
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmplified vibrational circular dichroism as a manifestation of the interaction between a water soluble gold nanocluster and cobalt salt†
Sarita
Roy Bhattacharya
* and
Thomas
Bürgi
 *
*
Department of Physical Chemistry, University of Geneva, 30 Quai Ernest-Ansermet, 1211 Geneva 4, Switzerland. E-mail: Sarita.Bhattacharya@unige.ch; Thomas.Buergi@unige.ch
First published on 11th November 2019
Abstract
Vibrational circular dichroism (VCD) is a powerful tool for the structure determination of dissolved molecules. However, the application of VCD to nanostructures is limited up to now due to the weakness of the effect and hence the low signal intensities. Here we show that the addition of a small amount of cobalt(II) drastically enhances the VCD signals of a thiolate-protected gold cluster Au25(Capt)18 (Capt = captopril) in aqueous solution. An increase of VCD signal intensity of at least one order of magnitude is observed. The enhancement depends on the amount of CoCl2 added but almost an order of magnitude enhancement is already observed at a cluster![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CoCl2 ratio of 1:1. In contrast, circular dichroism (CD) and infrared spectra hardly change. The increase in VCD intensity goes along with a qualitative change of the spectrum and the enhancement increases with time reaching a stable state only after several hours. The enhancement is due to an interaction between the cobalt(II) and the cluster, which also leads to quenching of its fluorescence. The behaviour is completely different for free captopril, where the addition of cobalt(II) salt does not affect the VCD spectrum.
:CoCl2 ratio of 1:1. In contrast, circular dichroism (CD) and infrared spectra hardly change. The increase in VCD intensity goes along with a qualitative change of the spectrum and the enhancement increases with time reaching a stable state only after several hours. The enhancement is due to an interaction between the cobalt(II) and the cluster, which also leads to quenching of its fluorescence. The behaviour is completely different for free captopril, where the addition of cobalt(II) salt does not affect the VCD spectrum.
Introduction
Symmetry breaking is an important attribute of evolutionary processes. The higher abundance of particles over antiparticles is an attribute of cosmological evolution.1 Similarly one finds higher abundance of L-amino acids and D-sugars over D-amino acids and L-sugars in biological evolution. Enzymatic pathways in biology are generally highly enantiospecific. Chirality also exists at a higher scale (topological). For example one finds right handed helix (B-DNA) and not left handed helix (Z-DNA), and one finds only the negatively supercoiled state.2 In sharp contrast to the molecules of life, metals are inherently symmetric (in their bulk) unless they are perturbed with some chiral molecules.3 However, at the nanoscale regime, where surface effects play a dominant role, the scenario is totally different. Chirality in nanoclusters is often observed and can be attributed to different reasons: (1) capping of the cluster by chiral ligands, (2) dissymmetric distribution of ligands (chiral arrangement of ligands around the metal core), and (3) inherent chirality of the metal core of the cluster.4,5 Chiral metal nanoclusters have properties that make them attractive for applications in different domains like enantiomeric separation, enantioselective detection, asymmetric catalysis, pharmaceutical applications and others.6–11The chirality of metal nanoclusters can be probed by electronic circular dichroism (CD).12,13 CD spectra are a sensitive probe of the size and structure of the cluster. For example the CD spectra of Au38(SR)24 (SR: thiolate) and Au40(SR)24 are completely different (every atom counts).6,14 However, CD is not a good method to study the structure of ligands on the clusters for various reasons but mostly because the relevant electronic transitions of the ligands overlap with transitions of the cluster. In contrast, most vibrational transitions of organic ligands are spectrally well separated from the vibrations of the metal core, which are found at very low frequencies (<350 cm−1).15 Therefore, vibrational circular dichroism (VCD), which is very sensitive to structure, seems a promising method to study ligand layers. VCD measures the differential absorption of left- and right-circularly polarized light by a chiral sample. It is one of the powerful techniques to study the configuration as well as conformational state of molecules in solution.16 However, one of the major experimental challenges of VCD spectroscopy is the inherently small signals (differential absorption typically in the order of 10−4 to 10−6) and consequently the long measurement times. This makes VCD measurements of molecules adsorbed on metal structures particularly difficult. Hence techniques to enhance the VCD signal are required. Compared to flat surfaces and nanoparticles, clusters have a relatively high specific surface area, which makes VCD measurements feasible. Indeed the VCD spectra of monolayer-protected clusters have been reported. These studies give insight into the binding mode and the structure (conformation) of the ligand.13,17–19 Another method to increase the VCD intensity is based on the use of elliptically polarized ultrashort laser pulses as reported by Helbing and co-workers.20 The group has also previously applied time-resolved VCD spectroscopy by using a femtosecond laser system synchronised with a photoelastic modulator to measure VCD signals. In this way, they were able to measure the enhanced CH-stretching vibrations of a cobalt–sparteine complex.21 Important progress in the direction of VCD signal enhancement has been reported by Nafie and co-workers.22 They have used transition metals like Zn(II), Ni(II), Co(II) to form complexes with sparteine. Co(II) and Ni(II) form open-shell complexes whereas a closed-shell complex is obtained with Zn(II). In the case of open-shell complexes, the VCD signals were enhanced by approximately one order of magnitude. In addition, the sign of several VCD bands changed when enhancement was observed, whereas the un-polarized infrared (IR) absorption spectra were the same for the open-shell and the closed-shell systems. The enhancement of the VCD signal could be attributed to the presence of low-lying magnetic-dipole allowed d–d transitions vibronically coupled to the ground state.23
With recent advancement in technology VCD has found wide application in biology. For example one can study the configurational and conformational state and chiral microenvironment of bio-molecules. Domingos et al. have successfully shown that one can achieve an increase of at least two orders of magnitude in VCD signal intensities of amino acids and oligopeptides when they are coupled to paramagnetic metal ions.24 The group proposed that the enhancement in VCD signal is due to the presence of low-lying electronic states provided by the paramagnetic, high-spin Co2+ ion. By using this approach, they could probe the local structure of biomolecules like amino acids and sometimes even larger biomolecules like polypeptides surrounding a paramagnetic ion. Using this enhancement, the study of larger biomolecules or the formation and development of protein amyloid fibrils are now feasible.25–27
In contrast to biomolecules no such enhancement of VCD signals has been reported yet for nanoclusters. In the following we report for the first time enhancement of VCD signal for a water soluble nanocluster. To better understand the structure of their surface layer, enhanced VCD could be a potential technique, as such information is very hard to obtain especially for water soluble nanoclusters. For this study we have used captopril (Capt) as a thiol ligand for the synthesis of Au25(Capt)18. An enhancement of at least one order of magnitude in VCD signal was observed when these nanoclusters were mixed with CoCl2 solution, even at very low cluster:CoCl2 ratios. The increase in VCD signal of gold nanocluster depends on the amount of CoCl2 added. Interaction of the nanocluster and CoCl2 was further studied by UV/vis, fluorescence, gel electrophoresis and circular dichroism spectroscopy. The interaction of Co2+ with the Au25(Capt)18 cluster was distinctly different from the one with captopril. In the latter case no enhancement at all has been observed.
