′-bis(ditertbutylphosphino)ferrocene (DtBPF) ligand and at high temperature in a polar solvent, yielding an indole scaffold fused to five- to seven-membered rings (122). Authors proposed that the reaction begins with the formation of the Pd-containing zwitterion 124 by oxidative addition of Pd(0) to 121 (via123) followed by the formation of the bicyclic palladacycle 125 by syn amidopalladation of the acetylene into 124 and finally the reductive elimination of the 125 to afford the indole ring (122, Scheme 20).
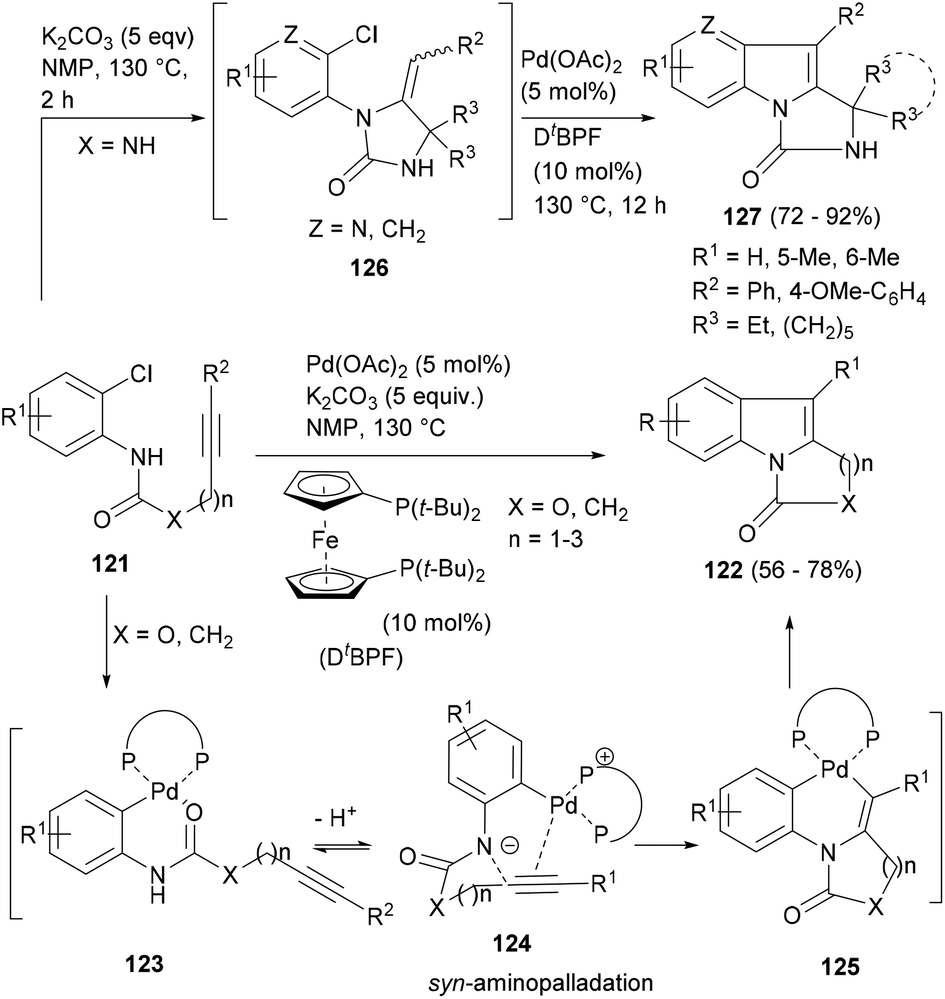 | ||
| Scheme 20 A tandem hydroamination–palladium-catalysed indolization sequence to polycyclic indoles. | ||
Although this reaction worked satisfactorily in making cyclic amides and carbamates (X = O, CH2), the yields for the products with the urea moiety (X = NH) were disappointingly low. Lu and co-workers98 found another solution for this challenge and reported a robust one-pot process that worked through a consecutive sequence of base mediated hydroaminations of the substrates (121) to afford intermediates 126 which upon palladium-catalysed annulation delivered the indole polycycles (127, Scheme 20) in very high yields. The tandem process also afforded a number of aza-indole polycycles.
Werz's group99 made exquisite use of a domino reaction wherein a formal anti-carbopalladation100 is followed by a β-silyl-directed Heck reaction to provide a key intermediate to NP Lysergol. Thus, treatment of the designed substrate (128) with PdCl2 and Xphos ligand in the polar N,N-dimethyl-acetamide (DMA) solvent afforded the desired product (129) wherein two six-membered rings of the ergot scaffold were formed in a completely stereospecific manner (Scheme 21).
 | ||
| Scheme 21 A palladium-catalysed cascade synthesis of a key indole intermediate (122) used in the synthesis of (+)-Lysergol. | ||
The palladium(II)-catalysed functionalization of alkynes can form multiple carbon–carbon/carbon–heteroatom bonds in one step and due to its broader functional-group compatibility and air- and moisture-tolerance, it has emerged as a powerful tool in synthetic chemistry. The group of Liang101 developed an interesting tandem approach for palladium catalysed cyclization/C–H functionalization of two alkynes to build a series of polycyclic functionalized indoles. Employing oxidative reaction conditions (Scheme 22) that could regenerate the Pd(II) from Pd(0), differently substituted acetylenes reacted with o-(1-alkynyl)-anilines (130) to afford indole tetracycles (131). The cascade reaction tolerated a range of internal alkynes bearing synthetically useful functional groups. When alkyl substituted alkynes were used, the reaction observed a good regioselectivity and a single product was obtained (Scheme 22). Mechanistically, a 5-endo-dig anti-addition of the tethered N,N-dimethylaniline (130) to the palladium activated triple bond affords intermediate 133 that gets demethylated by the pivolate anion to form key intermediate 134. Insertion of the latter into the less hindered alkyne (Rs) resulted in vinylic palladium(II) intermediate 135. Arylation of Pd(II) species to phenyl units afforded the seven-membered palladacycle 136. A reductive elimination in the latter generated the cyclic product (131) and a Pd(0) complex which was reoxidized to the Pd(II) species by copper salt and O2 for the next catalytic cycle.
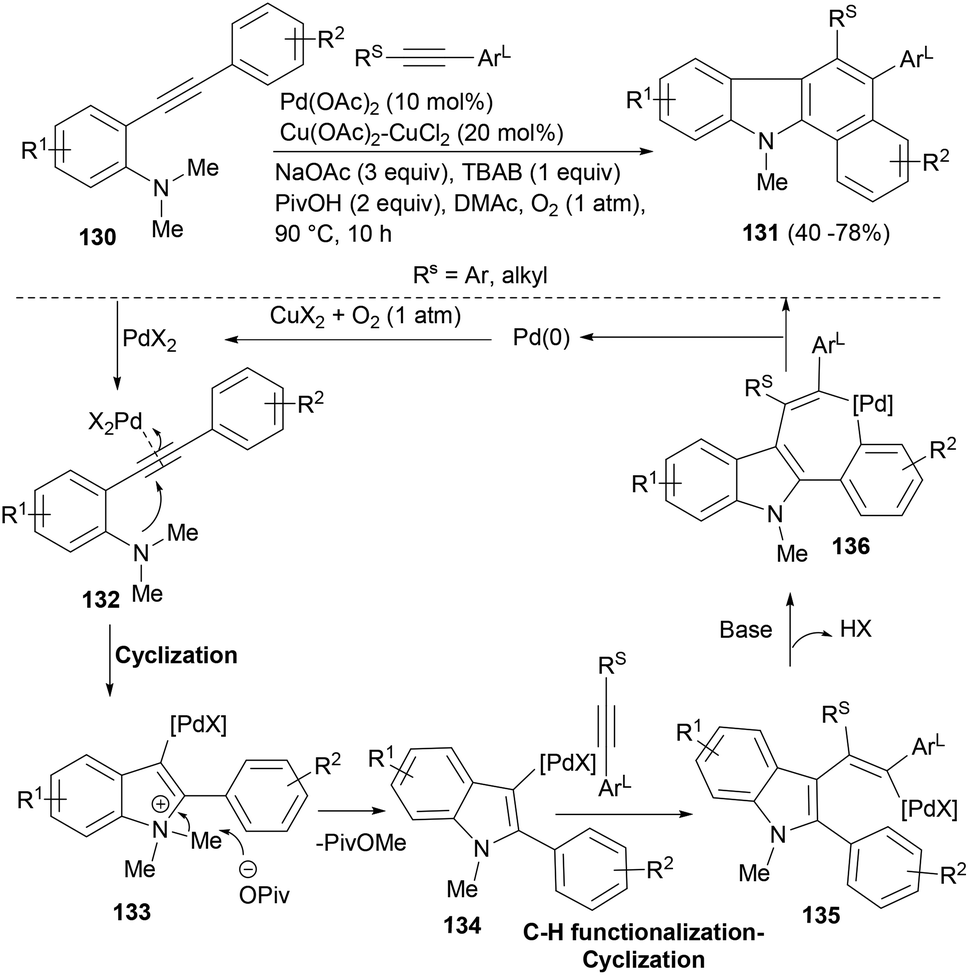 | ||
| Scheme 22 A palladium-catalysed tandem cyclization/C–H functionalization of alkynes to polycyclic indoles. | ||
One of the research themes in our group is to develop efficient synthetic access to the core scaffolds of NPs so that a compound set based on these privileged structures can be quickly built up for biological screening.111–113 In this regard, a silver-catalysed cascade reaction sequence was developed affording different indole polycyclic scaffold classes in an efficient one-pot process. The cascade reaction design was based on the formation of an imine (140) from aniline with a pendant nucleophile (137) and acetylenic benzaldehyde (138) which would undergo cycloisomerization upon silver mediated activation of the alkyne to yield the isoquinolinium intermediate (141).114 A nucleophilic attack of the pendant 1,3-dicarbonyl on this cation forms the intermediate 142, which under optimized reaction conditions quickly decarboxylates to form indolo[2,1-a]isoquinolines (139, Scheme 23a). The cascade reaction worked nicely albeit under microwave heating and using lutidine as a base and delivered the desired adducts in very high yields. Moreover, the strategy also could be used to employ different heterocyclic aldehydes to form the corresponding indoles (143).
