
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Plant defensin PvD1 modulates the membrane composition of breast tumour-derived exosomes†
Julia
Skalska
 a,
Filipa D.
Oliveira
a,
Tiago N.
Figueira
a,
Érica O.
Mello
b,
Valdirene M.
Gomes
b,
Grant
McNaughton-Smith
c,
Miguel A. R. B.
Castanho
*a and
Diana
Gaspar
a
a,
Filipa D.
Oliveira
a,
Tiago N.
Figueira
a,
Érica O.
Mello
b,
Valdirene M.
Gomes
b,
Grant
McNaughton-Smith
c,
Miguel A. R. B.
Castanho
*a and
Diana
Gaspar
a
aInstituto de Medicina Molecular, Faculdade de Medicina, Universidade de Lisboa, Lisbon, Portugal. E-mail: macastanho@medicina.ulisboa.pt
bLaboratório de Fisiologia e Bioquímica de Microrganismos do Centro de Biociências e Biotecnologia da Universidade Estadual do Norte Fluminense Darcy Ribeiro, Rio de Janeiro, Brazil
cCEAMED - Centro Atlántico del Medicamento, S.A., San Cristobal de La Laguna, S/C Tenerife, Spain
First published on 3rd December 2019
Abstract
One of the most important causes of failure in tumour treatment is the development of resistance to therapy. Cancer cells can develop the ability to lose sensitivity to anti-neoplastic drugs during reciprocal crosstalk between cells and their interaction with the tumour microenvironment (TME). Cell-to-cell communication regulates a cascade of interdependent events essential for disease development and progression and can be mediated by several signalling pathways. Exosome-mediated communication is one of the pathways regulating these events. Tumour-derived exosomes (TDE) are believed to have the ability to modulate TMEs and participate in multidrug resistance mechanisms. In this work, we studied the effect of the natural defensin from common bean, PvD1, on the formation of exosomes by breast cancer MCF-7 cells, mainly the modulatory effect it has on the level of CD63 and CD9 tetraspanins. Moreover, we followed the interaction of PvD1 with biological and model membranes of selected composition, by biophysical and imaging techniques. Overall, the results show that PvD1 induces a dual effect on MCF-7 derived exosomes: the peptide attenuates the recruitment of CD63 and CD9 to exosomes intracellularly and binds to the mature exosomes in the extracellular environment. This work uncovers the exosome-mediated anticancer action of PvD1, a potential nutraceutical agent.
Introduction
Breast cancer is the second leading cause of cancer-related deaths worldwide, accounting for 11.6% of total globally predicted terminal cancer cases in 2018 and reflected by over 1 million deaths.1 Despite the significant progress that has been made in the development of targeted treatment strategies such as hormone therapy2 or immunotherapy,3 which increased the overall survival of breast cancer patients, a successful method to overcome the obstacles that lead to therapy failure is still needed.Multidrug-resistance mechanisms represent a major limitation to therapy and are the main cause associated with disease recurrence.4 In fact, nearly one-third of women were diagnosed with early stage breast cancer relapse and the probability of resistance development increases in advanced cases.5 The mechanism of resistance to therapeutics involves events impeding drug accumulation inside the cell which include reduced uptake, capture inside intracellular vesicles and enhanced clearance of drugs.6,7 These processes are associated with alterations in the lipid composition and biophysical properties of the cell membrane.8 Therefore, it is important to understand cell membrane changes occurring during therapy at the biophysics level so the process of drug development can be optimized.
It has been shown recently that multidrug-resistance can be also induced by tumour-derived exosomes (TDE) through epigenetic regulation or transfer of specific proteins.9,10 Exosomes are nano-sized membrane-embedded vesicles produced by reverse budding of the endosomal membrane.11 These extracellular vesicles (EVs) have been reported to facilitate the crosstalk between cells by transporting a unique composition of proteins, lipids and RNA species12 contributing to the regulation of the tumour microenvironment (TME).13 Exosomes isolated from docetaxel-resistant MCF-7 cells, for instance, have upregulated levels of proteins involved in multidrug-resistance14–16 and can induce resistance in MCF-7 drug-sensitive cells.17 Moreover, TDE are directly involved in anti-neoplastic drugs’ elimination from cancer cells.18–20 Therefore, the development of anticancer molecules that use novel mechanisms of action and target TDE can potentially help bypass resistance development in cancer cells.
Antimicrobial peptides (AMPs) have attracted growing attention for the development of anticancer chemotherapies due to their cytotoxic effects.21,22 The mechanism of action of AMPs with anticancer activity (anticancer peptides, ACPs), is assigned to the electrostatic interactions between the peptides and the cellular membrane, which is correlated with reduced probability of resistance development.23–25 Our group has previously investigated two representative ACPs, belonging to the defensin family: HNP-1,26 and PvD1.27 HNP-1 originates from the human azurophilic granules of neutrophils28 and has been shown to exert cytotoxic effects on several eukaryotic and neoplastic cells.26,29,30PvD1 is a natural plant defensin isolated from the seeds of common beans.31–34 In our previous studies, PvD1 showed inhibitory effect on the growth and metastatic spread of breast cancer cells.27 Additionally, other authors revealed a significant contribution of the membrane lipid composition to PvD1 activity.32
In this work, we examined whether the active ACP PvD1 can interfere with exosomal membranes and modulate their biophysical properties and/or structure. Additionally, we investigated the regulatory effect of the peptide on exosomes’ formation by screening changes in the expression of tetraspanins CD63 and CD9. These proteins are well-established hallmarks of exosomes and contribute to exosomal formation from the endosomal membrane and uptake by the cells.35,36 Tetraspanins play a very important role in processes such as cell adhesion, tumour growth, migration37 and chemo-resistance development.38 Here, we show that the membrane active peptide PvD1 can target exosomes, which could lead to a disturbed communication between cells in the primary breast tumour and in the TME, resulting in affected tumour growth and survival mechanisms.
Experimental
Reagents
Adherent cell line MCF-7 was purchased from American Type Culture Collection (ATCC, HTB-22). RPMI 1640 media, heat-inactivated fetal bovine serum (FBS), exosome-depleted FBS (dFBS), penicillin and streptomycin solution, trypsin (TrypLE Express enzyme) and Total Exosome Isolation (TEI) reagent were obtained from Life Technologies. Dimethyl sulfoxide (DMSO), 3-(4,5-di-methylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), ethylenediaminetetraacetic acid (EDTA) and 4-(2-[6-(dioctylamino)-2-naphthalenyl]ethenyl)-1-(3-sulfopropyl)pyridinium inner salt (di-8-ANEPPS) probe, were purchased from Sigma-Aldrich. The MicroBCA assay kit and protein ladder were obtained from Thermofisher Scientific. Western Blot reagents 10× TGS and 10× TG, APS, bromophenol blue and acrylamide 30% were obtained from Bio-Rad. Amersham™ ECL kit and Amersham™ nitrocellulose membranes were obtained from GE Healthcare.Primary antibodies anti-CD63 (clone RFAC4, CBL553), anti-CD9 (clone MM2/57, CBL162) and secondary anti-mouse antibody (AP181P) were purchased from Merck Millipore. Anti-Calnexin (clone 37/Calnexin, 610524) was purchased from BD Bioscience and Anti-β-actin (clone AC-15, A5441) was purchased from Sigma-Aldrich. Fluorescent antibodies anti-CD63-Alexa Fluor 647 (clone H5CS, 561924), anti-CD105-PerCp-Cy™5.5 (clone 266, 560819), anti-EpCAM-PE (clone EBA-1, 347198) and PE Mouse IgG1, κ isotype control (clone MOPC-21, 559320) as well as carboxyfluorescein diacetate succinimidyl ester (CFSE) were purchased form BD Bioscience. Nanobead Calibration Kit was obtained from Bangs Laboratories, Inc.
Cholesterol (Chol) was purchased from Sigma Aldrich and 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), sphingomyelin (SM) and glucosylceramide (GluCer) were obtained from Avanti Polar Lipids (Alabaster, AL, USA).
Biacore sensor chip regeneration reagents, 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate (CHAPS), octyl-β-glucopyranoside and methanol were purchased from Sigma. Sodium dodecyl sulfate (SDS) was obtained from GE Healthcare (Little Chalfont, United Kingdom).
PvD1 peptide
For protein extraction, 30 grams of Phaseolus vulgaris cotyledons were milled and after this process 150 mL of extraction buffer (Na2HPO4 10 mM, NaH2PO4 15 mM, KCl 100 mM, EDTA 1.5%) pH 5.4 was added to the flour and the mixture was constantly stirred for 2 h at 4 °C. This homogenate was centrifuged at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 20 min at 4 °C and the precipitate was discarded and the supernatant was subjected to ammonium sulfate precipitation at 70% saturation and centrifuged as before. The resulting precipitate was resuspended in distilled water and warmed at 80 °C for 15 min and centrifuged once more at 10000g for 8 min at 4 °C. The supernatant was dialyzed against distilled water, concentrated by freeze drying resulting in a protein-rich extract (PRE). The purification of the PvD1 peptide from the PER of P. vulgaris was accomplished by chromatographic methods which were performed as described by Games et al. 2008.34 The retention time to recover the PvD1 defensin during reversed-phase chromatography in the μRPC C2/C18 column (ST 4.6/100) (GE Healthcare) was 32 min.
000g for 20 min at 4 °C and the precipitate was discarded and the supernatant was subjected to ammonium sulfate precipitation at 70% saturation and centrifuged as before. The resulting precipitate was resuspended in distilled water and warmed at 80 °C for 15 min and centrifuged once more at 10000g for 8 min at 4 °C. The supernatant was dialyzed against distilled water, concentrated by freeze drying resulting in a protein-rich extract (PRE). The purification of the PvD1 peptide from the PER of P. vulgaris was accomplished by chromatographic methods which were performed as described by Games et al. 2008.34 The retention time to recover the PvD1 defensin during reversed-phase chromatography in the μRPC C2/C18 column (ST 4.6/100) (GE Healthcare) was 32 min.
Cell culture
MCF-7 adherent human breast cancer cells were cultured as a monolayer in RPMI 1640 medium supplemented with 10% FBS and 1% penicillin–streptomycin. The culture was maintained in 75 cm2 cell culture flasks (ThermoFisher™) at 37 °C and 5% CO2 in a humidified environment.MTT toxicity assay
MCF-7 cells were seeded in triplicate at 5000 cells per 200 μL per well, in a 96-well plate and incubated for 24 h. Cells were then washed with phosphate buffer saline (PBS, pH 7.4) and new serum-free media for the control and serum-free media containing PvD1 (0.01–10 μM) were added to the wells. After 24 h of incubation, 10 μL of 6 mg mL−1 MTT solution was added to each well following a 2 h incubation. The mixture medium, peptide and MTT were removed and 150 μL of DMSO (spectrophotometric grade) was added. Absorbance was measured at 595 nm. Serum-free medium was used as the control for 100% viability, and serum-free medium containing 10 μM doxorubicin was used as the positive control.Viability (%) was determined as:
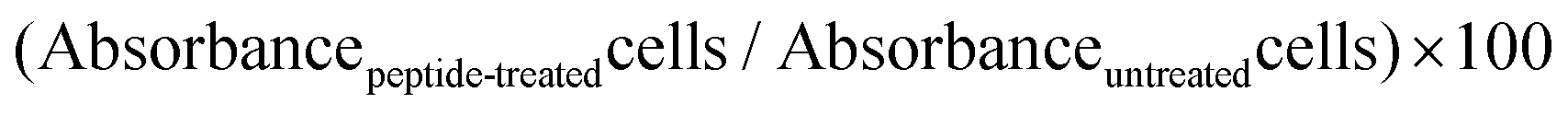
Hemolysis assay
Human blood samples were collected from healthy blood donors, after obtaining written informed consent, following a protocol established by the Portuguese Blood Institute (Lisbon), approved by the Ethics Committee of the Faculty of Medicine of the University of Lisbon. The blood samples were collected directly into K3-EDTA coated tubes to prevent coagulation. For isolating human red blood cells (hRBCs) the blood was washed three times with PBS buffer (pH 7.4) and centrifuged at 4000 rpm for 10 min at 4 °C. In a 96-well round bottom polypropylene plate, 100 μL of the peptide solution (from 0.01 μM to 30 μM final concentration in PBS buffer (pH 7.4)) was added to the wells and then mixed with 100 μL of 0.25% v/v hRBCs in PBS buffer (pH 7.4). The 0% hemolysis and 100% hemolysis controls consisted of hRBCs treated with PBS buffer (pH 7.4) and 0.5% Triton X-100 in PBS buffer (pH 7.4), respectively. The plates were incubated for either 1 or 24 h at 37 °C. The solution was then centrifuged at 4000 rpm for 10 min at 4 °C, and the supernatant was transferred into a new 96-well flat bottom polystyrene plate. Absorbance was measured at 415 nm using a microplate reader and per cent hemolysis was calculated using the following equation: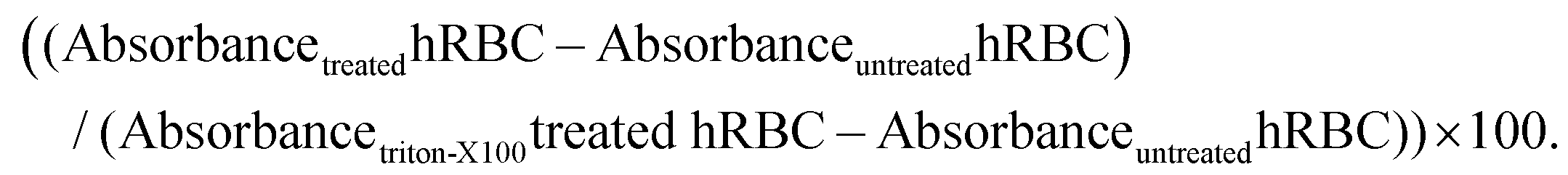
Each experiment was performed in duplicate, and the dose–response curve is an average from two independent donors.
