
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalogen bond-assisted self-assembly of gold nanoparticles in solution and on a planar surface†
Kavitha
Buntara Sanjeeva
 a,
Claudia
Pigliacelli
*b,
Lara
Gazzera
a,
Valentina
Dichiarante
a,
Francesca
Baldelli Bombelli
*a and
Pierangelo
Metrangolo
ab
a,
Claudia
Pigliacelli
*b,
Lara
Gazzera
a,
Valentina
Dichiarante
a,
Francesca
Baldelli Bombelli
*a and
Pierangelo
Metrangolo
ab
aLaboratory of Supramolecular and BioNano Materials (SupraBioNanoLab), Department of Chemistry, Materials, and Chemical Engineering “Giulio Natta”, Politecnico di Milano Via L. Mancinelli 7, 20131 Milan, Italy. E-mail: francesca.baldelli@polimi.it
bHyber Center of Excellence, Department of Applied Physics, Aalto University, Puumiehenkuja 2, FI-00076 Espoo, Finland. E-mail: claudia.pigliacelli@aalto.fi
First published on 24th September 2019
Abstract
Halogen bonding (XB) has been shown to be a powerful tool for promoting molecular self-assembly in different fields. The use of XB for noncovalent assembly of inorganic nanoparticles (NP) is, instead, quite limited, considering how extensively other interactions (i.e., electrostatic forces, hydrophobic effect, hydrogen bonding, etc.) have been exploited to modulate and program NP self-assembly. Here, we designed and synthesized XB-capable organic ligands that were efficiently used to functionalize the surface of gold NPs (AuNPs). XB-assisted AuNP self-assembly was attained in solution mixing AuNPs bearing XB-donor ligands with ditopic XB-acceptor molecules and AuNPs functionalized with XB-acceptor moieties. Likewise, a preliminary study of XB-driven adsorption of these AuNPs on surface was performed via Quartz Crystal Microbalance with Dissipation Monitoring (QCM-D), used as an in situ tool for measuring mass changes upon XB-driven self-assembly.
Introduction
Self-assembly of ligand-protected gold nanoparticles (AuNPs) is a growing research area aiming at the design of specific superstructures bearing new functionalities.1–4 Customizing the building blocks with a ligand of choice, a wide repertoire of different interactions can be employed to govern AuNP organization.5–7 Noncovalent interactions such as electrostatic forces,8 protein pairing,9 hydrophobic and fluorophobic interactions,10,11 metal ligand coordination,12 and hydrogen bonding13 (HB) have already been recognized as efficient tools to modulate and program AuNP self-assembly. However, the ability to direct NP–NP interaction and achieve in-depth control of the self-assembly process has remained highly challenging, with consequent lack, in most cases, of structural specificity in the resulting assemblies.14–16The least explored noncovalent interaction in the design and development of AuNP assemblies is the halogen bond (XB), which takes place when an attractive interaction between an electrophilic region, associated with an halogen atom in a molecular entity, and a nucleophilic region, on another or the same molecular entity, occurs.17,18 Being similar to hydrogen bond (HB), XB has emerged as a new noncovalent interaction of choice in constructing well-organized supramolecular architectures and has established its role as a powerful tool in various research fields such as crystal engineering,19,20 anion sensing,21,22 organic reactivity,23,24 functional materials25,26 and biological systems.27 Despite its primary role in molecular self-assembly, XB application for the construction of NP assemblies is, to date, very limited. In particular, van der Boom and co-workers reported the surface functionalization of AuNPs with XB-donating iodotetrafluorobenzene derivatives, which formed either chain-like structures or large, dense assemblies, upon addition to bidentate XB-acceptors depending on the concentration of the bipyridyl cross-linker.28 Similar strategy was employed by the same research group for the preparation of surface-confined nanostructures, achieved by binding the XB-donor NPs onto silicon and glass substrates functionalized with pyridine moieties.29 Recently, we showed the synthesis of novel thioctic acid coupled XB-donor ligands, which, bound to AuNPs, could efficiently drive the dispersion and/or assembly of NPs in solvents having XB-acceptor sites.30 Compared to HB, XB provides unique directionality31 and interaction strength tunability,32,33 varying the halogen atom and/or the motif to which it is bound. These features might represent a valuable paradigm in NP self-assembly to achieve directional interactions among the NPs.
In this study, we designed and synthesized a series of ligands to functionalize Au nanosurfaces to obtain AuNP assemblies mediated by XB in solution and on planar surfaces. Following a stepwise approach, a series of XB ligands based on the thioctic acid structure was synthesized and employed to functionalize and stabilize AuNPs (Fig. 1a and b). XB-assisted AuNP assembly in solution was attained mixing AuNPs bearing XB donor ligands with either ditopic XB-acceptor molecules or AuNPs functionalized with XB-acceptor moieties (Fig. 1c). Likewise, a preliminary study of XB-driven adsorption of these AuNPs on surfaces was studied via QCM-D technique, which was used as an in situ tool for measuring mass changes at the QCM electrodes upon self-assembly occurrence (Fig. 1c).
