
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntibody-targeting of ultra-small nanoparticles enhances imaging sensitivity and enables longitudinal tracking of multiple myeloma†
Alexandre
Detappe
 abcd,
Mairead
Reidy
ab,
Yingjie
Yu
c,
Clelia
Mathieu
ab,
Hung V.-T.
Nguyen
e,
Thibaud P.
Coroller
bf,
Fred
Lam
cl,
Petr
Jarolim
bg,
Peter
Harvey
h,
Andrea
Protti
i,
Quang-De
Nguyen
i,
Jeremiah A.
Johnson
e,
Yannick
Cremillieux
j,
Olivier
Tillement
k,
Irene M.
Ghobrial
*ab and
P. Peter
Ghoroghchian
*abc
abcd,
Mairead
Reidy
ab,
Yingjie
Yu
c,
Clelia
Mathieu
ab,
Hung V.-T.
Nguyen
e,
Thibaud P.
Coroller
bf,
Fred
Lam
cl,
Petr
Jarolim
bg,
Peter
Harvey
h,
Andrea
Protti
i,
Quang-De
Nguyen
i,
Jeremiah A.
Johnson
e,
Yannick
Cremillieux
j,
Olivier
Tillement
k,
Irene M.
Ghobrial
*ab and
P. Peter
Ghoroghchian
*abc
aDepartment of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA 02215, USA. E-mail: Irene_Ghobrial@dfci.harvard.edu; Tel: +1 617-632-4198
bHarvard Medical School, 25 Shattuck Street, Boston, MA 02115, USA
cDavid H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA. E-mail: ppg@mit.edu; Tel: +1 (617) 715-4470
dCentre Paul Strauss, 3 rue de la porte de l'hôpital, 67000 Strasbourg, France
eDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
fDepartment of Radiation Oncology, Dana-Farber Cancer Institute, Boston, MA 02215, USA
gDepartment of Pathology, Brigham and Women's Hospital, Boston, Massachusetts 02115, USA
hDepartment of Biological Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA
iLurie Family Imaging Center, Department of Radiology, Dana-Farber Cancer Institute and Harvard Medical School, Boston, MA, USA
jInstitut des Sciences Moléculaires, Université de Bordeaux, UMR CNRS 5255, 33076 Bordeaux, France
kInstitut Lumière Matière, UMR 5306 Université Lyon1-CNRS, Université de Lyon, 69622 Villeurbanne Cedex, France
lClinical Scholar Program, Division of Neurosurgery, McMaster University, 237 Barton St East, Hamilton General Hospital, Hamilton ON, L8L 2X2, Canada
First published on 16th October 2019
Abstract
Monitoring malignant progression and disease recurrence post-therapy are central challenges to improving the outcomes of patients with multiple myeloma (MM). Whereas current detection methods that rely upon bone marrow examination allow for precise monitoring of minimal residual disease and can help to elucidate clonal evolution, they do not take into account the spatial heterogeneity of the tumor microenvironment. As such, they are uninformative as to the localization of malignant plasma cells and may lead to false negative results. With respect to the latter challenge, clinically-available imaging agents are neither sufficiently sensitive nor specific enough to detect minute plasma cell populations. Here, we sought to explore methods by which to improve detection of MM cells within their natural bone marrow environment, using whole-animal magnetic resonance imaging to longitudinally monitor early-stage disease as well as to enhance tumor detection after systemic therapy. We conducted a proof-of-concept study to demonstrate that ultra-small (<5 nm) gadolinium-containing nanoparticles bound to full-length antibodies against the B-cell maturation antigen (BCMA) exhibit rapid tumor uptake followed by renal clearance, improving the signal-to-noise ratio for MM detection beyond levels that are currently afforded by other FDA-approved clinical imaging modalities. We anticipate that when combined with bone marrow or blood biopsy, such imaging constructs could help to augment the effective management of patients with MM.
Introduction
Currently-available diagnostic methods to detect the malignant plasma cells of multiple myeloma (MM) and to follow either their therapeutic responses or proliferation rely upon frequent serologic studies and serial bone marrow examinations. Imaging modalities help in the further evaluation of systemic disease burden; they are employed to localize malignant plasma cells that are missed during bone marrow aspiration and biopsy, which are otherwise used to detect minimal residual disease and which are performed in only one foci of bony pelvis.1 Despite efforts to combine modalities, the ability to detect MM at its earliest stages (e.g., smoldering myeloma) or post-remission is still very limited.2 Establishment of novel imaging methods could have a transformative impact on the care of MM patients and those with other hematologic malignancies, enabling noninvasive and repetitive testing to visualize malignant cells at earlier time points and when present even in focal distribution patterns that would otherwise preclude identification.Techniques for magnetic resonance imaging (MRI) with conventional FDA-approved agents are being developed and have shown to be more reliable for assessing disease burden,3 for enabling accurate disease prognostication,4 as well as for following therapeutic responses in MM patients when compared to computed tomography (CT), single-photon emission computed tomography (SPECT), or 18-fluorodeoxyglucose (18F-FDG)-based positron emission tomography (PET).5 While MRI can better distinguish between benign and malignant lesions and can detect bone marrow infiltration at earlier time points,6 current protocols are time-consuming, expensive, and rely on the passive accumulation of non-targeted contrast agents within the tumor microenvironment,7 which has, hitherto, limited both their detection specificity and sensitivity. Changes seen by conventional MRI are slow to normalize after treatment; and, current techniques are unable to distinguish between remodeling bone and residual disease.6,8,9 While SPECT and 18F-FDG PET are able to accurately identify plasma cell populations,1 they utilize ionizing radiation that prevents repetitive testing in short intervals. 18F-FDG PET also displays poor detection sensitivity for malignant plasma cells in the residual disease state, which are more slowly proliferative.10,11
To improve upon current imaging modalities, we explored the development of an MM-targeted contrast agent that utilizes short MRI sequences to identify minute tumor cell populations with high spatial localization. Gadolinium (Gd)-containing nanoparticles, including ones conjugated to antibodies, have previously been utilized for imaging of solid tumors.12 These constructs, which have ranged in size from 10–400 nm in diameter,13,14 have generally demonstrated modest contrast enhancement in subcutaneously-implanted tumor models, a preponderance for liver accumulation, and minimal renal clearance,15 raising concerns for potentially longer-term side effects related to Gd exposure. We have previously demonstrated that ultra-small (<5 nm) Gd-containing nanoparticles enhance tumor uptake, improve contrast imaging, and exhibit complete renal clearance in small animal models16 and in nonhuman primates.17 To facilitate clinical translation, we have further modified a process for their batch manufacturing that is being employed in an ongoing phase I trial (NCT02820454, NCT03308604), assessing their biodistribution and safety.
In the present work, whole-animal imaging of ultra-small (<5 nm) Gd-containing nanoparticles conjugated to monoclonal antibodies was pursued in order to enable rapid detection of clonal plasma cells in the bone marrow environment. While the use of antibodies has long been proposed for the targeting of nanoparticle-based contrast agents by binding receptors that are overexpressed on tumor cell surfaces,18,19 the sizes of these constructs (30–200 nm in diameter)20 have been much larger than those of full monoclonal antibodies or of their molecular-conjugates (10–15 nm × 3–5 nm in dimension).21 As such, to date their pharmacology has been largely dictated by the nanoparticle rather than by the antibody. Moreover, conventional nanoparticle-antibody constructs have demonstrated circulation times on the order of hours, have achieved minimal tumor-uptake via passive modes of targeting, and have been shown to be largely distributed to organs of the reticuloendothelial system due to uptake by resident macrophages that clear them from the circulation.22 Most preclinical studies with such agents have further been conducted in subcutaneous xenograft models23,24 that do not recapitulate the vascular patterns found in the natural tumor environment;25,26 not surprisingly, many of these constructs have exhibited no differences when compared to their untargeted counterparts with respect to their in situ relaxivity values, their required systemic doses, and/or their ability to achieve tumor localization,13 which has stymied their further clinical development. To circumvent these challenges and to develop antibody-targeted nanoparticles that exhibit high sensitivity and specificity for malignant plasma cells, we fabricated ultra-small, sub-5 nm nanoparticles (NPs) that were conjugated to antibodies against the signaling lymphocytic activation molecule-F7 (SLAMF7) or the B-cell maturation antigen (BCMA). Both antigens are highly expressed and almost exclusively present on the surfaces of B-cells;27,28 and, BCMA plays an important role in plasma cell transformation and MM progression29,30 (Fig. 1A), making it an attractive and specific biomarker for MM detection.