Atomic force microscopy (AFM) imaging
AFM images were acquired with a JPK Nano Wizard IV (Berlin, Germany) mounted on a Zeiss Axiovert 200 inverted microscope (Carl Zeiss, Germany). The AFM head is equipped with a 15 μm z-range linearized piezoelectric scanner and an infrared laser. Human breast cancer cells MCF-7 were seed at 6 × 104 cells per mL into 40 mm culture dishes (TPP) and cultured for 24 h. Cells were washed with PBS buffer (pH 7.4) before adding PvD1 peptide diluted in serum-free media. For the control images, cells were incubated with the medium supplemented without serum. After the 24 h incubation period cells were washed with PBS buffer (pH 7.4) and incubated with 1% glutaraldehyde solution for 10 minutes at room temperature, washed with PBS buffer (pH 7.4) and sterile Milli-Q water and air-dried. Images were obtained in air using uncoated silicon ACL cantilevers from AppNano with typical resonance frequencies ranging between 145 and 230 kHz and an average spring constant of 45 N m−1 in intermittent contact mode. Scan speeds were lower than 1 Hz. Total areas with 100 μm × 100 μm were scanned with a 512 × 512 pixel resolution.Cell height was assessed using the JPK SPM Data Processing (version 6.0.55) by drawing a height profile for each cell. The difference between the bottom and the highest point of the cell corresponds to the presented cell height. The total number of analysed cells was 70. This number includes 17 cells for the control and 20 cells for PvD1 at 0.03 μM, 20 cells for PvD1 at 0.24 μM and 13 cells for PvD1 at 1 μM.
The surface area of the tumour cells was determined through AFM height images using ImageJ software (version 1.39k). The number of cells analyzed for control images was 21 while that for peptide treatment was 24 (0.03 μM PvD1), 21 (0.24 μM PvD1) and 12 (1 μM PvD1).
Cell membrane roughness was defined as the mean square roughness (Sq) and determined using Gwyddion software (version 2.50) from AFM height images. Final Sq values were obtained as the average of 2.5 μm × 2.5 μm squared areas from different areas of the cell (nucleus and cytoplasm). The total number of cells included in this analysis was 29 for the control and that in the presence of PvD1 at 0.03 μM – 24 cells, 0.24 μM – 22 cells and for 1 μM – 19 cells. Cultures were independently grown and observed during two to three different days.
Di-8-ANNEPS labelling of MCF-7 cells, MCF-7 exosomes, hRBC and fluorescence spectroscopy study
Breast tumour cells were washed and suspended in PBS buffer (pH 7.4) before incubation with 30 μM of di-8-ANEPPS for 1 h at room temperature at constant rotation. The cells were then washed to remove the non-incorporated probe before being suspended to a final concentration of 5 × 105 cells per mL. hRBCs were purified as described above and incubated at 1% (v/v) with 10 μM of di-8-ANEPPS in PBS buffer (pH 7.4) supplemented with 0.5% (m/v) of Pluronic F-127. After washing with Pluronic F-127 supplemented PBS buffer (pH 7.4), hRBCs were diluted to 0.1% (v/v) final concentration. MCF-7 exosomes were incubated with 10 μM of di-8-ANEPPS at the concentration of 25 μg mL−1 total protein overnight to ensure incorporation of the dye. Membrane dipole potential variations in the presence of PvD1 were evaluated by di-8-ANEPPS fluorescence excitation spectrum shifts. Di-8-ANEPPS fluorescence excitation spectra were recorded on an Edinburgh FLS920 spectrofluorimeter (Livingston, UK). The emission wavelength, λemi, was set to 670 nm, avoiding membrane fluidity artefacts.40 Prior to the measurement the cells and exosomes were incubated with PvD1 for 15 min at room temperature. Controls were prepared without peptide addition. Fluorescence excitation spectra were recorded between 380 and 580 nm and corrected for background intensity. Differential spectra were recorded after subtraction of the normalized (to the spectrum integral) excitation spectra of the labelled cells incubated with PvD1 from the control spectra (no peptide present). For the quantification of di-8-ANEPPS spectral shifts, excitation intensity ratios (R) were calculated through the relationship R = [Iexc(λa)]/[Iexc(λb)], where Iexcλa and Iexcλb correspond to the maximum and minimum excitation spectrum intensity, respectively. Variations in the cell membrane dipole potential were represented by intensity ratios normalized to the control (Rnorm). The experiment was repeated on two different days using independent cell samples.Isolation of exosomes from human breast cancer cells
MCF-7 derived exosomes were isolated with the Total Exosome Isolation (TEI) reagent according to the manufacturer's instructions. Briefly, the cells were cultured in 175 cm2 cell culture flasks (ThermoFisher™). After reaching 60–70% confluence, cells were washed with PBS buffer (pH 7.4) and medium with dFBS was added to the culture. Conditioned cell culture medium was collected after 24–48 hours of additional cell growth and centrifuged at 2000g for 30 minutes at room temperature for removing debris and dead cells. The supernatant was incubated with TEI overnight at 4 °C and the precipitated exosomes were recovered by centrifugation at 10000g for 60 minutes at 4 °C. Finally, the pellet was resuspended directly in PBS buffer (pH 7.4) for further analysis.
Exosome protein quantification
Exosomes’ total protein concentration was quantified with a MicroBCA protein assay kit according to the manufacturer's instructions. Briefly, 150 μL of exosome samples were mixed with 150 μL of freshly prepared working reagent in flat, transparent 96-well plates in duplicate. The reaction was carried out for 2 h at 37 °C. The absorbance was measured at 570 nm. The total protein concentration in μg mL−1 was calculated based on a BSA standard curve prepared freshly for each quantification.Dynamic light scattering (DLS)
Confluent human breast cancer MCF-7 cells were washed after trypsinization and diluted in PBS buffer (pH 7.4) to 1 × 105 cells per mL. Purified hRBCs were diluted to 0.35% (v/v). Exosomes isolated from MCF-7 cells were diluted to 25 μg mL−1 in PBS buffer (pH 7.4).Size and zeta potential measurements were performed on a Zetasizer Nano ZS from Malvern (Worcestershire, UK). For zeta potential measurements, cell suspensions and exosomes with and without PvD1 were incubated in disposable polystyrene folded capillary cells with gold electrodes for 30 minutes, at 37 °C. Experiments consisted of 10–15 measurements with 40–70 runs performed at a constant voltage of 40 V for MCF-7 cells and exosomes and 30 V for hRBCs. The obtained electrophoretic mobility was used for the zeta potential calculation through the Henry and Smoluchowski equations.41 Each represented group value is an average of at least two independent measurements carried out with independent samples.
For size measurements, samples were prepared as 25 μg mL−1 exosome dilutions in PBS buffer (pH 7.4) and allowed to equilibrate for 5 min at 37 °C. Measurements consisted of 15 individual runs, each corresponding to an averaged autocorrelation curve obtained from at least 12 repeated sample scans. Normalized autocorrelation functions were analysed using the cumulant and CONTIN methods, used to determine the diffusion constants (D).42 Associated size distribution profiles were calculated using the Stokes–Einstein–Sutherland relationship.43 Experiments were carried out with at least two independent samples.
Transmission electron microscopy (TEM) imaging
TEM observations were performed on a Hitachi H-7000 instrument at an acceleration voltage of 100 kV; acquisitions and measurements were performed using a Megaview III side mount camera and iTEM software (Olympus®). To prepare the TEM samples, 5 μL of aqueous exosomes isolated from MCF-7 cells were deposited onto formvar (Agar Scientific®)/carbon-coated 400 mesh copper grids (Agar Scientific®). After 5 minutes the grid was washed in pure water and negatively stained with 2% (w/v) aqueous uranyl acetate (Agar Scientific®) for approximately 2 min. The copper grid was air dried prior to visualization. Experiments were repeated on different days using exosomes isolated from independent cell cultures.Immunolabelling of exosomes using gold particles
For immunolabelling, 5 μL of an aqueous suspension containing exosomes was deposited onto a formvar (Agar Scientific)/carbon-coated 400 mesh copper grid (Agar Scientific). After 5 minutes the grid was washed in pure water and quickly rinsed in PBS. The grids were then blocked in a solution containing 1% of gelatin from cold water fish skin (Sigma Aldrich) and incubated in anti-CD63 (1:50) and anti-CD9 (1:50) for 30 minutes at room temperature. Efficient labelling was achieved using goat anti-mouse secondary antibody coupled to 6 nm gold particle (1:50, Abcam). Grids were negatively stained with 2% (w/v) aqueous uranyl acetate (Agar Scientific) for approximately 2 min and air dried prior to visualization. TEM observations were performed as described above.
SDS-PAGE electrophoresis and immunoblotting
MCF-7 human breast cancer cells were seeded at 3 × 106 cells per 175 cm2 cell culture flasks (ThermoFisher™) and cultured until they reached 60%–70% confluence. Exosomes were isolated from the culture medium with TEI. Tumour cells were lysed with NP-40 buffer (150 mM NaCl, 1% Triton X-100, 50 mM Tris, pH 8.0) in the presence of a proteinase inhibitor cocktail and 1 mM EDTA for 30 minutes at 4 °C. Lysates were centrifuged at 12000 rpm for 20 minutes at 4 °C and the supernatant was quantified by the MicroBCA assay as described above. Cell lysates and exosomes were mixed with 4× Laemmli Buffer and denatured for 10 minutes at 70 °C. Equal amounts of proteins (2 μg per lane) were separated on 12% SDS-PAGE44 gel and transferred to a nitrocellulose membrane Amersham™. After blocking with 5% milk in PBS buffer (pH 7.4) with 0.1% Tween 20® (PBS-T) the membrane was incubated overnight with anti-CD63 antibody (1:500), anti-CD9 (1:5000), and anti-calnexin (1:2000) followed by washing with PBS-T and incubation with HRP-conjugated anti-mouse secondary antibody.45 Chemiluminescence was induced with Amersham™ ECL kit and blots were visualized using Chemidoc XRS + (Bio-Rad). Images were analysed with ImageLab (Bio-Rad) software.
For evaluating PvD1 impact on the exosomal membrane composition and directly on breast tumour cells, cells were incubated with PvD1 at 0.24 μM and 1 μM in serum-free medium for 24 h after media removal and washing with PBS (pH 7.4). Cells incubated in serum-free medium were used as a control. After peptide treatment, the culture media were collected for exosome isolation. Isolated exosomes and cell lysates for immunoblotting analysis were prepared as described above.
To assess direct PvD1 interaction with exosomes under cell-free conditions, exosomes isolated from non-treated cells were incubated with the peptide at 150 μg mL−1 for 30 min at 37 °C. All samples were diluted in 4× Laemmli Buffer and denatured for 10 min at 70 °C. Equal amounts of proteins (1 μg of per lane) were separated on 12% SDS-PAGE gel, blocked with 5% milk in PBS-T following incubation overnight at 4 °C with primary anti-CD63 (1:500), anti-CD9 (1:5000) and β-actin (1:10000) antibodies. Membranes were washed with PBS-T, incubated with secondary anti-mouse antibody and visualized on Chemidoc XRS + (Bio-Rad) after the induction of chemiluminescence as described above.
CFSE labelling of breast tumour cells
Cells were labelled with BD Horizon™ CFSE dye according to manufacturer's instruction with minor modifications. Briefly, cells were diluted in DPBS (pH 7.4) at 2 × 107 cell per mL and incubated with CFSE at 1:100 dilution for 15 min at 37 °C. Labelling was quenched by adding a 9× initial volume of DPBS buffer and the cell dilution was spun down at 120g for 5 min. Labelled and control MCF-7 cells were seeded into 175 cm2 cell culture flasks and cultured for exosome isolation. Exosomes were isolated following the standard procedure as described above.
