
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDibenzochrysene enables tightly controlled docking and stabilizes photoexcited states in dual-pore covalent organic frameworks†
Niklas
Keller‡
 ,
Torben
Sick‡
,
Nicolai N.
Bach
,
Andreas
Koszalkowski
,
Julian M.
Rotter
,
Dana D.
Medina
* and
Thomas
Bein
*
,
Torben
Sick‡
,
Nicolai N.
Bach
,
Andreas
Koszalkowski
,
Julian M.
Rotter
,
Dana D.
Medina
* and
Thomas
Bein
*
Department of Chemistry and Center for NanoScience (CeNS), University of Munich (LMU), Butenandtstraße 5-13, 81377 Munich, Germany. E-mail: bein@lmu.de
First published on 8th November 2019
Abstract
Covalent organic frameworks (COFs), consisting of covalently connected organic building units, combine attractive features such as crystallinity, open porosity and widely tunable physical properties. For optoelectronic applications, the incorporation of heteroatoms into a 2D COF has the potential to yield desired photophysical properties such as lower band gaps, but can also cause lateral offsets of adjacent layers. Here, we introduce dibenzo[g,p]chrysene (DBC) as a novel building block for the synthesis of highly crystalline and porous 2D dual-pore COFs showing interesting properties for optoelectronic applications. The newly synthesized terephthalaldehyde (TA), biphenyl (Biph), and thienothiophene (TT) DBC-COFs combine conjugation in the a,b-plane with a tight packing of adjacent layers guided through the molecular DBC node serving as specific docking site for successive layers. The resulting DBC-COFs exhibit a hexagonal dual-pore kagome geometry, which is comparable to COFs containing another molecular docking site, namely 4,4′,4′′,4′′′-(ethylene-1,1,2,2-tetrayl)-tetraaniline (ETTA). In this context, the respective interlayer distances decrease from about 4.6 Å in ETTA-COFs to about 3.6 Å in DBC-COFs, leading to well-defined hexagonally faceted single crystals sized about 50–100 nm. The TT DBC-COF features broad light absorption covering large parts of the visible spectrum, while Biph DBC-COF shows extraordinary excited state lifetimes exceeding 10 ns. In combination with the large number of recently developed linear conjugated building blocks, the new DBC tetra-connected node is expected to enable the synthesis of a large family of highly correlated and ordered 2D COFs with promising optoelectronic properties.
Introduction
Covalent organic frameworks (COFs) are crystalline polymers formed by covalently connecting relatively rigid and geometrically defined organic building blocks yielding larger molecular assemblies. Usually, the covalent linkages are achieved through slightly reversible condensation reactions forming boronic esters,1 boroxines,2 imines,3 imides4 or others.5–7 The high porosity, great structural diversity, tunability, and thermal stability of these frameworks make them attractive candidates for applications in energy storage,8–10 gas storage,11–13 proton conduction,14–16 optoelectronics,17–22 molecular separations,23–25 and catalysis.26–28 While in 3D COFs the backbones consist of building blocks covalently connected in all dimensions, in 2D COFs individual polymer layers stack in a defined manner to form molecular columns and 1D pores. This mode of layer assembly is governed by weak dispersive interactions between adjacent layers. These crystalline and porous materials offer numerous options regarding structural design, as tailoring of the pore size, shape and molecular interface is mainly governed by the selection of the building blocks. In the context of optoelectronics, this involves the potential ability to precisely manipulate the COFs’ electronic properties such as the optical absorption, charge carrier mobility29–33 or photoluminescence.2,7,34,35 The incorporation of heteroatoms within the structures enables the chemical linkage of different building blocks via strong covalent bonds, for example boron atoms in the case of boronic esters and boroxines or nitrogen atoms in the case of imines, imides or others. However, heteroatoms exhibit electrostatic repulsion in an eclipsed stacking of adjacent layers, which results in slight lateral offsets.36 Especially large and polarizable heteroatoms within adjacent COF layers prefer a stacking with lateral offsets with respect to one another.37,38 This phenomenon can also have a strong effect on the stacking distance of adjacent layers and thereby the overall long-range order in the framework.39 Furthermore, the polarization of certain chemical bonds may affect the stability of the frameworks.40 Strategies aimed at controlling stacking interactions through steric considerations include the implementation of sterically demanding pyrene building blocks for enforcing predefined offset stacking angles41 or the usage of propeller-like building blocks in imine-based frameworks.42 In the latter case, the propeller-like building blocks define specific molecular docking sites for adjacent COF sheets, thereby enforcing a perfectly eclipsed stacking of adjacent layers. However, due to the steric demands of molecular docking sites such as 4,4′,4′′,4′′′-(ethylene-1,1,2,2-tetrayl)-tetraaniline (ETTA),43 the stacking distance of adjacent layers of the resulting dual-pore kagome COF structures is comparably large. The enlarged stacking distance in combination with out-of-plane rotated building blocks may have an influence on the π-electron overlap of adjacent COF layers and on the conjugation within each COF layer.Dibenzochrysenetetraamine (DBCTA) is a rigid molecular building block consisting of four benzene rings fused to a naphthyl unit. This molecular configuration can be envisioned as a fused ETTA analogue which enables a more subtle rotation of the attached phenyl rings out of plane compared with ETTA.44 Therefore, the incorporation of DBCTA into a framework is expected to drastically reduce the steric constraints for stacking, allowing for more closely packed molecular assemblies.45 In addition, the DBC core and its derivatives exhibit attractive photophysical properties and charge carrier mobilities.44,46,47 This combination of properties is promising for the realization of highly ordered materials based on the DBC nodes with encoded features for optoelectronic applications. In the context of framework materials, COF-based optoelectronic devices are still rare48 and further breakthroughs in molecular design are anticipated to enable the construction of efficient devices.