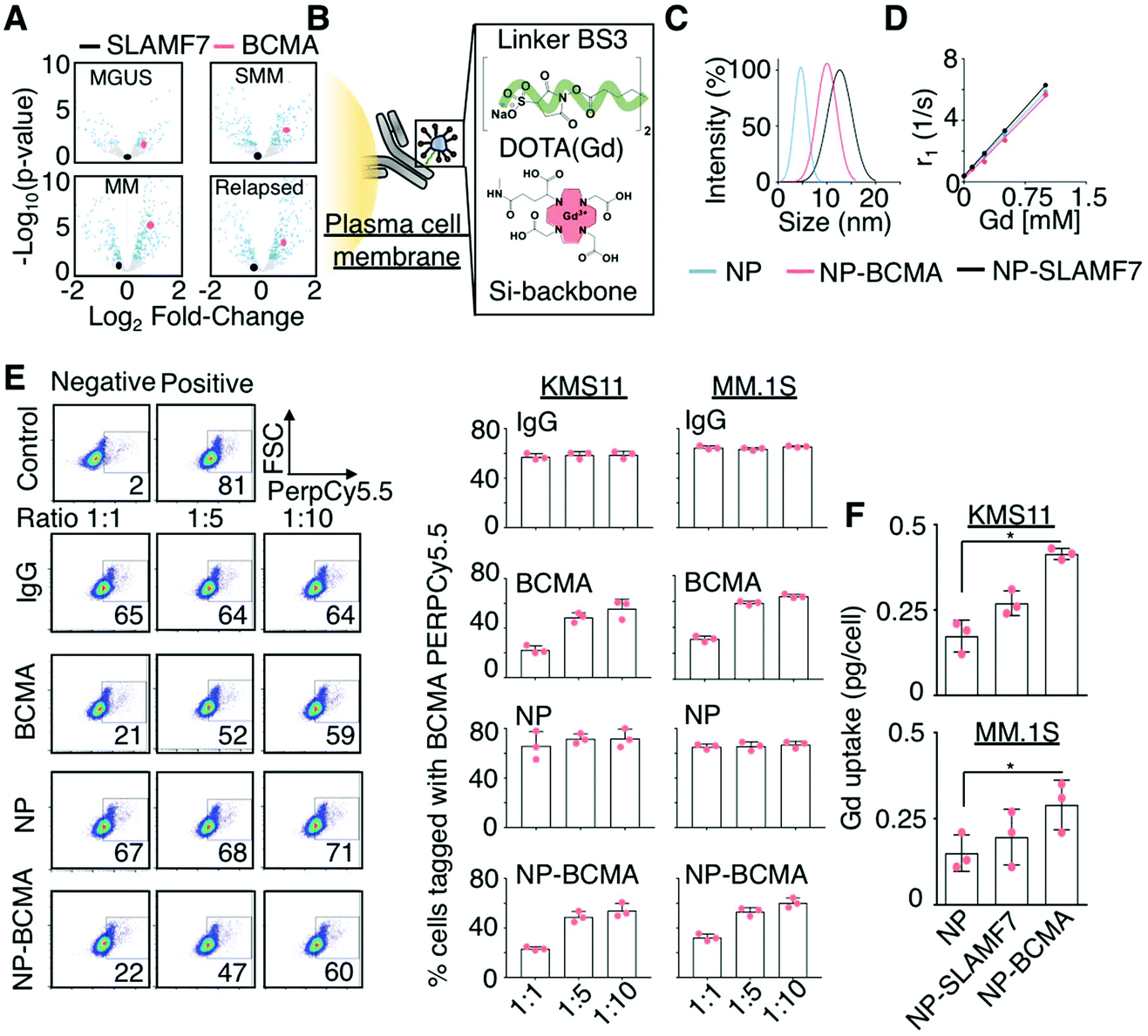 | ||
Fig. 1 Rational design of a targeted contrast agent for multiple myeloma. (A) Volcano plot comparing the expression levels of SLAMF7 and BCMA as a function of disease stage for patients with plasma cell dyscrasias identified from the Achilles dataset.31 MGUS: Monoclonal gammopathy of undetermined significance; SMM: smoldering multiple myeloma. (B) Schematic representation of the conjugation of Gd-bound, DOTA-functionalized (red), and silica-based nanoparticles (NPs; blue) to monoclonal antibodies (grey) targeting malignant plasma cells (yellow), using a homobifunctional linker (green). (C) Hydrodynamic sizes of the NP-antibody complexes of anti-SLAMF7 (NP-SLAMF7) and anti-BCMA antibodies (NP-BCMA) and (D) their corresponding relaxivity (r1) values. (E) Competitive labeling assay32 in which KMS11 and MM.1S cells were first incubated with a combination consisting of either NP-BCMA, anti-BCMA, or an IgG antibody along with a PerCP/Cy5.5-conjugated anti-BCMA antibody at different molar ratios and then subjected to flow cytometry. The lower signal observed at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of unlabeled anti-BCMA antibody or NP-BCMA to PerCP/Cy5.5-conjugated anti-BCMA antibody confirms that the NP conjugate competes with the fluorophore-conjugated antibody for cellular labeling and does not affect the antibody's binding ability. (F) Uptake of various Gd-containing NPs as assessed by ICP-MS at 30 min after cellular incubation. * p-Value <0.05, Mann Whitney test. :1 ratio of unlabeled anti-BCMA antibody or NP-BCMA to PerCP/Cy5.5-conjugated anti-BCMA antibody confirms that the NP conjugate competes with the fluorophore-conjugated antibody for cellular labeling and does not affect the antibody's binding ability. (F) Uptake of various Gd-containing NPs as assessed by ICP-MS at 30 min after cellular incubation. * p-Value <0.05, Mann Whitney test. | ||
To generate our MM-targeted contrast agents, Gd-containing NPs that possessed free amino groups on their surfaces were conjugated to anti-SLAMF7 or anti-BCMA antibodies, using a bissulfosuccinimidyl suberate crosslinker (Fig. 1B). We subsequently evaluated the targeting efficiencies of the resultant ultra-small nanoparticle-antibody complexes both in vitro and in vivo prior to performing a comparative study to determine their detection capabilities. We demonstrate that these constructs are able to effectively target malignant plasma cells, to improve longitudinal detection of disease burden, and to more accurately determine early therapeutic responses when compared to CT, 18F-FDG PET/CT, and the serologic free light-chain assay in an orthotopic murine xenograft model of MM that recapitulates its genetic and histologic features.
Results
Development of antibody-conjugated ultra-small Gd-containing nanoparticles
The rational selection of SLAMF7 and BCMA as targets for molecular imaging in MM was based on the results of the Achilles dataset.31Z-Score standardized expression values of various genes were tested for differential expression between normal and several disease states (i.e., monoclonal gammopathy of undetermined significance (MGUS), smoldering, new, and relapsed MM), using DESeq2 to calculate log-fold changes in expression and corresponding p-values, which were adjusted for multiple hypothesis testing; note that log-fold changes were considered significant when adjusted p-values were less than 0.05 (i.e., −log10(p) < 1.3) (Fig. 1A). Chemical coupling of mouse anti-human SLAMF7 and BCMA antibodies to NPs, using EDC/NHS chemistry, led to the generation of their respective antibody-conjugated nanoparticle constructs, which are denoted as NP-SLAMF7 and NP-BCMA, respectively; their structures were validated by high-performance liquid chromatography, by polysaccharide analysis using carbohydrate gel electrophoresis, and by matrix-assisted laser desorption/ionization mass spectrometry (ESI Fig. 1A–C†). Dynamic light scattering (DLS) measurements confirmed that the average hydrodynamic diameters of both NP-SLAMF7 (12.9 ± 2.3 nm by intensity-weighted DLS; 8.11 ± 0.83 nm by number-weighted DLS) and NP-BCMA (10.01 ± 2.03 nm by intensity-weighted DLS; 7.85 ± 0.85 nm by number-weighted DLS) were larger than that of the unmodified NP (4.4 ± 1.4 nm by intensity-weighted DLS; 2.93 ± 1.14 nm by number-weighted DLS; Fig. 1C). NP-antibody complexes remained stable over time and even under acidic suspension conditions (ESI Fig. 1C and D†). The relaxivity values remained constant before and after NP conjugation to different antibodies (r1 = 5.90, 5.49, 5.33 mM−1 s−1 for NP, NP-BCMA, and NP-SLAMF7, respectively); and, these values were similar to those of the clinically-available agent Mangevist™ (r1 = 4.73 mM−1 s−1, Fig. 1D).
The enhanced in vitro targeting efficiency of NP-BCMA was subsequently verified by employing human MM cell lines (MM·1S and KMS11) and by using a competitive binding assay,32 wherein cells were incubated with a combination consisting of purified human IgG, an anti-BCMA antibody, unmodified NPs, or NP-BCMA along with PerCP/Cy5.5-conjugated anti-BCMA antibodies at 3 different molar ratios. The results obtained with NP-BCMA were similar to those seen with the anti-BCMA antibody alone (Fig. 1E), confirming that antibody specificity was maintained after NP conjugation. Flow cytometry analyses demonstrated that 74.1 ± 2.9% of MM.1S cells were labeled with NP-BCMA complexes by 30 min post-incubation; in contrast, unmodified NPs labeled only 20 ± 4.9% of the cellular populations under identical conditions (p-value <0.001). The exact concentrations of Gd in the final cellular suspensions were determined by inductively-coupled plasma mass spectrometry (ICP-MS), which verified a nearly two-fold increase in labeling of both MM cells lines (MM·1S and KMS11) by using NP-BCMA as compared to unmodified NPs (Fig. 1F). Cellular viability assays on the same cultured cell lines demonstrated that neither the unmodified NPs, the free anti-SLAMF7 or anti-BCMA antibodies, nor the nanoparticle-antibody complexes imparted any in vitro toxicities at protein (10000 μg mL−1) and NP concentrations (1 μg mL−1) that were 125× and 4× higher, respectively, than those that would be expected in the blood stream after intravenous (IV) administration (ESI Fig. 2†).