Flow cytometry
To verify the intracellular origin of the isolated vesicles and identify respective exosomal markers, exosomes isolated from CFSE-labeled and non-labelled (control) cells were analyzed in a flow cytometer. Exosomes were incubated with anti-CD63-Alexa Fluor 647, anti-CD105-PerCp-Cy™5.5, anti-EpCAM-PE and PE Mouse IgG1, κ isotype control antibodies for 20 minutes at room temperature with gentle agitation following the addition of 1 mL of PBS buffer (pH 7.4). The experiments were performed on a BD LSR Fortessa (BD Bioscience) equipped with the FACSDiva 6.2 software. Prior to the experiment the system was primed 3 times and cleaned extensively to ensure the highest quality detection of small particles. 100 nm calibration beads labelled with manufacturer's YG fluorophore, permitting gating via SSC/green fluorescence were used to define the target population. The threshold of the detection was set at 200 nm in the FITC channel excited by blue laser 488 nm (530/30, 505LP). The results were analyzed with FlowJo 10.4.2 software. The calculated percentage of population positive to fluorescence staining is an average of three independent experiments.Small unilamellar vesicles (SUVs) preparation
Small unilamellar vesicle (SUV) suspensions were prepared as described previously.46 The lipid mixture was dissolved in chloroform in a round bottom flask. Chloroform was then evaporated under a constant nitrogen flow and the resulting lipid film was dried in a vacuum, overnight. A multilamellar vesicle suspension (MLV) was obtained after lipid film rehydration with PBS buffer (pH 7.4) and a series of 10 freeze/thaw cycles. MLV suspensions were extruded through a 50 nm track-etched polycarbonate membrane from Whatman (GE Healthcare). Extrusion was performed in a LiposoFast-Basic plus Stabilizer setup (Avestin). POPC, POPC:POPS (9:1) POPC:Chol (9:1), POPC:POPS:Chol (8:1:1). POPC:POPE (9:1) and POPC:POPS:Chol:POPE:SM (2:2:1:2:4) and POPC:GluCer (9:1) mixtures were prepared.
Surface plasmon resonance (SPR)
SPR experiments were carried out in a Biacore X100 apparatus from GE Healthcare. The temperature was set at 37 °C for all experiments. In all experiments, a PBS solution (pH 7.4) was used as a running buffer. The flow system was primed 3 times before initiating an experiment. Prior to exosome/SUV deposition, the L1 sensor chip surface was rinsed with three injections of 20 mM CHAPS. MCF-7 exosomes at concentrations ranging from 1 μg mL−1 to 50 μg mL−1 were injected over the L1 sensor chip for 2400 s, at a 2 μL min−1 flow speed. RU values >6000, considered as indicating the coverage of the sensor chip, were obtained for 50 μg mL−1 of exosomes. SUVs were immobilized from samples at 1 mM concentration. Loose vesicles or exosomes were removed after a 36 s injection of 10 mM NaOH at 50 μL min−1. PvD1 dilutions ranging from 0.1 μM to 30 μM were injected over pre-formed exosome/liposome-coated surfaces at 5 μL min−1, for a total of 200 s (association phase). PvD1 was allowed to dissociate for 800 s. L1 sensor chip surface regeneration after the experiment with exosomes was performed with 5 repeated sequential injections of 80 mM octyl-β-glucopyranoside (5 μL min−1 for 60 s) and 0.5% (w/v) SDS (5 μL min−1 for 60 s) followed by the injection of 20 mM CHAPS (5 μL min−1 for 60 s) and 10 mM NaOH containing 20% (v/v) methanol (50 μL min−1 for 36 s). After the experiment with liposomes the L1 sensor chip was regenerated with sequential injections of 20 mM CHAPS (5 μL min−1 for 60 s), 0.5% (w/v) SDS (5 μL min−1 for 60 s), 10 mM NaOH containing 20% (v/v) methanol (50 μL min−1 for 36 s) and 10 mM NaOH (50 μL min−1 for 36 s). The system was allowed to stabilize for 300 s with the running buffer. Baseline response values were compared before and after each experiment to evaluate surface regeneration. Raw SPR sensorgram data were collected for both exosome/lipid immobilization and solute binding. Exosome/SUV deposition response values were collected from sensorgrams upon reaching a stable response. The associated steady-state response values for PvD1 were collected from individual sensorgrams at t = 250 s. The data were analysed using the partition relationship previously developed by our group.47Statistical analysis
All data points are presented as mean ± standard deviation (SD) of results obtained on different days and repeated at least two times. Statistical significance was calculated by applying one-way ANNOVA followed by Dunnett's multiple comparison test, ***0.0001 < p-value <0.001; **0.001 < p-value <0.01, *0.01 < p-value <0.05. GraphPad Prism 7.0 software package and Excel 2013 (Microsoft, USA) were used for quantitative data processing.Results
Cytotoxic activity of the PvD1 peptide
As previously reported, PvD1 shows high toxicity toward the invasive MDA-MB-231 breast cancer cell line with no significant impact on the viability of the non-malignant MCF 10A cell line.27 In the present study we have used an estrogen positive, luminal A molecular subtype human breast cancer cell line, MCF-7.48,49 MCF-7 is a non-invasive cancer cell line and is a well described and consecrated model system of breast cancer.48–50PvD1 induces strong growth inhibition of MCF-7 cells (Fig. 1; IC50 = 0.24 ± 0.08 μM). The hemolysis assay performed with hRBCs purified from the blood of healthy donors revealed no disruptive effect of PvD1 on the hRBC after 1 h (data not shown) or 24 h incubation with the peptide (Fig. 1).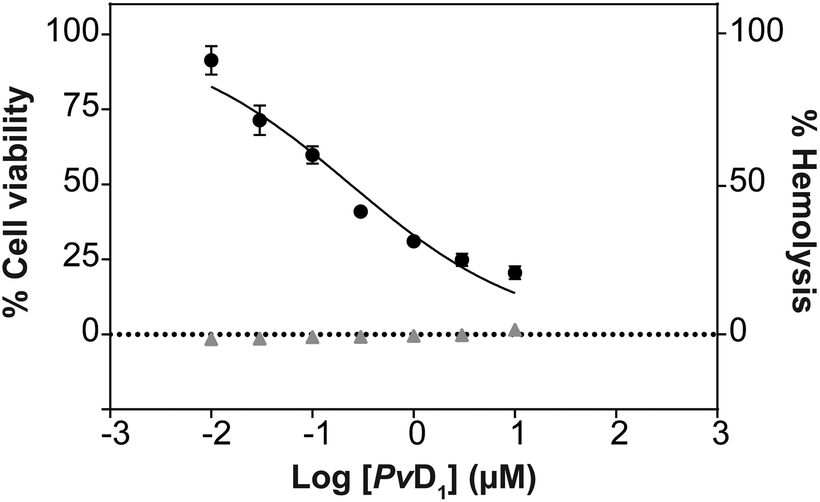 | ||
| Fig. 1 In vitro toxicity of PvD1. Cytotoxicity towards human breast cancer MCF-7 cells (black circles) and hemolytic activity towards human red blood cells (hRBCs, gray triangles) after 24 h incubation with the PvD1 peptide. All the experiments were performed using independently grown cultures and independent healthy blood donors on different days. | ||
PvD1 interaction with cellular membranes from human breast cancer and red blood cells
The cationic residues of PvD1 can have an important role in the interaction of the peptide with negative charges distributed on the interface of the eukaryotic membranes. Therefore, we investigated the electrostatic affinity of PvD1 towards human breast cancer MCF-7 cells and hRBCs using zeta potential measurements.51 The determination of this parameter allows to track the electrostatic interaction between dispersed entities such as bacteria and peptides.41,51,52 The results show a gradual increase in the zeta potential of MCF-7 cells towards less negative values, from −25.23 ± 2.8 mV to −13.73 ± 2.4 mV, after exposure to PvD1 even at very low concentrations (Fig. 2A). In the case of hRBCs, only peptide concentrations above 10 μM have been shown to have an impact on the zeta potential of the cell membrane, even though these concentrations do not induce hemolysis (Fig. 1 and 2B).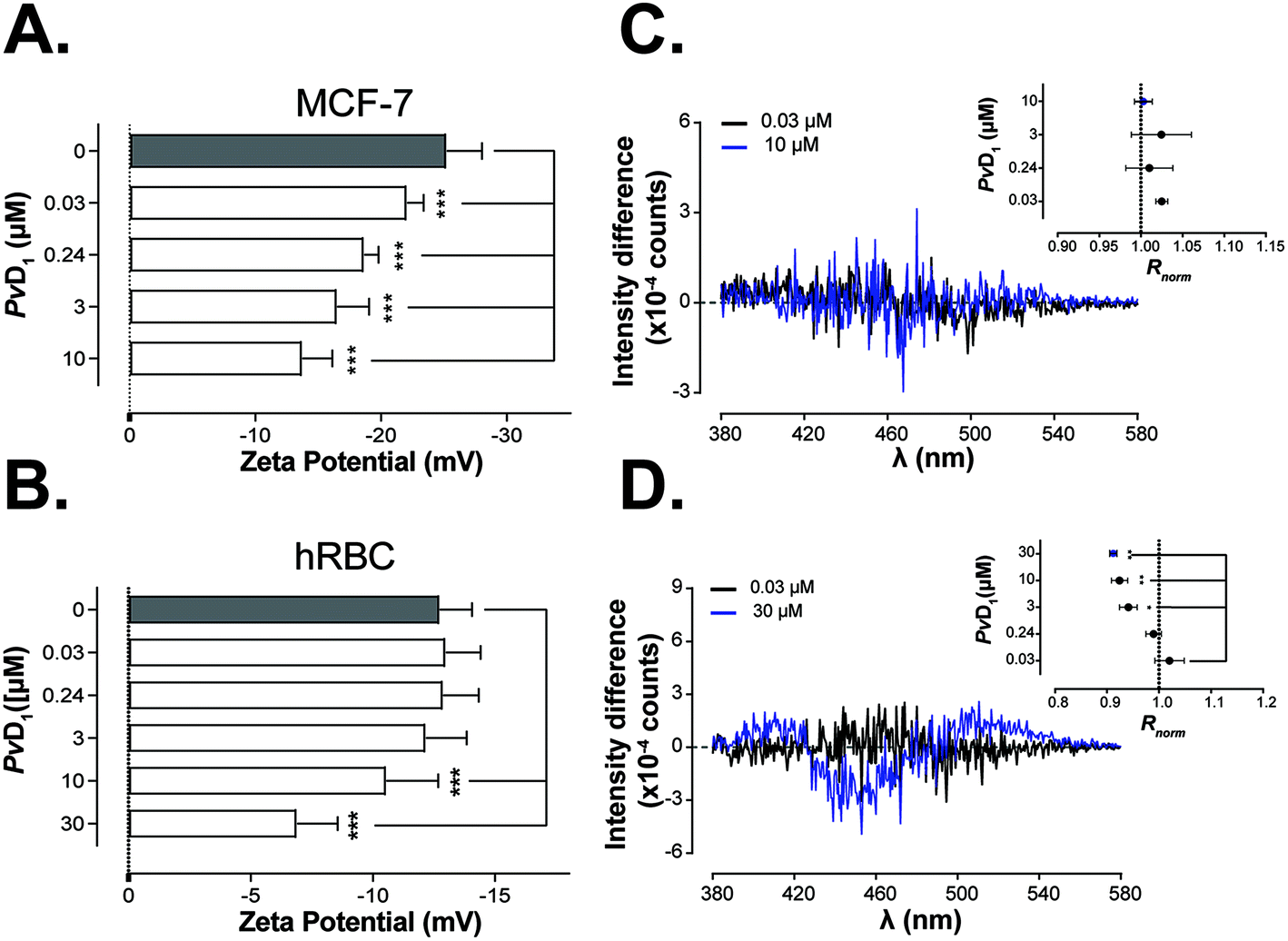 | ||
| Fig. 2 Interaction of PvD1 with cellular membranes of human breast tumour and red blood cells. Changes in the membrane charge density of human breast cancer MCF-7 cells (A) and hRBCs (B) induced by PvD1 were measured by the zeta potential technique. Statistical significance was evaluated applying one-way ANNOVA followed by Dunnett's multiple comparison test. ***0.0001 < p-value < 0.001. Peptide-membrane interactions were additionally followed by fluorescence spectroscopy of di-8-ANEPPS dye introduced in the cell membranes. Differential di-8-ANEPPS excitation spectra and respective normalized intensity ratio, Rnorm are presented for human breast cancer MCF-7 cells (C) and hRBCs (D). Statistical significance was calculated applying one-way ANNOVA followed by Dunnett's multiple comparison test. **0.001 < p-value < 0.01, *0.01 < p-value < 0.05. Data points are an average of results obtained on different days and using independently grown cultures and independent healthy blood donors. | ||
By using fluorescence spectroscopy and after labelling both tumour and hRBCs with the di-8-ANEPPS probe, it was possible to evaluate the membrane perturbations induced by PvD1. Fig. 2C shows that PvD1 does not perturb the membrane of human breast cancer MCF-7 cells as no variation in the differential excitation spectra was observed. In contrast, the differential excitation spectra are PvD1 concentration-dependent for hRBCs and Rnorm decreases significantly at peptide's concentration greater than 3 μM (Fig. 2D).