Experimental
Reagents
The following chemicals and reagents are commercially available: hydrogen tetrachloroaurate(III) trihydrate (Alfa Aesar, ACS grade, 99.99%), tetra-n-octyl ammonium bromide (TOABr, Alfa Aesar, >98%), N-[(S)-3-mercapto-2-methylpropionyl]-L-proline (captopril) (Sigma Aldrich, ≥98% (HPLC)), methanol and ethanol analytical grade (Fisher chemical), CoCl2·6H2O (Fluka), D2O (Acros Organic, 99.8 atom % D), alpha-cyano-4-hydroxycinnamic acid (Sigma Aldrich, ≥99.0%, HPLC), acrylamide (Acros Organic, >99%, electrophoresis grade), N,N′-methylenebis(acrylamide) (Acros Organic, >99%, electrophoresis grade), glycine (Acros Organic, >99%, for analysis), tris(hydroxymethyl)aminomethane (Acros Organic, 99.8%, ACS reagent), ammonium persulfate (APS, Acros Organic, 98%, extra pure), N,N,N′,N′-tetramethylethylenediamine (TEMED, 99.5%, Acros Organic), and sodium chloride (Sigma-Aldrich, >99.5%, for analysis). Water was purified with a Milli-Q system (≥18 MΩ cm). All the chemicals were used as received except CoCl2. CoCl2 was obtained by heating CoCl2·6H2O at 150 °C under vacuum overnight.Synthesis of captopril (Capt) stabilized Au25(Capt)18 nanoclusters
Synthesis of water soluble Au25(Capt)18 was done according to a previously reported method.28 In brief, 0.2 mmol HAuCl4·3H2O (78.7 mg) was mixed vigorously with 0.23 mmol TOABr (126.8 mg) in 10 ml of methanol at room temperature. After approximately 25 min, 1 mmol captopril (217.2 mg) in 5 ml of MeOH was rapidly added into the reaction mixture under constant stirring and the reaction was allowed to proceed for another 30 min. After that 2 mmol NaBH4 (75.6 mg) was dissolved in 5 ml of ice cold water and rapidly injected into the reaction mixture. The reaction was continued for at least 8 to 10 h to get a reddish brown color which indicates the formation of the Au25 nanocluster. Later the solution was centrifuged to remove unreacted, insoluble gold–thiolate polymers. The supernatant was collected and concentrated by rotary evaporation. The clusters were precipitated by ethanol and extracted with minimum amounts of methanol. This step was repeated at least two more times to obtain the Au25(Capt)18 cluster in pure form. Finally, the cluster was dried under vacuum, re-dissolved in milliQ water and further lyophilized (Telstar Cryodos) to obtain a dry powder of pure nanocluster. The sample was stored at 4 °C for further experiments.Characterization methods
UV–Vis spectra were recorded on a Varian Cary 50 spectrophotometer, using a quartz cuvette of 1 cm path length. All the spectra were recorded in Milli-Q water. The spectra were normalized at 400 nm when required.Mass spectra were recorded in positive mode using a Bruker Autoflex mass spectrometer equipped with a nitrogen laser. Alpha-cyano-4-hydroxycinnamic acid (α-CHCA) was used as the matrix with a 1:10, 1:100 and 1:1000 analyte:matrix ratio. 25 mM α-CHCA was prepared in 50% acetonitrile:H2O with 0.1% trifluoroacetic acid. After preparing the matrix solution it was sonicated for 5 to 10 min and then centrifuged to exclude any undissolved matrix. A volume of 2 μL of the analyte/matrix mixture was applied to the target and air-dried.
IR and vibrational circular dichroism (VCD) spectra were recorded on a Bruker PMA 50 accessory coupled to a Tensor 27 Fourier transform infrared spectrometer. A photoelastic modulator (Hinds PEM 90) set at quarter wave retardation was used to modulate the handedness of the circular polarized light. Demodulation was performed by using a lock-in amplifier (SR830 DSP). An optical low-pass filter (<1800 cm−1) placed before the photoelastic modulator was used to enhance the signal/noise ratio.
For VCD measurements 0.2 mg μl−1 of Au25(Capt)18 was dissolved in D2O. CoCl2 was added to obtain solutions with defined cluster:CoCl2 molar ratios (1:1, 1:3, 1:5). All spectra were recorded at room temperature with a resolution of 4 cm−1 in a cell equipped with CaF2 windows and a 12 μm Teflon spacer. VCD spectra of the nanocluster and nanocluster mixed with different amounts of CoCl2 were corrected with the reference VCD spectrum of D2O. Each sample and reference were measured for a minimum of 8 hours in time slices of one hour, corresponding to about 32000 scans, unless otherwise stated.
Polyacrylamide gel electrophoresis (PAGE) was used to confirm the purity of the cluster and to study the binding of cobalt. It was performed with a Biorad protean II XI system with a gel thickness of approximately 3 mm. For the stacking and the separating, the percentage of gels was 5% and 20% respectively (T = 30% and C = 3.33%, where T is the mass-volume percentage of the monomer including the cross-linker, N,N′-methylene-bis-acrylamide (Bis), and C is the proportion of the cross-linker as a percentage of the total monomer). The stacking and the separating gels were buffered at pH = 6.8 and 8.8, respectively, with Tris-HCl solution. Running buffer consisted of 25 mM tris-HCl and 192 mM glycine. The gels were run at a constant voltage of 150 V for a minimum of 8 h. Approximately, 1 mg of the (Au25(Capt)18 sample and Au25(Capt)18 nanoclusters mixed with different amounts of CoCl2 in 5% glycerol solution were loaded in each well.
The hydrodynamic diameters of Au25(Capt)18 nanoclusters and its complex with CoCl2 were measured at room temperature using dynamic light scattering (DLS) technique (Nano-ZS; Malvern Instruments, Malvern, United Kingdom).29
CD spectra were recorded on a JASCO J-715 CD spectrometer using a quartz cuvette of 1 mm path length. The spectra were recorded in milliQ water and the signal of the blank solvent was subtracted from the CD signal of the sample. CD spectra were obtained by averaging three scans with a scanning speed of 100 nm min−1 and a data pitch of 0.1 nm.
The fluorescence spectra were recorded at room temperature on a Fluorolog 3 (Horiba) spectrometer using a 10 mm × 2 mm quartz cuvette. The excitation wavelength was fixed at 514 nm (from a 450 W Xe lamp) in emission measurements. The emission was measured between 550 and 850 nm and identical excitation and emission slit widths of 5.0 nm were used.