In vivo targeting of plasma cells using nanoparticle-antibody complexes
We next evaluated the targeting efficiency of NP-SLAMF7 and NP-BCMA to detect plasma cells in a murine model of MM that was established via intravenous (IV) dissemination of luciferase (LUC) and green fluoresence protein (GFP)-expressing MM·1S cells (LUC+-MM·1SGFP) followed by their bone marrow engraftment within immunocompromised SCID-beige mice. Tumor burden was followed by bioluminescence imaging (BLI) at bi-weekly intervals, starting on day 19 after tumor xenotransplantation (ESI Fig. 3†). An MRI study was undertaken to compare (at an equivalent systemic level of ∼175 μmol Gd per kg; see Materials and methods and ESI Fig. 4†) the efficiencies of the various nanoparticle constructs (NP, NP-IgG, NP-SLAMF7, or NP-BCMA) to identify identical plasma cell burdens; the results were further evaluated against those that could be achieved with a clinically-available MRI contrast agent (i.e., Magnevist™), which was administered at a standard dose (200 μmol Gd per kg). Note that Magnevist™ is a molecular probe made of gadopentate dimeglumine chelates; it exhibits a nanometric size, a short circulation time (half-life <10 min), and complete renal clearance that underlie its established safety profile. As such, Magnevist is routinely used in the clinic as a preferred MRI contrast agent.To compare our experimental constructs to Magnevist™, Gd uptake was visualized in the spines (Fig. 2A and ESI Fig. 5†) and femurs of treated animals (ESI Fig. 6A†), using a 7T Bruker Biospin MRI scanner and by employing a T1-gradiant echo (GRE) sequence at 5 and 30 min after systemic injection as well as various T2 sequences with and without fat suppression. The specificity of each of the administered contrast agents to target MM cells was confirmed by animal sacrifice immediately after MRI; the vertebral tissues of each animal were harvested for histologic assessment after staining with H&E (ESI Fig. 7†); and, fluorescence microscopy of parallel sections was conducted in order to determine the spatial localization of various NP formulations (by the near-infrared fluorescence of Cy5.5, which was used for their labeling at the time of preparation) with respect to MM·1S cells (which were visualized via their intrinsic GFP emission) (Fig. 2B).
 | ||
| Fig. 2 Selection of the optimal antibody-conjugated nanoparticle for malignant plasma-cell targeting. (A) MRI of tumor deposits comprised of LUC+-MM·1SGFP cells (yellow arrows) in mice on day 19 after tumor cell implantation and after the administration of various contrast formulations (n = 5 mice per group). (B) Representative fluorescence micrographs of bone marrow taken from the spines of the same mice treated with each of the different NP formulations, confirming that uptake of NPs in the tumor microenvironment is driven by specific binding of their associated anti-SLAMF7 and anti-BCMA antibodies to receptors on cellular surfaces. Plasma-cells are visualized via the fluorescence of their intrinsic GFP reporter (green) and the NP formulations by the emission of Cy5.5 bound to their surfaces (red). Scale bar = 100 μm. (C) Signal-to-noise ratio (SNR) of the spine over time normalized to that of the baseline acquisition; note that NP, NP-IgG, NP-SLAMF7 and NP-BCMA were administered at an equivalent dose level based on Gd (∼175 μmol kg−1) while Magnevist™ was injected at a standard dose (200 μmol Gd per kg). A two-tailed t-test with Welch's correction was performed to compare groups. * p < 0.05, ** p < 0.005, and *** p < 0.001. | ||
For quantitative comparisons of MRI sensitivity, the in vivo signal-to-noise ratio (SNR) for the detection of plasma cell populations was enumerated in each image taken at different time points after the administration of the various Gd-containing contrast agents; signal intensities were quantified after a 3D segmentation of the spines (Fig. 2C) and femurs of each animal (ESI Fig. 6†). This quantification demonstrated the enhanced sensitivity of NP-SLAMF7 and NP-BCMA, when compared to the passive targeting agents (NP, NP-IgG, and Magnevist™), to detect plasma cell populations. As soon as 30 min after IV injection, animals that had been administered NP-SLAMF7 demonstrated a 1.76-fold increase in the SNR for plasmacytomas in the spine while those that had received NP-BCMA exhibited a 2.98-fold enhancement. The decrease in MRI contrast observed from 30 min to 3 h after systemic administration could be attributable to bioprocessing of the nanoparticle-antibody complexes (ESI Fig. 8†), which would be expected to result in their breakdown with subsequent loss of T1 signal as has been demonstrated with other constructs.33
ICP-MS was used to measure the Gd levels in excised organs taken from MM·1S tumor-bearing mice that were sacrificed at various time points after the administration of untargeted NP; the results confirmed that 4.11 ± 3.44 μmol Gd per kg, which equates to 0.11 ± 0.9% of the injected dose of Gd per gram (%ID g−1), localized to the spine at 30 min post-injection (Fig. 3A and ESI Fig. 9†). The corresponding values from the spines of mice sacrificed at 30 min after the administration of NP-SLAMF7 (65.82 ± 23.29 μmol Gd per kg; 1.89 ± 0.67%ID g−1) or NP-BCMA were significantly greater (153.54 ± 25.40 μmol Gd per kg; 4.4 ± 0.7%ID g−1). Together, these findings support rapid washout of unbound contrast reagent and the ability of the antibody-targeted NPs to bind resident plasma cells. With an average disease burden comprising 6.79 ± 0.58% of the spinal volume (as determined by MRI; ESI Fig. 10†), NP-BCMA is, therefore, able to achieve a localized tumor concentration of Gd that is significantly higher than that found throughout the spinal volume at an equivalent dose of untargeted NP, which is otherwise a vascular pooling agent.
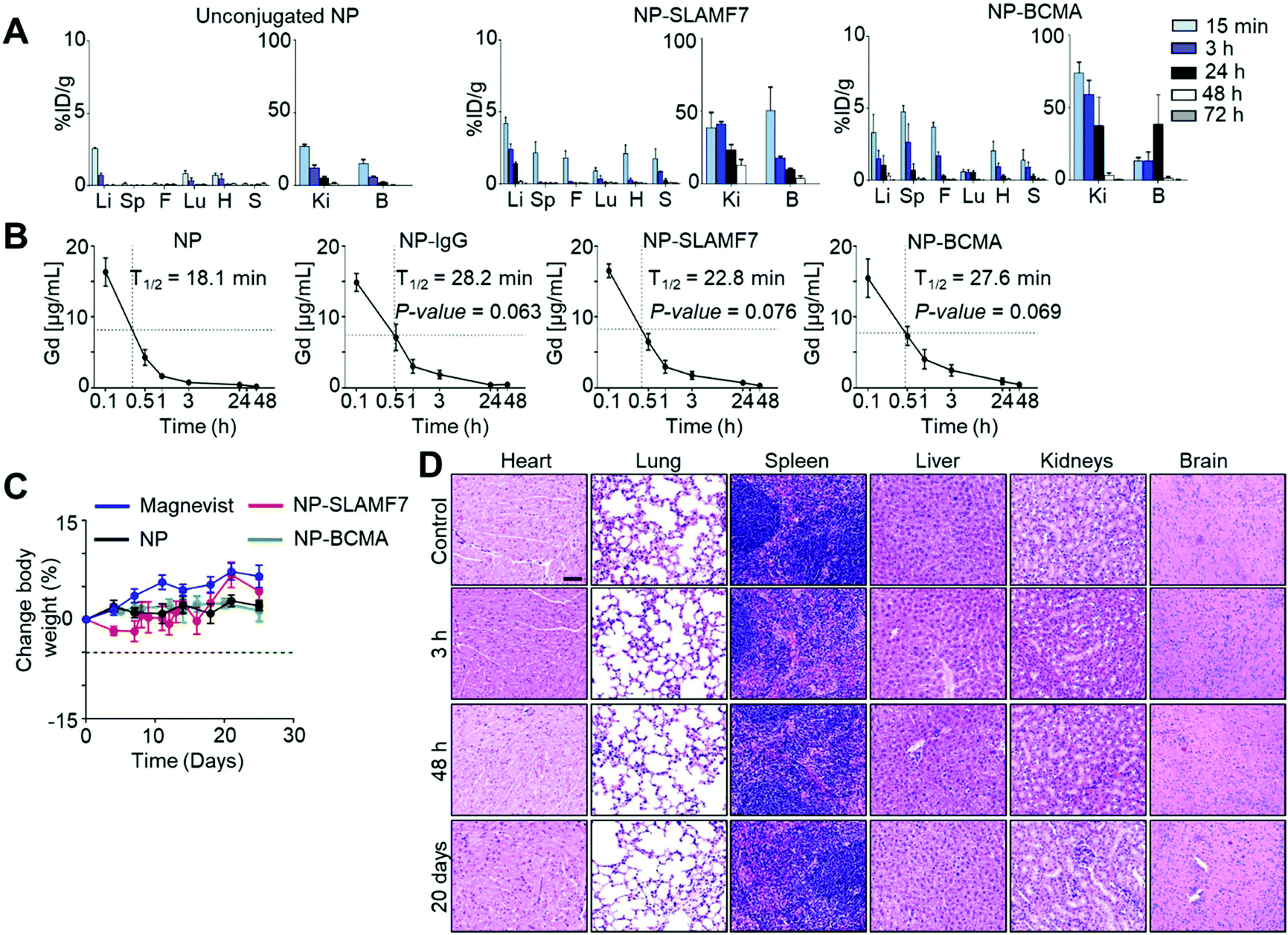 | ||
| Fig. 3 Biodistribution, pharmacokinetic and toxicity evaluation of gadolinium-containing nanoparticles and their antibody complexes. (A) Tissue distribution of unconjugated (NP), anti-SLAMF7 antibody-conjugated (NP-SLAMF7), and anti-BCMA antibody-conjugated (NP-BCMA) Gd-containing nanoparticles in MM·1S tumor-bearing mice on day 19 after tumor cell implantation; quantification of the percentages of the injected dose of Gd per gram tissue (%ID g−1) in various organs was performed by ICM-MS (n = 5 mice per time point). Li: Liver, Sp: spine, F: femur, Lu: lung, H: heart, S: spleen, Ki: kidneys, B: bladder. (B) Pharmacokinetic study demonstrating changes in the Gd-concentrations in blood from healthy balb/c mice that had been administered NP, NP-IgG, NP-SLAMF7, or NP-BCMA as determined by ICM-MS measurements, which were performed on serial blood samples drawn from the same animals (n = 5 mice per time point). Statistical analyses were performed by using a two-tailed t-test with Welch's correction to compare the NP group to others. No significant differences were observed between the antibody-conjugated NP groups (one-way ANOVA). (C) Changes in the body weights of healthy balb/c mice as a function of time after administration of a single dose of various Gd-containing contrast agents (n = 5 mice per group for Magnevist, NP, and NP-SLAMF7; n = 8 mice per group for NP-BCMA). No significant differences were observed between mice in the NP-BCMA and NP groups (repeated measurements ANOVA test). (D) H&E stained micrographs of tissue sections from healthy balb/c mice sacrificed at various time points after the administration of a single dose of NP-BCMA. | ||