Morphology and topography of MCF-7 cells observed with atomic force microscopy
AFM images obtained for breast tumour cells before and after incubation with PvD1 allowed us to observe morphological and topographical changes induced by the peptide on human cells. Fig. 3A and B are representative height and three-dimensional (3D) projections, respectively, of MCF-7 cells in the absence and presence of PvD1. MCF-7 cells grow in an epithelial-like multi-layer forming dense colonies (Fig. 3A). This observation agrees with previous reports.50,53 The regular membrane surface observed for control cells changes after contact with the peptide. The images reveal differentiated sub-membrane structures and nuclear collapse (Fig. 3A and B). Quantitative analyses of cells’ height and surface area are shown with representative height profiles obtained for cells under the effect of the peptide (Fig. 4). We observed a negative correlation between the height and surface area of MCF-7 cells after treatment with the peptide at the IC50, with a 43% decrease in height and 148% increase in the surface area. Cell membrane roughness suffered no significant changes after the incubation with the plant defensin (Fig. S1†), similar to what we have described in previous studies.27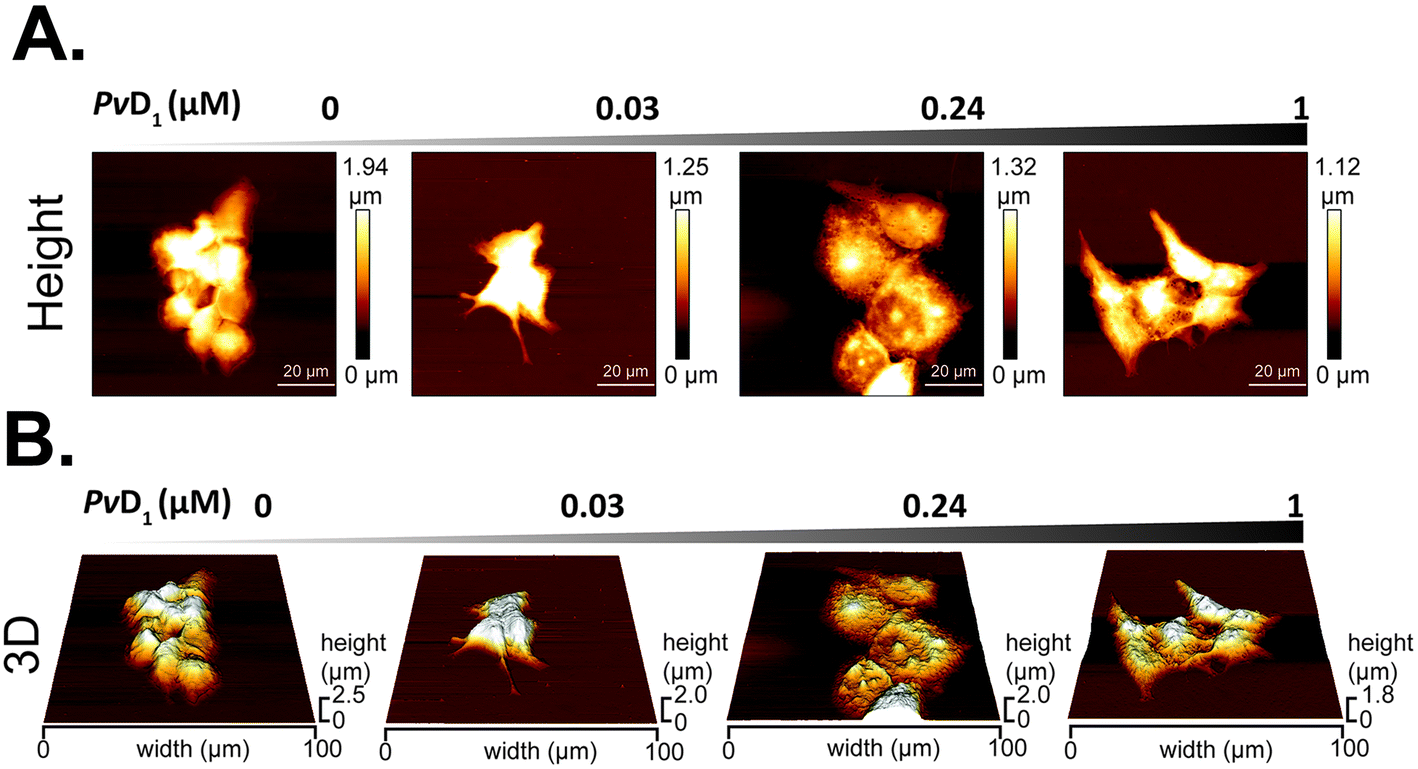 | ||
| Fig. 3 Morphology of MCF-7 cells observed with atomic force microscopy (AFM). Representative AFM height images (A) and respective 3D projections (B) show changes in MCF-7 cells’ morphology after incubation with increasing concentration of PvD1. All experiments were repeated on different days using independently grown cell cultures. Statistical significance was calculated with one-way ANNOVA followed by Dunnett's multiple comparison test. **0.001 < p-value < 0.01. | ||
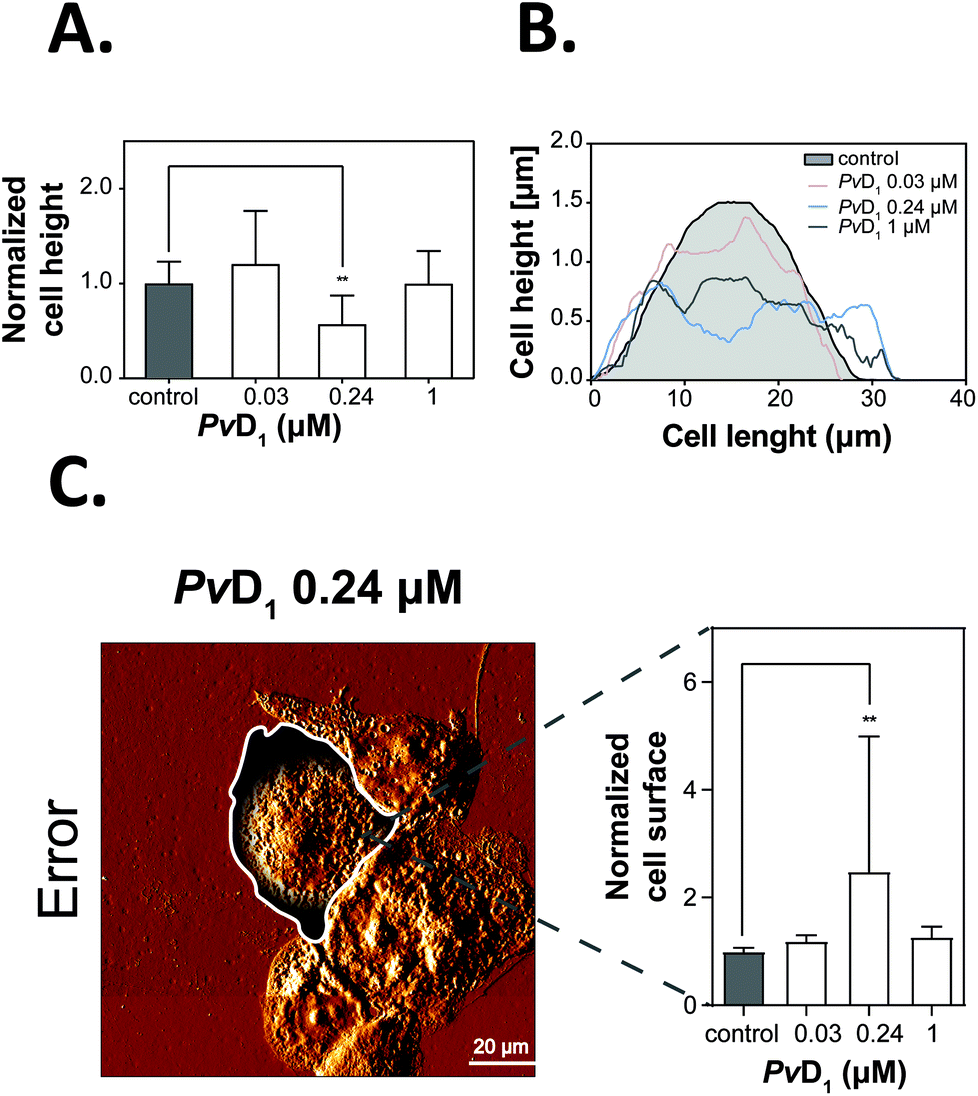 | ||
| Fig. 4 Cell height and surface area examination using atomic force microscopy (AFM). The average height of MCF-7 cells was calculated from the highest point of the cell normalized to the control (A) after drawing representative height profiles of the tumour cells (B) before and after incubation with PvD1. The total number of analysed cells was 70. This includes 17 cells for the control condition while in the presence of PvD1 the number of cells analyzed was 20 cells (PvD1 at 0.03 μM), 20 cells (PvD1 at 0.24 μM) and 13 cells (PvD1 at 1 μM). The surface area of human breast cancer MCF-7 cells (control and after the incubation with increasing concentrations of PvD1) was determined using ImageJ version 1.39k software (C). Cell surface areas were selected manually by drawing a line around each cell. Error trace images were used for accurate visualization and individualization of single cell boundaries. Bars represent the average values obtained for the surface area of MCF-7 cells normalized to the control. The number of cells used for this analysis was 21 for the control and that in the presence of PvD1 at the concentration of 0.03 μM – 24 cells, 0.24 μM – 21 cells and for 1 μM – 12 cells. All experiments were repeated on different days using independently grown cell cultures. Statistical significance was calculated using one-way ANNOVA followed by Dunnett's multiple comparison test. **0.001 < p-value < 0.01. | ||
Interactions of PvD1 with model membranes followed by surface plasmon resonance (SPR)
For the identification of the main factors that govern PvD1 peptide interaction with lipid membranes, we prepared a panel of SUVs of different lipid compositions and performed SPR binding studies. Liposomes were prepared with neutral POPC, the major component of eukaryotic cells, and additional components known to be enriched in cancer cells: POPC:POPS (9:1), POPC:Chol (9:1), POPC:POPS:Chol (8:1:1) and POPC:POPE (9:1).54,55 We also prepared vesicles containing glucosylceramide (GlucCer), proposed to be important for PvD1's cytotoxic activity.31 Applying the steady-state model developed by our group,47 we retrieved partition coefficients, (Kp), for quantifying the relative solute (peptide) distribution in the lipid and aqueous phases, and the lipid:solute molar ratio at the membrane saturation (σ) for each SUVs’ type.47 The obtained Kp and σ values are presented in Table 1 and the fits of the partition formalism to experimental data are depicted in Fig. 5. The highest Kp was obtained for negatively charged and fluid liposomes of POPC:POPS (9:1) and the lowest for neutral and rigid POPC:GluCer (9:1).
