
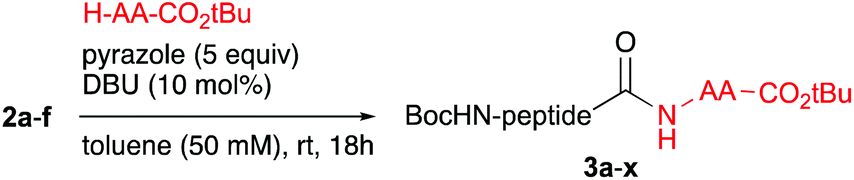
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoselective C-terminal peptide elongation from Chan–Lam–Evans reaction generated isopropenyl esters†
Luuk
Steemers
and
Jan H.
van Maarseveen
 *
*
Van ‘t Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: j.h.vanmaarseveen@uva.nl
First published on 4th February 2019
Abstract
C-Terminal dipeptide isopropenyl esters were synthesised by a Cu(II)-mediated Chan–Lam–Evans enol esterification of peptide carboxylic acids and isoprenyl boroxine. These shelf stable peptide esters could be coupled stereoselectively with a variety of amino acid and dipeptide nucleophiles in high yield and purity in the presence of pyrazole/DBU as the catalyst.
In current pharmaceutic research peptides play an increasingly important role.1 As a result, in recent years several peptides and analogs thereof were registered as drugs.2 By using both solid and solution phase techniques peptides are synthesized in the C → N direction exclusively. In other words, the N-terminus is elongated by consecutive coupling of single N-protected amino acids. Commonly employed carboxyl group activating reagents, for example uronium, phosphonium and carbodiimide based, form a highly reactive intermediate, that is usually transformed in situ into less reactive HOBt or HOAt esters to avoid racemisation.3 The mesomerically stabilised and, as compared to an amide group, less nucleophilic N-carbamate protective group suppresses racemisation of the stereogenic center by hampering enolisation or oxazolone formation. Especially for peptide fragment couplings or peptide cyclisation C-terminal peptide activation is required. However, coupling of two peptide fragments or cyclisation with traditional coupling reagents inevitably leads to epimerisation of the α-carbon of the C-terminally carboxyl-activated residue. As a result, in current ligation or cyclisation strategies, usually a C-terminal glycine is chosen as the site of carboxyl activation.4 In stark contrast, both ribosomally and non-ribosomally synthesised peptides are elongated in the N → C direction, via aminolysis of the growing peptide chain of the activated C-terminus. Nature uses very mild carboxyl group activation such as the connecting ester bond between the amino acid and tRNA, or as a thioester while linked at the peptidyl carrier protein in the case of non-ribosomal peptide synthesis. Aminolysis of the rather unreactive secondary ester at the tRNA occurs due to the proximity of the incoming amine nucleophile to this ester, induced by the ribosome, underscoring the power of pre-organisation for certain chemical transformations.
Synthetic strategies employing elongation in the N → C direction are scarce. Katritzky has shown that peptide benzotriazole esters may be prepared in the cold using SOCl2/HOBt and subsequently coupled to other peptide fragments.5 More recently, the Bode group has shown that unprotected peptides bearing a C-terminal keto acid ligate with peptides modified with a hydroxylamine at the N-terminus.6 Another viable method used for fragment couplings is the synthesis of peptides that deliver a stable C-terminal ester serving as a substrate for chemoenzymatic coupling.7 In the search for catalytic carboxyl group activation methods to substitute the traditional stoichiometric coupling reagents we have reported earlier the use of mildly activated 4-methylsulfonylphenyl esters, obtained via a Cu(II)-catalysed Chan–Lam–Evans (CLE) reaction8 and subsequent oxidation, as suitable substrates for epimerisation-free coupling (Scheme 1).9