Results and discussion
Synthesis of Au25(Capt)18 nanoclusters was confirmed by UV/Vis spectroscopy. In Fig. 1(a), the presence of peaks in the UV-vis spectrum at 670 nm and 450 nm with some shoulder peaks around 550 nm and 400 nm is in good agreement with the literature,30 confirming the synthesis of the Au25(Capt)18 nanocluster. We furthermore performed matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) to confirm the presence of Au25(Capt)18. Note that MALDI analysis of water-soluble nanoclusters is challenging but we successfully recorded the spectra of the Au25(Capt)18 nanocluster using α-CHCA as a matrix. Fig. 1b shows the molecular ion peak at ∼8826 m/z (theoretical: ∼8818 m/z) with the predominant fragment at ∼7168 m/z (for Au21(Capt)14, theoretical: ∼7165 m/z) after the loss of ∼1658 m/z i.e. Au4(Capt)4 unit (theoretical: ∼1653 m/z). Several previous reports also showed that the strongest fragment peak of the Au25(SR)18 cluster is Au21(SR)14.31,32 Although, there were a lot of fragment peaks, we could assign each peak as shown in Fig. 1(b). The numbers (n = 1…4) in the figure mark the fragment peaks obtained by the loss of Aun(Capt)n from the Au25(Capt)18 nanocluster. The peaks between these marked peaks correspond to the loss of one additional gold atom or one captopril ligand.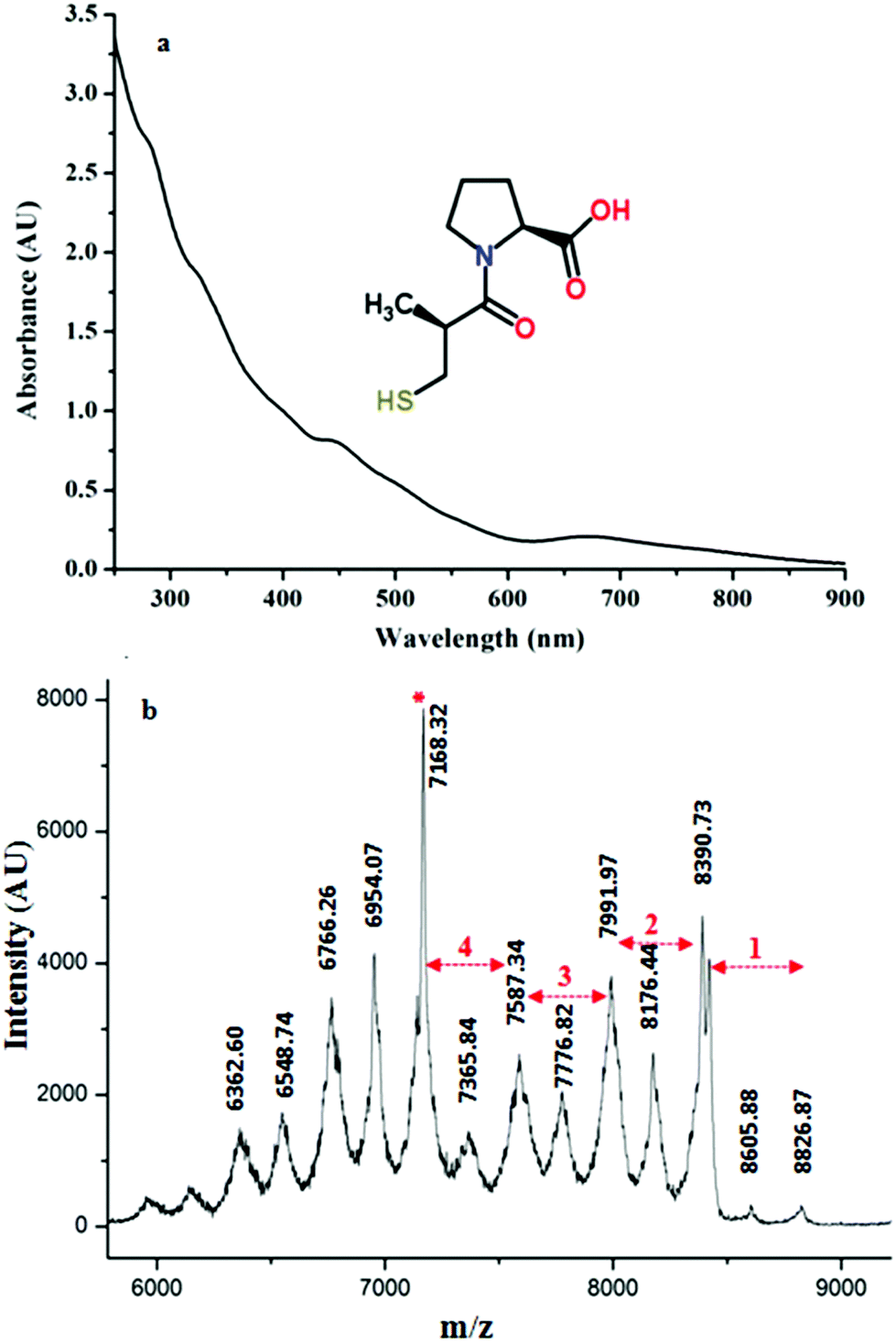 | ||
| Fig. 1 (a) UV/Vis spectrum of the synthesized Au25(Capt)18 nanocluster. The inset of the figure shows the structure of the captopril ligand used for the synthesis of Au25(Capt)18. (b) MALDI mass spectrum of Au25(Capt)18 recorded in positive mode with α-CHCA as the matrix. The numbers (n = 1…4) mark the fragment peaks obtained by the loss of Aun(Capt)n from the parent cluster. The peak marked with an asterisk corresponds to the most important fragment Au21(Capt)14. | ||
Fig. 2a represents the VCD spectra of water soluble Au25(Capt)18 nanoclusters both in the absence (black line) and presence of CoCl2 at different cluster:cobalt ratios (1:1, 1:3 and 1:5 indicated by red, blue and cyan color, respectively). The VCD spectrum of the Au25(Capt)18 cluster (0.2 mg μl−1) in the absence of cobalt salt is very weak. The major VCD feature ranging from 1620–1560 cm−1 is a bi-polar peak (positive–negative from high to low wavenumbers) with the intensity of 1.35 × 10−5 to −8.35 × 10−5. Several very weak peaks are hardly distinguishable from the noise of the measurement which was typically ΔA ∼ 7 × 10−6 in this spectral range. Interestingly, the VCD spectrum of captopril in water was distinctly different from the one of Au25(Capt)18. In fact, captopril showed only one weak negative peak in the region around 1600 cm−1 (Fig. 2b) in contrast to the bi-polar band observed for captopril adsorbed on the Au25 cluster. This could be due to a structural change upon adsorption of captopril on the cluster, which is further reflected in the infrared spectra (see below).