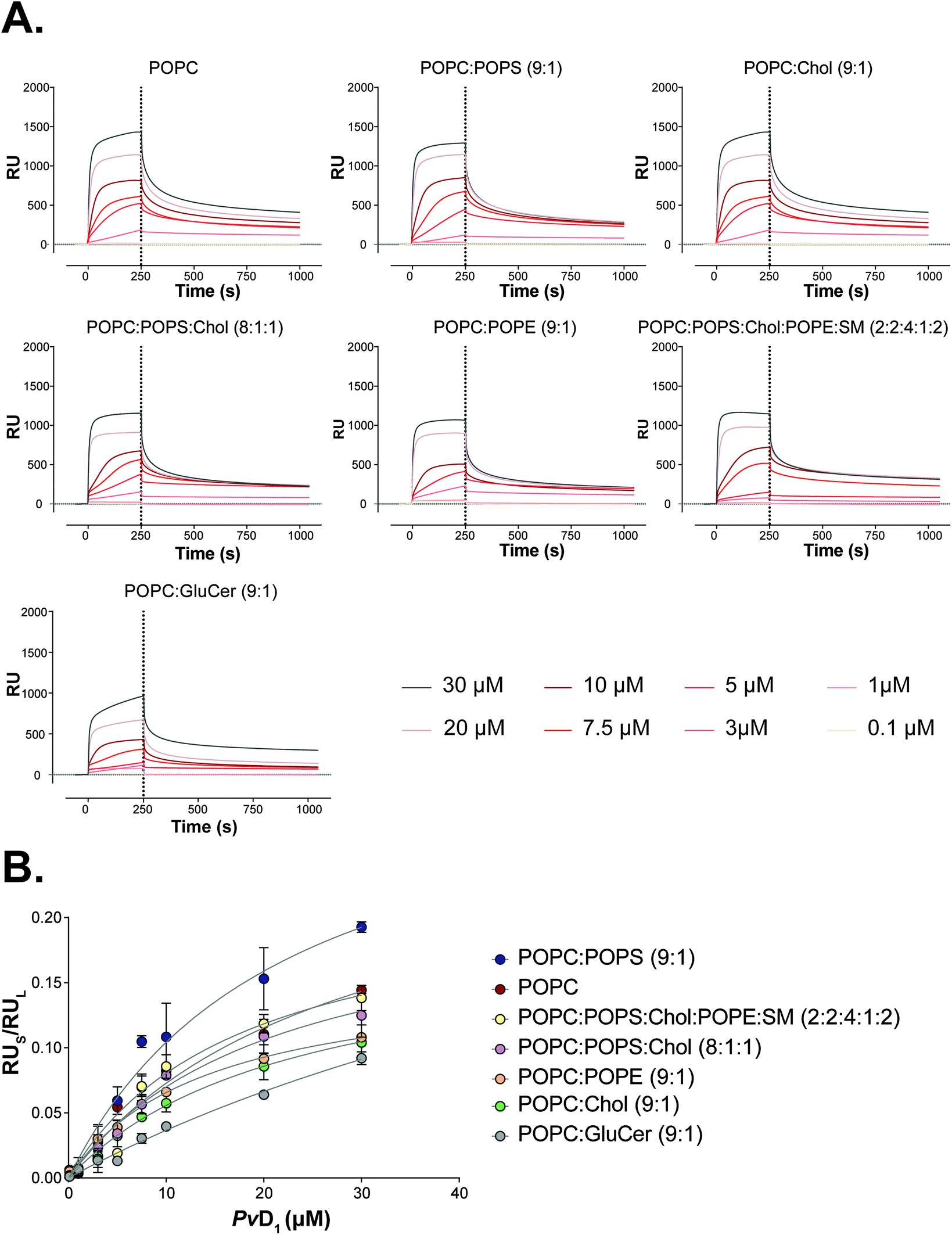 | ||
| Fig. 5 Partition of the PvD1 peptide into model membranes studied by surface plasmon resonance (SPR). SPR sensograms representing the binding of PvD1 to small unilamellar vesicles (SUVs) of different lipid composition (A). The association and dissociation phases for the peptide were followed for 250 s and 800 s, respectively. PvD1 partition into SUVs (B). RU values for PvD1 (RUs) were collected at 250 s for each peptide concentration. Data were fitted to the steady state model described elsewhere.47 Each data point represents an average from experiments performed on different days using independently prepared SUVs. | ||
:peptide membrane saturation ratios (σ) obtained for the interaction of PvD1 with small unilamellar vesicles (SUVs)
| SUVs | K p × 103 (±SD) | σ (±SD) |
|---|---|---|
| POPC | 2.082 (±0.19) | 35.25 (±3.2) |
| POPC:POPS (9:1) |
2.514 (±0.35) | 23.63 (±3.8) |
| POPC:Chol (9:1) |
1.316 (±0.12) | 42.24 (±4.7) |
| POPC:POPS:Chol (8:1:1) |
1.703 (±0.19) | 45.93 (±4.7) |
| POPC:POPE (9:1) |
1.857 (±0.13) | 49.88 (±2.9) |
| POPC:GluCer (9:1) |
0.677 (±0.05) | 22.04 (±6.2) |
| POPC:POPS:Chol:POPE:SM (2:2:4:1:2) |
1.635 (±0.12) | 28.37 (±6.0) |
Targeting human breast cancer exosomes with PvD1
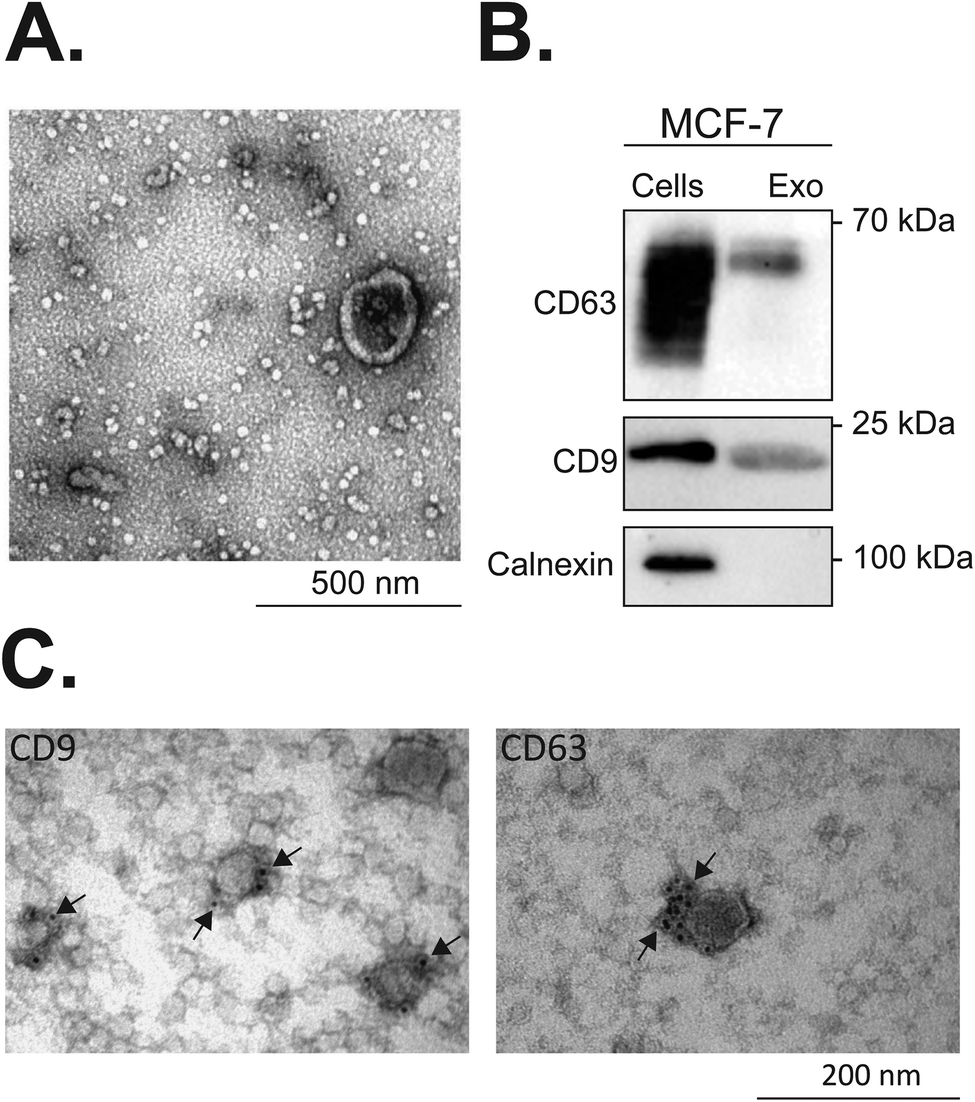 | ||
| Fig. 6 Characterization of exosomes isolated from MCF-7 breast tumour cells. Transmission electron microscopy (TEM) was used for observing isolated exosomes (A). Western blot identification of exosomal markers in MCF-7 exosomes and cell lysates is shown in (B). The total protein content of exosomes and cell lysates was quantified by a MicroBCA protein assay kit. Equal protein loading of 2 μg was resolved by SDS-PAGE electrophoresis and subjected to western blot analysis. CD63 and CD9 belong to the tetraspanin family and are enriched in exosomes, whereas calnexin is a control protein localized in the endoplasmic reticulum, and is not recruited to exosomes. Localization of CD63 and CD9 exosomal markers in MCF-7 exosomes was additionally confirmed by immunolabelling and TEM imaging (C). All the experiments were repeated on different days using exosomes isolated from independently grown cell cultures. | ||
The interaction of PvD1 with exosomal membranes was followed by zeta potential, fluorescence spectroscopy and surface plasmon resonance (SPR) measurements.
SPR experiments were performed to evaluate the affinity of PvD1 for MCF-7 exosomes. Fig. S4† shows the typical response obtained for the deposition of MCF-7 derived exosomes as well as for synthetic vesicles prepared for mimicking the exosomal membrane – SUVs composed of POPC – 18%, POPS – 18%, POPE – 9%, SM – 18% and cholesterol in high percentage, Chol – 36%: POPC:POPS:Chol:POPE:SM (2:2:4:1:2).57,58 The steady response retained after the injection of NaOH confirmed that the exosomes formed a stable layer. Fig. 7A shows the direct binding of PvD1 to exosomes. The ratio between the response units at the point of peptide binding and exosome deposition (RUs/RUL), initially increases proportionally in the function of the peptide concentration. PvD1 binds to MCF-7 derived exosomes with a slightly higher affinity than it does synthetic vesicles mimicking the exosomal membrane (Fig. S5A†).
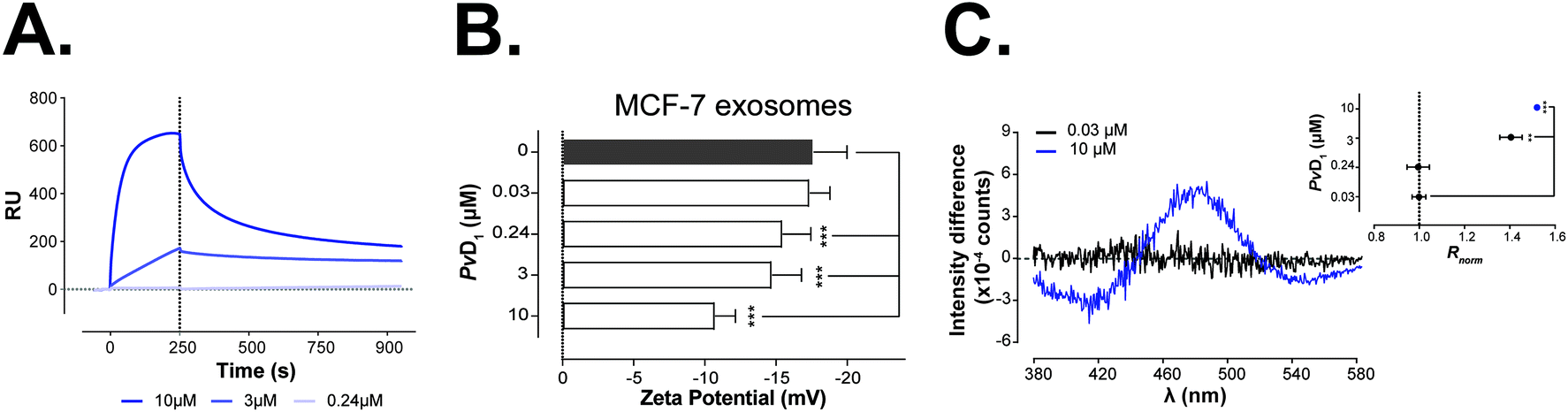 | ||
| Fig. 7 PvD1-induced changes on exosomes’ surface charge and binding to exosome and synthetic model membranes. SPR sensogram representing PvD1 binding to MCF-7 derived exosomes (A). The association and dissociation phases for the peptide were followed for 250 s and 800 s respectively. Changes in the membrane surface charge density of human breast cancer MCF-7 derived exosomes induced by PvD1 (B). Differential di-8-ANEPPS excitation spectra and respective normalized intensity ratio, Rnorm are presented for MCF-7 derived exosomes (C). Each data point represents an average from experiments performed on different days using exosomes isolated from independent cell cultures. Statistical significance was calculated using one-way ANNOVA followed by Dunnett's multiple comparison test. ***0.0001 < p-value < 0.001, **0.001 < p-value < 0.01. | ||
To assess if PvD1 interacts with the exosomal membranes similarly as it does with the membrane of MCF-7 cells, the surface charge density of the vesicles exposed to increased concentrations of the peptide was evaluated with zeta potential measurements. The exosomes’ rich membrane composition in anionic molecules confers to control samples (vesicles with no peptide added) a negative zeta potential: −17.6 ± 2.4 mV (Fig. 7B). This value is roughly 30% higher than the obtained zeta potential of MCF-7 cells (−25.2 ± 2.8 mV) (Fig. 2A). The presence of PvD1 shifted the exosomes’ zeta potential towards neutrality; however, this effect was less prominent than the one observed on breast cells. The peptide exhibits lower affinity to exosomes increasing their zeta potential approximately by 17% at the IC50 (0.24 μM of PvD1) and by 39% at the highest peptide concentration tested, while on cells these increments corresponded to 35% and 46%, respectively.