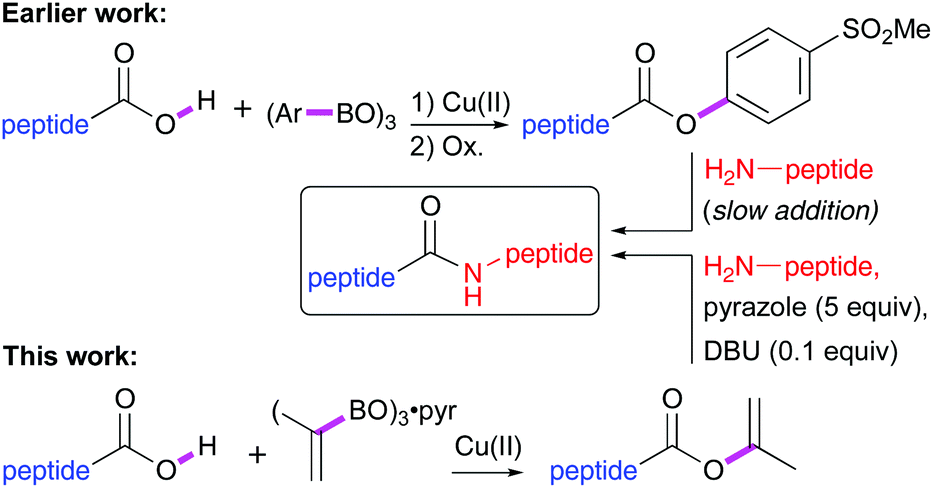 | ||
| Scheme 1 CLE-mediated peptide esterification and follow-up elongation by aminolysis. | ||
It was found that the CLE-type aryl esterification of peptides proceeded with complete stereoretention. However, to obtain high de's in the aminolysis reaction the amine had to be added slowly to avoid too basic conditions. Recently, we disclosed the efficient enol esterification of carboxylic acids via the CLE reaction and demonstrated the compatibility with all proteogenic amino acids, except methionine.10 Furthermore, no racemisation of the C-terminus was observed during the CLE-type enol esterification of amino acids. This prompted us to study the possibility of stereoselective elongation of peptides at the C-terminus employing isopropenyl esters as mild activating groups. As compared to 4-methylsulfonylphenyl esters, isopropenyl esters are less reactive and only react slowly in direct aminolysis reactions. Recently, the group of Birman reported anionic azoles as efficient catalysts for the aminolysis of both phenyl and isopropenyl esters.11 Remarkably, in a landmark paper that was far ahead of its time, the group of Beyerman reported back in the 1960s the use of neutral azoles as additives for the stereoselective aminolysis of peptide vinyl esters that were made via Pd-catalysis.12 Although vinyl esters can be efficiently synthesized via the CLE reaction from commercially available trivinylboroxine13 but also by Pd(II)14 or Hg(II)15 catalysed transesterification with vinyl acetate, the acetaldehyde side product released after aminolysis is far more reactive than acetone, the side product resulting from amide bond formation from isopropenyl esters.
For our studies, six model dipeptide acids were synthesized, i.e. Boc-Phe-Ala-OH (1a), Boc-Phe-Phe-OH (1c) and Boc-Phe-Val-OH (1e) and, to allow facile monitoring of the stereointegrity, their diastereomers 1b, 1d and 1f, (see Table 1).
|
|
||||
|---|---|---|---|---|
| Starting dipeptide | Starting peptide acid product | Yield (%) | de (%) | |
| 1a | Boc-Phe-Ala-OH | 2a | 92 | nd |
| 1b | Boc-Phe-D-Ala-OH | 2b | 91 | nd |
| 1c | Boc-Phe-Phe-OH | 2c | 90 | >99 |
| 1d | Boc-Phe-D-Phe-OH | 2d | 97 | >99 |
| 1e | Boc-Phe-Val-OH | 2e | 80 | nd |
| 1f | Boc-Phe-D-Val-OH | 2f | 91 | nd |
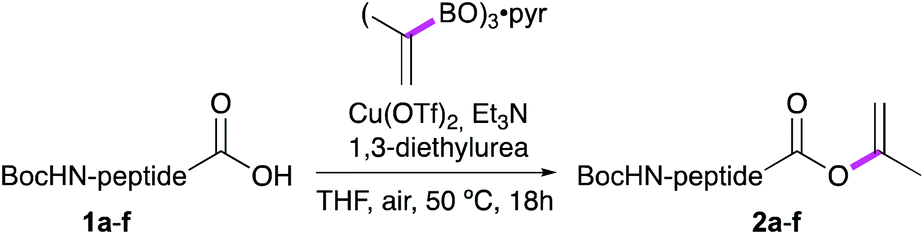
Ala, Phe and Val were selected as the C-terminal residues due to their increasing steric properties. Transformation of the peptide acids 1a–f into the isopropenyl esters 2a–f was carried out smoothly by the previously reported CLE conditions, using tri(isopropenyl)boroxine pyridine complex16 as the coupling partner. All peptide isoprenyl esters were obtained in excellent yields as shelf-stable solid compounds. To determine the stereointegrity, the most epimerisation sensitive peptide esters 2c and 2d, bearing a C-terminal phenylalanine, were subjected to chiral HPLC analysis showing no detectable loss of stereointegrity. For the same reason, for our optimisation studies of the aminolysis reaction, Boc-Phe-Phe-OC(CH3)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 (1c) was chosen as the C-terminally activated peptide to deliver, by using Ala-OtBu as the nucleophile, Boc-Phe-Phe-Ala-OtBu (3k) as the target tripeptide (Table 2).
CH2 (1c) was chosen as the C-terminally activated peptide to deliver, by using Ala-OtBu as the nucleophile, Boc-Phe-Phe-Ala-OtBu (3k) as the target tripeptide (Table 2).
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Azole (equiv.) | Equiv. H-Ala-OtBu | dea (%) |
| a Determined by chiral HPLC. | ||||
| 1 | CH2Cl2 | 1,2,4-Triazole (1.2) | 1.2 | 69 |
| 2 | CH2Cl2 | Pyrazole (1.2) | 1.2 | 72 |
| 3 | CH2Cl2 | 1,2,4-Triazole (5) | 1.2 | 79 |
| 4 | CH2Cl2 | 1,2,4-Triazole (5) | 2 | 89 |
| 5 | PhCH3 | 1,2,4-Triazole (1.2) | 1.2 | 84 |
| 6 | PhCH3 | 1,2,4-Triazole (5) | 2 | 95 |
| 7 | PhCH3 | Pyrazole (5) | 2 | 97 |
| 8 | PhCH3 | Pyrazole (5) | 4 | 96 |
| 9 | PhH | Pyrazole (5) | 2 | 95 |
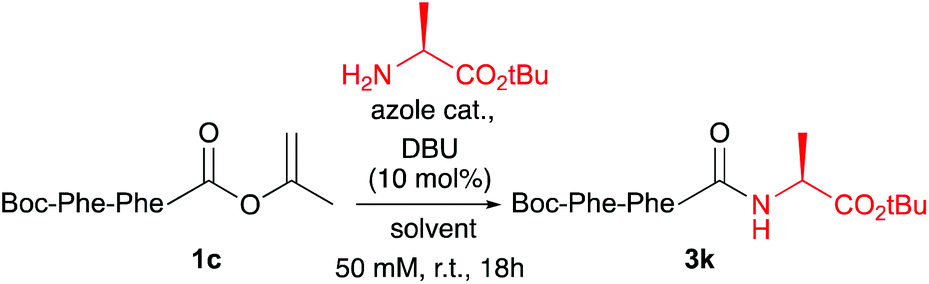
The optimisation studies started using CH2Cl2 as solvent and by adding a slight excess of 1,2,4-triazole as a catalyst and 10 mol% DBU as the base to generate the active anionic azolate species. Although the aminolysis reaction was clean and did not require further purification besides a simple acidic washing step, only a moderate de of 69% was found (Table 2, entry 1). By using pyrazole as the catalyst, a similar outcome of the reaction was found (entry 2). A significant improvement of the de was achieved by increasing the amount of azole to five equiv. (entry 3). By adding two equiv. of the amine nucleophile (entry 4) the de went further up to 89%. Switching to toluene as a more apolar solvent helped to improve the de even further to 97% (entry 5 vs. 1 and entry 6 vs. 4). Also in this case pyrazole performed slightly better than 1,2,4-triazole (entry 7 vs. 6). Raising the amount of amine or using an even more apolar solvent did not lead to a further increase of the de (entries 8 and 9). Also 1,2,3-triazole and benzotriazole were screened as catalysts, however these reactions either did not reach full conversion, had lower de and/or produced unidentified byproducts (not shown).
The proposed reaction mechanism for the pyrazole-catalysed aminolysis reaction starts by deprotonation of pyrazole I by DBU to form the highly nucleophilic azole anion II (Scheme 2). Nucleophilic attack of the anionic nitrogen at the isopropenyl ester provides acylpyrazole III, thereby irreversibly releasing acetone and regenerating DBU. The incoming amine nucleophile can coordinate to the pyrazole 2-nitrogen atom via H-bonding forming complex IV showing a favourable 5-membered transition state for the aminolysis step to give the amide product and releasing pyrazole I.
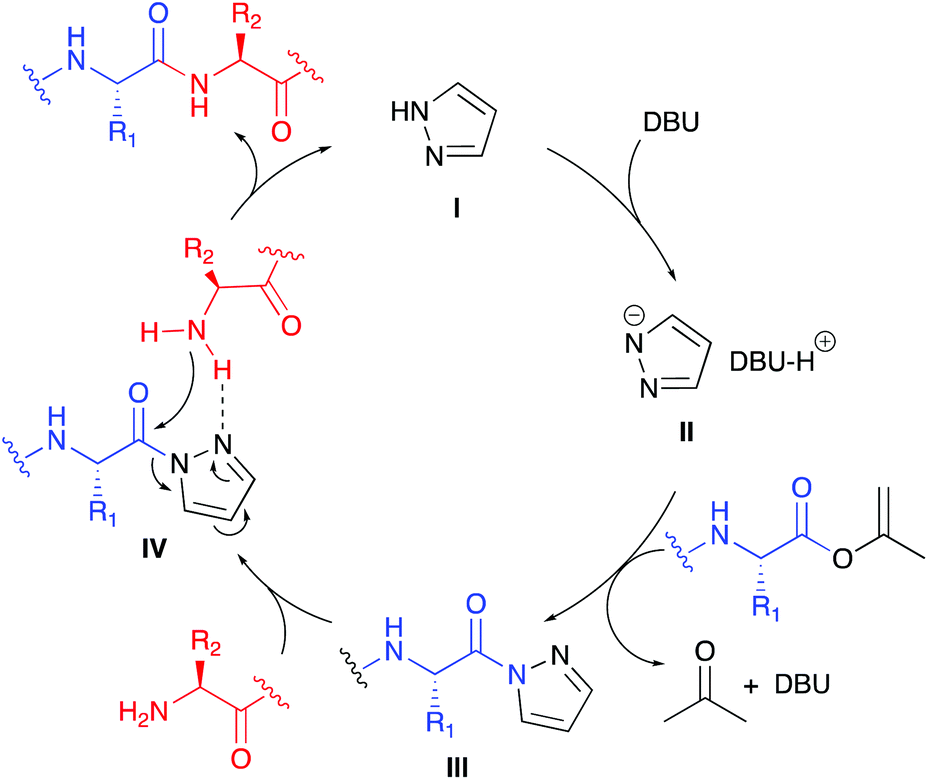 | ||
| Scheme 2 Proposed catalytic cycle for the pyrazole catalysed aminolysis of isoprenyl esters. | ||
Some observations in Table 1 can be explained by this proposed mechanism. At first, in polar solvents DBU is more likely to deprotonate the α-H within III to give an enolate resulting in epimerisation. Secondly, the proposed H-bonds in IV are stronger in apolar solvents, thereby increasing the rate of the aminolysis reaction thus suppressing epimerisation. The positive influence of using an excess of pyrazole on the de may be explained by a buffering effect lowering the basicity of the reaction mixture. With the optimised aminolysis conditions in hand we set out to investigate the scope of the reaction. Because we have shown earlier the compatibility of the CLE-reaction with appropriately protected canonical amino acids, we aimed at a limited but representative set of target tripeptides.10 As the C- and N-terminal residues Gly, Ala, Phe and Val, representing a gradual increase in steric hindrance, were selected of which the outcome may be extrapolated to the other canonical amino acids.17 At the C-terminus also the D-configured analogs were included to allow precise HPLC-analysis to determine the stereointegrity of the aminolysis reaction. The results of this screening by reacting the six dipeptide isopropenyl esters 2a–f with Gly-OtBu, Ala-OtBu, Phe-OtBu and Val-OtBu as the model amino acid nucleophiles are summarised in Table 3. It is worth mentioning that for all reactions a simple acidic extraction was sufficient to remove all non-volatiles such as excess amine nucleophile, pyrazole, and DBU. The tripeptides 3a–x thus obtained were virtually pure and needed no further purification, as determined by 1H- and 13C-NMR spectroscopy. The screening showed that all products were obtained in good to excellent yields. The diastereomeric excess for all peptides 3a–u is >96% and in most cases exceeds 98%. For the aminolysis of dipeptides 2e and 2f with the sterically challenging valine at the C-terminus the reaction gave either incomplete or no conversion at all at room temperature. For Gly-OtBu or Ala-OtBu as nucleophiles, heating to 50 °C was sufficient to complete the reaction, giving tripeptides 3q–t in excellent yields and diastereomeric excess. However, in the case of using Phe-OtBu or Val-OtBu as nucleophiles heating to 80 °C was required to ensure completion of the reaction. Although the yields of products 3v–x were still very good, we had to accept that the de's showed a small but significant drop. Presumably this is caused by the longer living epimerisation-prone acylpyrazole intermediate III (see Scheme 2).
|
|
||||
|---|---|---|---|---|
| Starting dipeptide | Tripeptide product | Yield (%) | dea (%) | |
| a Determined by chiral HPLC. b Reaction run at 50 °C. c Reaction run at 80 °C. | ||||
| 2a | Boc-Phe-Ala-Gly-OtBu | 3a | 96 | 97 |
| 2b | Boc-Phe-D-Ala-Gly-OtBu | 3b | 98 | 98 |
| 2a | Boc-Phe-Ala-Ala-OtBu | 3c | 99 | 99 |
| 2b | Boc-Phe-D-Ala-Ala-OtBu | 3d | 93 | 99 |
| 2a | Boc-Phe-Ala-Phe-OtBu | 3e | 95 | 99 |
| 2b | Boc-Phe-D-Ala-Phe-OtBu | 3f | 95 | 99 |
| 2a | Boc-Phe-Ala-Val-OtBu | 3g | 92 | 99 |
| 2b | Boc-Phe-D-Ala-Val-OtBu | 3h | 98 | 98 |
| 2c | Boc-Phe-Phe-Gly-OtBu | 3i | 95 | 97 |
| 2d | Boc-Phe-D-Phe-Gly-OtBu | 3j | 93 | >99 |
| 2c | Boc-Phe-Phe-Ala-OtBu | 3k | 90 | 97 |
| 2d | Boc-Phe-D-Phe-Ala-OtBu | 3l | 88 | 98 |
| 2c | Boc-Phe-Phe-Phe-OtBu | 3m | 88 | 98 |
| 2d | Boc-Phe-D-Phe-Phe-OtBu | 3n | 94 | 97 |
| 2c | Boc-Phe-Phe-Val-OtBu | 3o | 90 | 99 |
| 2d | Boc-Phe-D-Phe-Val-OtBu | 3p | 94 | 97 |
| 2e | Boc-Phe-Val-Gly-OtBub | 3q | 92 | 99 |
| 2f | Boc-Phe-D-Val-Gly-OtBub | 3r | 90 | 99 |
| 2e | Boc-Phe-Val-Ala-OtBub | 3s | 96 | 97 |
| 2f | Boc-Phe-D-Val-Ala-OtBub | 3t | 82 | 97 |
| 2e | Boc-Phe-Val-Phe-OtBuc | 3u | 90 | 98 |
| 2f | Boc-Phe-D-Val-Phe-OtBuc | 3v | 79 | 94 |
| 2e | Boc-Phe-Val-Val-OtBuc | 3w | 77 | 90 |
| 2f | Boc-Phe-D-Val-Val-OtBuc | 3x | 85 | 95 |
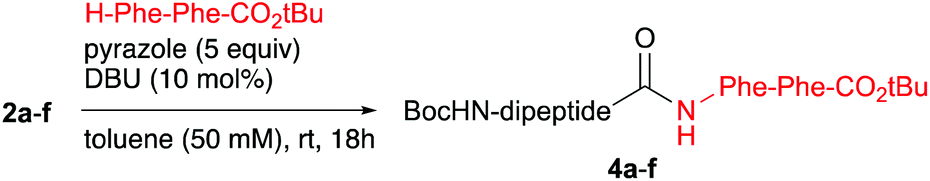
To mimic more closely a true peptide ligation reaction, the dipeptide isopropenyl esters 2a–f were subjected to pyrazole-catalysed aminolysis by reaction with dipeptide H-Phe-Phe-OtBu as the nucleophile.18 By application of the optimised conditions, Boc-Phe-Ala-Phe-Phe-OtBu (4a) and Boc-Phe-D-Ala-Phe-Phe-OtBu (4b) were isolated in yields of 68% and 79%, respectively. This drop of the yield as compared to using amino acid esters as the nucleophile was caused by the fact that the excess of apolar H-Phe-Phe-OtBu could not be removed by simply acidic washing. Moreover, chromatographic purification was unsuccessful due to the similar RF values of H-Phe-Phe-OtBu and the tetrapeptide products. The problem was overcome by reacting the crude mixture with Boc2O to transform the excess of unreacted nucleophile into Boc-Phe-Phe-OtBu, which could be separated via column chromatography. Fortunately, the de's of tetrapeptide 4a and its diasteromer 4b were both 99%, which is comparable to the tripeptide series. Similarly, tetrapeptides Boc-Phe-Phe-Phe-Phe-OtBu 4c and Boc-Phe-D-Phe-Phe-Phe-OtBu 4d were both isolated in a yield of 69% and gave de's of 93% and 95%, respectively. For obtaining tetrapeptide 4e and its diastereomer 4f, resulting from a coupling at C-terminal valine, the reaction mixture had to be warmed up to 70 °C to reach full conversion. Although a moderate yield had to be accepted, we were pleased that only little loss of de had occurred (Table 4).
|
|
||||
|---|---|---|---|---|
| Starting dipeptide | Tetrapeptide product | Yield (%) | dea (%) | |
| a Reaction run at 70 °C. | ||||
| 2a | Boc-Phe-Ala-Phe-Phe-OtBu | 4a | 68 | 99 |
| 2b | Boc-Phe-D-Ala-Phe-Phe-OtBu | 4b | 79 | 99 |
| 2c | Boc-Phe-Phe-Phe-Phe-OtBu | 4c | 69 | 93 |
| 2d | Boc-Phe-D-Phe-Phe-Phe-OtBu | 4d | 69 | 95 |
| 2e | Boc-Phe-Val-Phe-Phe-OtBua | 4e | 47 | 95 |
| 2f | Boc-Phe-D-Val-Phe-Phe-OtBua | 4f | 52 | 97 |
Conclusions
Robust, high yielding and scalable methodology was developed to transform the C-terminal carboxylic acid of dipeptides with complete stereoretention into isopropenyl esters via a CLE reaction. Further elongation into tri- and tetrapeptides was carried out via a mild pyrazole catalysed aminolysis reaction liberating acetone as an inert side product. For the tripeptide series, high yields of crude product were obtained, which required no further purification. Excellent de's (>96%) were observed for almost all tested tripeptides. Only hindered couplings, such as for making the connecting peptide bond within Val-Phe and Val–Val showed a small drop in de. For the tetrapeptide series similar results were observed, however in this case the Phe–Phe coupling also showed a small drop in de. Future work will focus on applications in peptide cyclizations and the ligation of larger, more diverse peptide fragments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank The Netherlands Organization for Scientific Research (NWO-CW, ECHO grant number 711.014.010 to J. v. M). E. Zuidinga and J. M. Ernsting (University of Amsterdam) are acknowledged for mass spectrometry and NMR assistance.Notes and references
- (a) A. Henninot, J. C. Collins and J. M. Nuss, J. Med. Chem., 2018, 61, 1382–1414 CrossRef CAS PubMed; (b) J L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700–2707 CrossRef CAS PubMed.