Herein, we investigate the possibility of generating highly ordered COF structures by means of a molecular node intended to induce close π-stacking distances and specific docking sites. For this purpose we utilized dibenzo[g,p]chrysene, DBC, as a structure-directing node. We synthesized a series of DBC-containing imine COFs with linear dialdehydes, namely terephthalaldehyde (TA), 4,4′-biphenyldicarbaldehyde (Biph) and thieno[3,2-b]thiophene-2,5-dicarboxaldehyde (TT). We present a detailed structural analysis showing the formation of highly crystalline and porous frameworks featuring outstanding crystal quality with hexagonal faceting. Furthermore, the photophysical properties of the DBC-based COFs, i.e. UV-vis absorption as well as steady-state and time-resolved photoluminescence (PL) were studied. Here, TT DBC-COF featured broad light absorption covering large parts of the visible spectrum. Strikingly, the Biph DBC-COF powder showed extended PL lifetimes as high as 2–14 ns with 62% of the overall signal.
Results and discussion
The DBC-based COF series was synthesized by the (slightly) reversible reaction of DBCTA with the respective linear dialdehydes TA, Biph, and TT to form imine-based COFs (Fig. S10–S12†). Briefly, 30 μmol of the linear dialdehyde and 15 μmol of the fourfold amine functionalized DBC (DBCTA) were suspended in a mixture of benzyl alcohol and mesitylene (500 μL) in a culture tube. After the addition of 50 μL of acetic acid (6 M), the sealed tubes were kept in an oven at 120 °C for 72 h. The resulting precipitate was isolated by filtration, rinsed and finally Soxhlet extracted with anhydrous THF for 2 h. After vacuum-drying, the COF powders were examined concerning their crystallinity, porosity and photophysical properties (for more details on the synthesis procedures see ESI†).X-ray diffraction (XRD) analysis of the obtained powders revealed highly crystalline materials with sharp and intense reflections at small diffraction angles and multiple additional high order reflections at higher diffraction angles (Fig. 1b–d). To confirm the formation of a kagome dual-pore hexagonal structure, simulations of the possible crystal structures were carried out assuming an AA model layer arrangement in the hexagonal space group P6 (see section 7 in the ESI†). Using this model, the experimental diffraction pattern was fully indexed and the observed reflections were attributed to hkl 100, 110, 200, 210, 120, 300, 220, 310, 400 and 500 (by order of appearance) indicating the excellent crystallinity and the formation of a kagome structure in all the examined samples (Fig. 1). For all the DBC-COFs, the observed reflection at around 23.9° 2θ is attributed to the 001 plane, which directly corresponds to the π-stacking distance. Accordingly, a d-spacing of about 3.6 Å was calculated for the examined DBC-COFs.
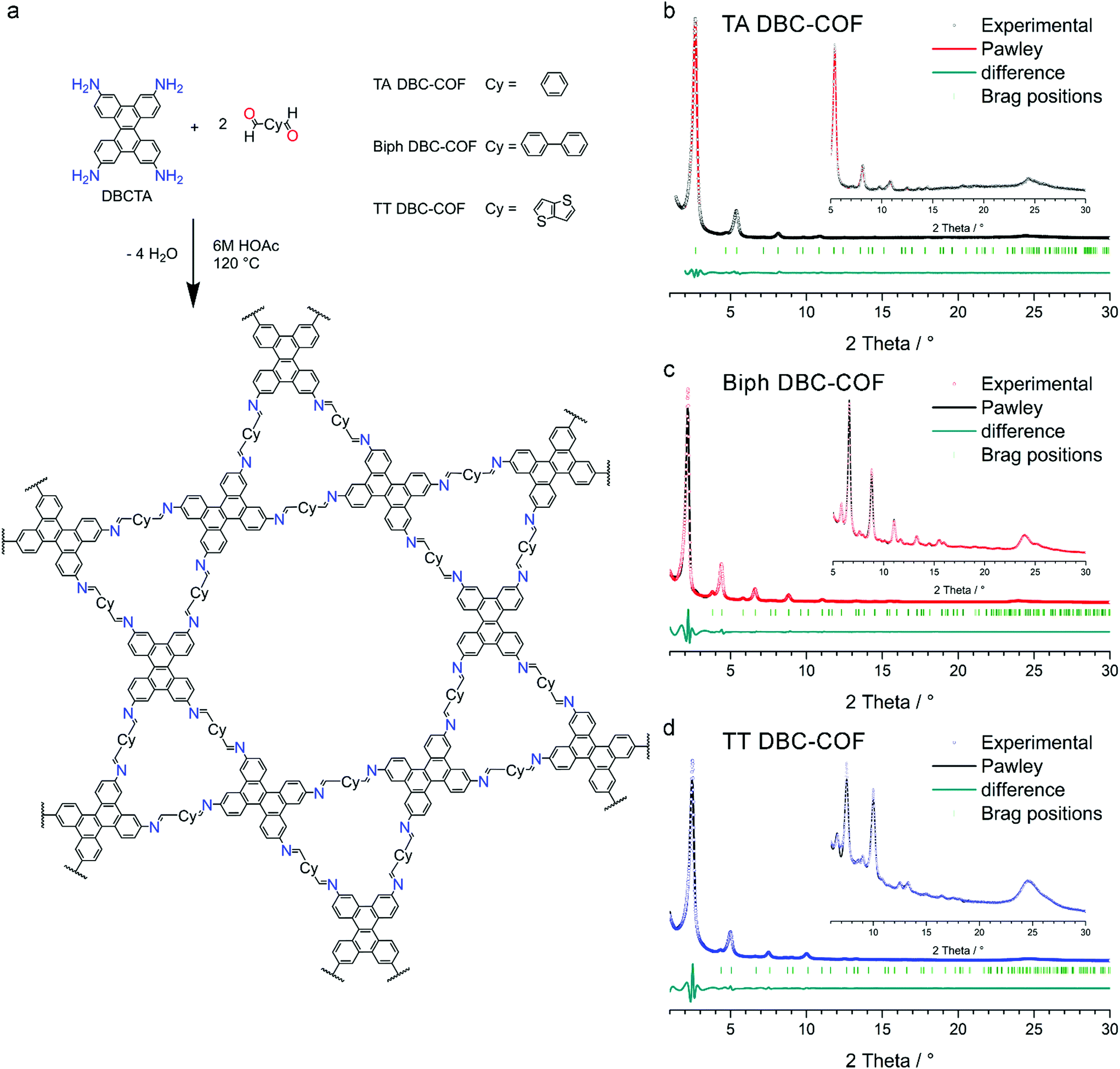 | ||
| Fig. 1 (a) Structure of the DBC-containing COFs (schematic). (b)–(d) PXRD patterns of the TA, Biph, and TT DBC-COFs with Pawley-refined simulations. The insets show higher order reflections in magnification. | ||
Nitrogen physisorption isotherms of the COFs exhibit a mixture of type I and type IVb isotherm shapes with two steep nitrogen uptakes, the one at low partial pressures indicates the presence of a microporous system and the one at relatively higher partial pressures characteristic for mesoporous systems (<5 nm) (Fig. 2a and Fig. S6–S8†).49 This confirms that DBC-COFs adopt a dual-pore structure similar to their ETTA-COF analogs.50 Calculated pore size distributions based on a QSDFT carbon kernel for cylindrical pores show excellent agreement with the structurally modeled pore sizes (Fig. S6–S8†). The DFT-calculated pore sizes were 1.50 nm and 2.89 nm (simulated 1.5 nm and 2.9 nm), 1.73 nm and 3.68 nm (simulated 1.7 nm and 3.7 nm) and 1.45 nm and 3.10 nm (simulated 1.5 nm and 3.1 nm) for TA DBC-COF, Biph DBC-COF, and TT DBC-COF, respectively. The BET surface areas and pore volumes of 2050 m2 g−1 and 1.23 cm3 g−1 (TA DBC-COF), 2170 m2 g−1 and 1.39 cm3 g−1 (Biph DBC-COF) as well as 1230 m2 g−1 and 0.67 cm3 g−1 (TT DBC-COF) can be compared to predicted accessible Connolly surface areas and pore volumes of 2090 m2 g−1 and 0.90 cm3 g−1 (TA DBC-COF), 2490 m2 g−1 and 1.31 cm3 g−1 (Biph DBC-COF) and 2260 m2 g−1 and 1.12 cm3 g−1 (TT DBC-COF), respectively.