The effect of PvD1 on exosomes’ membranes was additionally investigated by fluorescence spectroscopy. Exosomes were labeled with di-8-ANEPPS dye, similar to MCF-7 cells and hRBCs. The differential excitation spectra and changes in Rnorm of MCF-7 exosomes after treatment with PvD1 are presented in Fig. 7C.
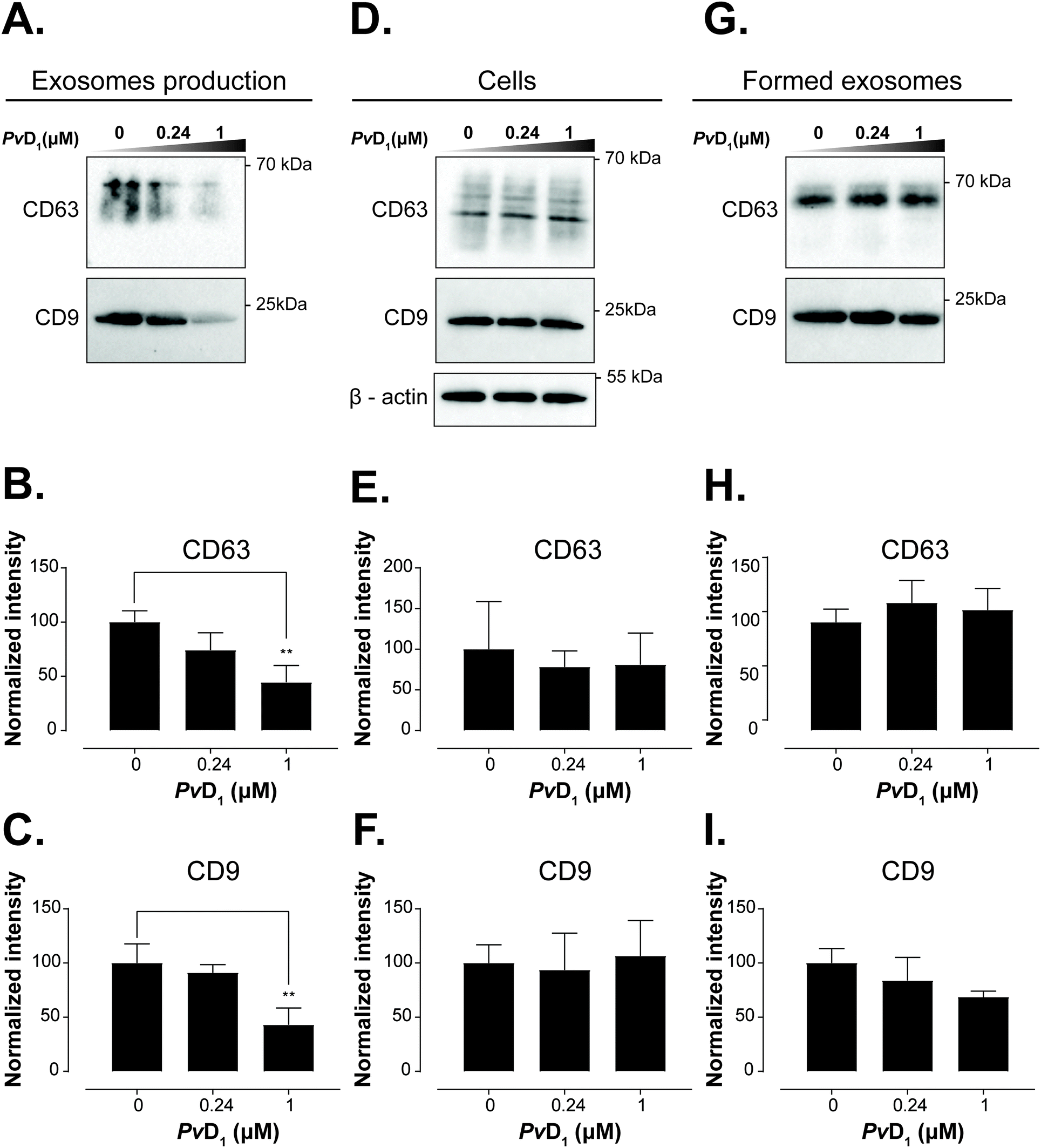 | ||
| Fig. 8 Effect of PvD1 on the recruitment of tetraspanins CD9 and CD63 to the membrane of MCF-7 exosomes. MCF-7 breast cells were incubated with PvD1 at 0.24 μM and 1 μM or with serum-free media for 24 h. The total protein content of exosomes and cell lysates was quantified by the MicroBCA protein assay kit. An equal protein loading of 1 μg was resolved by SDS-PAGE electrophoresis and subjected to western blot analysis. The expression level of tetraspanins CD63 and CD9 in exosomes isolated from human breast cancer MCF-7 conditioned media (A–C) and cell lysates (D–F) is shown. Additionally, PvD1 was incubated with the MCF-7 exosomes for 24 h at 37 °C under cell-free conditions (G–I). Densitometry analysis of the expression of CD63 and CD9 in exosomes isolated from MCF-7 cells exposed to PvD1 (B, C), cell lysates (H, I) and exosomes under cell-free conditions (E, F). The band densities were normalized to the sum of all signals and compared with the control. Bars represent average values of experiments performed on different days using independently grown cell cultures. Exosome samples were prepared from different batches obtained from independent cell cultures. Statistical significance was calculated applying one-way ANNOVA followed by Dunnett's multiple comparison test. **0.001 < p-value < 0.01. | ||
Discussion
Many studies report on the important role of cell-to-cell communication for the growth and development of tumours.59,60 The crosstalk between cellular and non-cellular components of the TME is also a key factor in the tumour responsiveness to treatment.9 In addition to hormones, growth factors and cell–cell junctions, exosomes have emerged as mediators of intercellular communication.10,59,61–63 TDE are significantly involved in the stimulation of the TME and participate in the loss of drug-sensitivity of tumour cells.10,64,65 Envisioning the target of TDE for arresting tumour growth and invasiveness, and reducing the development of resistance, represents an alternative and innovative strategy for treating cancer, either as a therapy alone or as a co-adjuvant in conventional treatments.Recently, our group has unraveled the multifaceted anticancer action of the plant defensin PvD1.27 The peptide attacks breast tumour cells at the primary site but also modulates the cells’ membrane properties and abrogates the adhesion of breast tumour cells to endothelial cells from the human blood–brain barrier (BBB). Therefore, this natural peptide is a promising template for undergoing further pharmacological development. In this work, we investigated the role of PvD1 in exosome-mediated cell-to-cell communication. We aimed at understanding the impact of PvD1 on the exosomes’ formation process by breast cancer cells. However, the peptide-membrane interactions are crucial in peptides’ mode of action and can result in a strong effect either on the tumour growth and/or on the resistance of tumour cells to treatments. Therefore, our studies focused on the direct interaction of PvD1 with tumour, erythrocyte and exosomal membranes.
The studies were performed using the MCF-7 human breast cancer cell line. The behaviour of this cell line in the laboratory agrees with what is clinically observed in patients over time, its genomic instability being responsible for the adaptation and evolution of the cells in culture.48 Human red blood cells (hRBCs) were used as a non-tumoural membrane model system. PvD1 shows nanomolar activity against MCF-7 breast tumour cells without compromising the integrity of hRBCs (Fig. 1). Detailed analysis of PvD1 interaction with MCF-7 cell membranes revealed the substantial role of the cell membrane composition in the peptide's mode of action (Fig. 2). Variations of the tumour cells’ zeta potential values with no changes in the membrane dipole potential that contrast with what is observed for hRBCs, do not allow us to exclude PvD1-hRBCs’ membrane interaction. PvD1 is likely penetrating the tumour cell membrane rather than accumulating at the cell surface. Interestingly the hRBC membrane dipole potential perturbations caused by PvD1 indicate that the peptide remains in contact with the cells’ surface being partially inserted in the membrane. These results led to the hypothesis that the activity of PvD1 relates to senescence induction in cancer cells. Decreased formation of formazan crystals evaluated by the MTT assay suggests the arrest of cells’ metabolic activity, reflected by lower NAD + production, one of the hallmarks of senescence.66 The increased zeta potential of MCF-7 cells does not likely arise from the interaction with PvD1 only, but also from the secretion of senescence-associated-secretory-phenotype (SASP) factors, including vimentin. This protein is evidenced to accumulate on the surface of senescent cells67 and is known to have preferential and strong affinity to negatively charged phospholipids.68 Therefore, vimentin, when presented on the cell surface of senescent cells, associates with anionic POPS and phosphatidylinositol (PI) shielding their negative charge and thus contributing to the partial neutralization of the membrane charge density. Since the predicted net charge of PvD1 is +1.4,27 only a high peptide concentration will lead to changes in the zeta potential of hRBCs, albeit peptide-membrane interaction occurrs already with 3 μM of PvD1 (Fig. 2D). These results are in line with the morphological alterations observed for MCF-7 cells and depicted in the AFM images. After incubation with 0.24 μM PvD1, cells become flat with a significantly expanded surface, another characteristic of cell senescence (Fig. 3 and 4).69 Higher concentrations of PvD1 induce severe damage to the tumour cells resulting in a heterogeneous population that is very difficult to image. Unchanged membrane roughness after the treatment with the plant defensin up to 1 μM additionally supports the maintenance of membrane integrity and lipid bilayer organization (Fig. S1†), expected for senescent cells.67,70
These observations led us to seek further evidence of PvD1 affinity to the lipid bilayer. We therefore performed SPR experiments with liposomes (SUVs) mimicking cell membranes of different compositions and features (fluid and neutral – POPC, POPC:POPE; anionic – POPC:POPS; and with increased membrane rigidity by the addition of cholesterol, Chol, sphingomyelin, SM, and glucosylceramide, GluCer); exosomes isolated from the tumour cells and liposomes prepared with a lipid mixture that closely resembles the natural isolated exosomes. The PvD1 peptide has the highest affinity to anionic fluid membranes containing POPS (Fig. 5 and Table 1). POPS, an anionic phospholipid, is abundant in the exosomal outer membrane where an increased content of Chol and SM is also found.71 The composition and organization of the lipid bilayer surrounding exosomes has not been fully characterized yet and is known to vary depending on the physiological state of the cell.72 Therefore, the constants necessary to extrapolate partition coefficients, Kp, such as the molar volume and certain lipids’ percentage, as well as the contribution and content of membrane proteins, remain elusive. Consequently, Kp was determined only for synthetic SUVs (Table 1).47 Our SPR approach also demonstrates that PvD1 is able to bind to MCF-7 exosomes and to the synthetic membranes composed of the major lipids represented in the exosomal membrane (Fig. 7A and S5†). The slightly higher affinity of PvD1 to the natural exosomes suggests that PvD1 is attracted to these vesicles by a synergistic combination of different factors. Proteins and minor lipids not exposed on the surface of simplified synthetic vesicle models, but present in natural exosomes, seem to account for the interaction with the peptide.
PvD1's binding to the exosome membrane was further analyzed by zeta potential determinations (Fig. 7B). Similar to their cells of origin, MCF-7 derived-exosomes show a net negative surface charge that gradually increases with PvD1 addition. These observations support SPR results by suggesting an interaction between both peptide and exosomes. However, variations of the zeta potential of exosomes after exposure to the peptide are distinct from the ones observed for MCF-7 cells. We hypothesize that PvD1 can penetrate the cellular membrane while accumulating on the surface of the nanovesicles, resulting in higher variations of the exosomes’ zeta potential values when compared with the ones observed for the tumour cells. This hypothesis was additionally reinforced by the results of fluorescence spectroscopy measurements which point towards substantial perturbation of the exosomal membrane by PvD1. The interaction of PvD1 with the surface of the cell membrane is limited due to the peptide's entrance into the cells. It seems that the peptide is spontaneously incorporated into the cells’ interior through a complex intracellular network. Contrarily, when in contact with hRBCs or exosomes PvD1 remains anchored in the membrane.