 | ||
| Fig. 2 (a) VCD spectra of Au25(Capt)18 (black line) and of Au25(Capt)18:CoCl2 mixtures at 1:1, 1:3 and 1:5 molar ratios (red, blue and cyan color respectively). The inset represents an enlarged area from 1500 to 1000 cm−1. (b) VCD spectra of captopril (black) and captopril:CoCl2 mixture (1:18 ratio, red solid line). The signal around 1200 cm−1 is an artifact due to the strong absorption of the solvent (D2O) in this region. | ||
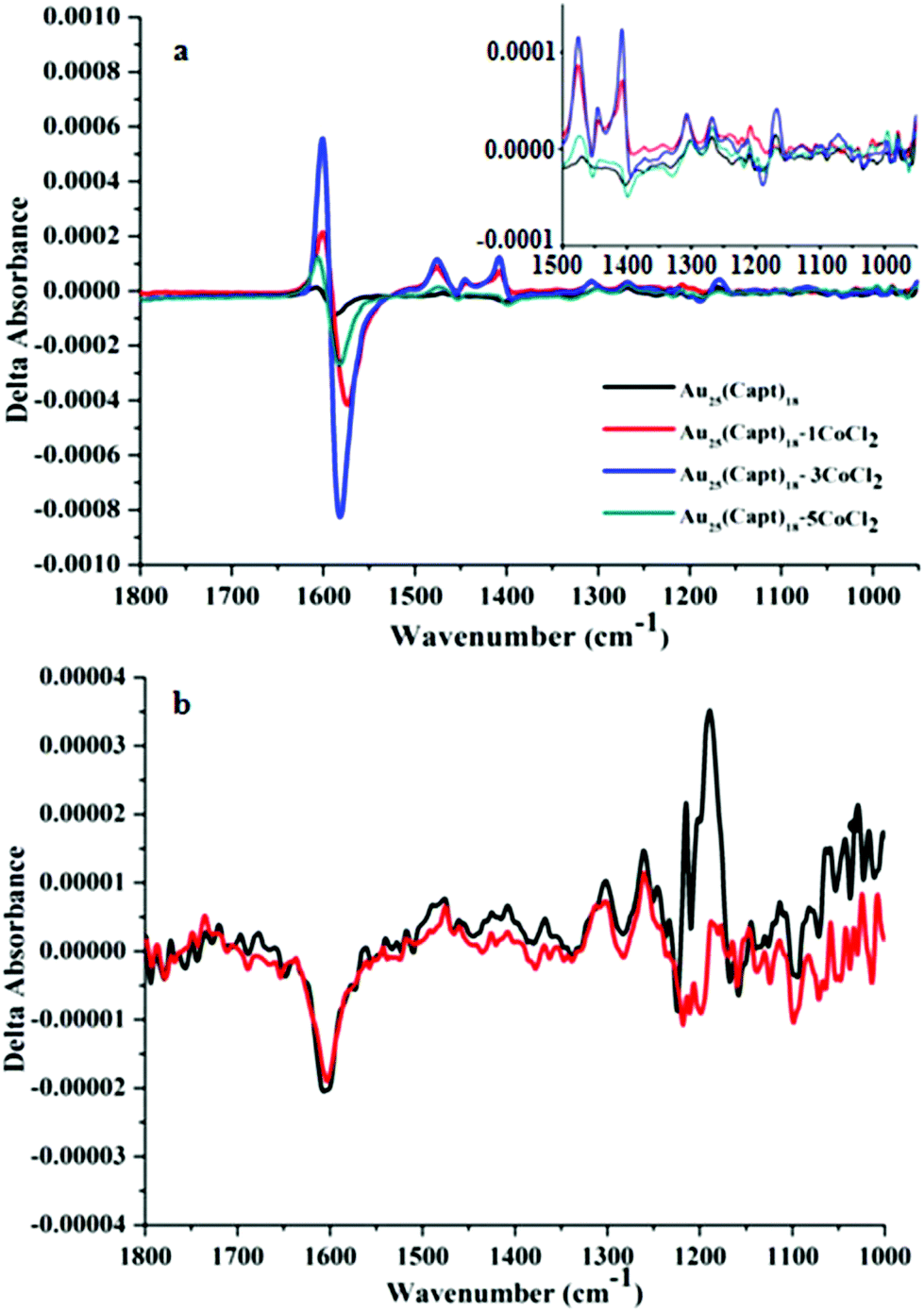
The addition of CoCl2 is expected to lead to complexation of Co2+ by captopril. Fig. S1(a) and (b)† show the IR spectra of the Au25(Capt)18 cluster and captopril, respectively and the effect of CoCl2 addition. In general, it is known33 that ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of free carboxylic groups (–COOH) arises at 1725–1700 cm−1. It is also well known33 that in metal carboxylates ν(COO−) asymmetric and symmetric stretching vibrations are typically found at 1600 cm−1 (overlapping with the amide I stretching vibration) and 1400 cm−1, respectively. It is evident from Fig. S1† that the peak at ∼1713 cm−1 is present in free captopril and captopril mixed with CoCl2 (Fig. S1(b)†) but the peak is absent in the spectra of the cluster (Fig. S1(a)†). This shows that the acid group of captopril is deprotonated in the cluster. As the ν(CO) band gets suppressed the symmetric and asymmetric carboxylate stretching bands (centered around 1400 cm−1 and 1600 cm−1) are more pronounced. A possible explanation for the different IR spectra of free captopril and captopril ligand on the cluster is that in the latter case the carboxyl groups also interact with the gold, similar to what was proposed for N-acetyl cysteine.34,35 In general, the addition of CoCl2 to captopril and Au25(Capt)18, respectively, did not have a pronounced effect on the infrared spectra except for a slight shift in the position of peaks (1–2 cm−1). However, the VCD spectrum of Au25(Capt)18 changed drastically, as can be seen in Fig. 2(a).
O) of free carboxylic groups (–COOH) arises at 1725–1700 cm−1. It is also well known33 that in metal carboxylates ν(COO−) asymmetric and symmetric stretching vibrations are typically found at 1600 cm−1 (overlapping with the amide I stretching vibration) and 1400 cm−1, respectively. It is evident from Fig. S1† that the peak at ∼1713 cm−1 is present in free captopril and captopril mixed with CoCl2 (Fig. S1(b)†) but the peak is absent in the spectra of the cluster (Fig. S1(a)†). This shows that the acid group of captopril is deprotonated in the cluster. As the ν(CO) band gets suppressed the symmetric and asymmetric carboxylate stretching bands (centered around 1400 cm−1 and 1600 cm−1) are more pronounced. A possible explanation for the different IR spectra of free captopril and captopril ligand on the cluster is that in the latter case the carboxyl groups also interact with the gold, similar to what was proposed for N-acetyl cysteine.34,35 In general, the addition of CoCl2 to captopril and Au25(Capt)18, respectively, did not have a pronounced effect on the infrared spectra except for a slight shift in the position of peaks (1–2 cm−1). However, the VCD spectrum of Au25(Capt)18 changed drastically, as can be seen in Fig. 2(a).
The addition of one equivalent of CoCl2 with respect to the Au25(Capt)18 cluster boosted the intensity of the VCD bands drastically. For example, the bi-polar band (1620–1560 cm−1) changed from 1.35 × 10−5 to 2.14 × 10−4 and from −8.35 × 10−5 to −4.16 × 10−4 (red solid line in Fig. 2a). By increasing the concentration of the cobalt salt to three equivalents the intensity of the VCD spectrum was further enhanced (blue solid line in Fig. 2a) by 2–3 times with respect to one equivalent of salt. With respect to the VCD spectrum of the Au25(Capt)18 cluster in the absence of cobalt salt the intensity increased by more than an order of magnitude when adding three equivalents of CoCl2. The strongest peak almost reached ΔA of 10−3, which is a huge VCD signal. Interestingly, a further increase of the cobalt salt concentration (1:5 cluster:Co2+ ratio) resulted in a decrease of the VCD intensity (cyan color spectrum in Fig. 2). Fig. S2,† depicts the intensity of the VCD signal at 1620–1560 cm−1 (difference between positive and negative band maxima) as a function of the cluster:CoCl2 ratio. The increase can be explained by increased complexation. The reason for the decrease of the VCD signal at a higher Co2+ concentration is surprising and more difficult to explain. We noted that at even higher cobalt concentrations (1:7 ratio) the cluster precipitated, as evidenced by the solid at the bottom of the cell. However, at a cluster:Co2+ ratio of 1:5 no precipitation was observed, which is also confirmed by the strong IR signal of the sample (Fig. S1†), since the IR signal should decrease if the cluster precipitates out of the solution.