 | ||
| Fig. 2 (a) Nitrogen physisorption isotherm of Biph DBC-COF. (b) SEM and (c) TEM micrographs of Biph DBC-COF. | ||
Thermogravimetric analysis under synthetic air flow revealed that DBC-COFs are thermally stable, while significant weight loss associated with structural degradation is detected at around 450 °C, and the degradation process terminates at around 600 °C with a complete weight loss (Fig. S20†).
Morphological investigations of the DBC-COFs using scanning electron microscopy (SEM) revealed well-defined crystallites of different sizes, ranging from 10 to 150 nm, forming large intergrown objects (Fig. S1†). In the case of Biph DBC-COF, the agglomerated particles consist of faceted hexagonally shaped crystals. This is in excellent agreement with a parallel report illustrating the high crystallinity of Biph-DBC COF.45,51 Transmission electron microscopy (TEM) micrographs show the highly crystalline nature of the COFs (Fig. S2†). Strikingly, the Biph DBC-COF exhibits large numbers of hexagonally faceted single crystals sized about 50 to 100 nm throughout the sample (Fig. 2).
In comparison to the previously reported ETTA systems with the same linear dialdehydes, namely TA, Biph, and TT, the reflections associated with the stacking distances in PXRD pattern are shifted from about 19.5° 2θ to 23.9° 2θ, corresponding to a reduction of stacking distances of about 4.6 Å (for TA ETTA-COF, Biph ETTA-COF, and TT ETTA-COF)42 to 3.6 Å (TA and TT DBC-COFs), and 3.7 Å (Biph DBC-COF). This strongly decreased stacking distance (by almost 1.0 Å) for each DBC-containing COF is attributed to the more planar (gently curved) shape of the chrysene building block compared to the more sterically demanding ETTA node.45 The influence of the linear dialdehyde on the stacking distance is apparently less dominant, though being still detectable.
Reported single crystal structures of the core elements of the different 4-connected COF building blocks, i.e., ETTA and DBC, clearly indicate the influence on the bond angles and steric requirements of the differently linked aromatic groups. In ETTA, an average dihedral angle of 47.3°![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 52 was determined, while in DBC the average angle reduces to 24.7°.53 Here, the angles were determined for two intersecting planes created, in ETTA, by the ethylene bond and the sequentially connected two carbon atoms in the aromatic substitution. The dihedral angle for the corresponding chrysene molecule was determined in the same manner by considering the same atom connectivity (Fig. S9†). Therefore, using a DBC node results in a COF kagome structure and connectivity similar to the ETTA-COF analogs, but the presence of a rigid naphthyl unit strongly reduces the torsion/rotation of the attached aromatic groups compared to the aromatic groups connected to the ethylene unit in ETTA. These findings are in line with the reported dihedral angles for ETTA and DBC.45 As a result, the π-stacking distance between adjacent COF layers is expected to be reduced while the existing DBC torsion angle is still sufficient to enable specific docking effects for the successive layers. This is expected to enable defined π–π-interactions in the stacks and improved π-orbital overlap within the individual layers allowing for extended electron delocalization.