Exosome uptake may depend on the exposure of anionic POPS on the surface of vesicles.73 Hindered access to POPS and the increased overall charge of MCF-7 exosomes caused by PvD1 binding could affect exosomes’ recognition by recipient cells and concurrently affect exosome uptake. We also observed that PvD1 binds to vesicles containing GlucCer (Fig. 5 and Table 1).74 Phuyal and coworkers presented evidence that an inhibited synthesis of GluCer accounts for a modified composition of exosomes produced by cancer cells.75 Motivated by these results and considering the cell-penetrating potential of PvD1, we sought to investigate whether this plant defensin is able to target the intracellular sorting machinery involved in vesicular trafficking, i.e., if the activity of the peptide may result in a distorted exosomal biogenesis, which could be reflected in an altered protein composition of the produced vesicles.
Tetraspanins CD63 and CD9 are important in the regulation of protein sorting into exosomes and cargo delivery. It has been shown that tetraspanin CD63 can either trigger endosomal membrane deformation or recruit other components of this budding pathway.76 The same study shows that CD63 is not only implied in the membrane budding but also associated with the cargo recruitment to intraluminal vesicles (ILVs), which are precursors of exosomes.76 Also, Miki et al. reported a substantial contribution of CD9 in the amplification of cancer invasiveness77 and Emam and coworkers evidenced the importance of this tetraspanin in the cellular uptake of exosomes.78 As exosomes originate from the inversely budded membrane of the endosome and are highly enriched in CD9 and CD63 tetraspanins, we investigated the effects of the PvD1 peptide on the content of CD9 and CD63 in MCF-7 exosomal membranes. MCF-7 isolated vesicles were characterized by TEM, western blot, DLS, and flow cytometry (Fig. 6, S2 and S3†). The average size of the vesicles with a diameter < 100 nm and detection of CD63 and CD9 tetraspanins on their surface (Fig. 6) underpin the presence of exosomes. The observed decrease in the signal intensity of both anti-CD63 and anti-CD9 antibodies in MCF-7 exosomes isolated from cells treated with the peptide evidence the impact of PvD1 on exosome formation (Fig. 8). Our data suggest that PvD1 modulates the protein composition of exosomes, which is illustrated by the relatively lower expression of CD63 and CD9 to total protein quantified in the isolated vesicles (Fig. 8A, B and C). No changes were detected in the expression of CD63 and CD9 by cells treated with PvD1 or in the isolated mature exosomes that were directly incubated with PvD1 (Fig. 8D, E, F, G, H and I), showing that the proteins are not directly disrupted by the peptide. These observations support alterations in the content of CD63 and CD9 in the exosomes’ membrane arising from the modulated process of exosomes biogenesis by PvD1. The direct incubation with the peptide did not affect the structure, size and the total protein content of exosomes isolated from conditioned media of MCF-7 cells (Fig. S6 and S7†) implying that the binding of PvD1 to natural exosomes does not have a harmful effect on the vesicles. However, the peptide is able to regulate the composition of the exosomal membrane proteins acting intracellularly. The maintained total protein content of exosomes isolated from PvD1-treated MCF-7 cells is in agreement with published data showing that exosomes’ production is not constrained in senescent cells.79 However, the detailed mechanism involved in the altered expression of CD63 and CD9 on the surface of MCF-7 exosomes caused by PvD1 requires further elucidation.
Conclusions
Our study reveals, for the first time, the modulatory effect of PvD1 defensin on the exosomes’ biogenesis in human breast tumour cells. The peptide is attracted to the negative surface membrane of the tumour cells and reaches the intracellular space, as previously reported for a different tumour cell line. Once inside the cell, PvD1 can bind to intracellular membranes, such as the endosomal membrane, and modulate the intraluminal budding of exosomes and their composition. Our data support this hypothesis by showing a decreased expression of both tetraspanins CD63 and CD9 in exosomes isolated from MCF-7 cells previously exposed to PvD1. Exosomes’ membrane composition might then be compromised in consequence of the peptide activity. Not only the modulated surface protein composition but also the extracellular interaction of PvD1 with the exosomes can be expected to affect vesicles’ fate and function in cellular communication, influencing tumour growth and resistance. The treatment of primary breast cancer with the PvD1 peptide would also benefit from the use of low peptide concentrations that could induce cell senescence, improving the therapeutic outcome and decreasing the severe side effects resulting from anticancer therapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Fundação para a Ciência e a Tecnologia (FCT I.P., Portugal) for funding – PTDC/BBB-BQB/1693/2014, and also acknowledge financial support from the Brazilian agencies CNPq, CAPES, and FAPERJ (E-26/203.090/2016; E-26/202.132/2015). Julia Skalska, Filipa D. Oliveira, Tiago N. Figueira and Diana Gaspar acknowledge FCT I.P. for fellowships PD/BD/114177/2016, PD/BD/135046/2017, SFRH/BD/5283/2013 and SFRH/BPD/109010/2015 respectively. Marie Skłodowska-Curie Research and Innovation Staff Exchange (RISE) is also acknowledged for funding: call H2020-MSCA-RISE-2014, Grant agreement 644167, 2015–2019. Additionally, the authors thank Andreia Pinto from the Histology and Comparative Pathology Laboratory from iMM for expert Electron Microscopy assistance.References
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries, Ca-Cancer J. Clin., 2018, 1–31 Search PubMed.
- J. Baselga, M. Campone, M. Piccart, H. A. Burris III, H. S. Rugo and T. Sahmoud, et al., Everolimus in Postmenopausal Hormone- Receptor–Positive Advanced Breast Cancer, N. Engl. J. Med., 2012, 366(6), 520–529 CrossRef CAS PubMed.
- R. Nahta, M.-C. Hung and F. J. Esteva, The HER-2-Targeting Antibodies Trastuzumab and Pertuzumab Synergistically Inhibit the Survival of Breast Cancer Cells, Cancer Res., 2004, 64(7), 2343–2346 CrossRef CAS PubMed.
- B. Mansoori, A. Mohammadi, S. Davudian, S. Shirjang and B. Baradaran, The different mechanisms of cancer drug resistance: A brief review, Adv. Pharm. Bull., 2017, 7(3), 339–348 CrossRef CAS PubMed.
- H. Martin, L. Smith and D. Tomlinson, Multidrug-resistant breast cancer: current perspectives, Breast Cancer: Targets Ther., 2014, 4, 1–13 Search PubMed.
- M. Moloudizargari, M. H. Asghari and M. Abdollahi, Modifying exosome release in cancer therapy: How can it help?, Pharmacol. Res., 2018, 134(4), 246–256 CrossRef CAS PubMed.
- M. H. Asghari, E. Ghobadi, M. Moloudizargari, M. Fallah and M. Abdollahi, Does the use of melatonin overcome drug resistance in cancer chemotherapy?, Life Sci., 2018, 196(December 2017), 143–155 CrossRef CAS PubMed.
- C. Peetla, S. Vijayaraghavalu and V. Labhasetwar, Biophysics of Cell Membrane Lipids in Cancer Drug Resistance: Implications for Drug Transport and Drug Delivery with Nanoparticles, Adv. Drug Delivery Rev., 2013, 65(0), 1–29 Search PubMed.
- H. Patel, P. Nilendu, D. Jahagirdar, J. K. Pal and N. K. Sharma, Modulating secreted components of tumor microenvironment: A masterstroke in tumor therapeutics, Cancer Biol. Ther., 2018, 19(1), 3–12 CrossRef CAS PubMed.
- C. Zhang, Q. Ji, Y. Yang, Q. Li and Z. Wang, Exosome: Function and Role in Cancer Metastasis and Drug Resistance, Technol. Cancer Res. Treat., 2018, 17, 1–12 Search PubMed.
- C. Théry, L. Zitvogel and S. Amigorena, Exosomes: Composition, biogenesis and function, Nat. Rev. Immunol., 2002, 2(8), 569–579 CrossRef.
- A. S. Azmi, B. Bao and F. H. Sarkar, Exosomes in cancer development, metastasis, and drug resistance: A comprehensive review, Cancer Metastasis Rev., 2013, 32(3–4), 623–642 CrossRef CAS PubMed.
- C. Roma-Rodrigues, A. R. Fernandes and P. V. Baptista, Exosome in tumour microenvironment: Overview of the crosstalk between normal and cancer cells, BioMed. Res. Int., 2014, 2014, 1–10 CrossRef PubMed.
- M. Lv, X. Zhu, W. Chen, S. Zhong, Q. Hu and T. Ma, et al., Exosomes mediate drug resistance transfer in MCF-7 breast cancer cells and a probable mechanism is delivery of P-glycoprotein, Tumor Biol., 2014, 35(11), 10773–10779 CrossRef CAS PubMed.
- K. Ning, T. Wang, T. Sun, P. Zhang, Y. Chen and J. Jin, et al., UCH-L1-containing exosomes mediate chemotherapeutic resistance transfer in breast cancer, J. Surg. Oncol., 2017, 115(8), 932–940 CrossRef CAS PubMed.
- W. Wang, L. Zou, D. Zhou, Z. Zhou, F. Tang and Z. Xu, et al., Overexpression of ubiquitin carboxyl terminal hydrolase-L1 enhances multidrug resistance and invasion/metastasis in breast cancer by activating the MAPK/Erk signaling pathway, Mol. Carcinog., 2016, 55(9), 1329–1342 CrossRef CAS PubMed.
- M. Giallombardo, S. Taverna, R. Alessandro, D. Hong and C. Rolfo, Exosome-mediated drug resistance in cancer: the near future is here, Ther. Adv. Med. Oncol., 2016, 8(5), 320–322 CrossRef CAS PubMed.
- F. M. Khan, E. Saleh, H. Alawadhi, R. Harati, W. H. Zimmermann and R. El-Awady, Inhibition of exosome release by ketotifen enhances sensitivity of cancer cells to doxorubicin, Cancer Biol. Ther., 2018, 19(1), 25–33 CrossRef CAS PubMed.
- C. Federici, F. Petrucci, S. Caimi, A. Cesolini, M. Logozzi and M. Borghi, et al., Exosome release and low pH belong to a framework of resistance of human melanoma cells to cisplatin, PLoS One, 2014, 9(2), 1–11 CrossRef PubMed.
- V. Ciravolo, V. Huber, G. C. Ghedini, E. Venturelli, F. Bianchi and M. Campiglio, et al., Potential role of HER2-overexpressing exosomes in countering trastuzumab-based therapy, J. Cell. Physiol., 2012, 227(2), 658–667 CrossRef CAS PubMed.
- M. R. Felício, O. N. Silva, S. Gonçalves, N. C. Santos and O. L. Franco, Peptides with Dual Antimicrobial and Anticancer Activities, Front. Chem., 2017, 5(February), 1–9 Search PubMed.
- J. M. Freire, D. Gaspar, A. S. Veiga and M. A. R. B. Castanho, Shifting gear in antimicrobial and anticancer peptides biophysical studies: From vesicles to cells, J. Pept. Sci., 2015, 21(3), 178–185 CrossRef CAS PubMed.
- D. W. Hoskin and A. Ramamoorthy, Studies on anticancer activities of antimicrobial peptides, Biochim. Biophys. Acta, Biomembr., 2008, 1778(2), 357–375 CrossRef CAS PubMed.
- K. C. L. Mulder, L. A. Lima, V. J. Miranda, S. C. Dias and O. L. Franco, Current scenario of peptide-based drugs: the key roles of cationic antitumor and antiviral peptides, Front. Microbiol., 2013, 4(October), 1–23 Search PubMed.
- S. Al-Benna, Y. Shai, F. Jacobsen and L. Steinstraesser, Oncolytic Activities of host defense peptides, Int. J. Mol. Sci., 2011, 12(11), 8027–8051 CrossRef CAS PubMed.
- D. Gaspar, J. M. Freire, T. R. Pacheco, J. T. Barata and M. A. R. B. Castanho, Apoptotic human neutrophil peptide-1 anti-tumor activity revealed by cellular biomechanics, Biochim. Biophys. Acta, Mol. Cell Res., 2015, 1853(2), 308–316 CrossRef CAS PubMed.
- T. N. Figueira, F. D. Oliveira, I. Almeida, É. O. Mello, V. M. Gomes and M. A. R. B. Castanho, et al., Challenging metastatic breast cancer with the natural defensin PvD1, Nanoscale, 2017, 9(43), 16887–16899 RSC.
- L. Hazlett and M. Wu, Defensins in innate immunity, Cell Tissue Res., 2011, 343(1), 175–188 CrossRef CAS PubMed.
- C. A. Müller, J. Markovic-Lipkovski, T. Klatt, J. Gamper, G. Schwarz and H. Beck, et al., Human alpha-defensins HNPs-1, -2, and -3 in renal cell carcinoma: influences on tumor cell proliferation, Am. J. Pathol., 2002, 160(4), 1311–1324 CrossRef.
- N. Xu, Y. Wang, W. Pan, B. Xiao, Y. Wen and X. Chen, et al., Human alpha-defensin-1 inhibits growth of human lung adenocarcinoma xenograft in nude mice, Mol. Cancer Ther., 2008, 7(June), 1588–1597 CrossRef CAS PubMed.