The increased intensity of the VCD signals is due to an interaction of Co2+ with the cluster. One possibility is the formation of a complex with the captopril ligands on the cluster (one or more) and possibly water.22,36 It has been shown that paramagnetic metal ions like Co2+ can enhance the VCD signals of amino acids.24 Interestingly the addition of Co2+ to solutions of captopril did not affect the VCD spectra at all (Fig. 2(b)). Whereas the addition of Co2+ to Au25(Capt)18 resulted in changes in the VCD signal, and the IR spectrum remained basically unchanged as is evident from Fig. S1.† This finding can be well explained by referring to the work by Nafie and Barnett.22,23,36 They have measured the IR and VCD spectra of (+)-sparteine complexes with Zn(II), Co(II) and Ni(II) and observed similar IR spectra for all the complexes but different VCD spectral patterns. The complex formed with Zn(II) exhibited typical VCD spectral features whereas in the case of Co(II) and Ni(II) an enhancement of at least one order of magnitude in VCD intensity was observed. The observation that the IR spectra of all these complexes were virtually the same was explained by the strongly electric-dipole forbidden (magnetic-dipole allowed) d–d transitions, which make irrelevant contribution to the electric-dipole transition moment.
Interestingly, we noticed a time-dependence of the VCD spectra (Fig. 3(a), cluster:CoCl2 ratio of 1:3). A gradual change in the ΔA from −4.3 × 10−4 to −9.9 × 10−4 of the most prominent negative band at 1582 cm−1 was observed after 1 h of addition of CoCl2. Whereas ΔA for only the nanocluster i.e. without any addition of CoCl2 was 9.87 × 10−5 (star mark in Fig. 3b). Fig. 3b represents a plot of the absorbance difference between the positive and the negative bands around 1600–1590 cm−1 as a function of time. After about 6 h the VCD signal was close to stable.
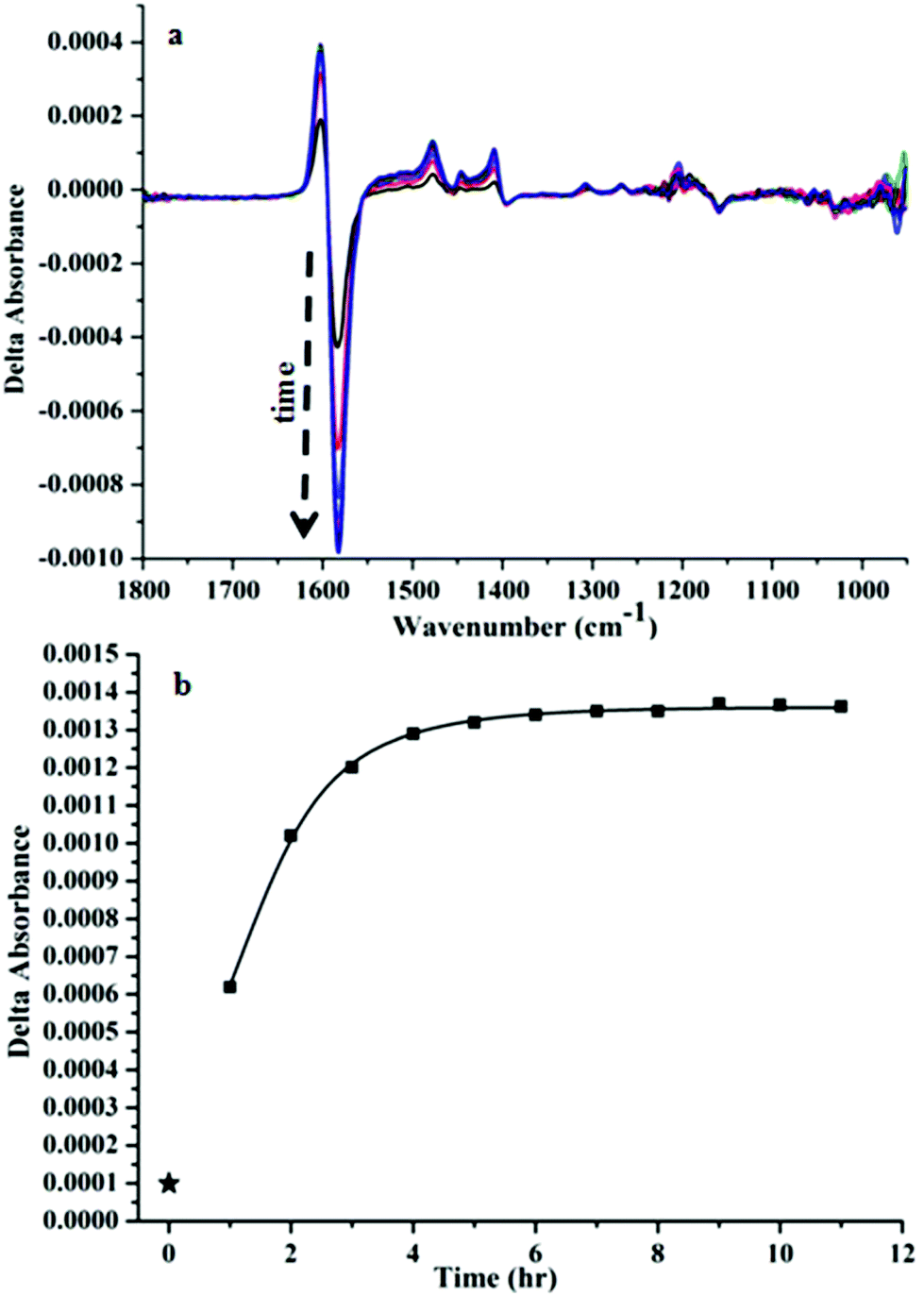 | ||
| Fig. 3 (a) Time dependent change in the VCD signal of Au25(Capt)18 after the addition of CoCl2 (1:3 ratio). (b) Plot of the intensity difference of the positive–negative band around 1620–1560 cm−1 (maximum–minimum) as a function of time (star mark represents the point in the absence of CoCl2). | ||
Co2+ in aqueous environment exhibits absorbance maxima around 520 nm and 470 nm,37 which fits well with our absorption spectra of CoCl2 both in water (Fig. S3(a)†) and in captopril solution (Fig. S3(b)†). This further confirms the presence of Co2+ in our system. Note that Co3+, when complexed with ethylenediamine, absorbs around 550 nm and sometimes the absorption is even more red shifted.38 The observation that the spectra of Co2+ are basically identical with and without captopril indicates that Co2+ does not form strong complexes with captopril, in agreement with the IR and VCD spectra of captopril–CoCl2 solutions. In solutions containing the cluster the absorption due to Co2+ is masked by the strong absorption of the cluster in this spectral range.