52 was determined, while in DBC the average angle reduces to 24.7°.53 Here, the angles were determined for two intersecting planes created, in ETTA, by the ethylene bond and the sequentially connected two carbon atoms in the aromatic substitution. The dihedral angle for the corresponding chrysene molecule was determined in the same manner by considering the same atom connectivity (Fig. S9†). Therefore, using a DBC node results in a COF kagome structure and connectivity similar to the ETTA-COF analogs, but the presence of a rigid naphthyl unit strongly reduces the torsion/rotation of the attached aromatic groups compared to the aromatic groups connected to the ethylene unit in ETTA. These findings are in line with the reported dihedral angles for ETTA and DBC.45 As a result, the π-stacking distance between adjacent COF layers is expected to be reduced while the existing DBC torsion angle is still sufficient to enable specific docking effects for the successive layers. This is expected to enable defined π–π-interactions in the stacks and improved π-orbital overlap within the individual layers allowing for extended electron delocalization.
Next, we investigated the photophysical properties of the DBC-COFs by means of UV-vis, PL and time-correlated single photon counting (TCSPC) spectroscopies. The influence of the respective linear building block on the optical absorption can be clearly seen in the absorbance spectra recorded for the DBC-COF powders (see Fig. 3 and Fig. S13†). While the phenyl-based linear linkers TA and Biph exhibit light brown color, comparable absorption features and absorption onsets at around 550 to 580 nm, the incorporation of thienothiophene into the framework shifts the steep onset to longer wavelengths at around 650 nm. Consequently, the TT DBC-COF exhibits dark red color and a comparably smaller band gap, which was calculated by Tauc plot. Assuming a direct optical band gap for the DBC-COFs, we obtained a band gap of 2.00 eV for the TT DBC-COF. In comparison, the direct optical band gaps of TA DBC-COF and Biph DBC-COF were calculated to be 2.30 eV and 2.38 eV, respectively (Fig. S14†). In the context of extended conjugation within the COF layers, the TT DBC-COF exhibits a clear red-shifted absorption onset compared to its ETTA analogue. This extended absorbance can be attributed to the effect of closely packed molecular aggregates and larger π-electron delocalization attained by the geometry of the DBC node.
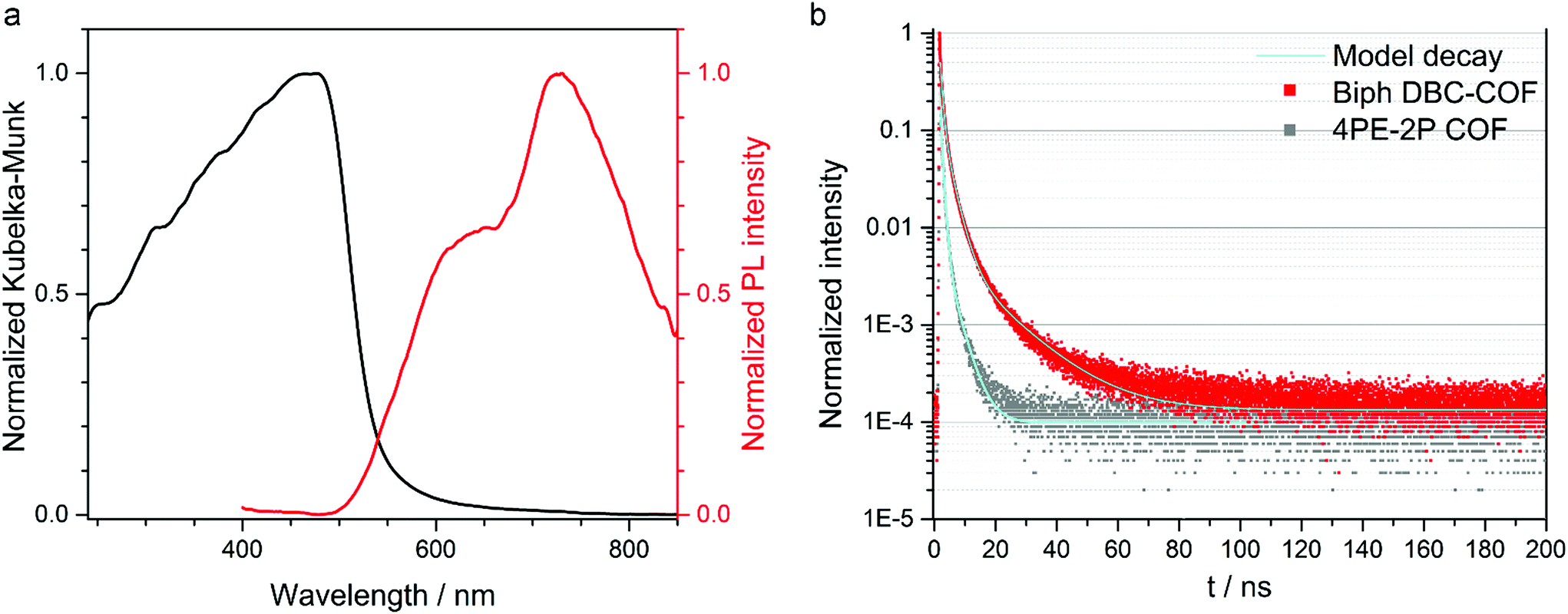 | ||
| Fig. 3 (a) Optical absorption spectrum (black) measured in diffuse reflectance mode, and PL (λexc = 378 nm, red) spectrum of Biph DBC-COF powder. (b) PL decay curve of Biph DBC-COF (red) and the ETTA-based 4PE-2P COF (grey). The samples were illuminated at 378 nm with a pump fluence of ∼0.99 nJ cm−2; the emission was monitored at 724 and 643 nm, respectively (model decay in cyan). | ||
TA and Biph DBC-COFs exhibit similar PL spectra consisting of two distinct emission bands at around 640 and 720 nm, where the latter features stronger emission intensity. TT DBC-COF is slightly red-shifted and reveals a broader PL maximum at 760 nm with a slight shoulder at 660 nm. For all the investigated DBC-COFs, the PL covers a broad spectral range from the visible to the near-infrared (see Fig. 3 and Fig. S16†). In comparison to the individual emission of the pristine, molecular components, the PL maxima of all DBC-COFs are significantly red-shifted, which points toward the emergence of new electronic structure through the formation of the framework (Fig. S15†).