- E. D. O. Mello, I. S. dos Santos, A. D. O. Carvalho, L. S. de Souza, G. A. de Souza-Filho and V. V. do Nascimento, et al., Functional expression and activity of the recombinant antifungal defensin PvD1r from Phaseolus vulgaris L. (common bean) seeds, BMC Biochem., 2014, 15(7), 1–13 Search PubMed.
- V. V. do Nascimento, E. D. O. Mello, L. P. Carvalho, E. J. T. de Melo, A. D. O. Carvalho and K. V. S. Fernandes, et al., PvD1 defensin, a plant antimicrobial peptide with inhibitory activity against Leishmania amazonensis, Biosci. Rep., 2015, 35(5), 1–7 CrossRef PubMed.
- E. O. Mello, S. F. F. Ribeiro, A. O. Carvalho, I. S. Santos, M. Da Cunha and C. Santa-Catarina, et al., Antifungal activity of PvD1 defensin involves plasma membrane permeabilization, inhibition of medium acidification, and induction of ROS in fungi cells, Curr. Microbiol., 2011, 62(4), 1209–1217 CrossRef CAS PubMed.
- P. D. Games, I. S. dos Santos, É. O. Mello, M. S. S. Diz, A. O. Carvalho and G. A. de Souza-Filho, et al., Isolation, characterization and cloning of a cDNA encoding a new antifungal defensin from Phaseolus vulgaris L. seeds, Peptides, 2008, 29(12), 2090–2100 CrossRef CAS PubMed.
- G. van Niel, S. Charrin, S. Simoes, M. Romao, L. Rochin and P. Saftig, et al., The Tetraspanin CD63 Regulates ESCRT-Independent and -Dependent Endosomal Sorting during Melanogenesis, Dev. Cell, 2011, 21(4), 708–721 CrossRef CAS PubMed.
- A. E. Morelli, A. T. Larregina, W. J. Shufesky, M. L. G. Sullivan, D. B. Stolz and G. D. Papworth, et al., Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells, Blood, 2004, 104(10), 3257–3266 CrossRef CAS PubMed.
- F. Vences-Catalán and S. Levy, Immune targeting of tetraspanins involved in cell invasion and metastasis, Front. Immunol., 2018, 9(Jun), 1–7 Search PubMed.
- S. Kohmo, T. Kijima, Y. Otani, M. Mori, T. Minami and R. Takahashi, et al., Cell surface tetraspanin CD9 mediates chemoresistance in small cell lung cancer, Cancer Res., 2010, 70(20), 8025–8035 CrossRef CAS.
- T. C. Chou, Derivation and properties of Michaelis-Menten type and Hill type equations for reference ligands, J. Theor. Biol., 1976, 59(2), 253–276 CrossRef CAS.
- R. J. Clarke and D. J. Kane, Optical detection of membrane dipole potential: Avoidance of fluidity and dye-induced effects, Biochim. Biophys. Acta, Biomembr., 1997, 1323(2), 223–239 CrossRef CAS.
- M. Kaszuba, J. Corbett, F. M. N. Watson and A. Jones, High-concentration zeta potential measurements using light-scattering techniques, Philos. Trans. R. Soc., A., 2010, 368(1927), 4439–4451 CrossRef CAS PubMed.
- D. E. Koppel, Analysis of macromolecular polydispersity in intensity correlation spectroscopy: The method of cumulants, J. Chem. Phys., 1972, 57(11), 4814–4820 CrossRef CAS.
- A. Einstein, Über die von der molekularkinetischen Theorie der Wärme geforderte Bewegung von in ruhenden Flüssigkeiten suspendierten Teilchen, Ann. Phys., 1905, 322, 549–560 CrossRef.
- U. K. Laemmli, F. Beguin and G. Gujer-Kellenberger, A factor preventing the major head protein of bacteriophage T4 from random aggregation, J. Mol. Biol., 1970, 47(1), 69–85 CrossRef CAS PubMed.
- H. Towbin, T. Staehelint and J. Gordon, Towbin Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets, Proc. Natl. Acad. Sci. U. S. A., 1979, 76(9), 4350–4354 CrossRef CAS PubMed.
- A. S. Veiga, L. K. Pattenden, J. M. Fletcher, M. A. R. B. Castanho and M. I. Aguilar, Interactions of HIV-1 antibodies 2F5 and 4E10 with a gp41 epitope prebound to host and viral membrane model systems, ChemBioChem, 2009, 10(6), 1032–1044 CrossRef CAS PubMed.
- T. N. Figueira, J. M. Freire, C. Cunha-Santos and M. A. R. B. Castanho, Quantitative analysis of molecular partition towards lipid membranes using surface plasmon resonance, Sci. Rep., 2017,(November 2016), 1–10 Search PubMed.
- A. V. Lee, S. Oesterreich and N. E. Davidson, MCF-7 Cells - Changing the Course of Breast Cancer Research and Care for 45 Years, J. Natl. Cancer Inst., 2015, 107(7), 1–4 CrossRef.
- Ş. Comşa, A. M. Cîmpean and M. Raica, The story of MCF-7 breast cancer cell line: 40 Years of experience in research, Anticancer Res., 2015, 35(6), 3147–3154 Search PubMed.
- S. Leporatti, D. Vergara, A. Zacheo, V. Vergaro, G. Maruccio and R. Cingolani, et al., Cytomechanical and topological investigation of MCF-7 cells by scanningforce microscopy, Nanotechnology, 2009, 20(5), 1–6 CrossRef PubMed.
- M. M. Domingues, P. S. Santiago, M. A. R. B. Castanho and N. C. Santos, What can light scattering spectroscopy do for membrane-ctive peptide studies, J. Pept. Sci., 2008, 14(10), 1084–1095 CrossRef PubMed.
- M. Freire, M. M. Domingues, J. Matos, M. N. Melo, A. Salome and N. C. Santos, et al., Using zeta-potential measurements to quantify peptide partition to lipid membranes, Eur. Biophys. J., 2011, 40, 481–487 CrossRef PubMed.
- Q. S. Li, G. Y. H. Lee, C. N. Ong and C. T. Lim, AFM indentation study of breast cancer cells, Biochem. Biophys. Res. Commun., 2008, 374(4), 609–613 CrossRef CAS PubMed.
- S. Zalba and T. L. M. ten Hagen, Cell membrane modulation as adjuvant in cancer therapy, Cancer Treat. Rev., 2017, 52, 48–57 CrossRef CAS PubMed.
- B. Sharma and S. S. Kanwar, Phosphatidylserine: A cancer cell targeting biomarker, Semin. Cancer Biol., 2018, 52, 17–25 CrossRef CAS PubMed.
- G. Raposo and W. Stoorvogel, Extracellular vesicles: Exosomes, microvesicles, and friends, J. Cell Biol., 2013, 200(4), 373–383 CrossRef CAS PubMed.
- M. Lu, X. Zhao, H. Xing, Z. Xun, S. Zhu and L. Lang, et al., Comparison of exosome-mimicking liposomes with conventional liposomes for intracellular delivery of siRNA, Int. J. Pharm., 2018, 550(1–2), 100–113 CrossRef CAS PubMed.
- T. Skotland, K. Sandvig and A. Llorente, Lipids in exosomes: Current knowledge and the way forward, Prog. Lipid Res., 2017, 66, 30–41 CrossRef CAS PubMed.
- G. D. Carystinos, A. Bier and G. Batist, The role of connexin-mediated cell-cell communication in breast cancer metastasis, J. Mammary Gland Biol. Neoplasia, 2001, 6(4), 431–440 CrossRef CAS PubMed.
- A. R. Chin and S. E. Wang, Cancer-derived extracellular vesicles: the “soil conditioner” in breast cancer metastasis?, Cancer Metastasis Rev., 2016, 35(4), 669–676 CrossRef CAS PubMed.
- A. Becker, B. K. Thakur, J. M. Weiss, H. S. Kim, H. Peinado and D. Lyden, Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis, Cancer Cell, 2016, 30(6), 836–848 CrossRef CAS PubMed.
- A. S. Azmi, B. Bao and F. H. Sarkar, Exosomes in Cancer Development, Metastisis and Drug Resistance: A Comprehensive Review, Cancer Metastasis Rev., 2014, 32(0), 1–33 Search PubMed.
- A. Latifkar, R. A. Cerione and M. A. Antonyak, Probing the mechanisms of extracellular vesicle biogenesis and function in cancer, Biochem. Soc. Trans., 2018, 1–10 Search PubMed.
- Z. Wang, J. Q. Chen, J. Liu and L. Tian, Exosomes in tumor microenvironment: Novel transporters and biomarkers, J. Transl. Med., 2016, 14(1), 1–9 CrossRef PubMed.
- J. Skog, T. Würdinger, S. van Rijn, D. H. Meijer, L. Gainche and W. T. Curry, et al., Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers, Nat. Cell Biol., 2008, 10(12), 1470–1476 CrossRef CAS PubMed.
- J. Birch, P. J. Barnes and J. F. Passos, Mitochondria, telomeres and cell senescence: Implications for lung ageing and disease, Pharmacol. Ther., 2018, 183(October 2017), 34–49 CrossRef CAS PubMed.
- D. Frescas, C. M. Roux, S. Aygun-Sunar, A. S. Gleiberman, P. Krasnov and O. V. Kurnasov, et al., Senescent cells expose and secrete an oxidized form of membrane-bound vimentin as revealed by a natural polyreactive antibody, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(9), E1668–E1677 CrossRef CAS PubMed.
- S. Horkovics-Kovats and P. Traub, Specific Interaction of the Intermediate Filament Protein Vimentin and Its Isolated N-Terminus with Negatively Charged Phospholipids As Determined by Vesicle Aggregation, Fusion, and Leakage Measurements, Biochemistry, 1990, 29(37), 8652–8657 CrossRef CAS PubMed.
- M. Y. Terzi, M. Izmirli and B. Gogebakan, The cell fate: senescence or quiescence, Mol. Biol. Rep., 2016, 43(11), 1213–1220 CrossRef CAS PubMed.
- J. M. Vicencio, L. Galluzzi, N. Najeddine, C. Ortiz, A. Criollo and E. Tasdemir, et al., Senescence, Apoptosis or Autophagy?, Gerontology, 2008, 54(2), 92–99 CrossRef.
- L. A. Mulcahy, R. C. Pink and D. R. F. Carter, Routes and mechanisms of extracellular vesicle uptake, J. Extracell. Vesicles, 2014, 3(1), 1–14 Search PubMed.
- L. Barile and G. Vassalli, Exosomes: Therapy delivery tools of diseasses, Pharmacol. Ther., 2017, 174, 63–78 CrossRef CAS PubMed.
- S. Keller, A.-K. König, F. Marmé, S. Runz, S. Wolterink and D. Koensgen, et al., Systemic presence and tumor-growth promoting effect of ovarian carcinoma released exosomes, Cancer Lett., 2009, 278(1), 73–81 CrossRef CAS PubMed.
- V. S. G. Amaral, C. M. Fernandes, M. R. Felício, A. S. Valle, P. G. Quintana and C. C. Almeida, et al., Psd2 pea defensin shows a preference for mimetic membrane rafts enriched with glucosylceramide and ergosterol, Biochim. Biophys. Acta, Biomembr., 2019, 1861(4), 713–728 CrossRef CAS PubMed.
- S. Phuyal, N. P. Hessvik, T. Skotland, K. Sandvig and A. Llorente, Regulation of exosome release by glycosphingolipids and flotillins, FEBS J., 2014, 281(9), 2214–2227 CrossRef CAS PubMed.
- J. R. Edgar, E. R. Eden and C. E. Futter, Hrs- and CD63-Dependent Competing Mechanisms Make Different Sized Endosomal Intraluminal Vesicles, Traffic, 2014, 15(2), 197–211 CrossRef CAS PubMed.
- Y. Miki, M. Yashiro, T. Okuno, K. Kitayama, G. Masuda and K. Hirakawa, et al., CD9-positive exosomes from cancerassociated fibroblasts stimulate the migration ability of scirrhous-type gastric cancer cells, Br. J. Cancer, 2018, 118(6), 867–877 CrossRef CAS PubMed.
- S. E. Emam, H. Ando, A. S. A. Lila, T. Shimizu, K. Okuhira and Y. Ishima, et al., Liposome co-incubation with cancer cells secreted exosomes (extracellular vesicles) with different proteins expressions and different uptake pathways, Sci. Rep., 2018, 8(1), 1–11 CrossRef CAS PubMed.
- M. Takasugi, R. Okada, A. Takahashi, D. V. Chen, S. Watanabe and E. Hara, Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2, Nat. Commun., 2017, 8(15729), 1–11 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr07843f |
| This journal is © The Royal Society of Chemistry 2019 |