NP-BCMA further demonstrates better tumor uptake than NP-SLAMF7 (p = 0.0045, one-sided paired t-test), which may be attributed to the greater numbers of surface BCMA antigens per MM·1S cell. The observed SNRs for plasmacytomas visualized by MRI (Fig. 2C and ESI Fig. 5†) correlate well with simulations of the expected SNRs (ESI Fig. 11†), demonstrating that nanoparticle-antibody complexes are able to achieve Gd concentrations that are >100 μM throughout the measured tumor volume. While we have previously observed effective contrast of subcutaneous tumors after the administration of untargeted NP (via 7T MRI),34,35 NP-BCMA specifically binds plasmacytomas and enables visualization of minute tumor populations in their natural environment. Note that only trace amounts of Gd were otherwise observed in the liver, kidney, lungs, or in any other organ at 72 h after the administration of each of the agents (i.e., NP, NP-SLAMF7, or NP-BCMA; Fig. 3A), supporting their systemic elimination. The pharmacokinetic profiles of NP-SLAMF7 and NP-BCMA were similar (Fig. 3B); and, their circulatory half-lives were longer than that of the unmodified NP (t1/2 = 16.1, 22.3, 25.2 and 30.3 min for NP, NP-IgG, NP-SLAMF7, and NP-BCMA, respectively). These enhancements in vascular persistence may be attributed to their slightly larger sizes and to the intrinsic properties of the selected antibodies.
Surprisingly, NP-SLAMF7 and NP-BCMA were found to exhibit rapid renal clearance, which could be expected to limit their long-term exposure to healthy organs. Urine was collected at various time points and assayed for its Gd content by ICP-MS, demonstrating that >95% and >93% of the injected dose was found in the urine at 72 h after administration of NP and NP-BCMA, respectively (ESI Fig. 12A†). While the complete renal clearance of the untargeted NP (<5 nm) has been previously demonstrated17,36 and is consistent with the known glomerular filtration threshold (∼6 nm),37 similar clearance of NP-BCMA was unexpected due to the relatively larger size of this bioconjugate (average diameter of ∼8 to 10 nm by number vs. intensity-weighted DLS, respectively). While in vivo decoupling of surface-bound antibody from NP-BCMA could potentially explain these observations, which would also be consistent with the loss of in vivo MRI contrast observed after 30 min (vide supra), dynamic light scattering measurements of both normalized number- and intensity-weighted size distributions confirmed that NP and NP-BCMA remained relatively stable from administration to systemic clearance (ESI Fig. 12B†). The exact mechanisms whereby NP-BCMA is filtered through the kidneys, thus, remain unknown and warrant further investigation.
Both the untargeted and antibody-conjugated NP constructs were tolerated by balb/c mice as evidenced by stable animal weights and the paucity of gross adverse phenomena (i.e., changes in general appearance, in skin and fur, in nose, mouse and head respirations, in urine, in feces and in locomotor factors) over a two-week period after a single-dose IV administration (Fig. 3C). Terminal blood studies confirmed normal basic metabolic panels (BMPs; ESI Fig. 13A†), complete blood counts (CBCs; ESI Fig. 13B†), and white blood cell differential counts at the end of this period (ESI Fig. 13C†). Pathology review of H&E stained tissue sections uncovered no evidence of microarchitectural distortion (Fig. 3D). As such, the NP-antibody complexes were deemed to exhibit no disqualifying short-term toxicities that could compromise the findings of their enhanced imaging performance or that could preclude their continued preclinical development.
Comparisions of in vivo imaging sensitivity and specificity
Having validated the analytical capabilities of our novel constructs, we subsequently sought to compare MRI of NP-BCMA with respect to each of the clinical imaging modalities that are routinely used for the longitudinal detection of plasma cells. We again employed a murine model of MM, consisting of the intravascular dissemination of LUC+-MM·1SGFP cells in mice; therapeutic debulking was subsequently conducted at 21 days after xenotransplatation, using three doses of bortezomib (0.5 mg kg−1) and one dose of melphalan (5.5 mg kg−1). Tumor growth was monitored by weekly BLI as the gold standard for preclinical detection (Fig. 4A) as well as by whole-body CT (Fig. 4B), 18F-FDG PET/CT (Fig. 4C), and by MRI of BCMA-NP (Fig. 4D). This post-treatment model was validated by obtaining a negative BLI signal at day 25, which corresponded to the completion of bortezomib and melphalan administration. Changes in the signal intensities (BLI), SNRs (CT and MRI), and standard uptake values (SUVs; 18F-FDG PET/CT) of the spine over time were subsequently used to track disease re-expansion (Fig. 4E). In addition, the levels of serum λ light-chains were measured at the same time points (Fig. 4F) and served as an independent biomarker of disease activity. Note that MM·1S cells only express the λ light-chain; they do not express the kappa light-chain nor exhibit a spike in their levels of M-protein.38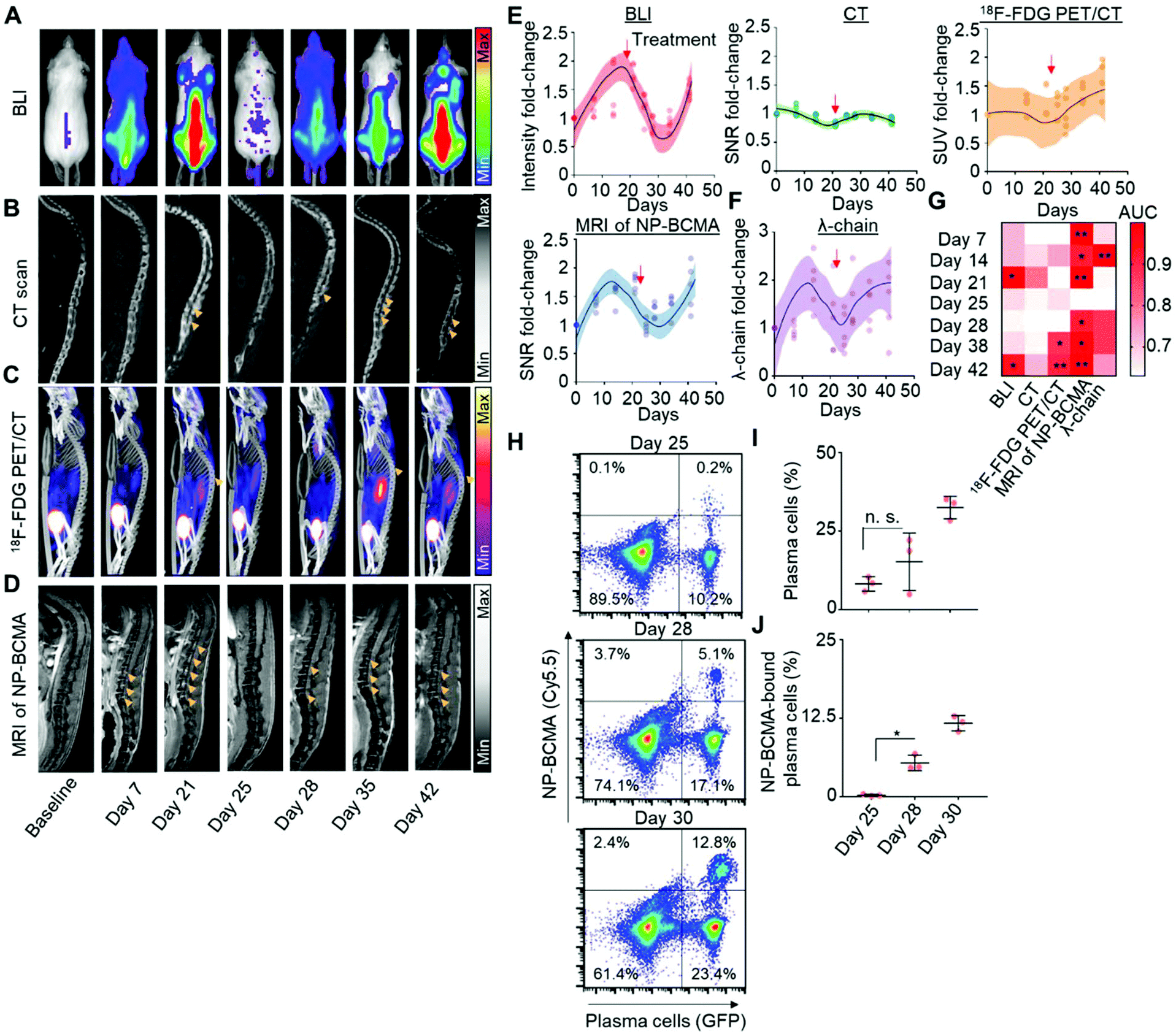 | ||
| Fig. 4 MRI of NP-BCMA as a novel biomarker for longitudinal tracking of MM activity. LUC+-MM·1SGFP tumor-bearing mice were imaged weekly by (A) bioluminescence imaging (BLI), (B) CT, (C) 18F-FDG PET/CT, or (D) MRI at 30 min after injection of NP-BCMA to visualize tumor burden (yellow arrows). On day 21 after tumor cell implantation, treatment with bortezomib (0.5 mg kg−1 ×3) and melphalan (5.5 mg kg−1 ×1) ensued. Thereafter, longitudinal imaging was conducted at weekly intervals to follow disease burden. Changes in (E) BLI signal intensities, the signal-to-noise ratio (SNR) for CT and MRI, and the standard uptake value (SUV) for 18F-FDG PET/CT were quantified to determine the sensitivity of each modality to detect tumor cells at various time points. The red arrows correspond to the first time points after systemic treatment. (F) Lambda light-chain levels were similarly quantified by immunoassay. Shadowing demarcates the 90% confidence interval; note that all scans and light chain assays were performed on the same animals (n = 5 mice). (G) Receiver operator characteristic (ROC) curve at week 5, comparing the sensitivity and specificity of the 4 modalities to detect the presence of residual disease. From there, a comparison of the area under the curve (AUC) over the course of treatment for the 4 detection modalities was calculated. (H) Flow cytometry histograms depicting the percentages of total plasma cells (GFP signal) and NP-BCMA-bound plasma cells (Cy5.5 signal) at each time point. (I) The total percentages of plasma cells and (J) the percentages of NP-BCMA-bound plasma cells in the bone marrow were enumerated and compared (n = 3 mice per group). * p-value < 0.05, Mann Whitney test. | ||