To study the PL decay dynamics, time-correlated single photon counting (TCSPC) was employed using a pulsed 378 nm laser under nitrogen atmosphere (section 12 in the ESI†), revealing the lifetimes of the photoexcited singlet states generated in the DBC-COF powders. The obtained decay curves were fitted with a tri-exponential function. The longest lifetimes were observed for the Biph DBC-COF, which features a long-time decay constant of 14.1 ns (12%) (see Fig. 3). The additional two exponential time constants indicate 2.8 ns (50%) and 0.7 ns (38%). For the TA DBC-COF lifetimes of up to 7 ns were detected and the TT DBC-COF showed the fastest decay times for this series of DBC-COFs of up to 5 ns (for more details see ESI†).
As illustrated, small modifications in the molecular structure of the building blocks can strongly impact the structural properties of the COF, here, a stacking distance reduced by about 1.0 Å was obtained with a DBC node. In addition, indications for enhanced photophysical properties in DBC-COF were observed. To study the differences in the photophysical properties of these two COF families, we compared the PL decay dynamics of DBC-COFs featuring tight molecular stacks with the ones of their ETTA-based COF analogues.42 For all the examined COFs the decay profiles were fitted with tri-exponential decay functions. In general, the decay times obtained for the DBC-COFs were significantly longer, leading to overall longer PL lifetimes (see Fig. 3 and section 12 in the ESI†). The Biph DBC-COF exhibits the longest lifetimes, of more than 10 ns longer compared to its corresponding 4PE-2P COF analog. 4PE-2P COF features PL decay times of 3.6 ns (11%) as the long-time constant, the rest of the PL signal (90%) arises from fast decay channels below 1 ns (0.8 ns (50%) and 0.14 ns (39%)). We postulate that in DBC-COFs the structural features greatly influence the PL decay and induce a stabilization of the photoexcited species in the COF. The impact of stacking on the electronic structure of COFs has been recently reported.54 In that study, well-stacked COF layers were calculated to yield smaller band gaps than isolated COF sheets. This agrees with our observations for the DBC-COFs compared to the ETTA-based COF analogs. We therefore attribute the decrease of the bandgap (and the possibly resulting changes of the band dispersions) to the better π-orbital overlap between the stacked chrysene docking sites due to lower dihedral angles resulting in short π-distances. The proximity of the layers in the DBC-COFs may allow for enhanced delocalization of the excited states across the π-stacked chrysene columns and thereby extend their relative lifetimes.
Conclusions
In this study, we have established dibenzo[g,p]chrysene (DBC) as a novel structural node for the construction of 2D COFs, serving as a specific docking site for the synthesis of flattened structural analogues of the well-studied ETTA-based COFs. Different DBC-COFs were synthesized with different linear linkers to not only vary the pore sizes but more importantly the photophysical properties. DBC-COFs show extraordinarily high crystallinity, porosity and structural precision, which is reflected in the formation of hexagonally faceted single crystals. Strikingly, in comparison with ETTA-based COF analogues the π-stacking distance of adjacent layers was drastically reduced by 1 Å, from about 4.6 Å in ETTA-COFs to about 3.6 Å in DBC-COFs. Among the newly synthesized, tightly π-stacked DBC-COFs, TT DBC-COF is viewed to be a promising candidate for photovoltaic applications, as it combines a relatively small band gap and a steep absorption onset with a broad optical absorption ranging from the UV deep into the visible. The time-resolved photoluminescence profile of Biph DBC-COF shows remarkably long lifetimes, partially exceeding 10 ns. In general, the DBC-COFs showed significantly slower decay dynamics compared to their ETTA analogues. Further mechanistic investigations in our laboratories regarding the charge carrier dynamics in these intriguing molecular frameworks are expected to shed light on the impact of the building blocks and the nature of the layer stacking on the optoelectronic properties. In view of the above, dibenzochrysene-based COFs represent a promising platform of tuneable molecular framework materials with intriguing prospects for optoelectronic applications.Experimental
Synthetic procedures
Synthesis of TA DBC-COF
In a 6 mL culture tube, terephthalaldehyde (4.02 mg, 30.0 μmol) and DBCTA (5.83 mg, 15.0 μmol) were suspended in a mixture of BnOH and mesitylene (500 μL, v:v 9:1). After adding 50 μL of acetic acid (aqueous, 6 M) the tube was sealed and heated at 120 °C for 72 h. The resulting precipitate was filtered and rinsed with anhydrous THF before Soxhlet extraction with anhydrous THF for 12 h. The final product was vacuum-dried, to yield 5.07 mg (58%) as a light brown powder.