To further study the interaction between Co2+ and the cluster additional analysis was performed. MALDI mass spectra of samples at different cluster:CoCl2 ratios (1:1, 1:3, 1:5) were recorded. We did not find any significant changes in the mass spectra neither in the position of the molecular ion peak (∼8826 Da) nor in the fragment peaks (Fig. 4). This indicates that the cobalt–cluster complex is not strong enough to survive the MALDI process.
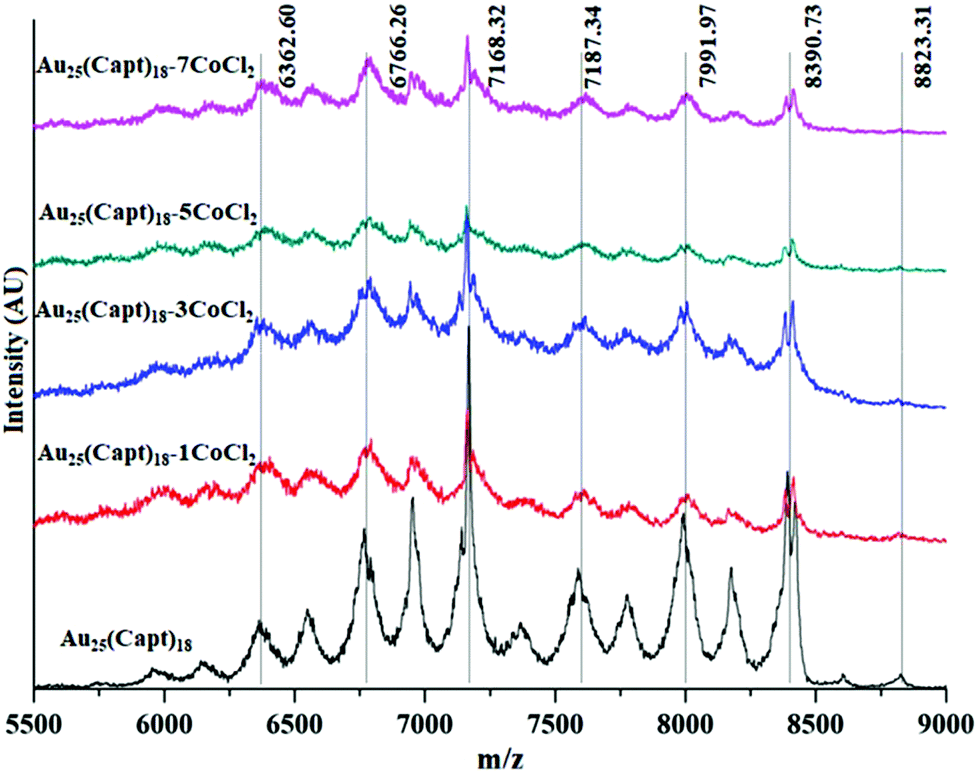 | ||
| Fig. 4 Comparative MALDI mass spectra of Au25(Capt)18 and mixtures with different amounts of CoCl2 (cluster Co2+ ratios 1:1, 1:3, 1:5, 1:7). | ||
Since our nanocluster system is water soluble, we took advantage of this to characterize it with gel electrophoresis (PAGE) and to further confirm the MALDI result. The PAGE experiment allowed us to evaluate the purity of nanoclusters and to verify whether any other cluster population arises due to interaction with cobalt chloride. From Fig. S4,† it is evident that the addition of CoCl2 to Au25(Capt)18 did not have an influence on the mobility of the cluster in PAGE. In this experiment the mobility of Au25(Capt)18 was compared to the same sample mixed with CoCl2 (1:1, 1:3 and 1:5 ratio). In fact, only one clear band due to Au25(Capt)18 was observed in all lanes of the PAGE experiment. A slight impurity with lower mobility was observed in all samples (also without the addition of CoCl2). The intensity and width of the Au25 nanocluster band was basically identical in all the four lanes. Therefore the cluster was migrating at the same speed irrespective of interaction with cobalt chloride. Hence the PAGE experiment confirmed the rather weak interaction between Co2+ and the cluster.
Fig. 5 shows the time dependent UV-vis spectra of the Au25(Capt)18 cluster after the addition of CoCl2 at 1:1, 1:3 and 1:5 cluster salt ratios. For each sample the spectrum was recorded at different times i.e. 0 (immediately after mixing the cluster and CoCl2), 60, 120 and 240 min. It is evident that CoCl2 did not impart significant changes to the Au25(Capt)18 spectral features even after 240 minutes. Hence the interaction between Co2+ and the cluster did not influence the optical properties of Au25(Capt)18. This finding further confirms the MALDI data (Fig. 4) and also the PAGE experiment (Fig. S4†). All these experiments indicate that the interaction between Co2+ and the cluster is rather weak. The hydrodynamic size of the Au25(Capt)18 nanocluster and its mixtures at different cluster:CoCl2 ratios (1:0 (only nanocluster), 1:1, 1:3 and 1:5) are shown in Fig. S5.† From the DLS data we can confirm that there are no large cluster aggregates and no aggregation effect due to CoCl2 addition could be observed. All the hydrodynamic diameters were 3 nm or below for the cluster–CoCl2 mixtures.
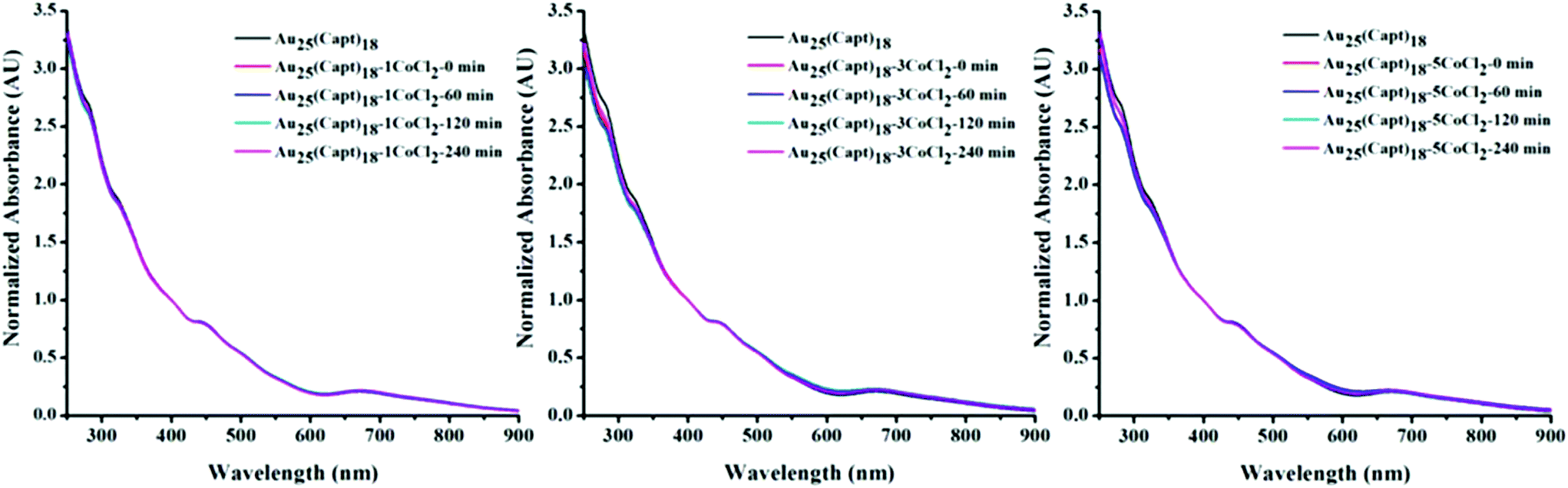 | ||
| Fig. 5 Time dependent UV/Vis spectra of Au25(Capt)18 containing different amounts of CoCl2 (left: cluster:CoCl2 ratio 1:1, middle: 1:3, right: 1:5). Spectra were recorded 0, 60, 120 and 240 min after addition of the salt to the cluster solution. | ||
Circular dichroism is a sensitive probe for structure and therefore CD spectra could shed light on the interaction between the cluster and Co2+, since this interaction would likely lead to changes of the structure (conformation) and/or of the electronic structure. CD spectra of captopril and Au25(Capt)18 solutions in the presence and absence of cobalt salt are shown in Fig. S6.† Panel (a) shows the effect of different cluster:Co2+ ratios on the CD spectra, which was very small, if present at all.
Possibly the negative band around 325 nm was slightly shifting to a higher energy upon the addition of Co2+. Note that these bands are not due to the captopril but due to the electronic structure of the cluster. Fig. S6(b)† also shows no changes in the electronic CD of captopril upon the addition of CoCl2. There are slight changes in the intensity of CD bands, which can be attributed to a dilution effect when adding the salt.
Fluorescence spectroscopy is another sensitive probe for molecular interactions. Au25(Capt)18 possesses a well characterized fluorescence emission peak in the range of 600–800 nm when excited at 514 nm.28 As is evident from Fig. S7,† the addition of CoCl2 leads to a decrease of the fluorescence of the cluster. The effect was more pronounced for a higher Co2+ concentration and increased with time. Hence, Co2+ acts as a quencher of Au25(Capt)18 fluorescence. It has also been reported previously that heavy metal ions have the tendency to quench the fluorescence of water soluble nanoclusters.39,40 The observation that the fluorescence is not completely quenched is either due to incomplete complexation (presence of free Au25(Capt)18, with no Co2+ bound) or due to the weaker fluorescence of the Au25(Capt)18–Co2+ complex compared to Au25(Capt)18. The fluorescence seems to decrease on a time-scale of hours, which is similar to the increase of the VCD signal intensity.
The drastic increase in the VCD signal of the Au25(Capt)18 cluster upon interaction with Co2+ could be due to intensity borrowing from low lying electronic transitions, similar to that observed for metal complexes.22–24 The Au25(SR)18 cluster has a band gap of 1.3 eV41 and therefore no low-lying electronic transitions. Therefore, such transitions in our system should be associated with Co2+. As mentioned above, no enhancement of VCD signals was observed for solutions of CoCl2 and captopril (CoCl2:captopril ratio of 1:18, Fig. 2b and ESI Fig. S1b†). The dissimilar behaviour of the free ligand and the ligand-protected cluster could be due to different complex formation with cobalt in the two cases. Possibly complexation of cobalt on the cluster involves more than one captopril ligand leading to stronger complex formation (chelate effect) and different complex geometry compared to captopril. Another explanation for the different behaviour of the cluster and captopril could be a different geometry (and composition) of the respective complexes (number of captopril and water molecules in the coordination sphere of Co2+), leading to altered electronic properties of Co2+ in the two cases. Hence the complex with captopril possibly does not have suitable low-lying electronic transitions that would lead to VCD enhancement. Based on the observation that both the IR and VCD spectra do not change upon the addition of CoCl2 to captopril solution42 we prefer the first interpretation namely that complexes with captopril are much weaker (or even absent).
Despite the drastic effect of Co2+ on the VCD spectra of the Au25(Capt)18 cluster, UV-vis, IR, MALDI and PAGE do not provide evidence of cluster–Co2+ interaction. The only other clear evidence for the interaction of Co2+ with the cluster is the fluorescence measurements. Based on these findings only, it is difficult to draw conclusions about the nature of the “complex” between Co2+ and the cluster. In this context it is important to note that both VCD and fluorescence showed similar time-dependence, which was very slow in both cases. The formation of a complex between Co2+ and one or two captopril ligands on the cluster surface and possibly some water molecules should be fast. This could involve some conformational change of the captopril, but this should be a fast process. Possibly the slow kinetics is associated with another process, like the formation of an Au25(Capt)18–Co2+ adduct, similar to that detected for Cu.43
Finally, it is worth mentioning that the enhancement of VCD signals due to interaction with paramagnetic metal ions like Co2+ has been shown to be a local effect.24 Only the functional groups in close proximity to the metal ion give rise to enhanced signals. For our case this means that locally the enhancement is larger than one order of magnitude. The enhancement furthermore opens the possibility to study reactions taking place within the ligand shell of thiolate-protected metal clusters.
Conclusions
The present study shows an enhancement of at least one order of magnitude in VCD signal when Au25(Capt)18 nanoclusters interact with CoCl2. The increase in the VCD signal of the gold nanocluster increases with the amount of Co(II) added and is maximum when the ratio of Au25(Capt)18:CoCl2 is around 1:5. The interaction of the nanocluster and CoCl2 is further studied by UV/vis, fluorescence, circular dichroism spectroscopy and PAGE. Besides VCD only the fluorescence measurements provide clear evidence of an interaction between Co2+ and the cluster, whereas the other methods are blind for this interaction. The behaviour of Au25(Capt)18 is completely different from the one of captopril, which shows no enhancement of VCD signals at all. Possibly the interaction between the cluster and Co2+ involves not only the captropril ligands but also the staple units of the cluster, thus forming a Au25(Capt)18–Co2+ adduct. This could explain the slow kinetics of the VCD signal enhancement. The increase in VCD signals could be explained by the presence of low lying electronic excited states.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Swiss National Science Foundation (grant number 200020_172511) and the University of Geneva is kindly acknowledged. We also acknowledge Rossella Grillo for helping us with DLS measurements.Notes and references
- E. Vangioni and M. Cassé, Front. Life Sci., 2017, 10, 84–97 CrossRef CAS.
- D. G. Blackmond, Cold Spring Harbor Perspect. Biol., 2019, 11, a032540 CrossRef PubMed.
- H. Behar-Levy, O. Neumann, R. Naaman and D. Avnir, Adv. Mater., 2007, 19, 1207–1211 CrossRef CAS.