Results obtained by BLI, MRI, CT, 18F-FDG PET/CT, and by the serum λ light-chain assay were compared at 1 week after therapeutic debulking (i.e., 5 weeks after initial tumor cell implantation). A receiver operator characteristic (ROC) curve was generated to assess the sensitivity and specificity of each of the 5 detection modalities to identify disease recurrence and confirmed the superiority of MRI using NP-BCMA. Comparisons of area-under-the-curve (AUC) for the SNR observed with each modality and over the entire duration of the experiment (i.e., from initial tumor cell implantation to therapeutic debulking to eventual animal demise from tumor regrowth) further supported these findings (Fig. 4G, ESI Fig. 14†). To determine the analytical sensitivity of MRI of NP-BCMA, additional mice were sacrificed on day 25 (i.e., immediately after tumor debulking) as well as on days 28 and 30 post tumor cell implantation, which corresponded to the time points after which plasma cells were first visible (in the spine) by MRI. Flow cytometry experiments on the entire cellular populations recovered from each animal were conducted (Fig. 4H); the results were enumerated to confirm that MRI with NP-BCMA had a detection threshold of 2200 ± 450 plasma cells (in the spine) per mouse. As expected, the percentages of plasma cells amongst the total cellular populations (Fig. 4I), as well as the percentages of NP-BCMA-bound plasma cells (Fig. 4J), increased as a function of time and were due to tumor regrowth.
Conclusions
We demonstrate a proof-of-concept method in which changes in the SNR obtained by serial MRI of an antibody-conjugated nanoparticle-based contrast agent may be used to follow MM activity throughout the whole-body, using highly-sensitive, specific, and non-invasive imaging measurements made at serial time points. Central to the success of these efforts was the utilization of ultra-small (<5 nm) nanoparticles bound to tumor-specific antibodies to enhance imaging contrast. Our constructs were able to circumvent the challenges seen with the first generation of antibody-bound nanoparticles that were much larger in size (30–200 nm) and that have been shown to accumulate in subcutaneous tumor models over the course of several hours.15,39 In comparison, our agents demonstrate maximal contrast enhancement as quickly as 30 min after injection, which could enable their utilization within the existing clinical work-flow.We further demonstrate that antibody-targeting of ultra-small Gd-containing NPs enables precise localization of malignant plasma cells in their natural microenvironment by MRI, which is due to the ability of these constructs to bind tumor cell populations and to dramatically enhance localized concentrations of Gd. Although similar systemic doses of Gd have been employed for imaging with other targeted contrast agents (via 7T MRI),13,14,40,41 many exhibited poor systemic clearance, thereby raising safety concerns. Moreover, while Gd-containing constructs are not suitable for use in the context of compromised kidney function, given the well-established risks of all Gd-containing contrast agents,42 targeted nanoparticle constructs comprised of nonmetallic materials43 may find utility in prompting early therapeutic discontinuation and/or re-initiation after prolonged periods of MM remission. With the increasing utilization of antibody-based therapies in MM (e.g., elotuzumab,27 BCMA-targeted chimeric antigen receptor T-cells (CAR-T),44 and daratumumab45), future formulations of SLAMF7, BCMA and CD38-based antibody-nanoparticle complexes may enable imaging to guide patient-specific therapeutic selection.
While our results with T1-weight MRI are promising, further investigations will be necessary in order to ascertain the safety of antibody-targeted ultra-small NPs as well as the benefits that may be expected when translating our findings from small animals to novel clinical imaging agents. As there is a known size limit for glomerular filtration, antibody decoupling and/or nanoparticle degradation could account for the phenomena of rapid renal clearance that was observed in our current study. DLS measurements, however, did not show appreciable changes in particle size upon renal filtration; serological and histologic studies of the kidney also did not yield evidence of acute compromise nor effacement of glomeruli. As such, a central focus of future investigations will be the verification and exploration of the mechanisms by which NP-BCMA may be renally cleared, which is a result that is inconsistent with previous literature.46,47
It should be noted that our experimental constructs introduce features of potential clinical novelty that bypass many of the limitations seen with other diagnostic agents used for the detection of MM. For instance, unlike with experimental immunoconjugates that are employed with PET/CT, our constructs are not reliant upon ionizing radiation and may enable longitudinal administration to the same subject. Alternatively, further incorporation of PET radioisotopes within antibody-conjugated ultra-small NPs may be pursued to enhance sensitivity as compared to bioconjugates of single chelators or to PET imaging with 18F-FDG. Such targeted agents could be expected to enable detection of heterogeneous disease foci that are routinely missed by bone marrow biopsy, which is prone to sampling error,48 or by blood biopsy, whose results may be reflective of the partial disease activity of select clones.49 In conclusion, our results may afford insights into the fabrication of other targeted and ultra-small NPs that could help to detect or treat residual disease, thereby improving the care and survival of MM patients.
Materials and methods
Cell lines
The human MM cell line MM·1S was purchased from ATCC (Manassas, VA, USA). LUC+-MM·1SGFP cells were generated via retroviral transduction, using the pGC-GFP/Luc vector, and were authenticated by short tandem repeat DNA profiling. MM·1S, OPM2, and KMS11 were cultured in RPMI media, containing 10% fetal bovine serum, 1% PenStrep, and 1% glutamine.Synthesis of antibody-conjugated gadolinium-containing nanoparticles (NPs)
Ultra-small, silica-comprised and Gd-containing nanoparticles (NPs) were provided by NH Theraguix, Inc. (Villeurbanne, France). In brief, the constructs are synthesized by a top-down process and are comprised of a polysiloxane shell surrounded by DOTA (1,4,7,10-tetra-azacyclododecane-1-glutaric anhydride-4,7,10-triacetic acid), which is covalently-bound to the inorganic matrix and which further chelates Gd.17,50–52 NP-antibody complexes were generated via conjugation of NPs with mouse anti-human SLAMF7 or anti-human BCMA monoclonal antibodies (Biolegend Inc., San Diego, CA), using a previously-reported homobifunctional linker chemistry.53 Briefly, NPs were diluted in UltraPure water to a final concentration of 50 nM. A 1:10 molar ratio of the bissulfosuccinimidyl suberate linker was then added and mixed with NP suspensions for 30 min at room temperature to promote the generation of linker-bound NPs. These surface-modified constructs were then incubated with the monoclonal antibodies at a 100:1 molar ratio; and, the suspensions were stirred for an additional 1 h at room temperature. The NP-antibody complexes were purified by centrifugation filtration, using a filtration device equipped with a 50 kDa molecular weight cutoff membrane (Milipore) that was spun at 15000 rcf, and were subsequently resuspended in PBS (1 M). This process was conducted in triplicate to assure removal of all excess free nanoparticles into the filtrate and to concentrate the suspensions of pure NP-SLAMF7 and NP-BCMA. The final concentrations of the NP-antibody complexes were determined by ICP-MS, using an Agilent 7900 instrument (Agilent Technologies, Inc., Santa Clara, CA).