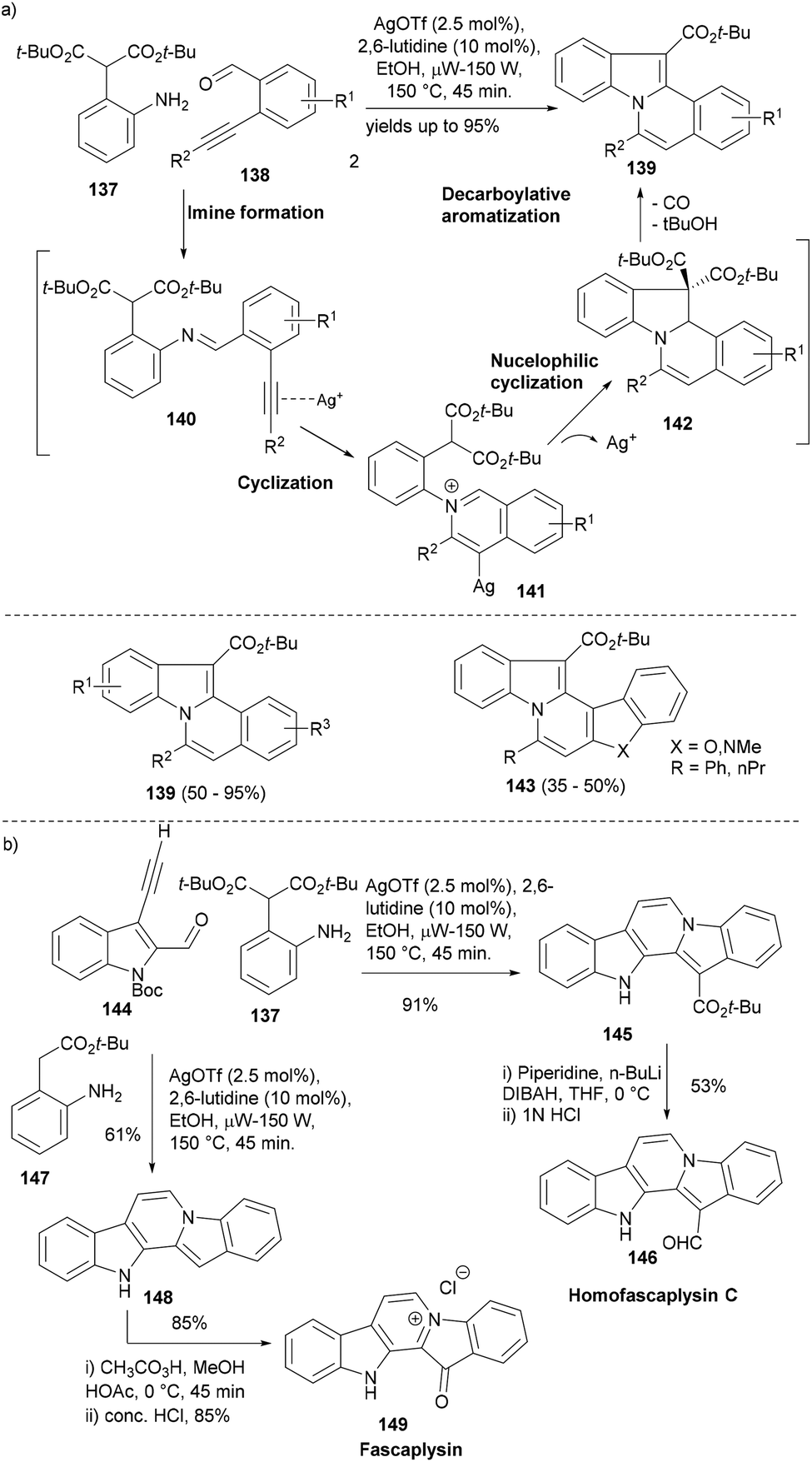 | ||
| Scheme 23 (a) A silver-catalysed cascade synthesis of indolo[2,1-a]isoquinolines and (b) application of the same cascade reaction in the synthesis of indole NPs. | ||
The silver mediated cascade synthesis was later employed to provide very concise access to indole NPs, homofascaplysin C 146 and fascaplysin 149 (Scheme 23b). The microwave assisted silver catalysed cascade cyclization of Boc-protected 3-ethynyl-indole-2-carbaldehyde (144) with aniline 137 yielded the pentacyclic core 145 in high yield. The latter was transformed into Homofascaplysin (146) in a further two steps. The silver catalysed cascade reaction of 144 also worked well with aniline 147 to form the unsubstituted pentacyclic core 148 that in a further two steps led to anticancer NP fascaplysin (149, Scheme 23b).
In recent years, a number of active gold-catalysts have been developed for the synthesis of heterocyclic scaffolds. Despite the challenges in controlling chemo-reactivity and regioselectivity issues in gold catalysed reactions as well as the purification issues due to the hydrophobic nature of the products formed and the substrates used, gold catalysis could provide a sustainable and green chemistry approach to heterocyclic scaffolds. In one such case, Feng et al. reported a gold-catalysed synthesis of fused polycyclic indoles (152, Scheme 24) from alkynoic acids (150) and substituted 2-(1H-indol-1-yl)alkyl amines (151) and in water under microwave irradiation.