Synthesis of Biph DBC-COF
In a 6 mL culture tube, 4,4′-biphenyldicarboxaldehyde (6.31 mg, 30.0 μmol) and DBCTA (5.83 mg, 15.0 μmol) were suspended in a mixture of BnOH and mesitylene (500 μL, v:v 1:1). After adding 50 μL of acetic acid (aqueous, 6 M) the culture tube was sealed and heated at 120 °C for 72 h. The resulting precipitate was filtered and rinsed with anhydrous THF before Soxhlet extraction with anhydrous THF for 12 h. The final product was vacuum-dried, to yield 5.47 mg (50%) as a brown powder.
Synthesis of TT DBC-COF
In a 6 mL culture tube, thieno[3,2-b]thiophene-2,5-dicarboxaldehyde (5.89 mg, 30.0 μmol) and DBCTA (5.83 mg, 15.0 μmol) were suspended in a mixture of BnOH and mesitylene (500 μL, v:v 9:1). After adding 50 μL of acetic acid (aqueous, 6 M) the tube was sealed and heated at 120 °C for 72 h. The resulting precipitate was filtered and rinsed with anhydrous THF before Soxhlet extraction with anhydrous THF for 12 h. The final product was vacuum-dried, to yield 5.87 mg (55%) as a red powder.
Synthesis of analogue 4PE COFs
The ETTA-based COFs, 4PE-1P, 4PE-2P and 4PE-TT, are well-studied COF systems. Their synthesis here was carried out under the reported conditions previously reported in the literature.42Materials and methods
All reagents and solvents were obtained from in house supply or commercial suppliers and used as received. Acetic acid (in house supply), benzyl alcohol (BnOH, anhydrous, Sigma-Aldrich), 4,4′-biphenyldicarboxaldehyde (Biph, >98%, TCI), 1,4-dioxane (anhydrous, Sigma Aldrich), dibenzo[g,p]chrysene (DBC, >98%, TCI), ethanol (anhydrous, Sigma Aldrich), hydrazine monohydrate (Sigma Aldrich), mesitylene (anhydrous, Sigma-Aldrich), nitrobenzene (anhydrous, Sigma Aldrich), RANEY® Nickel slurry (Sigma Aldrich), terephthalaldehyde (TA, 99%, Sigma-Aldrich), tetrahydrofuran (THF, extra dry, stabilized, Acros Organics). Thieno[3,2-b]thiophene-2,5-dicarboxaldehyde (TT, >93%, TCI) was recrystallized from N,N-dimethylformamide (DMF, anhydrous, 99.8%, Sigma-Aldrich).Nuclear magnetic resonance (NMR) spectra were recorded on Bruker AV 400 and AV 400 TR spectrometers. Proton chemical shifts are expressed in parts per million (δ scale) and are calibrated using residual non-deuterated solvent peaks as internal reference (e.g. DMSO-d6: 2.50 ppm).
Infrared (IR) spectra were recorded on a PerkinElmer Spectrum BX II FT-IR system and a Thermo Scientific Nicolet™ 6700 FT-IR spectrometer in transmission mode. IR data are reported in wavenumbers (cm−1).
UV-vis spectra were recorded using a PerkinElmer Lambda 1050 spectrometer equipped with a 150 mm integrating sphere. Diffuse reflectance spectra were collected with a Praying Mantis (Harrick) accessory and were referenced to barium sulfate powder as white standard.
Nitrogen sorption isotherms were recorded on a Quantachrome Autosorb 1 at 77 K within a pressure range of p/p0 = 0.001 to 0.98. Prior to the measurement of the sorption isotherms the samples were heated for 24 h at 120 °C under turbo-pumped vacuum. For the evaluation of the surface area the BET model was applied between 0.05 and 0.3 p/p0. Pore size distributions were calculated using the QSDFT equilibrium model (desorption branch) with a carbon kernel for cylindrical pores. Connolly surfaces were generated using an N2-sized probe (r = 0.184 nm) at a 0.025 nm grid interval.
Thermogravimetric analysis (TGA) measurements were performed on a Netzsch Jupiter ST 449 C instrument equipped with a Netzsch TASC 414/4 controller. The samples were heated from room temperature to 900 °C under a synthetic air flow (25 mL min−1) at a heating rate of 10 K min−1.
Powder X-ray diffraction (PXRD) measurements were performed using a Bruker D8 Discover with Ni-filtered Cu-Kα radiation and a LynxEye position-sensitive detector (scan speed of 4 s per 0.01° 2θ). Experimental XRD data were used for Pawley refinement to optimize the hypothetical structure.
The initial structure models of the COFs were built using the Forcite module of the Accelrys Materials Studio software package. We applied the space group with the highest possible symmetry, i.e. P6, considering the specific conformation of the DBC building blocks. Using this coarse model, we determined the unit cell parameters via Pawley refinement of our PXRD data.
Transmission electron microscopy (TEM) was performed on an FEI Titan Themis equipped with a field emission gun operated at 300 kV. Scanning electron microscopy (SEM) images were recorded with a JEOL 6500F and an FEI Helios NanoLab G3 UC scanning electron microscope equipped with a field emission gun operated at 3–5 kV.
Photoluminescence (PL) and time-correlated single photon counting (TCSPC) data were processed with a FluoTime 300 from PicoQuant GmbH. The samples were photo-excited using lasers with suitable wavelengths according to the sample absorption, i.e. 378 nm, 403 nm or 507 nm wavelength (LDH-P-C-375, LDH-P-C-405, and LDH-P-C-510, respectively, all from PicoQuant GmbH) pulsed at 500 kHz, with a pulse duration of ∼100 ps and fluence of ∼300 nJ per cm−2 per pulse. The samples were exposed to the pulsed light source set at 3 μJ per cm−2 per pulse fluence for ∼10 minutes prior to measurement to ensure stable sample emission. The PL was collected using a high-resolution monochromator and photomultiplier detector assembly (PMA-C 192-N-M, PicoQuant GmbH).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for funding from the German Science Foundation (DFG) for SPP 1613, the NIM and e-conversion Excellence Clusters (DFG), and from the Free State of Bavaria (research network ‘Solar Technologies Go Hybrid’). Research leading to these results received funding from the European Research Council under the European Union's Seventh Framework Programme (FP7/2007-3013)/ERC grant Agreement 321339. The authors thank Dr Markus Döblinger for electron microscopy.Notes and references
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2009, 48, 5439–5442 CrossRef CAS PubMed.