In vitro assays determining the specificity of NP-antibody complexes to bind MM cells
Flow cytometry analyses of different MM cell lines treated with various NP-antibody complexes were performed by employing a competitive binding assay.32 The cells (1 × 106 mL−1) were incubated with one of three different pool sets (for 1 h at 37 °C), consisting of free anti-IgG antibodies, anti-BCMA antibodies, or NPs (i.e., unmodified NPs or NP-BCMA; 0.5 mM), which were each combined with free PerCP/Cy5.5-labeled anti-human BCMA antibodies. The initial concentration of PerCP/Cy5.5-labeled anti-human BCMA antibody was 2.5 μg mL−1 (in the pool set consisting of a 1:1 molar ratio) and increased up to 25.5 μg mL−1 (in the pool set at a 1:10 molar ratio). A decrease in the PerCP/Cy5.5 signal corresponded to competition with the particular pooled agent for the same surface antigen on the cells; free anti-BCMA antibodies were used as a positive control (for competition with PerCP/Cy5.5-labeled anti-human BCMA antibodies) while free anti-IgG and unmodified NPs constituted the negative controls in the experiment. To cross-validate the results, ICP-MS was utilized to quantify the amounts of Gd bound per cell. To perform these later experiments, the treated MM cells were lysed with 0.3% Triton-X 100 solution prior to enumeration of the amounts of Gd in each sample, using ICP-MS.
Animals
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals as set forth by the Institutional Care and Use Committee (IACUC) of the Dana-Farber Cancer Institute (protocol 14-001). LUC+-MM·1SGFP cells were administered to SCID/beige mice (5 × 106 cells per mouse; n = 5 mice per group) via IV dissemination, establishing an orthotopic xenograft model of human MM. Tumor growth was monitored weekly by BLI, using an IVIS Spectrum-bioluminescence and fluorescence imaging system (Perkins Elmer). Tumor debulking was established by treating the mice with bortezomib (0.5 mg kg−1 daily ×3 doses) followed by melphalan (5.5 mg kg−1 ×1 dose). Preliminary toxicity studies were conducted in balb/c mice (n = 8 mice per group) after a single-dose IV administration of NP-BCMA (vide infra).Dosing with NP-BCMA
ICP-MS was used to confirm the presence of 4 NPs (each with 10 Gd atoms)36 per anti-BCMA antibody in a NP-antibody complex. Upon injection of 200 μL of a suspension of NP-antibody complexes, the dose equivalents were 0.0956 mg (4.8 mg kg−1) of antibody and ∼550 μg (∼175 μmol kg−1) of Gd per mouse (ESI Fig. 4†).In vivo and ex vivo imaging studies
MR image acquisition was conducted with a preclinical Bruker BioSpec 7T/30 cm USR horizontal bore Superconducting Magnet System (Bruker Corp., Billerica, MA). A 40 mm volume radiofrequency (RF) coil was used for both RF excitation and receiving. Animals were anesthetized with 1.5% isoflurane mixed in medical air at a flow rate of 2 L min−1. Body temperature was maintained at 37 °C, using a warm air fan. Animal respiration and temperature were monitored and regulated by the SAII monitoring and gating system model 1025T (Sa Instruments Inc., Stony Brook, NY). Neither cardiac nor respiration gating was applied. Bruker Paravision 6.0.1 was used for MRI data acquisition. A T1 GRE sequence, employing a repetition time (TR) of 87 ms, an echo time (TE) of 3.9 ms, and a flip angle of 60°, was utilized for imaging. Acquisition matrix = 256 × 192 pixels; reconstructed matrix = 256 × 256 pixels; slice thickness = 0.5 mm; FOV = 40 × 24 mm (spine) or 20 × 24 mm (femur); number of averages = 13; and, scanning time = ∼5 min for each image. When comparing imaging parameters obtained with the different Gd-containing contrast agents, MRI was performed at various time intervals after contrast administration; and, the results were compared to baseline images. For the early diagnostic and post-treatment quantification studies, MRI was performed 30 min after IV contrast injection.CT acquisitions were conducted on a preclinical Inveon CT scanner (Siemens) equipped with a 50 kVp source; the image resolution was 10.2 pixels per mm; and, a slice thickness of 0.1 mm was utilized. CT imaging was performed at various time intervals and before the injection of each MR contrast agent in order to compare changes in the SNR for different disease burdens detected via each imaging modality (vide infra).
As a method of validating the binding of NP-antibody complexes to MM cells, confocal microscopy was performed to visualize the co-localization of NP-antibody complexes, which had been labeled with a separate fluorophore, on the surfaces of LUC+-MM·1SGFP cells. For these experiments, NPs were first conjugated with Cy5.5–NHS at 1:1000 molar ratio of fluorophore to NP, using EDC/NHS chemistry, purified (to remove unbound fluorophore), and subsequently injected into mice. MRI commenced at 30 min after administration of various NP-antibody constructs (i.e., NP, NP-IgG, NP-SLAMF7, or NP-BCMA); and, the animals were sacrificed immediately thereafter. Their spines and femurs were excised, flash frozen, and sectioned; bone marrow was mounted on cover slips coated with Dapi Fluoromount-G (SouthernBiotech). Confocal microscopy (Olympus FV12000, Olympus) was then used to verify co-localization of the two fluorophores on tumor cells.
18F-FDG PET/CT was performed on an Inveon Multimodality Systems (Siemens Medical Solutions USA Inc.). Low-dose CT scans were first acquired (80 kVp, 0.5 mA, 220 degree rotation, 600 ms per degree exposure time, 80 μm reconstruction pixel size) for anatomical reference and to provide guidance for the delineation of the selected tissue region/volume of interest (ROI/VOI). Following a bolus IV injection of approximately 6.5 MBq of 18F-FDG, static PET emission scans were acquired in list-mode format over 10 min (60 min post-radiotracer injection) and corrected for decay and dead time. The acquired data were then sorted into 0.5 mm sinogram bins and a time frame of 1 s was employed for image reconstruction, using ordered subset expectation maximization in 3 dimensions followed by MAP reconstruction (OSEM3D/MAP; 256 × 256 × 159 matrix size, 0.43 × 0.43 × 0.80 mm3 pixel size, 4 OSEM3D iterations, 18 MAP iterations, b = 0.1 optimized for uniform resolution, FWHM 1.29 mm). PET data analysis was performed with Siemens Inveon Research Workplace software. The radioactivity concentrations within selected tissues were obtained from mean voxel intensity values within the VOI and then converted to megabecquerels per milliliter, using the calibration factor determined for the Inveon PET system. These values were then divided by the administered activity in megabecquerels and animal body weight to obtain an image VOI-derived standardized uptake value (SUV).
Quantitative comparisons of imaging modalities
Evaluation of the relative detection sensitivity for plasma cells at different time points and/or via different imaging modalities was performed by conducting a signal-noise-ratio (SNR) calculation on each acquired image. These SNR values were obtained after first performing a 3D segmentation of the spine and a femur of each animal, using Fiji freeware (https://fiji.sc/). Each image was normalized to the same intensity level and a region of interest (ROI), including the whole examined organ (i.e., spine or femur), was segmented; the signal intensity in the ROI was recorded and compared to the background level, which was measured on each scan. SNR and normalized SNR values were calculated, according to eqn (1) and (2):| SNR = intensity/noise; | (1) |
| Normalized SNR(i) = SNR(i)/SNRbaseline. | (2) |
Absolute quantification of the uptake of various Gd-based contrast agents was determined, using ICP-MS (Agilent 7900) and by following previously described protocols.17,50,52 Briefly, animals were sacrificed at 30 min after contrast injection; their excised organs were dissolved in a 70% HCl solution; and, the Gd content of each organ was determined by ICP-MS measurement.
Lambda light-chain quantification
Mice were bled once per week and immediately before imaging. Serum was separated from blood samples and frozen at −80 °C until the end of the study. These samples were then diluted 1:10 v:v with PBS and a clinical-grade immunoturbidimetric assay, which is routinely performed in the clinical laboratories of the Brigham and Women's Hospital (Boston, MA), was used to quantify the amounts of lambda light chains in each sample.
Receiver operator characteristic (ROC) curve
The ROC curve was used to represent the ability of the SNR to discriminate the presence or absence of tumor cells. The SNR at 5 weeks post-tumor cell implantation was enumerated for each of the various imaging modalities and served as a metric by which to compare their detection sensitivities. The class was defined for each time point by using the following method: baseline measurements prior to tumor cell implantation served as the control (Ct0 = 0) and were compared against subsequent time points (Ct = 1) with the assumption that tumor cells were thereafter always present. To ensure that the prediction was not random, a two-sided Wilcoxon rank-sum test was used. A p-value below 0.05 indicated that the SNR value for a given class was significantly different than that of another class. The R code used for the AUC calculation and the statistical analysis are presented in ESI Fig. 15 and 16.†Preliminary toxicity evaluation
Female balb/c mice were administered (IV) a single dose (200 μL) of PBS (n = 8 mice), NP-BCMA (n = 8 mice; 174.49 μmol kg−1 of Gd; 4.8 mg kg−1 of anti-BCMA), anti-BCMA antibody (n = 5 mice; 4.8 mg kg−1), or unconjugated NPs (n = 5 mice; 183.97 μmol kg−1 of Gd). The body weights of the animals were monitored daily, starting on the day of injection. After 21 days, the mice were sacrificed and blood samples were collected by submandibular puncture to determine basic metabolic profiles, complete blood counts, and white blood cell differential counts. The major organs of the animals were also collected and stained with H&E prior to histological analyses by a board-certified veterinary pathologist (Dr. Roderick Bronson, D.V.M.).Statistical analyses
All in vitro statistical analyses were performed with GraphPad Prism software (V.7.1). The ability to discriminate the presence of MM cells, using each of the different detection modalities, was determined by using R version 3.3.3.Author Contributions
A.D., Q.D.N., I.M.G., and P.P.G. designed the research study. A.D., M.R., Y.Y, C.M., H.V.-T.N, P.H., Q-D.N., A.P. and Y.C. conducted experiments. A.D., C.M., H.V.-T.N., and J.A.J. designed the chemical conjugation process. A.D., C.M., and H.V.-T.N. performed the chemical conjugation and characterization tests. T.P.C. performed the statistical analyses. F.L. confirmed the presence of tumor cells on MRI and CT scans. P.J. performed the light chain assay analyses. O.T. provided the untargeted nanoparticles (NPs). A.D., I.M.G., and P.P.G. wrote the manuscript.Conflicts of interest
The authors declare that they have no competing financial interests.Acknowledgements
Imaging experiments were conducted in the Lurie Imaging Center at the DFCI. This work was supported in part by a grant from the NCI (R01CA181683-01A1; I. M. G.) as well as from the Charles W. and Jennifer C. Johnson Clinical Investigator Fund (P. P. G.). A. D. acknowledges support from the Fondation Française pour la Recherche contre le Myélome et les Gammapathies Monoclonales (FFRMG), the Multiple Myeloma Research Foundation (MMRF), and the Philippe Foundation. H.V.-T.N. acknowledges support from the National Science Foundation (graduate research fellowship). P. P. G. acknowledges support from the Kathryn Fox Samway Foundation.References
- M. Cavo, E. Terpos, C. Nanni, P. Moreau, S. Lentzsch, S. Zweegman, J. Hillengass, M. Engelhardt, S. Z. Usmani, D. H. Vesole, J. San-Miguel, S. K. Kumar, P. G. Richardson, J. R. Mikhael, F. L. da Costa, M. A. Dimopoulos, C. Zingaretti, N. Abildgaard, H. Goldschmidt, R. Z. Orlowski, W. J. Chng, H. Einsele, S. Lonial, B. Barlogie, K. C. Anderson, S. V. Rajkumar, B. G. M. Durie and E. Zamagni, Lancet Oncol., 2017, 18(4), e206–e217 CrossRef PubMed.
- S. V. Rajkumar, O. Landgren and M. V. Mateos, Blood, 2015, 125(20), 3069–3075 CrossRef CAS PubMed.
- C. Pawlyn, L. Fowkes, S. Otero, J. R. Jones, K. D. Boyd, F. E. Davies, G. J. Morgan, D. J. Collins, B. Sharma, A. Riddell, M. F. Kaiser and C. Messiou, Leukemia, 2016, 30(6), 1446–1448 CrossRef CAS PubMed.
- M. A. Dimopoulos, J. Hillengass, S. Usmani, E. Zamagni, S. Lentzsch, F. E. Davies, N. Raje, O. Sezer, S. Zweegman, J. Shah, A. Badros, K. Shimizu, P. Moreau, C. S. Chim, J. J. Lahuerta, J. Hou, A. Jurczyszyn, H. Goldschmidt, P. Sonneveld, A. Palumbo, H. Ludwig, M. Cavo, B. Barlogie, K. Anderson, G. D. Roodman, S. V. Rajkumar, B. G. Durie and E. Terpos, J. Clin. Oncol., 2015, 33(6), 657–664 CrossRef PubMed.
- P. Spinnato, A. Bazzocchi, A. Brioli, C. Nanni, E. Zamagni, U. Albisinni, M. Cavo, S. Fanti, G. Battista and E. Salizzoni, Eur. J. Radiol., 2012, 81(12), 4013–4018 CrossRef CAS PubMed.
- C. P. Shortt, T. G. Gleeson, K. A. Breen, J. McHugh, M. J. O'Connell, P. J. O'Gorman and S. J. Eustace, AJR, Am. J. Roentgenol., 2009, 192(4), 980–986 CrossRef PubMed.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46(12 Pt 1), 6387–6392 CAS.
- P. Moreau, M. Attal, D. Caillot, M. Macro, L. Karlin, L. Garderet, T. Facon, L. Benboubker, M. Escoffre-Barbe, A. M. Stoppa, K. Laribi, C. Hulin, A. Perrot, G. Marit, J. R. Eveillard, F. Caillon, C. Bodet-Milin, B. Pegourie, V. Dorvaux, C. Chaleteix, K. Anderson, P. Richardson, N. C. Munshi, H. Avet-Loiseau, A. Gaultier, J. M. Nguyen, B. Dupas, E. Frampas and F. Kraeber-Bodere, J. Clin. Oncol., 2017, 35(25), 2911–2918 CrossRef CAS PubMed.
- J. Hillengass and O. Landgren, Leuk. Lymphoma, 2013, 54(7), 1355–1363 CrossRef PubMed.
- J. R. Bading and A. F. Shields, J. Nucl. Med., 2008, 49(Suppl 2), 64S–80S CrossRef CAS PubMed.
- Y. Shou, J. Lu, T. Chen, D. Ma and L. Tong, J. Cancer Res. Ther., 2012, 8(1), 96–102 CrossRef PubMed.
- Y. Liu, Z. Chen, C. Liu, D. Yu, Z. Lu and N. Zhang, Biomaterials, 2011, 32(22), 5167–5176 CrossRef CAS PubMed.
- Z. Zhou and Z. R. Lu, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5(1), 1–18 CrossRef CAS.
- S. M. Vithanarachchi and M. J. Allen, Curr. Mol. Imaging, 2012, 1(1), 12–25 CrossRef CAS PubMed.
- M. Colombo, L. Fiandra, G. Alessio, S. Mazzucchelli, M. Nebuloni, C. De Palma, K. Kantner, B. Pelaz, R. Rotem, F. Corsi, W. J. Parak and D. Prosperi, Nat. Commun., 2016, 7, 13818 CrossRef CAS PubMed.
- S. Kotb, A. Detappe, F. Lux, F. Appaix, E. L. Barbier, V. L. Tran, M. Plissonneau, H. Gehan, F. Lefranc, C. Rodriguez-Lafrasse, C. Verry, R. Berbeco, O. Tillement and L. Sancey, Theranostics, 2016, 6(3), 418–427 CrossRef CAS PubMed.
- A. Detappe, S. Kunjachan, L. Sancey, V. Motto-Ros, D. Biancur, P. Drane, R. Guieze, G. M. Makrigiorgos, O. Tillement, R. Langer and R. Berbeco, J. Controlled Release, 2016, 238, 103–113 CrossRef CAS PubMed.
- K. Ulbrich, K. Hola, V. Subr, A. Bakandritsos, J. Tucek and R. Zboril, Chem. Rev., 2016, 116(9), 5338–5431 CrossRef CAS PubMed.
- J. J. Mulvey, C. H. Villa, M. R. McDevitt, F. E. Escorcia, E. Casey and D. A. Scheinberg, Nat. Nanotechnol., 2013, 8(10), 763–771 CrossRef CAS PubMed.
- M. Arruebo, M. Valladares and A. González-Fernández, J. Nanomater., 2009, 2009, 439389 Search PubMed.
- M. Reth, Nat. Immunol., 2013, 14(8), 765–767 CrossRef CAS PubMed.
- S. A. MacParland, K. M. Tsoi, B. Ouyang, X. Z. Ma, J. Manuel, A. Fawaz, M. A. Ostrowski, B. A. Alman, A. Zilman, W. C. Chan and I. D. McGilvray, ACS Nano, 2017, 11(3), 2428–2443 CrossRef CAS PubMed.
- X. Qian, X. H. Peng, D. O. Ansari, Q. Yin-Goen, G. Z. Chen, D. M. Shin, L. Yang, A. N. Young, M. D. Wang and S. Nie, Nat. Biotechnol., 2008, 26(1), 83–90 CrossRef CAS PubMed.
- E. D. Hsi, R. Steinle, B. Balasa, S. Szmania, A. Draksharapu, B. P. Shum, M. Huseni, D. Powers, A. Nanisetti, Y. Zhang, A. G. Rice, A. van Abbema, M. Wong, G. Liu, F. Zhan, M. Dillon, S. Chen, S. Rhodes, F. Fuh, N. Tsurushita, S. Kumar, V. Vexler, J. D. Shaughnessy, Jr., B. Barlogie, F. van Rhee, M. Hussein, D. E. Afar and M. B. Williams, Clin. Cancer Res., 2008, 14(9), 2775–2784 CrossRef CAS PubMed.
- G. S. Mack and A. Marshall, Nat. Biotechnol., 2010, 28(3), 214–229 CrossRef CAS PubMed.
- S. T. Lwin, C. M. Edwards and R. Silbermann, BoneKEy Rep., 2016, 5, 772 CAS.
- S. Lonial, M. Dimopoulos, A. Palumbo, D. White, S. Grosicki, I. Spicka, A. Walter-Croneck, P. Moreau, M. V. Mateos, H. Magen, A. Belch, D. Reece, M. Beksac, A. Spencer, H. Oakervee, R. Z. Orlowski, M. Taniwaki, C. Rollig, H. Einsele, K. L. Wu, A. Singhal, J. San-Miguel, M. Matsumoto, J. Katz, E. Bleickardt, V. Poulart, K. C. Anderson, P. Richardson and E. Investigators, N. Engl. J. Med., 2015, 373(7), 621–631 CrossRef CAS PubMed.
- A. J. Novak, J. R. Darce, B. K. Arendt, B. Harder, K. Henderson, W. Kindsvogel, J. A. Gross, P. R. Greipp and D. F. Jelinek, Blood, 2004, 103(2), 689–694 CrossRef CAS PubMed.
- S. L. Nutt, P. D. Hodgkin, D. M. Tarlinton and L. M. Corcoran, Nat. Rev. Immunol., 2015, 15(3), 160–171 CrossRef CAS PubMed.
- V. Peperzak, I. Vikstrom, J. Walker, S. P. Glaser, M. LePage, C. M. Coquery, L. D. Erickson, K. Fairfax, F. Mackay, A. Strasser, S. L. Nutt and D. M. Tarlinton, Nat. Immunol., 2013, 14(3), 290–297 CrossRef CAS PubMed.
- G. S. Cowley, B. A. Weir, F. Vazquez, P. Tamayo, J. A. Scott, S. Rusin, A. East-Seletsky, L. D. Ali, W. F. Gerath, S. E. Pantel, P. H. Lizotte, G. Jiang, J. Hsiao, A. Tsherniak, E. Dwinell, S. Aoyama, M. Okamoto, W. Harrington, E. Gelfand, T. M. Green, M. J. Tomko, S. Gopal, T. C. Wong, H. Li, S. Howell, N. Stransky, T. Liefeld, D. Jang, J. Bistline, B. Hill Meyers, S. A. Armstrong, K. C. Anderson, K. Stegmaier, M. Reich, D. Pellman, J. S. Boehm, J. P. Mesirov, T. R. Golub, D. E. Root and W. C. Hahn, Sci Data, 2014, 1, 140035 CrossRef CAS PubMed.
- S. L. Chew, M. Y. Or, C. X. Chang, A. J. Gehring, A. Bertoletti and G. M. Grotenbreg, J. Biol. Chem., 2011, 286(32), 28466–28475 CrossRef CAS PubMed.
- G. Le Duc, S. Roux, A. Paruta-Tuarez, S. Dufort, E. Brauer, A. Marais, C. Truillet, L. Sancey, P. Perriat, F. Lux and O. Tillement, Cancer Nanotechnol., 2014, 5(1), 4 CrossRef PubMed.
- A. Bianchi, S. Dufort, F. Lux, A. Courtois, O. Tillement, J. L. Coll and Y. Cremillieux, MAGMA, 2014, 27(4), 303–316 CrossRef CAS PubMed.
- A. Bianchi, S. Dufort, F. Lux, P. Y. Fortin, N. Tassali, O. Tillement, J. L. Coll and Y. Cremillieux, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(25), 9247–9252 CrossRef CAS PubMed.
- L. Sancey, S. Kotb, C. Truillet, F. Appaix, A. Marais, E. Thomas, B. van der Sanden, J. P. Klein, B. Laurent, M. Cottier, R. Antoine, P. Dugourd, G. Panczer, F. Lux, P. Perriat, V. Motto-Ros and O. Tillement, ACS Nano, 2015, 9(3), 2477–2488 CrossRef CAS PubMed.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. Itty Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25(10), 1165–1170 CrossRef CAS PubMed.
- B. A. Walker, C. P. Wardell, A. Brioli, E. Boyle, M. F. Kaiser, D. B. Begum, N. B. Dahir, D. C. Johnson, F. M. Ross, F. E. Davies and G. J. Morgan, Blood Cancer J., 2014, 4, e191 CrossRef CAS PubMed.
- G. T. Tietjen, S. A. Hosgood, J. DiRito, J. Cui, D. Deep, E. Song, J. R. Kraehling, A. S. Piotrowski-Daspit, N. C. Kirkiles-Smith, R. Al-Lamki, S. Thiru, J. A. Bradley, K. Saeb-Parsy, J. R. Bradley, M. L. Nicholson, W. M. Saltzman and J. S. Pober, Sci. Transl. Med., 2017, 9(418), eaam6764 CrossRef PubMed.
- D. Shahbazi-Gahrouei, J. Res. Med. Sci., 2009, 14(3), 141–147 CAS.
- C. Curtet, C. Bourgoin, J. Bohy, J. C. Saccavini, P. Thedrez, S. Akoka, C. Tellier and J. F. Chatal, Int. J. Cancer, Suppl., 1988, 2, 126–132 CrossRef CAS.
- B. J. Barrett and P. S. Parfrey, N. Engl. J. Med., 2006, 354(4), 379–386 CrossRef CAS PubMed.
- H. V. Nguyen, A. Detappe, N. M. Gallagher, H. Zhang, P. Harvey, C. Yan, C. Mathieu, M. R. Golder, Y. Jiang, M. F. Ottaviani, A. Jasanoff, A. Rajca, I. Ghobrial, P. P. Ghoroghchian and J. A. Johnson, ACS Nano, 2018, 12(11), 11343–11354 CrossRef CAS PubMed.
- S. A. Ali, V. Shi, I. Maric, M. Wang, D. F. Stroncek, J. J. Rose, J. N. Brudno, M. Stetler-Stevenson, S. A. Feldman, B. G. Hansen, V. S. Fellowes, F. T. Hakim, R. E. Gress and J. N. Kochenderfer, Blood, 2016, 128(13), 1688–1700 CrossRef CAS PubMed.
- N. W. van de Donk, P. Moreau, T. Plesner, A. Palumbo, F. Gay, J. P. Laubach, F. Malavasi, H. Avet-Loiseau, M. V. Mateos, P. Sonneveld, H. M. Lokhorst and P. G. Richardson, Blood, 2016, 127(6), 681–695 CrossRef CAS PubMed.
- O. W. Press, J. F. Eary, F. R. Appelbaum, P. J. Martin, C. C. Badger, W. B. Nelp, S. Glenn, G. Butchko, D. Fisher and B. Porter, et al. , N. Engl. J. Med., 1993, 329(17), 1219–1224 CrossRef CAS PubMed.
- W. D. Hedrich, T. E. Fandy, H. M. Ashour, H. Wang and H. E. Hassan, Clin. Pharmacokinet., 2018, 57(6), 687–703 CrossRef CAS PubMed.
- N. Lee, S. Y. Moon, J. H. Lee, H. K. Park, S. Y. Kong, S. M. Bang, J. H. Lee, S. S. Yoon and D. S. Lee, Blood Cancer J., 2017, 7(2), e530 CrossRef CAS PubMed.
- G. Siravegna, S. Marsoni, S. Siena and A. Bardelli, Nat. Rev. Clin Oncol., 2017, 14(9), 531–548 CrossRef CAS PubMed.
- A. Detappe, S. Kunjachan, P. Drane, S. Kotb, M. Myronakis, D. E. Biancur, T. Ireland, M. Wagar, F. Lux, O. Tillement and R. Berbeco, Sci. Rep., 2016, 6, 34040 CrossRef CAS PubMed.
- A. Detappe, S. Kunjachan, J. Rottmann, J. Robar, P. Tsiamas, H. Korideck, O. Tillement and R. Berbeco, Cancer Nanotechnol., 2015, 6(1), 4 CrossRef PubMed.
- A. Detappe, E. Thomas, M. W. Tibbitt, S. Kunjachan, O. Zavidij, N. Parnandi, E. Reznichenko, F. Lux, O. Tillement and R. Berbeco, Nano Lett., 2017, 17(3), 1733–1740 CrossRef CAS PubMed.
- C. Schmidt and C. V. Robinson, Nat. Protoc., 2014, 9(9), 2224–2236 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S15. See DOI: 10.1039/c9nr06512a |
| This journal is © The Royal Society of Chemistry 2019 |