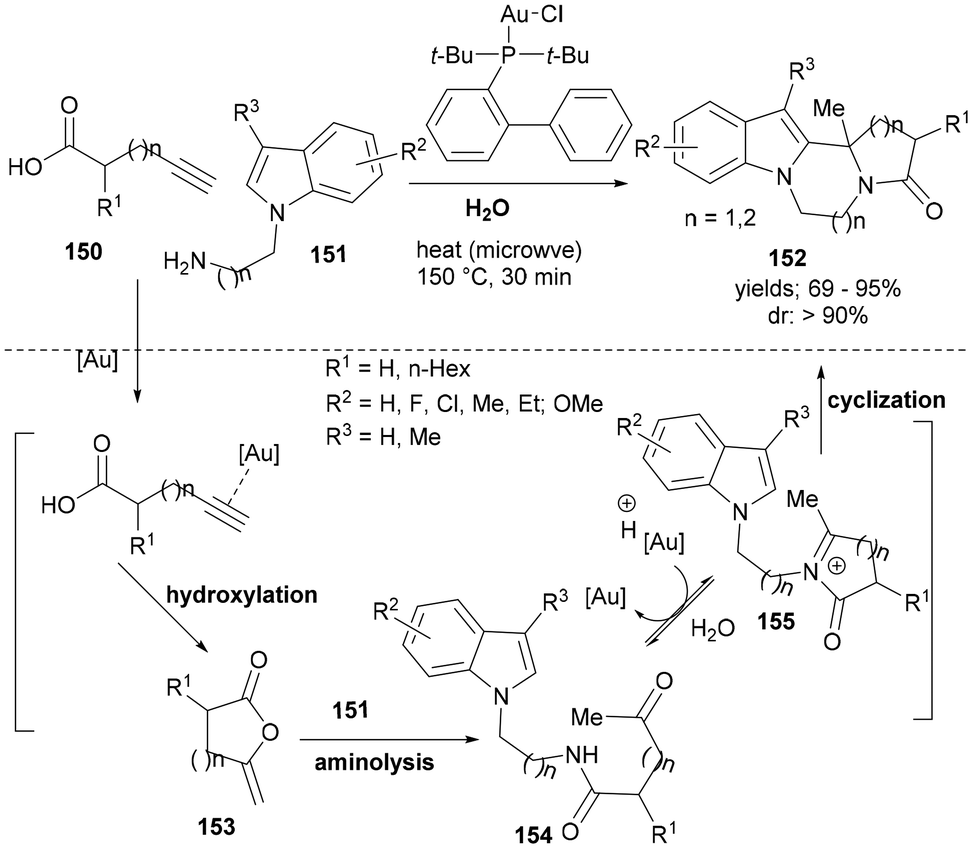 | ||
| Scheme 24 A one-pot tandem synthesis of various fused polycyclic indoles. | ||
The cascade synthesis was proposed to begin with a gold(I) catalysed intramolecular cyclization of alkynoic acid to form enol–lactone intermediate 153. A subsequent aminolysis with 151 formed 154. Under the optimized reaction conditions and using chloro[(1,1′-bi-phenyl-2-yl)di-tert-butylphosphine]-gold(I) as a catalyst, the resulting keto-amide 154 afforded the N-acyliminium ion (155) for the final nucleophilic attack by the C2 indoles to form the final product (152) in high to excellent yields and with very good diastereoselectivity (Scheme 24).
Among the synthetic approaches targeting polycyclic indoles, gold-catalysed cyclization/annulation of indole/ynes have proved their versatility due to their high efficiency and wider scope of reaction. For instance, Xie et al.115 developed an efficient cascade reaction based synthesis to form indole polycycles by using gold-catalysed electrophilic cyclization of enynones. In their reaction designs, diynone substrate i.e. 1,2-bis(alkynyl)-2-en-1-ones (156) under the influence of gold activation formed the carbocationic intermediate (158) following the intramolecular cyclization of alkynones and invited the first C3-attack of the indole (99) to generate furanyl indole intermediate (159) supporting an alkyne moiety. Another gold activation of the alkyne brought the C2-nucleophilic cyclization to furnish a substituted dihydrocyclohepta[b]-indole (157, Scheme 25). Silver triflate did not provide good yields and NaAuCl4·2H2O was found to be the best catalyst for this double annulation cascade affording indole polycycles in appreciable yields.
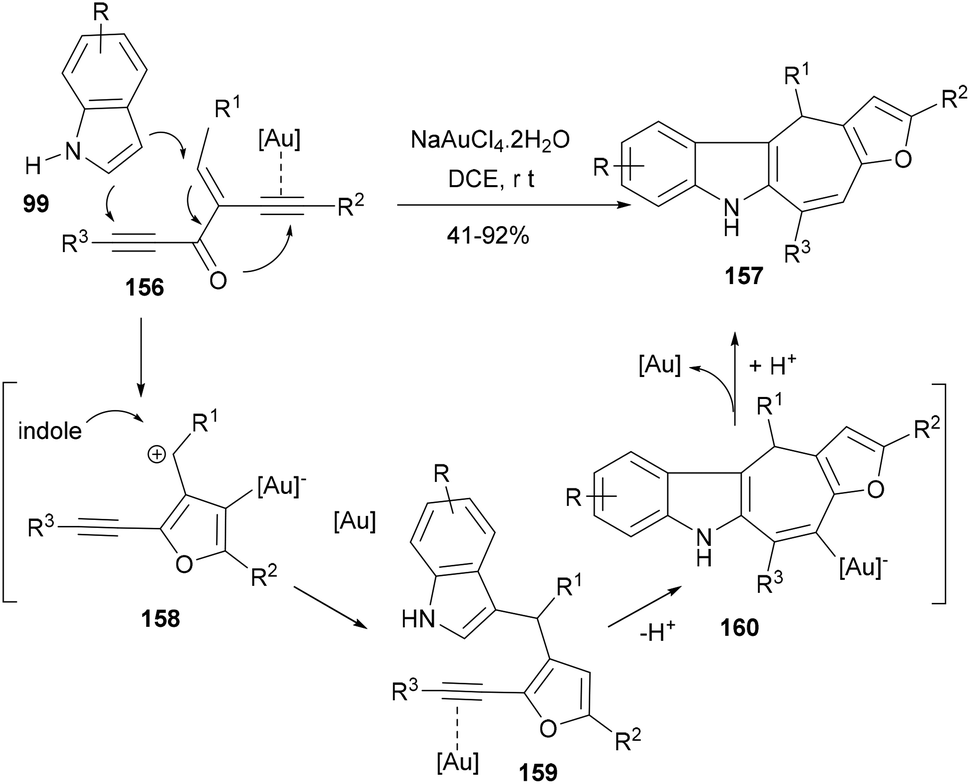 | ||
| Scheme 25 A one-pot synthesis of indole-fused scaffolds via gold-catalysed tandem annulation. | ||
Polycyclic indolines represent an important class of heterocycles and these scaffolds often mark their presence in biologically active NPs. Indoles having an appended acetylene moiety via different linkers can be used as general substrates (161, Scheme 26) for the synthesis of polycyclic indolines (163–164) via gold mediated tandem cyclization reactions. The first reaction in this cascade sequence is always the gold Lewis acid activation of the triple bond and an initial attack of C3-indole as a nucleophile resulting in an intermediate with an iminium indole moiety (162). The endo or exo mode of cyclization depends on the linker length as well as the gold complexes employed as catalyst and thus to some extent can be optimized. The presence of another nucleophile either on the indole ring (mode b) or on the linker having an acetylene moiety (mode a) brings in different modes of cyclization and consequently differently ring-fused indoline products (163–164). Liu et al.116 used the alkynylindole 165 in their gold catalysed tandem cyclization approach to tetracyclic indoline (166, Scheme 26). The reaction condition screening revealed that cationic gold(I) species and antimony hexafluoride as the counterion could provide the product (166) as single diastereomers in very high yields. For successful reactions, the indole required an electron-withdrawing substitution on its nitrogen. Importantly, the nucleophile for the second cyclization could be varied from alcohol to amines, thus yielding different heterocycles fused to indoline small molecules.
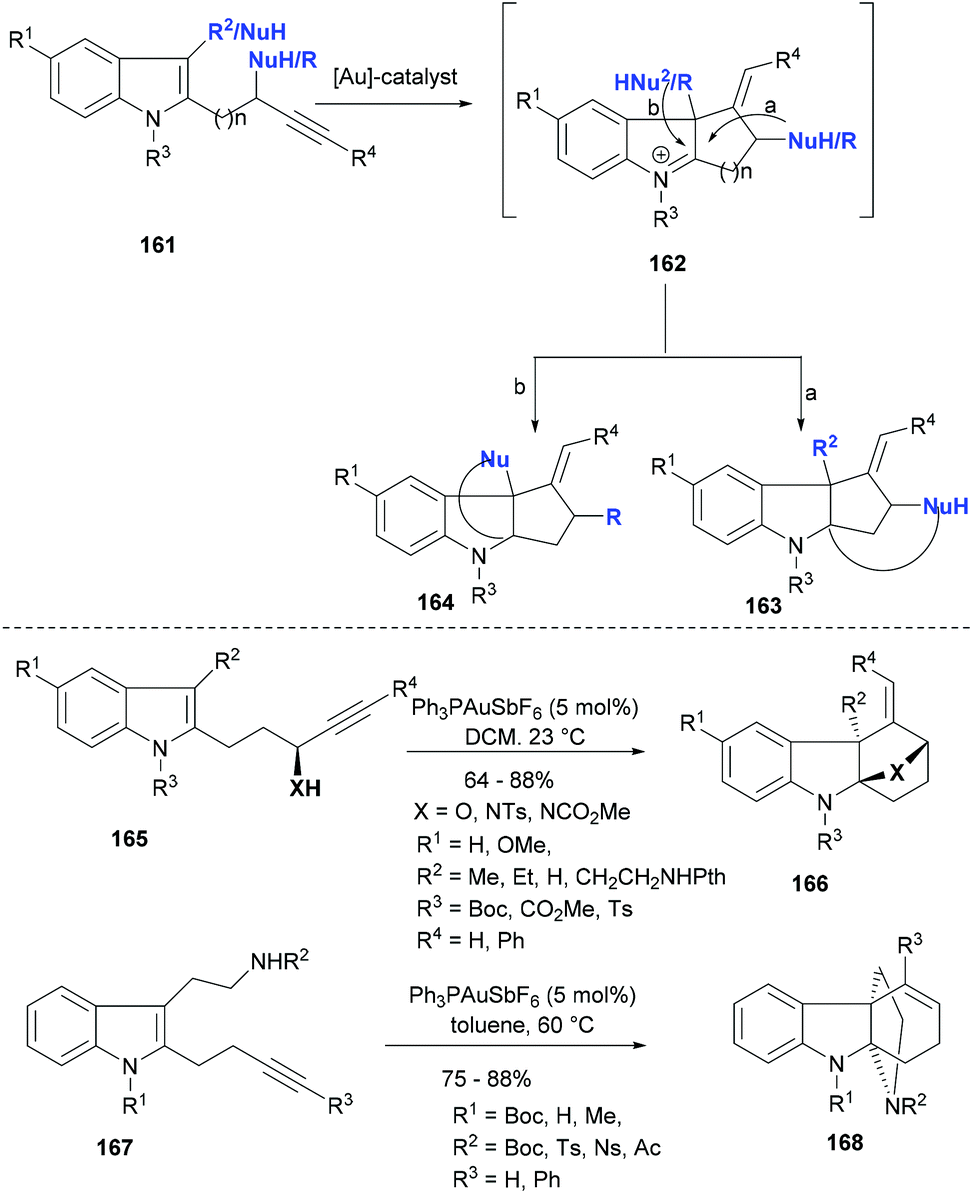 | ||
| Scheme 26 Gold catalysed tandem cyclizations to tetracyclic indolines. | ||
The same strategy was also explored for tryptamine derivatives 167 (Scheme 26), which contain a two-carbon linker between the indole and the alkyne. The gold(I) catalysed tandem cyclization proceeded smoothly using the same catalyst system in toluene at 60 °C and provided 6-endo-dig cyclization products 168 in high yields (Scheme 26).
Bandini's group reported a similar strategy of tandem cyclization reactions catalysed by cationic gold(I) complexes in 2011.117 In 2012, the same group elaborated this synthesis118 by developing the first gold(I)-catalysed enantioselective synthesis of differently ring-fused tetracyclic indolines (170–171) from the indole substrates having C3-linked propargylic alcohol (169, Scheme 27). The overall stereochemistry of the final product (170 or 171) was essentially controlled by the initial gold-catalysed hydroindolination of triple bonds leading to intermediate 172 or 173. A number of chiral gold complexes were screened to identify the best catalyst for the stereoselective tandem reaction leading to indolines bearing all-carbon quaternary stereocenters at the C(3) position. Interestingly, Bandini and co-workers could successfully optimize the reaction conditions for a completely regio-, diastereo and highly enantioselective synthesis of two different tetracyclic indolines (Scheme 27). While the [(R)-xylyl-BINAP(AuBF4)2] catalysed reaction of 169 delivered the dihydropyranyl indolines (170) in high yields and high enantioselectivity, a relatively bulkier ligand in the [(S)-DTBM-segphos(AuOTf)2] catalyst was required to selectively follow an initial 7-endo-dig cyclization leading to dihydrofuanyl indolines (171). Notably, unlike previous examples (Scheme 26), protection of indole NH was not required in this case.
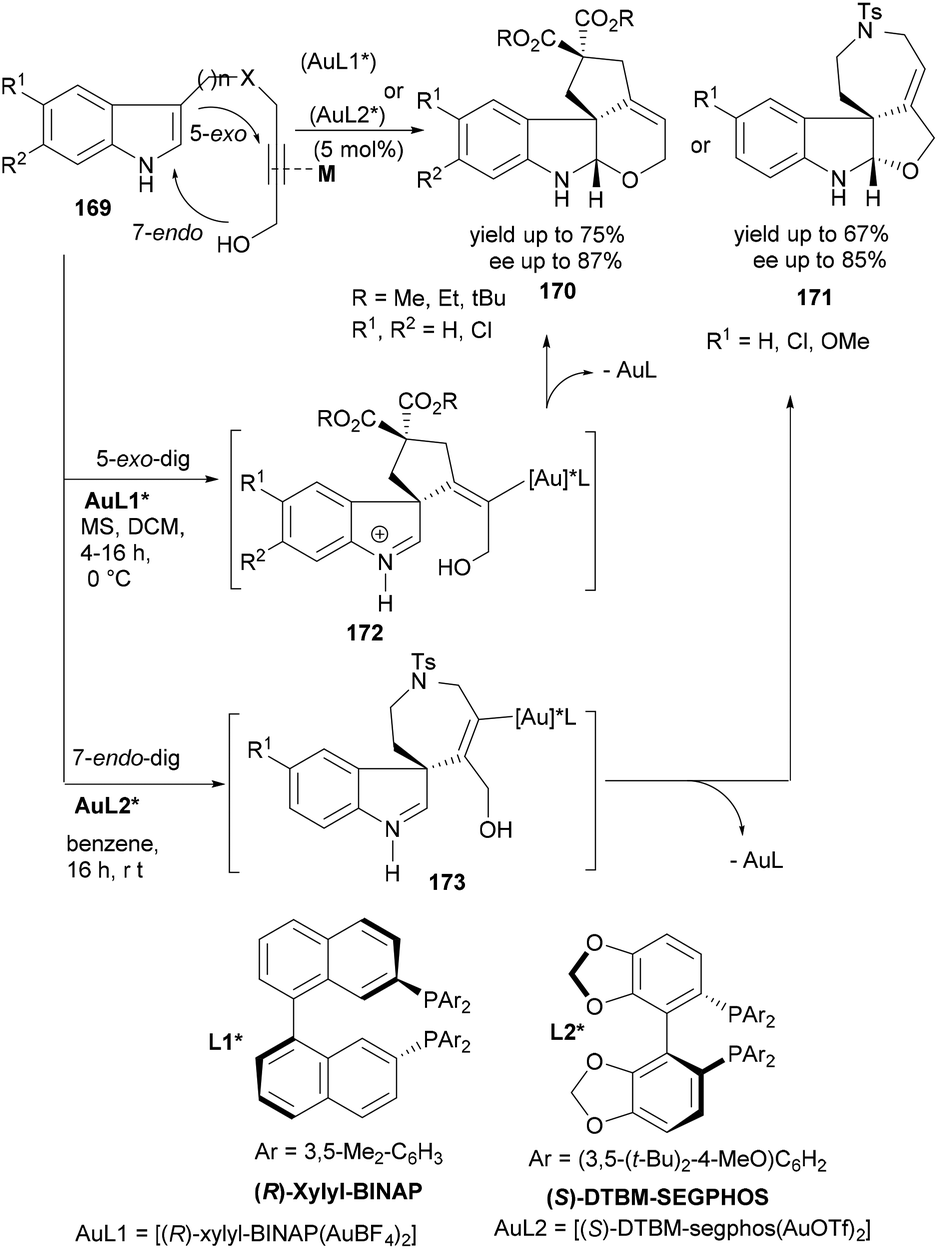 | ||
| Scheme 27 A gold-catalysed enantioselective tandem cyclization approach to tetracyclic indolines. | ||
Our lab has been interested in developing cascade synthesis of different indole polycyclic frameworks for the discovery of novel bioactive small molecules.113,114,119 One of the synthesis designs involved a two-step cascade reaction sequence wherein the first Pictet–Spengler cyclization of a designed acetylenic aldehyde (175) with tryptamines (174) forms the intermediate 176 having a secondary nucleophilic amine in proximity to the 1,5-enyne moiety for a gold-triggered polycyclization cascade.120 However, all our attempts to make this sequence work like a cascade or domino reaction failed and we had to settle with a one-pot two-step process. Thus, a mixture of 174 and 175 with Yb(OTf)3 in the presence of the ionic liquid [bmim]Cl-AlCl3 in dichloromethane was subjected to microwave irradiation at 120 °C for 1.0 h to yield the Pictet–Spengler cyclization product 176 (Scheme 28). The cationic gold(I) catalyst 177 efficiently catalysed the polycyclization to provide NP harmicine analogues in good yields albeit as mixtures of diastereoisomers (180). Notably, the reaction pathway preferred a 5-exo-tet cyclization mode to form a pyrrolidine ring in 180 over a possible 6-endo-tet cyclization.
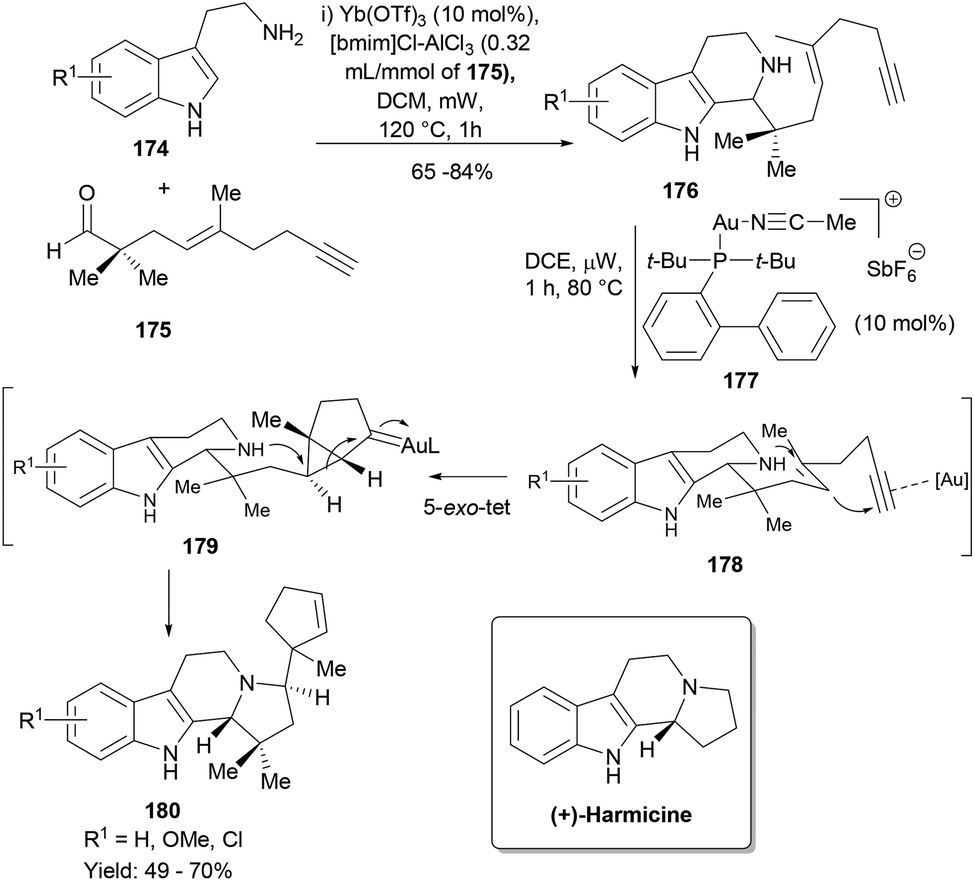 | ||
| Scheme 28 A gold-catalysed cascade synthesis of harmicine analogues. | ||
In most of the above synthetic strategies, the indole ring was already part of the substrates and the cascade reactions generated further molecular complexity as well as diversity around the indole core in the products. Cascade reactions that not only decorate the indole core with functional groups but also first generate the indole framework are highly challenging and have rarely been explored. Bandini's group reported a synthesis of densely functionalized tricyclic oxazino[4,3-a]indoles (182) from aniline diols (181) by means of the simultaneous construction of the indole and the N(1)–C(2)-fused ring (Scheme 29a).121,122
 | ||
| Scheme 29 A gold-catalysed cascade synthesis of tricyclic oxazino- and oxazepino-indoles. | ||
Mechanistically, the cascade reaction design involved a sequential hydroamination of the acetylene to form the initial indole core followed by a dehydrative sequence wherein a gold Lewis acid activated hydroxyl group is eliminated following the cyclization from the appended primary alcohol or phenol moiety forming the desired polycycles (183–185). Notably, the use of π-acid late-transition metal-catalysts was preferred over Brønsted and Lewis acids due to the unprotected amine diols (181) employed as substrates. Under the optimized conditions, the reaction at 50 °C in the presence of silver-free [XPhosAuNTf2] (5 mol%) afforded moderate to very high yields of a diverse set of oxazino- and oxazepino[4,3-a]indoles (183–185).
Further synthetic potential of the above cascade sequence was realized by the development of a triple–cascade reaction leading to tetracyclic indole (187). Exposure of 186 to AuIPrNTf2 (5 mol%) led to one-pot regioselective 5-endo-dig hydroamination of the C–C triple bond (188), alkoxylation of the carbynol carbon atom (189) followed by a 6-exo-trig hydroindolination of the appended olefin to form the final tetracyclic indole (187) in good yield (Scheme 29b).
The same group in their further efforts to build azepino[1,2-a]indoles (191) featuring a fused seven-membered ring through N1–C2 connection developed a gold-assisted cascade reaction for the target indoles using similar substrates (190, Scheme 29c) derived from readily available 2-alkynylanilines.123 The authors proposed a reaction sequence wherein an initial hydroamination yielded the indole intermediates (192–193) that under gold-activation led to the formation of the nucleophilic 2-vinylindole intermediates (194) through a gold-triggered 5-endo-dig hydroamination/dehydration sequence. The carbonyl group tethered to the aniline nitrogen atom underwent the second dehydrative cyclization reaction to produce the indole core (Scheme 29c).
Sharp et al.124 reported a consecutive hydroamination or cyclization strategy with anilinic diyne substrates (196) under the influence of gold catalysis to yield medicinally important pyrimido[1,6-a]indol-1(2H)-ones (197, Scheme 30). The first intramolecular 5-endo-dig cyclization formed the isolable intermediate 198, followed by another 6-endo-dig cyclization to deliver the product (197). Echavarren's gold(I) catalyst (177) was found to be efficient at this cascade cyclization to yield a number of substituted tricyclic indole product.
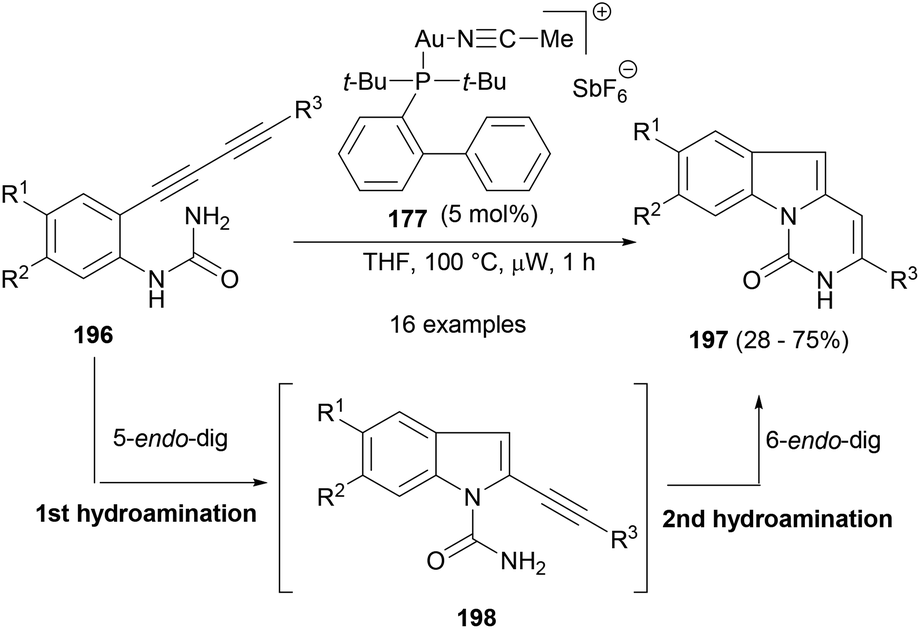 | ||
| Scheme 30 A gold-catalysed consecutive hydroamination/cyclization approach to pyrimido[1,6-a]indol-1(2H)-ones. | ||
:1). Further synthetic steps furnished (+)-cinchonidine (Scheme 31).
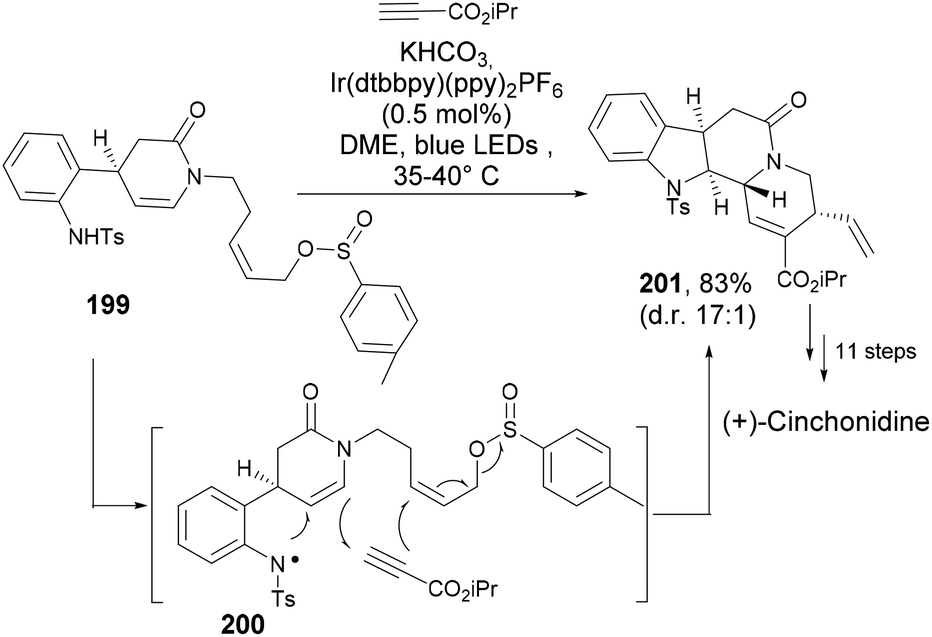 | ||
| Scheme 31 A cascade radical cyclization to tetracyclic indole (201). | ||
Copper catalysed coupling strategies have garnered a great deal of attention due to the low cost of the catalyst and often the good efficiency of the reactions. Xia et al.126 developed a mild and efficient Cu2O approach to generate benzoxazino[3,2-a]indol-12-ones (203, Scheme 32). The gem-dibromovinyl substrates (202) were designed to follow the cascade intramolecular C–N coupling and C–O bond formation through copper catalysis. Under the optimized reaction conditions moderate to very high yields of the indole polycycles (203) were obtained. Moreover, the protocol was found to be general and practical, and can also provide a range of different heterocycles fused to indole rings.
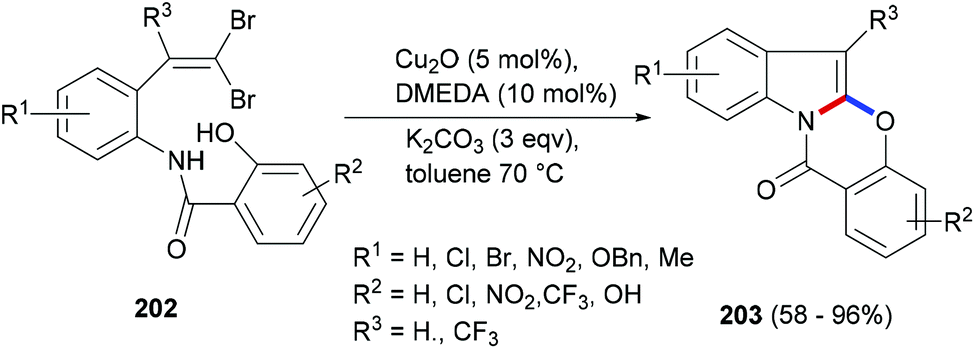 | ||
| Scheme 32 A copper catalysed domino cyclization to polycyclic indoles. | ||
4. Conclusions
The discipline of organic synthesis, though not in its infancy, is not yet mature enough to match up with Nature's ability to construct complex molecules. We have only limited chemical tools to construct and functionalize small molecules in an efficient and atom economical fashion. In fact, not only do structurally complex molecules require tedious multistep synthesis strategies, often a simple desired functionalization in small molecules does not find a straight forward protocol in the literature and can present a daunting synthetic challenge. Cascade and domino reactions are highly desired for their efficiency in building a number of bonds and rings and increasing the molecular complexity in the ensuing products. However, there is a dearth of chemical tools that one can employ in cascade synthesis designs. Owing to their interesting bioactivities, indole alkaloids have garnered a great deal of attention from organic and medicinal chemists. The cascade reaction designs presented in this review target indole polycyclic scaffolds and can further inspire the synthesis of new chemical entities based on indole frameworks. The combination of different key reactions in the design of cascade reactions to build structural complexity of NPs is a great challenge and needs to be explored further. Such endeavours unravel not only the new chemotypes but also offer new chemical transformations of wider synthetic utility. In particular, cascade reaction designs employing asymmetric synthesis of indole polycycles remain scarce and are highly desired as they can serve to deliver a good number of optically pure novel indole polycycles. In this era of medium to high-throughput screening requiring a compound library representing diverse molecular scaffolds, approaches like branching cascades127,128 that explore diverse cascade reactions on common substrates to build a number of scaffolds open new routes to construct indole polycycles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by funds from the Max-Planck-Gesellschaft. The authors are grateful to Prof. Dr H. Waldmann for his constant support and encouragement. Open Access funding provided by the Max Planck Society.Notes and references
- Molecules that Changed the World, ed. K. C. Nicolaou and T. Montagnon, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2008 Search PubMed.
- Classics in Total Synthesis II: More Targets, Strategies, Methods, ed. K. C. Nicolaou and S. A. Snyder, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2003 Search PubMed.
- Classics in Total Synthesis: Targets, Strategies, Methods, ed. K. C. Nicolaou and E. J. Sorensen, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 1996 Search PubMed.
- R. S. Bon and H. Waldmann, Acc. Chem. Res., 2010, 43, 1103–1114 CrossRef CAS.
- R. Breinbauer, I. R. Vetter and H. Waldmann, Angew. Chem., Int. Ed., 2002, 41, 2879–2890 Search PubMed.
- S. Rizzo and H. Waldmann, Chem. Rev., 2014, 114, 4621–4639 CrossRef CAS.
- K. Hubel, T. Lessmann and H. Waldmann, Chem. Soc. Rev., 2008, 37, 1361–1374 RSC.
- K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2009, 48, 3224–3242 CrossRef CAS.
- A. M. Walji and D. W. C. MacMillan, Synlett, 2007, 1477–1489 CAS.
- M. Garcia-Castro, S. Zimmermann, M. G. Sankar and K. Kumar, Angew. Chem., Int. Ed., 2016, 55, 7586–7605 CrossRef CAS.
- A. Pahl, H. Waldmann and K. Kumar, Chimia, 2017, 71, 653–660 CrossRef CAS.
- D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef CAS PubMed.
- M. Shiri, Chem. Rev., 2012, 112, 3508–3549 CrossRef CAS.
- P. J. Praveen, P. S. Parameswaran and M. S. Majik, Synthesis, 2015, 1827–1837 CAS.
- Y. Y. Zhang, T. Han, Q. L. Ming, L. S. Wu, K. Rahman and L. P. Qin, Nat. Prod. Commun., 2012, 7, 963–968 CAS.
- S. B. Bharate, S. Manda, N. Mupparapu, N. Battini and R. A. Vishwakarma, Mini-Rev. Med. Chem., 2012, 12, 650–664 CrossRef CAS.
- D. Bialonska and J. K. Zjawiony, Mar. Drugs, 2009, 7, 166–183 CrossRef CAS.
- K. Higuchi and T. Kawasaki, Nat. Prod. Rep., 2007, 24, 843–868 RSC.
- J. D. Podoll, Y. X. Liu, L. Chang, S. Walls, W. Wang and X. Wang, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15573–15578 CrossRef CAS.
- N. Chernyak, D. Tilly, Z. Li and V. Gevorgyan, ARKIVOC, 2011, 76–91, DOI:10.3998/ark.5550190.0012.509.
- Z. Wang, F. Song, Y. Zhao, Y. Huang, L. Yang, D. Zhao, J. Lan and J. You, Chem. – Eur. J., 2012, 18, 16616–16620 CrossRef CAS.
- R. Lock and H. Waldmann, Chem. – Eur. J., 1997, 3, 143–151 CrossRef CAS.
- P. M. Barbour, L. J. Marholz, L. Chang, W. Xu and X. Wang, Chem. Lett., 2014, 43, 572–578 CrossRef CAS.
- X. N. Xie and L. S. Zu, Synlett, 2018, 1008–1013 CAS.
- R. N. Rao, B. Maiti and K. Chanda, ACS Comb. Sci., 2017, 19, 199–228 CrossRef CAS.
- W. W. Zi, Z. W. Zuo and D. W. Ma, Acc. Chem. Res., 2015, 48, 702–711 CrossRef CAS PubMed.
- Z. Amara, J. Caron and D. Joseph, Nat. Prod. Rep., 2013, 30, 1211–1225 RSC.
- T. Gaich and P. S. Baran, J. Org. Chem., 2010, 75, 4657–4673 CrossRef CAS PubMed.
- J. B. Hendrickson, J. Am. Chem. Soc., 1975, 97, 5784–5800 CrossRef CAS.
- P. A. Wender and B. L. Miller, Nature, 2009, 460, 197–201 CrossRef CAS.
- L. F. Tietze, Chem. Rev., 1996, 96, 115–136 CrossRef CAS.
- L.-Q. Lu, J.-R. Chen and W.-J. Xiao, Acc. Chem. Res., 2012, 45, 1278–1293 CrossRef CAS.
- K. C. Nicolaou and J. S. Chen, Chem. Soc. Rev., 2009, 38, 2993–3009 RSC.
- M. Ihara, ARKIVOC, 2006, 416–438 Search PubMed.
- K. C. Nicolaou, T. Montagnon and S. A. Snyder, Chem. Commun., 2003, 551–564 RSC.
- F. Vetica, R. M. de Figueiredo, M. Orsini, D. Tofani and T. Gasperi, Synthesis, 2015, 2139–2184 CAS.
- M. Rueping, K. Haack, W. Ieawsuwan, H. Sunden, M. Blanco and F. R. Schoepke, Chem. Commun., 2011, 47, 3828–3830 RSC.
- N. J. Green and M. S. Sherburn, Aust. J. Chem., 2013, 66, 267–283 CrossRef CAS.
- L. F. Tietze, Chem. Ind., 1995, 453–457 CAS.
- L. F. Tietze and N. Rackelmann, Pure Appl. Chem., 2004, 76, 1967–1983 CAS.
- Catalytic Cascade Reactions, ed. P. F. Xu and W. Wang, John Wiley & Sons, Inc., Beijing, 2013 Search PubMed.
- C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef CAS.
- L. Hong and R. Wang, Adv. Synth. Catal., 2013, 355, 1023–1052 CrossRef CAS.
- R. Narayan, M. Potowski, Z. J. Jia, A. P. Antonchick and H. Wadmann, Acc. Chem. Res., 2014, 47, 1296–1310 CrossRef CAS.
- J. H. Ryan, ARKIVOC, 2015, 160–183 Search PubMed.
- V. A. Bakulev, T. Beryozkina, J. Thomas and W. Dehaen, Eur. J. Org. Chem., 2018, 262–294 CrossRef CAS.
- A. G. Meyer and J. H. Ryan, Molecules, 2016, 21 CrossRef CAS , 935.
- V. Nair and T. D. Suja, Tetrahedron, 2007, 63, 12247–12275 CrossRef CAS.
- L. P. Wakelin and M. J. Waring, in Comprehensive Medicinal Chemistry, ed. P. G. Sammes, Pergamon Press, Oxford, 1990, vol. 2, pp. 703–724 Search PubMed.
- A. Lauria, C. Patella, P. Diana, P. Barraja, A. Montalbano, G. Cirrincione, G. Dattolo and A. M. Almerico, Tetrahedron Lett., 2006, 47, 2187–2190 CrossRef CAS.
- S. E. Denmark and A. Thorarensen, Chem. Rev., 1996, 96, 137–165 CrossRef CAS.
- S. E. Denmark and J. I. Montgomery, Angew. Chem., Int. Ed., 2005, 44, 3732–3736 CrossRef CAS.
- S. E. Denmark and R. Y. Baiazitov, J. Org. Chem., 2006, 71, 593–605 CrossRef CAS.
- S. E. Denmark and E. A. Martinborough, J. Am. Chem. Soc., 1999, 121, 3046–3056 CrossRef CAS.
- I. Chataigner and S. R. Piettre, Org. Lett., 2007, 9, 4159–4162 CrossRef CAS.
- J. E. Sears, T. J. Barker and D. L. Boger, Org. Lett., 2015, 17, 5460–5463 CrossRef CAS.
- G. I. Elliott, J. Velcicky, H. Ishikawa, Y. K. Li and D. L. Boger, Angew. Chem., Int. Ed., 2006, 45, 620–622 CrossRef CAS.
- H. Ishikawa, G. I. Elliott, J. Velcicky, Y. Choi and D. L. Boger, J. Am. Chem. Soc., 2006, 128, 10596–10612 CrossRef CAS.
- G. D. Wilkie, G. I. Elliott, B. S. J. Blagg, S. E. Wolkenberg, D. R. Soenen, M. M. Miller, S. Pollack and D. L. Boger, J. Am. Chem. Soc., 2002, 124, 11292–11294 CrossRef CAS.
- B. Meunier, Acc. Chem. Res., 2008, 41, 69–77 CrossRef CAS.
- G. Mehta and V. Singh, Chem. Soc. Rev., 2002, 31, 324–334 RSC.
- L. F. Tietze, H. P. Bell and S. Chandrasekhar, Angew. Chem., Int. Ed., 2003, 42, 3996–4028 CrossRef CAS PubMed.
- R. D. R. S. Manian, J. Jayashankaran and R. Raghunathan, Tetrahedron Lett., 2007, 48, 1385–1389 CrossRef CAS.
- F. M. Moghaddam, M. Kiamehr, S. Taheri and Z. Mirjafary, Helv. Chim. Acta, 2010, 93, 964–973 CrossRef CAS.
- F. Palacios, C. Alonso, P. Amezua and G. Rubiales, J. Org. Chem., 2002, 67, 1941–1946 CrossRef CAS.
- S. B. Jones, B. Simmons, A. Mastracchio and D. W. C. MacMillan, Nature, 2011, 475, 183–188 CrossRef CAS.
- F. M. Moghaddam, Z. Mirjafary, H. Saeidian, S. Taheri, M. Doulabi and M. Kiamehr, Tetrahedron, 2010, 66, 134–138 CrossRef CAS.
- J.-L. Brochu, M. Prakesch, G. D. Enright, D. M. Leek and P. Arya, J. Comb. Chem., 2008, 10, 405–420 CrossRef CAS.
- Q. Cai, C. Zheng, J.-W. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2011, 50, 8665–8669 CrossRef CAS.
- J. R. Hummel, J. A. Boerth and J. A. Ellman, Chem. Rev., 2017, 117, 9163–9227 CrossRef CAS.
- Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. B. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef CAS.
- M. Tobisu and N. Chatani, Angew. Chem., Int. Ed., 2006, 45, 1683–1684 CrossRef CAS.
- P. Matyus, O. Elias, P. Tapolcsanyi, A. Polonka-Balint and B. Halasz-Dajka, Synthesis, 2006, 2625–2639 CrossRef CAS.
- M. C. Haibach, I. Deb, C. K. De and D. Seidel, J. Am. Chem. Soc., 2011, 133, 2100–2103 CrossRef CAS.
- Q. Wang, D. X. Wang, M. X. Wang and J. P. Zhu, Acc. Chem. Res., 2018, 51, 1290–1300 CrossRef CAS.
- A. Domling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168–3210 CrossRef CAS.
- X. Wang, S.-Y. Wang and S.-J. Ji, J. Org. Chem., 2014, 79, 8577–8583 CrossRef CAS.
- H. Duckert, V. Pries, V. Khedkar, S. Menninger, H. Bruss, A. W. Bird, Z. Maliga, A. Brockmeyer, P. Janning, A. Hyman, S. Grimme, M. Schurmann, H. Preut, K. Hubel, S. Ziegler, K. Kumar and H. Waldmann, Nat. Chem. Biol., 2012, 8, 179–184 CrossRef.
- V. Eschenbrenner-Lux, H. Duckert, V. Khedkar, H. Bruss, H. Waldmann and K. Kumar, Chem. – Eur. J., 2013, 19, 2294–2304 CrossRef CAS.
- H. Waldmann, V. Khedkar, H. Dückert, M. Schürmann, I. M. Oppel and K. Kumar, Angew. Chem., Int. Ed., 2008, 47, 6869–6872 CrossRef CAS.
- Y. C. Lee, K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2018, 57, 5212–5226 CrossRef CAS.
- C. G. Newton, S. G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908–8976 CrossRef CAS.
- B. M. Trost, Pure Appl. Chem., 1994, 66, 2007–2014 CAS.
- A. Duschek and S. F. Kirsch, Angew. Chem., Int. Ed., 2008, 47, 5703–5705 CrossRef CAS.
- L. F. Tietze, H. Ila and H. P. Bell, Chem. Rev., 2004, 104, 3453–3516 CrossRef CAS.
- H. F. Sore, W. R. Galloway and D. R. Spring, Chem. Soc. Rev., 2012, 41, 1845–1866 RSC.
- L. F. Tietze and T. Kinzel, Pure Appl. Chem., 2007, 79, 629–650 CAS.
- S.-S. Li, Y.-Q. Xia, F.-Z. Hu, C.-F. Liu, F. Su and L. Dong, Chem. – Asian J., 2016, 11, 3165–3168 CrossRef CAS PubMed.
- T.-R. Li, L.-Q. Lu, Y.-N. Wang, B.-C. Wang and W.-J. Xiao, Org. Lett., 2017, 19, 4098–4101 CrossRef CAS PubMed.
- D. A. Candito and M. Lautens, Org. Lett., 2010, 12, 3312–3315 CrossRef CAS.
- V. Eschenbrenner-Lux, P. Kuchler, S. Ziegler, K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2014, 53, 2134–2137 CrossRef CAS.
- T. Yokosaka, H. Nakayama, T. Nemoto and Y. Hamada, Org. Lett., 2013, 15, 2978–2981 CrossRef CAS.
- S. Lakhdar, M. Westermaier, F. Terrier, R. Goumont, T. Boubaker, A. R. Ofial and H. Mayr, J. Org. Chem., 2006, 71, 9088–9095 CrossRef CAS.
- J.-R. Chen, C.-F. Li, X.-L. An, J.-J. Zhang, X.-Y. Zhu and W.-J. Xiao, Angew. Chem., Int. Ed., 2008, 47, 2489–2492 CrossRef CAS.
- J. M. Mejia-Oneto and A. Padwa, Org. Lett., 2006, 8, 3275–3278 CrossRef CAS.
- N. Chernyak, D. Tilly, Z. Li and V. Gevorgyan, Chem. Commun., 2010, 46, 150–152 RSC.
- J. X. Liu, M. Shen, Y. D. Zhang, G. S. Li, A. Khodabocus, S. Rodriguez, B. Qu, V. Farina, C. H. Senanayake and B. Z. Lu, Org. Lett., 2006, 8, 3573–3575 CrossRef CAS PubMed.
- J. Liu, Y. Zhang, G. Li, F. Roschangar, V. Farina, C. H. Senanayake and B. Z. Lu, Adv. Synth. Catal., 2010, 352, 2667–2671 CrossRef CAS.
- B. Milde, M. Pawliczek, P. G. Jones and D. B. Werz, Org. Lett., 2017, 19, 1914–1917 CrossRef CAS.
- M. Pawliczek, T. F. Schneider, C. Maass, D. Stalke and D. B. Werz, Angew. Chem., Int. Ed., 2015, 54, 4119–4123 CrossRef CAS.
- X.-F. Xia, N. Wang, L. L. Zhang, X.-R. Song, X.-Y. Liu and Y.-M. Liang, J. Org. Chem., 2012, 77, 9163–9170 CrossRef CAS.
- J. W. Boyle, Y. C. Zhao and P. W. H. Chan, Synthesis, 2018, 1402–1416 CAS.
- P. M. Barbour, L. J. Marholz, L. Chang, W. Q. Xu and X. Wang, Chem. Lett., 2014, 43, 572–578 CrossRef CAS.
- C. Obradors and A. M. Echavarren, Acc. Chem. Res., 2014, 47, 902–912 CrossRef CAS.
- Y. Zhang, T. P. Luo and Z. Yang, Nat. Prod. Rep., 2014, 31, 489–503 RSC.
- D. Pflasterer and A. S. K. Hashmi, Chem. Soc. Rev., 2016, 45, 1331–1367 RSC.
- F. Bihelovic, B. Vulovic and R. N. Saicic, Isr. J. Chem., 2018, 58, 521–530 CrossRef CAS.
- Y. C. Lee and K. Kumar, Isr. J. Chem., 2018, 58, 531–556 CrossRef CAS.
- H. Pellissier, Chem. Rev., 2016, 116, 14868–14917 CrossRef CAS.
- G. C. Fang and X. H. Bi, Chem. Soc. Rev., 2015, 44, 8124–8173 RSC.
- B. Baskar, P. Y. Dakas and K. Kumar, Org. Lett., 2011, 13, 1988–1991 CrossRef CAS PubMed.
- L. Laraia, K. Ohsawa, G. Konstantinidis, L. Robke, Y. W. Wu, K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2017, 56, 2145–2150 CrossRef CAS.
- Y. C. Lee, S. Patil, C. Golz, C. Strohmann, S. Ziegler, K. Kumar and H. Waldmann, Nat. Commun., 2017, 8 Search PubMed , 14043.
- H. Waldmann, L. Eberhardt, K. Wittstein and K. Kumar, Chem. Commun., 2010, 46, 4622–4624 RSC.
- X. Xie, X. Du, Y. Chen and Y. Liu, J. Org. Chem., 2011, 76, 9175–9181 Search PubMed.
- Y. Liu, W. Xu and X. Wang, Org. Lett., 2010, 12, 1448–1451 CrossRef CAS.
- G. Cera, P. Crispino, M. Monari and M. Bandini, Chem. Commun., 2011, 47, 7803–7805 RSC.
- G. Cera, M. Chiarucci, A. Mazzanti, M. Mancinelli and M. Bandini, Org. Lett., 2012, 14, 1350–1353 CrossRef CAS.
- K. Wittstein, K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2011, 50, 9076–9080 CrossRef CAS PubMed.
- A. Danda, K. Kumar and H. Waldmann, Chem. Commun., 2015, 51, 7536–7539 RSC.
- M. Chiarucci, E. Matteucci, G. Cera, G. Fabrizi and M. Bandini, Chem. – Asian J., 2013, 8, 1776–1779 CrossRef CAS PubMed.
- M. Chiarucci, R. Mocci, L. D. Syntrivanis, G. Cera, A. Mazzanti and M. Bandini, Angew. Chem., Int. Ed., 2013, 52, 10850–10853 CrossRef CAS PubMed.
- G. Cera, S. Piscitelli, M. Chiarucci, G. Fabrizi, A. Goggiamani, R. S. Ramon, S. P. Nolan and M. Bandini, Angew. Chem., Int. Ed., 2012, 51, 9891–9895 CrossRef CAS PubMed.
- P. P. Sharp, M. G. Banwell, J. Renner, K. Lohmann and A. C. Willis, Org. Lett., 2013, 15, 2616–2619 CrossRef CAS PubMed.
- W. T. Liu, W. F. Qin, X. B. Wang, F. Xue, X. Y. Liu and Y. Qin, Angew. Chem., Int. Ed., 2018, 57, 12299–12302 CrossRef CAS PubMed.
- Z. Xia, K. Wang, J. Zheng, Z. Ma, Z. Jiang, X. Wang and X. Lv, Org. Biomol. Chem., 2012, 10, 1602–1611 RSC.
- W. Liu, V. Khedkar, B. Baskar, M. Schurmann and K. Kumar, Angew. Chem., Int. Ed., 2011, 50, 6900–6905 CrossRef CAS PubMed.
- M. Garcia-Castro, L. Kremer, C. D. Reinkemeier, C. Unkelbach, C. Strohmann, S. Ziegler, C. Ostermann and K. Kumar, Nat. Commun., 2015, 6 CAS , 6516.
| This journal is © The Royal Society of Chemistry 2019 |