- S. Knoppe and T. Bürgi, Acc. Chem. Res., 2014, 47, 1318–1326 CrossRef CAS PubMed.
- C. Zeng and R. Jin, Chem. – Asian J., 2017, 12, 1839–1850 CrossRef CAS PubMed.
- I. Dolamic, S. Knoppe, A. Dass and T. Bürgi, Nat. Commun., 2012, 3, 798 CrossRef PubMed.
- A. Gogoi, N. Mazumder, S. Konwer, H. Ranawat, N.-T. Chen and G.-Y. Zhuo, Molecules, 2019, 24, 1007 CrossRef CAS PubMed.
- E. Gross, J. H. Liu, S. Alayoglu, M. A. Marcus, S. C. Fakra, F. D. Toste and G. A. Somorjai, J. Am. Chem. Soc., 2013, 135, 3881–3886 CrossRef CAS PubMed.
- N. Shukla, N. Ondeck, N. Khosla, S. Klara, A. Petti and A. Gellman, Nanomater. Nanotechnol., 2015, 5, 1 CrossRef CAS.
- S. Roy, K. Bhattacharya, C. Mandal and A. K. Dasgupta, J. Mater. Chem. B, 2013, 1, 6634–6643 RSC.
- C. Zhang, Z. Zhou, X. Zhi, Y. Ma, K. Wang, Y. Wang, Y. Zhang, H. Fu, W. Jin and F. J. T. Pan, Theranostics, 2015, 5, 134 CrossRef PubMed.
- S. Roy, S. Basak and A. K. Dasgupta, J. Nanosci. Nanotechnol., 2010, 10, 819–825 CrossRef CAS PubMed.
- H. Yao, K. Miki, N. Nishida, A. Sasaki and K. Kimura, J. Am. Chem. Soc., 2005, 127, 15536–15543 CrossRef CAS PubMed.
- S. Knoppe, I. Dolamic, A. Dass and T. Bürgi, Angew. Chem., Int. Ed., 2012, 51, 7589–7591 CrossRef CAS PubMed.
- B. Nieto-Ortega and T. Burgi, Acc. Chem. Res., 2018, 51, 2811–2819 CrossRef CAS PubMed.
- L. A. Nafie and T. B. Freedman, Enantiomer, 1998, 3, 283–297 CAS.
- C. Gautier and T. Bürgi, J. Phys. Chem. C, 2010, 114, 15897–15902 CrossRef CAS.
- C. Gautier and T. Bürgi, J. Am. Chem. Soc., 2006, 128, 11079–11087 CrossRef CAS PubMed.
- I. Dolamic, B. Varnholt and T. Bürgi, Nat. Commun., 2015, 6, 7117 CrossRef CAS PubMed.
- J. Helbing and M. Bonmarin, J. Chem. Phys., 2009, 131, 174507 CrossRef PubMed.
- M. Bonmarin and J. Helbing, Opt. Lett., 2008, 33, 2086–2088 CrossRef PubMed.
- Y. He, X. Cao, L. A. Nafie and T. B. Freedman, J. Am. Chem. Soc., 2001, 123, 11320–11321 CrossRef CAS PubMed.
- L. A. Nafie, J. Phys. Chem. A, 2004, 108, 7222–7231 CrossRef CAS.
- S. R. Domingos, A. Huerta-Viga, L. Baij, S. Amirjalayer, D. A. E. Dunnebier, A. J. C. Walters, M. Finger, L. A. Nafie, B. de Bruin, W. J. Buma and S. Woutersen, J. Am. Chem. Soc., 2014, 136, 3530–3535 CrossRef CAS PubMed.
- S. Ma, X. Cao, M. Mak, A. Sadik, C. Walkner, T. B. Freedman, I. K. Lednev, R. K. Dukor and L. A. Nafie, J. Am. Chem. Soc., 2007, 129, 12364–12365 CrossRef CAS PubMed.
- D. Kurouski, X. Lu, L. Popova, W. Wan, M. Shanmugasundaram, G. Stubbs, R. K. Dukor, I. K. Lednev and L. A. Nafie, J. Am. Chem. Soc., 2014, 136, 2302–2312 CrossRef CAS PubMed.
- X. Lu, H. Li, J. W. Nafie, T. Pazderka, M. Pazderková, R. K. Dukor and L. A. Nafie, Appl. Spectrosc., 2017, 71, 1117–1126 CrossRef CAS PubMed.
- S. Kumar and R. Jin, Nanoscale, 2012, 4, 4222–4227 RSC.
- A. Baghdasaryan, R. Grillo, S. Roy Bhattacharya, M. Sharma, E. Reginato, H. Theraulaz, I. Dolamic, M. Dadras, S. Rudaz, E. Varesio and T. Burgi, ACS Appl. Nano Mater., 2018, 1, 4258–4267 CrossRef CAS.
- X. Kang, H. Chong and M. Zhu, Nanoscale, 2018, 10, 10758–10834 RSC.
- S. K. Katla, J. Zhang, E. Castro, R. A. Bernal and X. Li, ACS Appl. Mater. Interfaces, 2018, 10, 75–82 CrossRef CAS PubMed.
- A. Dass, A. Stevenson, G. R. Dubay, J. B. Tracy and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 5940–5946 CrossRef CAS PubMed.
- J. Oomens and J. D. Steill, J. Phys. Chem. A, 2008, 112, 3281–3283 CrossRef CAS PubMed.
- M. Bieri and T. Bürgi, J. Phys. Chem. B, 2005, 109, 22476–22485 CrossRef CAS PubMed.
- M. Bieri and T. Bürgi, ChemPhysChem, 2006, 7, 514–523 CrossRef CAS PubMed.
- C. J. Barnett, A. F. Drake, R. Kuroda, S. F. Mason and S. Savage, Chem. Phys. Lett., 1980, 70, 8–10 CrossRef CAS.
- N. Carmona, V. Bouzas, F. Jiménez, M. Plaza, L. Pérez, M. A. García, M. A. Villegas and J. Llopis, Sens. Actuators, B, 2010, 145, 139–145 CrossRef CAS.
- D. Jacewicz, J. Pranczk, D. Wyrzykowski, K. Żamojć and L. Chmurzyński, React. Kinet., Mech. Catal., 2014, 113, 321–331 CrossRef CAS.
- W. Chen, X. Tu and X. Guo, Chem. Commun., 2009, 1736–1738, 10.1039/B820145E.
- J. Xie, Y. Zheng and J. Y. Ying, Chem. Commun., 2010, 46, 961–963 RSC.
- H. Kawasaki, S. Kumar, G. Li, C. Zeng, D. R. Kauffman, J. Yoshimoto, Y. Iwasaki and R. Jin, Chem. Mater., 2014, 26, 2777–2788 CrossRef CAS.
- K. S. Siddiqi, S. Bano, A. Mohd and A. A. P. Khan, Chin. J. Chem., 2009, 27, 1755–1761 CrossRef CAS.
- R. Kazan, U. Müller and T. Bürgi, Nanoscale, 2019, 11, 2938–2945 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr07534h |
| This journal is © The Royal Society of Chemistry 2019 |