- F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klöck, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 4570–4571 CrossRef CAS.
- Q. Fang, Z. Zhuang, S. Gu, R. B. Kaspar, J. Zheng, J. Wang, S. Qiu and Y. Yan, Nat. Commun., 2014, 5, 4503 CrossRef.
- F. J. Uribe-Romo, C. J. Doonan, H. Furukawa, K. Oisaki and O. M. Yaghi, J. Am. Chem. Soc., 2011, 133, 11478–11481 CrossRef CAS.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed.
- S. Dalapati, S. Jin, J. Gao, Y. Xu, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2013, 135, 17310–17313 CrossRef CAS PubMed.
- H. Yang, S. Zhang, L. Han, Z. Zhang, Z. Xue, J. Gao, Y. Li, C. Huang, Y. Yi, H. Liu and Y. Li, ACS Appl. Mater. Interfaces, 2016, 8, 5366–5375 CrossRef CAS.
- C. R. DeBlase, K. Hernández-Burgos, K. E. Silberstein, G. G. Rodríguez-Calero, R. P. Bisbey, H. D. Abruña and W. R. Dichtel, ACS Nano, 2015, 9, 3178–3183 CrossRef CAS.
- Y. Hu, N. Dunlap, S. Wan, S. Lu, S. Huang, I. Sellinger, M. Ortiz, Y. Jin, S.-h. Lee and W. Zhang, J. Am. Chem. Soc., 2019, 141, 7518–7525 CrossRef CAS PubMed.
- G.-Y. Lee, J. Lee, H. T. Vo, S. Kim, H. Lee and T. Park, Sci. Rep., 2017, 7, 557 CrossRef.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS.
- D. B. Shinde, H. B. Aiyappa, M. Bhadra, B. P. Biswal, P. Wadge, S. Kandambeth, B. Garai, T. Kundu, S. Kurungot and R. Banerjee, J. Mater. Chem. A, 2016, 4, 2682–2690 RSC.
- H. Xu, S. Tao and D. Jiang, Nat. Mater., 2016, 15, 722 CrossRef CAS.
- S. Chandra, T. Kundu, S. Kandambeth, R. BabaRao, Y. Marathe, S. M. Kunjir and R. Banerjee, J. Am. Chem. Soc., 2014, 136, 6570–6573 CrossRef CAS PubMed.
- D. D. Medina, V. Werner, F. Auras, R. Tautz, M. Dogru, J. Schuster, S. Linke, M. Döblinger, J. Feldmann, P. Knochel and T. Bein, ACS Nano, 2014, 8, 4042–4052 CrossRef CAS.
- M. Calik, F. Auras, L. M. Salonen, K. Bader, I. Grill, M. Handloser, D. D. Medina, M. Dogru, F. Löbermann, D. Trauner, A. Hartschuh and T. Bein, J. Am. Chem. Soc., 2014, 136, 17802–17807 CrossRef CAS PubMed.
- D. Bessinger, L. Ascherl, F. Auras and T. Bein, J. Am. Chem. Soc., 2017, 139, 12035–12042 CrossRef CAS PubMed.
- S. Wuttke, D. D. Medina, J. M. Rotter, S. Begum, T. Stassin, R. Ameloot, M. Oschatz and M. Tsotsalas, Adv. Funct. Mater., 2018, 28, 1801545 CrossRef.
- A. C. Jakowetz, T. F. Hinrichsen, L. Ascherl, T. Sick, M. Calik, F. Auras, D. D. Medina, R. H. Friend, A. Rao and T. Bein, J. Am. Chem. Soc., 2019, 141, 11565–11571 CrossRef CAS PubMed.
- Y. Zhang, Y. Hu, J. Zhao, E. Park, Y. Jin, Q. Liu and W. Zhang, J. Mater. Chem. A, 2019, 7, 16364–16371 RSC.
- X. Han, J. Zhang, J. Huang, X. Wu, D. Yuan, Y. Liu and Y. Cui, Nat. Commun., 2018, 9, 1294 CrossRef PubMed.
- L.-H. Liu, C.-X. Yang and X.-P. Yan, J. Chromatogr., A, 2017, 1479, 137–144 CrossRef CAS PubMed.
- X. Guan, H. Li, Y. Ma, M. Xue, Q. Fang, Y. Yan, V. Valtchev and S. Qiu, Nat. Chem., 2019, 11, 587–594 CrossRef CAS PubMed.
- S.-Y. Ding, J. Gao, Q. Wang, Y. Zhang, W.-G. Song, C.-Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef CAS PubMed.
- X. Han, Q. Xia, J. Huang, Y. Liu, C. Tan and Y. Cui, J. Am. Chem. Soc., 2017, 139, 8693–8697 CrossRef CAS PubMed.
- S. Lu, Y. Hu, S. Wan, R. McCaffrey, Y. Jin, H. Gu and W. Zhang, J. Am. Chem. Soc., 2017, 139, 17082–17088 CrossRef CAS PubMed.