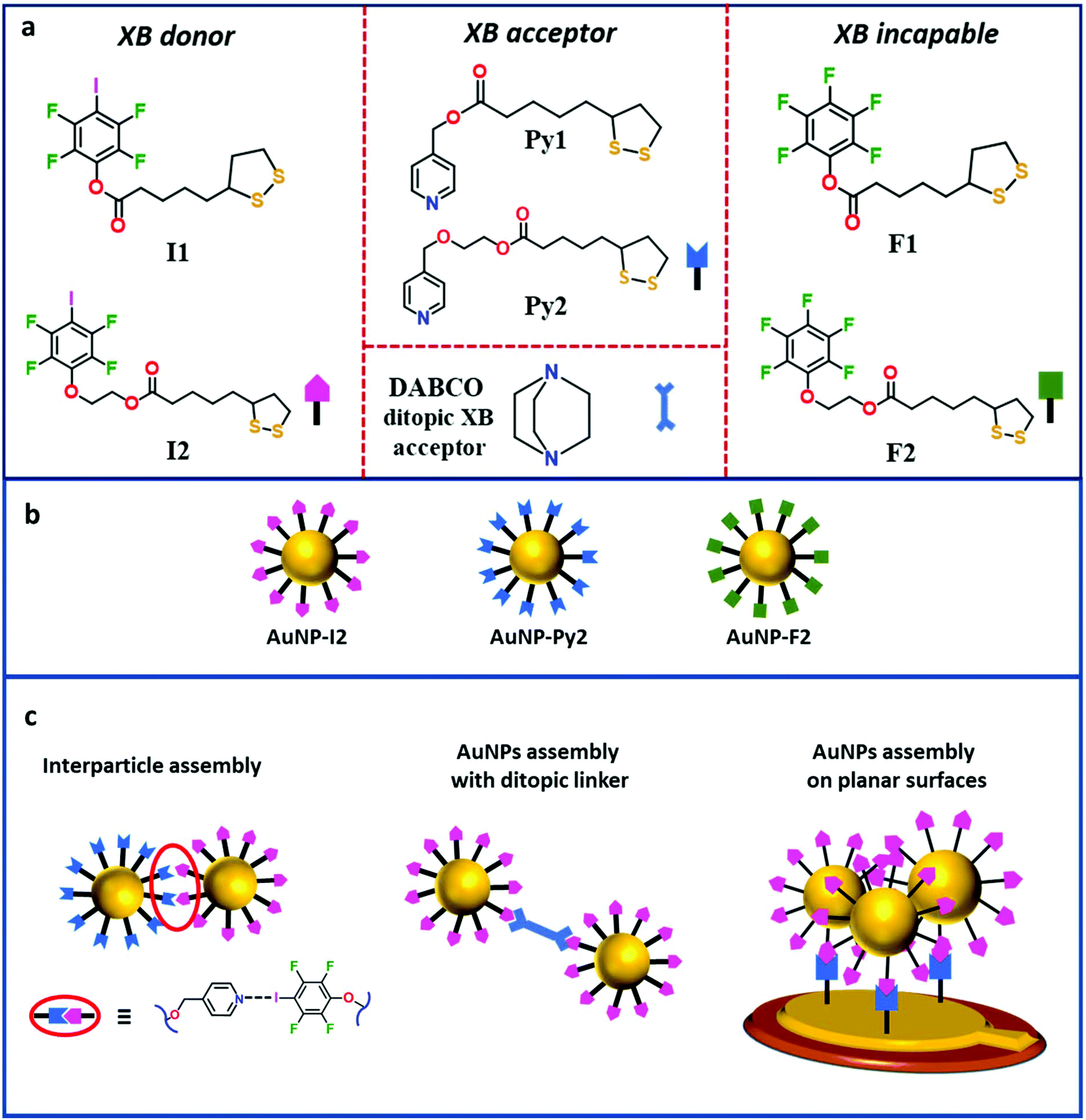 | ||
| Fig. 1 (a) Chemical structure of XB-donor ligands (left), XB-acceptor ligands and ditopic XB acceptor (middle), and control ligands unable to give XB (right) used in this study. (b) Schematic representation of AuNPs synthesized in this study. (c) Schematic representation of AuNPs assembly strategies employed in this study. | ||
Results and discussion
Synthesis and characterization of AuNPs stabilized by XB capable ligands
The protecting ligands used as precursors for the functionalization of AuNPs are listed in Fig. 1a. All ligands hold a thioctic motif as anchoring group to the AuNP, as well as a functional group bearing either an XB-donor site (iodotetrafluorobenzene) or an XB-acceptor group (pyridine) or a fully fluorinated benzene ring unable to give XB. Moreover, in ligands 2 the alkyl linkers between the two edges are decorated by an additional ethylene glycol unit, introduced by Steglich esterification reaction, to increase the distance between the NP surface and the ending functional group with respect to ligands 1. These precursors were prepared by multi-step synthesis with a reasonably good yield (60–80%). In particular, ligands Py1 and F1 were synthesised according to a previously reported procedure with slight modifications,34,35 while ligands I1, I2, Py2, and F2 were prepared following a new synthetic route (section S.1 in the ESI†). The partially fluorinated molecules, F1 and F2, have an isostructural backbone to the ligands functionalized with iodotetrafluorobenzene and pyridine groups, thus, they function as excellent control systems for assessing the role of XB in the assembly process of AuNPs functionalized with those ligands. Our previous studies demonstrated that the crystal packing of pure I1 is mainly driven by intermolecular S⋯I XB interactions, resulting in an infinite chain-like structure code TAWFUV. Upon mixing this ligand with the ditopic XB-acceptor 1,2-di(4-pyridyl)ethylene (1a), the formation of a trimeric halogen-bonded complex (ref. code: TAWCUS) was shown.30 As a preliminary gauge of the XB-donor ability, molecular electrostatic potential surfaces (MEPSs) for the XB-donor ligand I1 were calculated. MEPS of I1 clearly showed the existence of a relevant σ-hole (electropositive regions, dark blue) with a MEPS value of 162.649 kJ mol−1, along the extension of the C–I bond of the iodotetrafluorobenzene ring (Fig. 2a), in agreement with the values reported in literature.36–38 | ||
| Fig. 2 (a) Electrostatic potential surface with an isodensity of 0.02 a.u. of XB donor ligand I1 (B3LYP/3-21g). Blue indicates positive charge density (common scale was used to compare the surfaces visually). (b) Hirshfeld surfaces plotted against dnorm for the complex I1·1a: red colour highlights the area of XB formation. (c) Single crystal X-ray trimeric structure of complex I1·1b, colour code: grey, carbon; green, fluorine; red, oxygen; white, hydrogen; magenta, iodine; yellow, sulphur; XB interactions are shown as black dotted lines. | ||
The ability of ligand I1 to function as efficient XB-donor was further proved in this study by the formation of halogen-bonded adduct with 1,4-diazabicyclo[2.2.2]octane (DABCO; 1b; Fig. 2c and ESI section S.2†). Specifically, the X-ray studies revealed the construction of trimeric halogen-bonded complex I1–1b, which was mainly driven by the short and directional N⋯I XB synthon (I⋯N distances of 2.693(4) Å and 2.742(3) Å, and C–I⋯N angles 173.9(2)° and 175.4(2)°).39 In addition to XB, the crystal packing of the complex I1–1b was stabilized by several C–H⋯O, S⋯H, and H⋯F short contacts. The “molecule in crystal” approach inherent in Hirshfeld surface analysis40 facilitated the identification of individual interactions through the electron density weighted molecular surfaces or Hirshfeld surfaces and depicts XB formation. Hirshfeld surfaces of the cocrystals I1–1a (ref. code: TAWCUS) and I1–1b are mapped with dnorm and electrostatic potential plotted on the Hirshfeld surface, where the XB interactions are visualized in red (Fig. 2b).
Having established the ability of ligand I1 to act as efficient XB donor, the synthesis of AuNPs functionalized with I1 was performed via direct reduction of HAuCl4 in various reaction conditions.41 This synthetic procedure was not successful, due to the XB-donor chemical instability in the presence of the reducing agent, sodium borohydride (NaBH4), which triggered the cleavage of C–I bond in the iodotetrafluorobenzene ring with the consequent iodine replacement by a proton, as described in the ESI in section S.4.1 and Fig. S.1.† Synthesis via direct reduction of HAuCl4 was also attempted for AuNPs functionalized with Py1 ligands, employing a slightly modified Brust method (section S.4.2 in the ESI†). The obtained AuNP-Py1 were not dispersible in toluene but in THF, and showed to be quite extensively aggregated (Fig. S.2–S.4 in the ESI†).
Given the unfeasibility of the direct synthetic approach, 4 nm sized (diameter) AuNPs capped with dodecanethiol chains (AuNP-DT) were prepared through a slightly modified Brust method (section S.4.3, Fig. S.5 and S.6 in the ESI†) and dispersed in toluene. These NPs were used as pre-formed AuNPs where I1 ligands were introduced via an exchange reaction with a replacement yield of about 70%, as it was shown in our previous study.30 The morphology of the exchanged NPs was kept similar to the AuNP-DT as shown by UV-Vis, DLS, and TEM characterization (Fig. S.7 and S.8 in the ESI†). As it was done for molecular ligands, exchanged AuNP-I1 were tested with an increasing concentration of a bidentate XB acceptor, 1,2-di(4-pyridyl)ethylene in solution. UV-Vis spectra of these dispersions did not show either shift or broadening of the plasmon peak, although, indicating no interaction with the ditopic linkers (Fig. S.9 in the ESI†). This behaviour was attributed to unavailability of I atoms to interact with the environment, being the length of the linker too short and very similar to the length of the DT chain. Thus, only NPs functionalized with ligands 2 were used for further investigation on XB-driven inter-particle assembly, as longer lengths guarantee a better availability of the functional groups to interact with the surrounding environment.
AuNPs capped with I2, Py2, and F2 were synthesized via ligand exchange procedure (section S.4.4. in the ESI†) and the actual occurrence of exchange between DT and the chosen ligand was confirmed by FTIR and 1H and 19F NMR analysis (section S.4.4 in the ESI†). In particular, FTIR data revealed the presence of the exchanged ligands signals in the AuNPs spectra for AuNP-I2, AUNP-Py2 and AuNP-F2 (Fig. S.10–S.12 in section S.4.4†), while 1H and 19F NMR spectra showed broad signals having chemical shifts values (section S.4.4†) in agreement with those of the exchanged molecules, confirming the presence of functional ligands. Indeed, no sharp peak associated to the ligand free molecules could be observed. The morphology of the exchanged AuNPs was probed via TEM and no significant changes in shape and size (4 nm in diameter) could be observed with respect to the starting AuNP-DT. This data was confirmed by UV-Vis analysis, which revealed surface plasmon resonance (SPR) peak at λmax ∼ 513 nm for all exchanged AuNPs, in accordance with the size of the NPs (Fig. 3d).30 Moreover, DLS investigation of the AuNP dispersions showed the presence of two populations in the intensity-weighted size distributions for all exchanged AuNPs: a dominant population with a hydrodynamic radius distribution centred at about 4 nm related to single AuNPs, and a second smaller population with larger hydrodynamic radius related to NP agglomerates, probably induced by the additional centrifugation steps necessary to isolate the NPs from the excess of ligands (Fig. 3e).
 | ||
| Fig. 3 (a–c) TEM images and size distribution of AuNP-I2, AuNP-Py2, and AuNP-F2 samples. (d) UV-Vis spectra of dispersions of AuNP-I2, AuNP-Py2 and AuNP-F2 in toluene compared to that of AuNP-DT. (e) Intensity-weighed size distribution obtained by DLS of AuNP-I2, AuNP-Py2 and AuNP-F2 compared to that of AuNP-DT. | ||
Assembly of AuNP-I2 with ditopic linker 1b
In agreement with previous studies28–30 and our co-crystallisation experiments, AuNP-I2 were mixed with an excess of the ditopic XB-acceptor 1b, to investigate the formation of AuNP assemblies. Upon 1b addition, AuNP-I2 sample exhibited a gradual colour change from purple to blue and, overnight, the formation of a precipitate could be observed, indicating the formation of larger aggregates. In particular, UV-Vis spectra of the fresh dispersions showed that the SPR peak of the individual AuNP-I2 was shifted from 517 nm to 528 nm in the mixed dispersion (Fig. 4a). DLS experiments of the fresh mixture also revealed an increase in the auto-correlation function decay time, with respect to that of the AuNP-I2 sample, and yielded an intensity-weighted size distribution dominated by a single population of AuNP agglomerates having a hydrodynamic radius of about 180 nm (Fig. 4b). TEM micrographs of AuNP-I2·1b sample showed the formation of AuNPs assemblies with a chain-like structure,42 which have already been reported for similar systems.28 To verify the role of XB in the construction of the AuNP assemblies described above, we treated AuNP-F2 (similar molecular backbone to AuNP-I2 but devoid of the XB donor moieties) with 1b as a control experiment. Interestingly, no aggregation occurred even with addition of excess amount of 1b to AuNP-F2 solution. | ||
Fig. 4 (a) UV-Vis absorption spectra showing the variation in the SPR band on passing from a pure dispersion of AuNP-I2 (blue line) to a mixed dispersion of AuNP-I2, after the addition of 50 μL of 10 mM 1b solution (red line), with final AuNP-I2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1b ratio 1:100. (b) Intensity-weighed AuNP size distributions for AuNP-I2 in toluene before and after addition of 1b obtained through CONTIN analysis of DLS auto-correlation functions. (c, d) TEM images of AuNP-I2 after mixing with 1b. (e) Schematic representation of XB-driven formation of chain-like assemblies. :1b ratio 1:100. (b) Intensity-weighed AuNP size distributions for AuNP-I2 in toluene before and after addition of 1b obtained through CONTIN analysis of DLS auto-correlation functions. (c, d) TEM images of AuNP-I2 after mixing with 1b. (e) Schematic representation of XB-driven formation of chain-like assemblies. | ||
XB based inter-particle assembly
Given the availability of two different AuNP systems stabilized with XB-donor and acceptor ligands, the formation of inter-particle assemblies via XB was tempted by mixing AuNP-I2 and AuNP-Py2 in equimolar ratio. The occurrence of XB-driven interparticle assembly was confirmed by UV-Vis results, which showed a dramatic red-shift for the AuNP-I2/AuNP-Py2 sample when compared to those of the pure AuNP-I2 and AuNP-Py2 dispersions (λmax ∼ 550 nm). Similarly, DLS auto-correlation functions of the AuNP-I2/AuNP-Py2 sample showed a significant increase in the decay time, indicating AuNP aggregation (Fig. S.13 in the ESI†). Moreover, intensity-weighted size distributions, obtained using CONTIN analysis, revealed that, despite the presence of a small population of individual AuNPs, the dominant population of the mixed sample was related to AuNP agglomerates with a hydrodynamic radius of about 100 nm (Fig. 5b). The formation of AuNP-I2/AuNP-Py2 assemblies was further confirmed by TEM images, which showed the presence of spheroidal AuNP agglomerates of about 100 nm (Fig. 5c and d) together with single NPs. Such evidences suggest the occurrence of XB between the two AuNPs bearing donor and acceptor moieties and the formation of halogen-bonded AuNP assemblies, with a morphology quite different from that highlighted for AuNP-I2 mixed with the ditopic XB acceptor ligand 1b. Moreover, one month aged AuNP-I2/AuNP-Py2 sample showed a strong change of colour passing from purple to blue (Fig. S.14 in the ESI†) with formation of a precipitate, while pure AuNP-I2 and AuNP-Py2 dispersions exhibited a good colloidal stability over time, with no changes in UV-Vis spectrum and DLS auto-correlation functions for months. To further verify the role of XB in driving the assembly process, we also prepared an analogous mixed sample, adding AuNP-F2 to AuNP-Py2 in the same experimental conditions. Interestingly, no inter-particle assembly was observed, even after two months of aging (Fig. S.15 and S.16 in the ESI†). Therefore, the replacement of a XB donor ligand with an XB incapable ligand hampered the formation of AuNP assemblies, asserting the driving role of XB in the AuNP assembly process. | ||
| Fig. 5 (a) UV-Vis spectra showing the red shift of the SPR signal upon AuNP-I2/AuNP-Py2 assembly. (b) Intensity-weighed AuNP size distributions of AuNP-I2/AuNP-Py2 dispersion with respect to those of the pure components obtained through CONTIN analysis of DLS auto-correlation functions. (c–d) TEM images of the AuNP-I2/AuNP-Py2 sample showing grid areas with the spheroidal inter-particle assemblies. (e) Schematic representation of XB-driven formation of spheroidal inter-particle assemblies. | ||
AuNPs assembly on planar surface
Further evidence of the ability of the synthesized AuNPs to exert XB was given by specific adsorption of AuNP-I2 on a planar gold surface coated with a monolayer of Py1 ligands. To study this, we used quartz crystal microbalance dissipation (QCM-D) technique as in situ tool for monitoring the binding process at the surface. The employed strategy is schematically represented in Fig. 6a. First, QCM sensors were treated with ligand Py1, by immersing it into a solution containing XB-acceptor ligand Py1 (section S.6 in the ESI†). The obtained sensors functionalized with ligand Py1 were placed in the QCM chamber and exposed to a flux of AuNP-I2 solution (0.02 ms2 mL−1). Fig. 6b shows the adsorbed mass of AuNP-I2 on Py1 functionalized surfaces calculated from QCM-D measurements using the viscoelastic model plotted against time. It can be seen that, upon AuNP-I2 flux, the resonance frequency decreased, while a concomitant increase of dissipation occurred, indicating adsorption of AuNP-I2 on the QCM sensors. The frequency change dropped almost linearly with time for about 15 minutes and then gradually reached a plateau, probably due to saturation of the sensor by AuNPs (Fig. S.17 in the ESI†). Subsequently, the AuNP-I2 dispersion was re-fluxed for evaluating the possibility of further AuNPs adsorption. Another small drop in the frequency change could be observed before reaching a new plateau value, proving the ability of the AuNP-I2 to keep adsorbing on the chip. A negligible amount of weight loss was obtained upon solvent washing, indicating a quite strong interaction between the AuNP-I2 and the Py1 monolayer deposited on the QCM sensor. The viscoelastic model was used to estimate the exact mass of adsorbed AuNP-I2 and, as reported in Table 1, 637 ng cm−2 of AuNPs were adsorbed onto the QCM sensor. | ||
| Fig. 6 (a) Schematic representation of the XB-driven AuNPs assembly on QCM sensors. (b) Mass deposition of AuNP-I2 on the sensor functionalized with Py1 ligand (red line) and deposition of AuNP-F2 on the sensor functionalized with Py1 ligand (blue line) plotted as a function of time. | ||
| AuNP | Monolayer surface | Mass absorbed (ng cm−2) |
|---|---|---|
| AuNP-I2 | Py1 | 637 |
| AuNP-F2 | Py1 | 50 |
| AuNP-I2 | DT | 102 |
To verify the role of the XB in the AuNP adsorption on the Py1 monolayer, we fluxed the control AuNP-F2 on the same sensors, in the same experimental conditions. A much lower frequency change occurred after several cycles of deposition, in comparison to that observed with AuNP-I2 (Fig. 6b). Data treatment performed using the Sauerbrey relationship indicated that only 50 ng cm−2 of AuNP-F2 was deposited (Table 1). To further confirm this result, a reverse control experiment was performed. In fact, QCM sensors were functionalized with DT molecules and AuNP-I2 NPs were deposited using similar experimental conditions. Similarly to the experiment with AuNP-F2, much lower frequency changes were observed and the calculations performed applying Sauerbrey equation showed that only 102 ng cm−2 of AuNP-I2 were deposited (Fig. S.18 in the ESI†). Water contact angle measurements were performed to investigate the changes in the hydrophilic character of the QCM sensor after functionalization with Py1 ligands and adsorption of AuNP-I2 (Fig. S.19 in the ESI†). The values are presented in Fig. S.19†: the water contact angle for bare Au is about 79 ± 2°, as expected for a freshly cleaned gold surface.43,44 This value decreased to 50 ± 2° upon surface functionalization with the XB-acceptor ligand Py1, while, when AuNP-I2 were deposited on the Py1 functionalized surface, it became 78 ± 2°, confirming the binding of AuNP-I2via XB on the surface coated with the XB-acceptor. These results, although preliminary, highlighted the ability of AuNP-I2 to specifically interact by XB with an Au planar surface functionalized with Py1 (XB-acceptor), promoting a much more efficient coating of such a surface, with respect to similar NPs lacking the XB-donor moiety (AuNP-F2), and support the ability of these NPs to form strong XBs.
Conclusions
A series of XB capable ligands with a thioctic acid anchoring group were synthesized and studied to form supramolecular XB-assisted assemblies. These ligands were also efficiently exchanged on the surface of AuNP-DT forming colloidal stable dispersions in toluene. Ditopic XB-acceptor molecules mixed with AuNPs bearing XB-donor ligands showed the effective formation of large particle assemblies with a chain-like morphology. Likewise, the formation of inter-particle assemblies among complementary AuNPs functionalized with XB-donor and acceptor ligands, respectively, was reported for the first time. In the concentration range investigated (equimolar ratio among the two typologies of NPs, excess of I with respect Py ligands), the morphology of the formed assemblies was quite different from those formed in the presence of the ditopic ligand, appearing as single spheroidal clusters of AuNP of about 100 nm size. Although more extensive studies are necessary to optimize and control the formation of these inter-particle assemblies, these findings confirm that XB can be a new powerful tool for directing the formation of hybrid supraparticles in solution with an organized internal structure. Finally, the XB-donor ability of these AuNPs was also proven by adsorbing them onto Au planar surfaces functionalized with organic monolayers bearing the complementary XB-acceptor groups as revealed by QCM-D studies. Overall, our results indicate that, as it has been demonstrated in other fields, XB may represent a new paradigm in NP self-assembly to engineer well-defined hybrid superstructures with different functionalities.Experimental
Materials
Starting materials were purchased from Apollo Scientific, Sigma-Aldrich, Merck, TCI Europe. All solvents employed in this study were of reagent grade quality and used without further purification. For the synthesis of selective ligands, anhydrous solvents were employed. Thin layer chromatography (TLC) was performed on silica gel 60 F254 (E. Merck) and visualized under a UV lamp at 254 nm. Column chromatography was carried out on silica gel 60F (Merck 9385, 0.040–0.063 mm). Fourier transform infrared spectroscopy (FTIR) was performed by Nicolet iS50 FT-IR. The mass spectra were recorded on a GC-MS AGILENT GC-MSD5975. Differential Scanning Calorimetry (DSC) analyses were performed on a Mettler Toledo DSC823e instrument with 20 μL aluminium light sample pans. Melting points were also determined using Reichert instrument, by observing the melting and crystallizing process through an optical microscope. UV-Vis spectra were recorded on a JASCO V-630 spectrophotometer with double beam transmission mode. Transmission electron microscope (TEM) images were acquired by using a Philips CM200 TEM, equipped with a field emission gun and operating at 200 kV. TEM samples were prepared by depositing a drop of the solution on freshly glow discharged 400 mesh carbon coated grids. Particle size distributions of AuNPs were measured by Dynamic Light Scattering (DLS) instrument equipped with ALV/CSE-5004 light scattering electronics and multiple Tau digital correlator.Preparation of the AuNP dispersions
Details on the synthesis of all the AuNPs used in this work can be found in the ESI.† Preparation of the mixture samples containing AuNP-I2 (or AuNP-F2) and the ditopic ligand 1b were done adding 50 μL of a solution of 1b ([acceptor] = 3 × 1017 molecules per mL) to a dispersion of AuNP-I2 in toluene (at a concentration of 1.12 × 1014 NPs per mL, 2.9 × 1016 iodine atoms per mL). The mixture samples containing AuNP-I2 (or AuNP-F2) and AuNP-Py2 were prepared mixing 20 μL of a dispersion of AuNP-I2 (or AuNP-F2) at a concentration of 5.6 × 1015 Nps per mL in toluene with 20 μL of a dispersion of AuNP-Py2 at a concentration of 5.6 × 1015 Nps per mL in toluene, the mixture was then diluted up to 500 μL to have an equimolar ratio of the two NPs (2.24 × 1014 Nps per mL with [I]/[Py] ratio = 4).Single crystal X-ray structural analysis
Single crystal X-ray data were collected on a Bruker KAPPA APEX II diffractometer with Mo Kα radiation (λ = 0.71073) and CCD detector. Data were collected at room temperature (296 K). Data collection and reduction were performed by SMART and SAINT and absorption correction, based on multi-scan procedure, by SADABS. The structures were solved by SHELXS45 and refined by SHELXL46 programs, respectively. The refinement was carried on by full-matrix least-squares on F2. Hydrogen atoms were placed using standard geometric models and with their thermal parameters riding on those of their parent atoms. Molecular graphics were obtained with Mercury 3.8.47Nuclear magnetic resonance spectroscopy
1H NMR, 13C NMR and 19F NMR spectra were recorded at room temperature with a Bruker 400 and 500 MHz spectrometer. 1H and 13C chemical shifts were measured relatively to internal TMS. Chemical shift (δ) values are provided in ppm while coupling constants (J) values are in Hz. NMR spectra of ligand coated AuNPs were recorded from AuNP samples transferred in deuterated solvent. Each AuNP stock solution in toluene (1 mL) was placed in a Falcon tube with ethanol (10 mL) and centrifuged (8000 rpm = 6869 rcf; 30 min). After removing the supernatant, the pellet was dried with air and dissolved in deuterated toluene (PhCH3-d8, 0.7 mL). 1H and 19F NMR spectra were acquired at 302 K and 305 K, respectively, on a Bruker AV400 spectrometer. Proton chemical shifts are reported in ppm downfield from SiMe4, with the residual proton (PhCH3-d8: δ = 2.09 ppm) solvent resonance as internal reference. To confirm the complete removal of the physisorbed ligands on the AuNPs after purification, 1H NMR analysis was carried out on the supernatants.Attenuated total reflectance FTIR (ATR-FTIR)
Spectra were obtained with a Thermo Scientific Nicolet iS50 FTIR spectrometer, equipped with an iS50 ATR accessory (Thermo Scientific, Madison, USA). NP dispersions were deposited by drop casting on the ATR probe and the solvent was left evaporating before the measurement.Dynamic light scattering (DLS)
Measurements were performed on an ALV apparatus equipped with ALV-5000/EPP Correlator, special optical fiber detector and ALV/CGS-3 Compact goniometer. The light source is He–Ne laser (λ = 633 nm), 22 mW output power. Measurements were performed at 25 °C. Approximately 1 mL of sample solution was transferred into the cylindrical Hellma scattering cell. Data analysis has been performed according to standard procedures and auto-correlation functions were analyzed through a constrained regularization method (Laplace inversion of the time auto-correlation functions), CONTIN, for obtaining the particle size distribution.TEM analysis
TEM images were acquired using a Philips CM200 TEM, equipped with a field emission gun and operating at 200 kV. AuNP samples were prepared dropping the NP dispersion in toluene on carbon-coated copper grids letting the solvent to dry. Quantitative analysis of NP size distributions was performed with ImageJ software. Size distributions were fitted by a Gaussian equation using SigmaPlot.QCM-D experiments
A Q-Sense E4 instrument (Q-Sense) quartz crystal microbalance with dissipation monitoring (QCM-D) was used to measure the adsorbed mass of AuNPs on QCM gold crystals functionalized with different ligands. The QCM-D sensors were first treated for 10 minutes in an UV/ozone chamber and then immersed at 75 °C in a H2O/NH3/H2O2 (5:1:1 volume ratio) mixture for 10 minutes. The sensors were subsequently rinsed thoroughly with mQW and dried with nitrogen. After rinsing the QCM-D sensors were again placed in UV/ozone chamber for 10 minutes to remove the contaminants from the surfaces. Surface functionalization was carried out by immersing clean QCM-D sensors into 10 mM ligand solution (CH2Cl2) overnight. The ligand functionalized sensors were washed with pure CH2Cl2 and then with mQW, dried with nitrogen and mounted into the measurement chamber, which was maintained at 21 °C.
Formation of AuNP based assemblies on monolayer
Firstly, pure toluene was fluxed to establish a stable baseline. Then, 1 mL of the AuNP solution at a concentration of 0.02 m2 mL−1 was fluxed through the measurement chamber using a flow rate of 100 μL min−1. The sensors were then incubated for 15 minutes in zero-flow conditions. Subsequently, 1 mL of the AuNP solution was flowed through the chambers at a rate of 100 μL min−1, monitoring an eventual additional deposition. The sensors were then incubated for 15 additional minutes in zero-flow conditions and then rinsed for 40 minutes using pure solvent. Several cycles of AuNPs were fluxed and the unbound AuNPs were removed by fluxing the pure solvent. The mass of surface-bound material as well as the viscoelastic properties of the adsorbed layer were determined as explained in section S.6 in the ESI.†Contact angle measurements
Contact angle measurements of the QCM sensors with water were determined by an OCA 15 PLUS instrument (Dataphysics) using droplet volume of 1 μL for mQW. The average contact angles (Elliptic method) were calculated from a series of five independent measurements by the SCA20 software.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from the European Research Council for the Starting Grant ERC-2012-StG_20111012 FOLDHALO (Grant Agreement 307108) and the Proof-of-Concept Grant ERC-2017-PoCMINIRES (Grant Agreement 789815) are acknowledged. Funding from the Academy of Finland Center of Excellence in Molecular Engineering of Biosynthetic Hybrid Materials (HYBER 2014-2019) at Aalto University is acknowledged. We acknowledge the provision of facilities and technical support by Aalto University at OtaNano-Nanomicroscopy Center (Aalto-NMC).Notes and references
- Z. Xue, C. Yan and T. Wang, Adv. Funct. Mater., 2019, 29, 1807658 CrossRef.
- A. K. Boal, F. Ilhan, J. E. DeRouchey, T. Thurn-Albrecht, T. P. Russell and V. M. Rotello, Nature, 2000, 404, 746–748 CrossRef CAS PubMed.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- C. Pigliacelli, K. B. Sanjeeva, Nonappa, A. Pizzi, A. Gori, F. B. Bombelli and P. Metrangolo, ACS Nano, 2019, 13, 2158–2166 CAS.
- M. Grzelczak, L. M. Liz-Marzán and R. Klajn, Chem. Soc. Rev., 2019, 1342–1361 RSC.
- Y. Xia and Z. Tang, Chem. Commun., 2012, 48, 6320–6336 RSC.
- J. Il Park, T. D. Nguyen, G. de Queirós Silveira, J. H. Bahng, S. Srivastava, G. Zhao, K. Sun, P. Zhang, S. C. Glotzer and N. A. Kotov, Nat. Commun., 2014, 5, 3593–3613 CrossRef PubMed.
- J. Majoinen, J. Hassinen, J. S. Haataja, H. T. Rekola, E. Kontturi, M. A. Kostiainen, R. H. A. Ras, P. Törmä and O. Ikkala, Adv. Mater., 2016, 28, 5262–5267 CrossRef CAS PubMed.
- A. Urvoas, K. L. Gurunatha, P. Minard, E. Dujardin, A. C. Fournier, M. Valerio-Lepiniec and V. Marchi, ACS Nano, 2016, 10, 3176–3185 CrossRef PubMed.
- D. Maiolo, C. Pigliacelli, P. Sánchez Moreno, M. B. Violatto, L. Talamini, I. Tirotta, R. Piccirillo, M. Zucchetti, L. Morosi, R. Frapolli, G. Candiani, P. Bigini, P. Metrangolo and F. Baldelli Bombelli, ACS Nano, 2017, 11, 9413–9423 CrossRef CAS PubMed.
- C. Pigliacelli, D. Maiolo, Nonappa, J. S. Haataja, H. Amenitsch, C. Michelet, P. Sánchez Moreno, I. Tirotta, P. Metrangolo and F. Baldelli Bombelli, Angew. Chem., Int. Ed., 2017, 56, 16186–16190 CrossRef CAS PubMed.
- J. Guo, B. L. Tardy, A. J. Christofferson, Y. Dai, J. J. Richardson, W. Zhu, M. Hu, Y. Ju, J. Cui, R. R. Dagastine, I. Yarovsky and F. Caruso, Nat. Nanotechnol., 2016, 11, 1105–1111 CrossRef CAS PubMed.
- Nonappa, T. Lahtinen, J. S. Haataja, T. R. Tero, H. Häkkinen and O. Ikkala, Angew. Chem., Int. Ed., 2016, 55, 16035–16038 CrossRef CAS PubMed.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzán, ACS Nano, 2010, 4, 3591–3605 CrossRef CAS PubMed.
- J. Genest, T. E. Hébert and J. Martin, Nature, 2016, 491, 975–977 Search PubMed.
- Nonappa and O. Ikkala, Adv. Funct. Mater., 2018, 28, 1–14 CrossRef.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- T. Wang, D. Lamontagne, J. Lynch, J. Zhuang and Y. C. Cao, Chem. Soc. Rev., 2013, 42, 2804–2823 RSC.
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524 CrossRef CAS PubMed.
- Halogen Bonding, ed. P. Metrangolo and G. Resnati, Springer Berlin Heidelberg, Berlin, Heidelberg, 2008, vol. 126 Search PubMed.
- F. Zapata, A. Caballero, N. G. White, T. D. W. Claridge, P. J. Costa, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2012, 134, 11533–11541 CrossRef CAS PubMed.
- A. V. Jentzsch, D. Emery, J. Mareda, S. K. Nayak, P. Metrangolo, G. Resnati, N. Sakai and S. Matile, Nat. Commun., 2012, 3, 905–908 CrossRef PubMed.
- A. Milani, A. Pizzi, A. Gori, N. Demitri, M. I. Martinez Espinoza, L. Lascialfari, G. Bergamaschi and P. Metrangolo, Chem. Commun., 2018, 54, 10718–10721 RSC.
- R. Tepper and U. S. Schubert, Angew. Chem., Int. Ed., 2018, 57, 6004–6016 CrossRef CAS PubMed.
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686–2695 CrossRef CAS PubMed.
- J. C. Christopherson, F. Topić, C. J. Barrett and T. Friščić, Cryst. Growth Des., 2018, 18, 1245–1259 CrossRef CAS.
- E. Parisini, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Chem. Soc. Rev., 2011, 40, 2267–2278 RSC.
- T. Shirman, T. Arad and M. E. van der Boom, Angew. Chem., Int. Ed., 2010, 49, 926–929 CrossRef CAS PubMed.
- T. Shirman, R. Kaminker, D. Freeman and M. E. Van Der Boom, ACS Nano, 2011, 5, 6553–6563 CrossRef CAS PubMed.
- K. B. Sanjeeva, I. Tirotta, V. Kumar, F. B. Bombelli, G. Terraneo and P. Metrangolo, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2017, 73, 240–246 CrossRef CAS PubMed.
- S. M. Huber, J. D. Scanlon, E. Jimenez-Izal, J. M. Ugalde and I. Infante, Phys. Chem. Chem. Phys., 2013, 15, 10350–10357 RSC.
- K. E. Riley, J. S. Murray, J. Fanfrlík, J. Řezáč, R. J. Solá, M. C. Concha, F. M. Ramos and P. Politzer, J. Mol. Model., 2013, 19, 4651–4659 CrossRef CAS PubMed.
- J. S. Murray, P. Politzer, R. J. Solá, J. Fanfrlík, K. E. Riley, M. C. Concha, J. Řezáč and F. M. Ramos, J. Mol. Model., 2011, 17, 3309–3318 CrossRef PubMed.
- W. E. Ghann, O. Aras, T. Fleiter and M. C. Daniel, Langmuir, 2012, 28, 10398–10408 CrossRef CAS PubMed.
- A. Martí, A. M. Costero, P. Gaviña, S. Gil, M. Parra, M. Brotons-Gisbert and J. F. Sánchez-Royo, Eur. J. Org. Chem., 2013, 4770–4779 CrossRef.
- C. B. Aakeröy, M. Baldrighi, J. Desper, P. Metrangolo and G. Resnati, Chem. – Eur. J., 2013, 19, 16240–16247 CrossRef PubMed.
- P. Politzer, P. Lane, M. C. Concha, Y. Ma and J. S. Murray, J. Mol. Model., 2007, 13, 305–311 CrossRef CAS PubMed.
- P. Politzer, J. S. Murray, T. Clark and G. Resnati, Phys. Chem. Chem. Phys., 2017, 19, 32166–32178 RSC.
- C. B. Aakeröy, C. L. Spartz, S. Dembowski, S. Dwyre and J. Desper, IUCrJ, 2015, 2, 498–510 CrossRef PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- L. Chen, B. Su and L. Jiang, Chem. Soc. Rev., 2019, 48, 8–21 RSC.
- M. Ben Ali, F. Bessueille, J. M. Chovelon, A. Abdelghani, N. Jaffrezic-Renault, M. A. Maaref and C. Martelet, Mater. Sci. Eng., C, 2008, 28, 628–632 CrossRef CAS.
- S. Azzouzi, H. K. Patra, M. Ben Ali, M. N. Abbas, C. Dridi, A. Errachid and A. P. F. Turner, Sens. Actuators, B, 2016, 228, 335–346 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr07054k |
| This journal is © The Royal Society of Chemistry 2019 |