- S. S. Usmani, G. Bedi, J. S. Samuel, S. Singh, S. Kalra, P. Kumar, A. A. Ahuja, M. Sharma, A. Gautam and G. P. S. Raghave, PLoS One, 2017, 12, e0181748 CrossRef PubMed.
- (a) C. Griehl, A. Kolbe and S. Merkel, J. Chem. Soc., Perkin Trans. 2, 1996, 2525–2529 RSC; (b) S.-Y. Han and Y.-A. Kim, Tetrahedron, 2004, 60, 2447–2467 CrossRef CAS; (c) A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS PubMed.
- (a) Q. Wan, J. Chen, Y. Yuan and S. J. Danishefsky, J. Am. Chem. Soc., 2008, 130, 15814–15816 CrossRef CAS PubMed; (b) Q. Wan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 9248–9252 CrossRef CAS.
- A. Abdelmajeid, S. R. Tala, M. S. Amine and A. R. Katritzky, Synthesis, 2011, 2995–3005 CAS.
- (a) J. W. Bode, R. M. Fox and K. D. Baucom, Angew. Chem., Int. Ed., 2006, 45, 1248–1252 CrossRef CAS PubMed; (b) J. W. Bode, Acc. Chem. Res., 2017, 50, 2104–2115 CrossRef CAS PubMed.
- (a) H. Schröder, G. A. Strohmeier, M. Leypold, T. Nuijens, P. J. L. M. Quaedflieg and R. Breinbauer, Adv. Synth. Catal., 2013, 355, 1799–1807 CrossRef. For an overview of chemoenzymatic peptide ligations see: (b) M. Schmidt, A. Toplak, P. J. L. M. Quaedflieg, J. H. van Maarseveen and T. Nuijens, Drug Discovery Today, 2017, 26, 11–16 CrossRef PubMed.
- For a recent overview on the CLE-reaction see: (a) P. Y. S. Lam, Synthetic methods in drug discovery, 2016, ch. 7, vol. 1, p. 242 Search PubMed. See also: (b) J. Ohata, M. B. Minus, M. E. Abernathy and Z. T. Ball, J. Am. Chem. Soc., 2016, 138, 7472–7475 CrossRef CAS PubMed.
- S. Popovic, H. Bieräugel, R. J. Detz, A. M. Kluwer, J. A. A. Koole, D. E. Streefkerk, H. Hiemstra and J. H. van Maarseveen, Chem. – Eur. J., 2013, 19, 16934–16937 CrossRef CAS PubMed.
- L. Steemers, L. Wijsman and J. H. van Maarseveen, Adv. Synth. Catal. DOI:10.1002/adsc.201800914.
- X. Yang and V. B. Birman, Org. Lett., 2009, 11, 1499–1502 CrossRef CAS.
- H. C. Beyerman, W. M. van den Brink, F. Weygand, A. Prox, W. König, L. Schmidhammer and E. Nintz, Recl. Trav. Chim. Pays-Bas, 1965, 84, 213–231 CrossRef CAS.
- F. Kerins and D. F. O'Shea, J. Org. Chem., 2002, 67, 4968–4971 CrossRef CAS PubMed.
- S. Martinez-Montero, S. Fernandez, Y. S. Sanghvi, V. Gotor and M. Ferrero, Org. Biomol. Chem., 2011, 9, 5960–5966 RSC.
- X.-Q. Cai, N. Wang and X.-F. Lin, J. Mol. Catal. B: Enzym., 2006, 40, 51–57 CrossRef CAS.
- S. M. Tan, A. C. Willis, M. N. Paddon-Row and M. S. Sherburn, Angew. Chem., Int. Ed., 2016, 55, 3081–3085 CrossRef CAS PubMed.
- S. M. King and S. L. Buchwald, Org. Lett., 2016, 18, 4128–4131 CrossRef CAS PubMed.
- U. Filp, A. Pekosak, A. J. Poot and A. D. Windhorst, Eur. J. Org. Chem., 2017, 5592–5596 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and copies of the 1H- and 13C-NMR spectra and chiral HPLC traces. See DOI: 10.1039/c8ob02102c |
| This journal is © The Royal Society of Chemistry 2019 |