- D. D. Medina, M. L. Petrus, A. N. Jumabekov, J. T. Margraf, S. Weinberger, J. M. Rotter, T. Clark and T. Bein, ACS Nano, 2017, 11, 2706–2713 CrossRef CAS PubMed.
- S.-L. Cai, Y.-B. Zhang, A. B. Pun, B. He, J. Yang, F. M. Toma, I. D. Sharp, O. M. Yaghi, J. Fan, S.-R. Zheng, W.-G. Zhang and Y. Liu, Chem. Sci., 2014, 5, 4693–4700 RSC.
- S. Patwardhan, A. A. Kocherzhenko, F. C. Grozema and L. D. A. Siebbeles, J. Phys. Chem. C, 2011, 115, 11768–11772 CrossRef CAS.
- S. Wan, F. Gándara, A. Asano, H. Furukawa, A. Saeki, S. K. Dey, L. Liao, M. W. Ambrogio, Y. Y. Botros, X. Duan, S. Seki, J. F. Stoddart and O. M. Yaghi, Chem. Mater., 2011, 23, 4094–4097 CrossRef CAS.
- H. Li, J. Chang, S. Li, X. Guan, D. Li, C. Li, L. Tang, M. Xue, Y. Yan, V. Valtchev, S. Qiu and Q. Fang, J. Am. Chem. Soc., 2019, 141, 13324–13329 CrossRef CAS PubMed.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2008, 47, 8826–8830 CrossRef CAS PubMed.
- S. Dalapati, E. Jin, M. Addicoat, T. Heine and D. Jiang, J. Am. Chem. Soc., 2016, 138, 5797–5800 CrossRef CAS PubMed.
- B. Lukose, A. Kuc, J. Frenzel and T. Heine, Beilstein J. Nanotechnol., 2010, 1, 60–70 CrossRef CAS PubMed.
- F. Haase, K. Gottschling, L. Stegbauer, L. S. Germann, R. Gutzler, V. Duppel, V. S. Vyas, K. Kern, R. E. Dinnebier and B. V. Lotsch, Mater. Chem. Front., 2017, 1, 1354–1361 RSC.
- B. Lukose, A. Kuc and T. Heine, Chem. – Eur. J., 2011, 17, 2388–2392 CrossRef CAS PubMed.
- M. S. Lohse, J. M. Rotter, J. T. Margraf, V. Werner, M. Becker, S. Herbert, P. Knochel, T. Clark, T. Bein and D. D. Medina, CrystEngComm, 2016, 18, 4295–4302 RSC.
- H. Xu, J. Gao and D. Jiang, Nat. Chem., 2015, 7, 905–912 CrossRef CAS PubMed.
- F. Auras, L. Ascherl, A. H. Hakimioun, J. T. Margraf, F. C. Hanusch, S. Reuter, D. Bessinger, M. Döblinger, C. Hettstedt, K. Karaghiosoff, S. Herbert, P. Knochel, T. Clark and T. Bein, J. Am. Chem. Soc., 2016, 138, 16703–16710 CrossRef CAS PubMed.
- L. Ascherl, T. Sick, J. T. Margraf, S. H. Lapidus, M. Calik, C. Hettstedt, K. Karaghiosoff, M. Döblinger, T. Clark, K. W. Chapman, F. Auras and T. Bein, Nat. Chem., 2016, 8, 310–316 CrossRef CAS.
- T. Y. Zhou, S. Q. Xu, Q. Wen, Z. F. Pang and X. Zhao, J. Am. Chem. Soc., 2014, 136, 15885–15888 CrossRef CAS.
- Y. Ueda, H. Tsuji, H. Tanaka and E. Nakamura, Chem.– Asian J., 2014, 9, 1623–1628 CrossRef CAS PubMed.
- Z. Xie, B. Wang, Z. Yang, X. Yang, X. Yu, G. Xing, Y. Zhang and L. Chen, Angew. Chem., Int. Ed., 2019, 58, 15742–15746 CrossRef CAS PubMed.
- R. Khatua, S. R. Sahoo, S. Sharma, R. Thangavel and S. Sahu, J. Phys. Org. Chem., 2018, 31, e3859 CrossRef.
- X.-Y. Liu, X. Tang, Y. Zhao, D. Zhao, J. Fan and L.-S. Liao, Dyes Pigm., 2017, 146, 234–239 CrossRef CAS.
- D. D. Medina, T. Sick and T. Bein, Adv. Energy Mater., 2017, 7, 1700387 CrossRef.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051 CAS.
- Y. Jin, Y. Hu and W. Zhang, Nat. Rev. Chem., 2017, 1, 0056 CrossRef.
- N. Keller, T. Sick, N. N. Bach, A. Koszalkowski, J. M. Rotter, D. D. Medina and T. Bein, ChemRxiv, 2019, DOI:10.26434/chemrxiv.9638633.v2.
- G.-F. Zhang, Z.-Q. Chen, M. P. Aldred, Z. Hu, T. Chen, Z. Huang, X. Meng and M.-Q. Zhu, Chem. Commun., 2014, 50, 12058–12060 RSC.
- T. Hatakeyama, S. Hashimoto, S. Seki and M. Nakamura, J. Am. Chem. Soc., 2011, 133, 18614–18617 CrossRef CAS PubMed.
- M. Wang, M. Ballabio, M. Wang, H.-H. Lin, B. P. Biswal, X. Han, S. Paasch, E. Brunner, P. Liu, M. Chen, M. Bonn, T. Heine, S. Zhou, E. Cánovas, R. Dong and X. Feng, J. Am. Chem. Soc., 2019, 141, 16810–16816 CrossRef CAS PubMed.
- J. Lu and J. Zhang, J. Mater. Chem. A, 2014, 2, 13831–13834 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr08007d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |