
The application of nanotechnology in enhancing immunotherapy for cancer treatment: current effects and perspective
Yongjiang
Li†
 *ab,
Ciceron
Ayala-Orozco†
b,
Pradipta Ranjan
Rauta
b and
Sunil
Krishnan
*bc
*ab,
Ciceron
Ayala-Orozco†
b,
Pradipta Ranjan
Rauta
b and
Sunil
Krishnan
*bc
aDepartment of Medical Oncology, Cancer Center, and State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Chengdu 610041, China. E-mail: yongjiangcarl@yeah.net; yli56@mdanderson.org
bDepartment of Experimental Radiation Oncology, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA. E-mail: skrishnan@mdanderson.org
cRadiation Oncology, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA
First published on 22nd August 2019
Abstract
Cancer immunotherapy is emerging as a promising treatment modality that suppresses and eliminates tumors by re-activating and maintaining the tumor-immune cycle, and further enhancing the body's anti-tumor immune response. Despite the impressive therapeutic potential of immunotherapy approaches such as immune checkpoint inhibitors and tumor vaccines in pre-clinical and clinical applications, the effective response is limited by insufficient accumulation in tumor tissues and severe side-effects. Recent years have witnessed the rise of nanotechnology as a solution to improve these technical weaknesses due to its inherent biophysical properties and multifunctional modifying potential. In this review, we summarized and discussed the current status of nanoparticle-enhanced cancer immunotherapy strategies, including intensified delivery of tumor vaccines and immune adjuvants, immune checkpoint inhibitor vehicles, targeting capacity to tumor-draining lymph nodes and immune cells, triggered releasing and regulating specific tumor microenvironments, and adoptive cell therapy enhancement effects.
 Yongjiang Li | Yongjiang Li received his MD degree in Oncology after graduation from West China Medical School of Sichuan University in 2016. He is pursuing his Ph.D. degree in Oncology at West China Hospital, Sichuan University, under the supervision of Professor Cheng Yi. Currently, he is studying as a joint doctoral student in Dr Krishnan's lab in the Department of Experimental Radiation Oncology and the Department of Radiation Oncology at the University of Texas MD Anderson Cancer Center. His research interest is focused on the investigation of nanotechnology strategies to improve efficacy and minimize toxicity of radiation therapy. |
 Ciceron Ayala-Orozco | Ciceron Ayala-Orozco received his PhD in Chemistry from Rice University under the supervision of Professor Naomi J. Halas. His PhD work focused on the chemical synthesis of metallic multilayered nanoparticles with plasmonic optical properties tailored to nanomedicine applications in optical imaging of cancer and photothermal cancer therapy. Other areas of experience include the enhanced delivery of nanoparticles to tumor-sites, light-dependent activation of therapeutic action, cancer theranostics through the combination of diagnostics and therapeutics in MRI-guided and optical-imaging-guided photothermal cancer therapy. Currently he holds the NIH postdoctoral fellowship T32 in Cancer Nanotechnology working at the University of Texas MD Anderson Cancer Center under the mentorship of Professor Sunil Krishnan. His current research focuses on the light-activated molecular machines that kill cancer cells through nanomechanical action. |
 Pradipta Ranjan Rauta | Pradipta Ranjan Rauta is a Postdoctoral Research Fellow at the Center for Radiation Oncology Research, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA. He received his PhD degree in Life Science from National Institute of Technology, Rourkela, Odisha, India, MSc. in Microbiology from Orissa University of Agriculture and Technology, Bhubaneswar, Odisha, India and BSc in Zoology from Utkal University, Bhubaneswar, Orissa, India. His research interests include developments of nanoscale drug delivery systems, multifunctional nanotheranostics for advanced cancer and biomaterials for tissue engineering. |
 Sunil Krishnan | Sunil Krishnan is the Director of the Center for Radiation Oncology Research and the John E. and Dorothy J. Harris Professor of Gastrointestinal Cancer in the department of Radiation Oncology at MD Anderson Cancer Center. He received his medical degree from Christian Medical College, Vellore, India and completed a radiation oncology residency at Mayo Clinic, Rochester, MN. His laboratory focuses on the advancement of clinically relevant translational applications for nanoparticles in imaging/image-guided radiotherapy. He has co-authored over 200 scientific publications, 17 book chapters, and 3 books. |
1. Introduction
1.1. Basics of immunotherapy and the tumor microenvironment
The immune system acts as the “police force” to protect the organism from foreign invaders including bacteria, germs, viruses and parasites. The recognition and eradication of the foreign invaders rely on precise cooperation of two components of the immune system: the innate and adaptive immunity. The innate immunity acts as the first-line of defense through rapid activation of granulocytes (neutrophils, basophils, eosinophils and mastocytes) and phagocytes (macrophagocytes and dendritic cells), which recognize general molecular patterns on pathogens or danger signals derived from inflammation, infection and tissue damage.1,2 The adaptive immunity functions by recognizing specific antigens (specificity) instead of the general molecular patterns, and it is capable of responding rapidly to pathogens or antigens it has encountered before (memory).3 These characteristics are implemented by random rearrangement of massive sequences in gene cassettes encoding the antigen recognition receptor complex that is located on the surface of lymphocytes. Huge variations of T-cell and B-cell receptors (TCR, BCR) for antigens with theoretically limitless specificities of T-cells and B-cells can be generated by this random rearrangement. The immune system launches a series of essential activities after the initial encounter with pathogens, which include rapid recruitment of non-antigen-specific innate immune cells, activation of humoral immune responses, pathogen processing and presentation by antigen presenting cells (APCs) to T-cell and B-cell lymphocytes, followed by a more time-consuming and elaborate activation and expansion of antigen-specific lymphocytes, and finally lead to clearance of pathogens.4,5 The sequential implementation of the innate and adaptive immunity could also leave pools of longeval memory lymphocytes that are specific to the invading antigen that can respond much quicker at the subsequent encounters with the same pathogen.Cancer can be regarded as a genetic disease that involves genes with key roles in cell proliferation and differentiation, which gives the cells ability to proliferate out of control and invade regional or distant regions.6 The immune system is also responsible for recognizing and destroying the aberrant cells, and prevents the occurrence or development of cancer, which requires the immune cells to have the ability to effectively recognize cancer versus normal cells.7,8 Mature T-lymphocytes and B-lymphocytes are actually the remaining cells that have passed the negative clonal selection during their development in thymus and bone marrow, in which lymphocytes expressing high affinity antigen receptors to self-antigens are eliminated. Although the negative clonal selection process reduces the harm of autoimmunity to the body, it also eliminates the cell clones having the specificity that could potentially target and resist cancer cells. As a result, the minority proteins encoded by chromosomal aberrations or synthesized through altered post-translational modifications that are exclusively to tumor cells, also named tumor-associated antigens (TAAs), have become the crucial subject for immune cells to recognize cancer cells.9,10 In some cases, the immunogenic TAAs could be normal self-proteins that are not expressed in differentiated cells or only expressed at a low level, but expressed or overexpressed in cancer cells.
Rapid growth of tumors is often accompanied by vast tumor cell death due to hypoxia or accumulation of gene mutation, which will lead to the release of damage-associated molecular patterns (DAMPs) and TAAs.11 The DAMPs can stimulate and recruit phagocytes and APCs, which process and present TAA-derived peptides to tumor-specific lymphocytes through the major histocompatibility complex (MHC). The activated CD8+ cytotoxic T-lymphocytes specifically recognize and eliminate the cancer cells through TCR-mediated recognition of TAA epitopes displayed by tumor MHC-I molecules or through the combination of the NKG2D receptor on CD8+ cytotoxic T-lymphocytes and NKG2D ligands expressed on tumor cells. Activated CD4+ T-lymphocytes generate cytokines including tumor necrosis factor (TNF)-α and interferon-γ that are able to inhibit tumor growth while also upregulating MHC-I expression on tumor cells, thus assisting the target recognition by TAA-specific CD-8+ T-lymphocytes12 (Fig. 1).
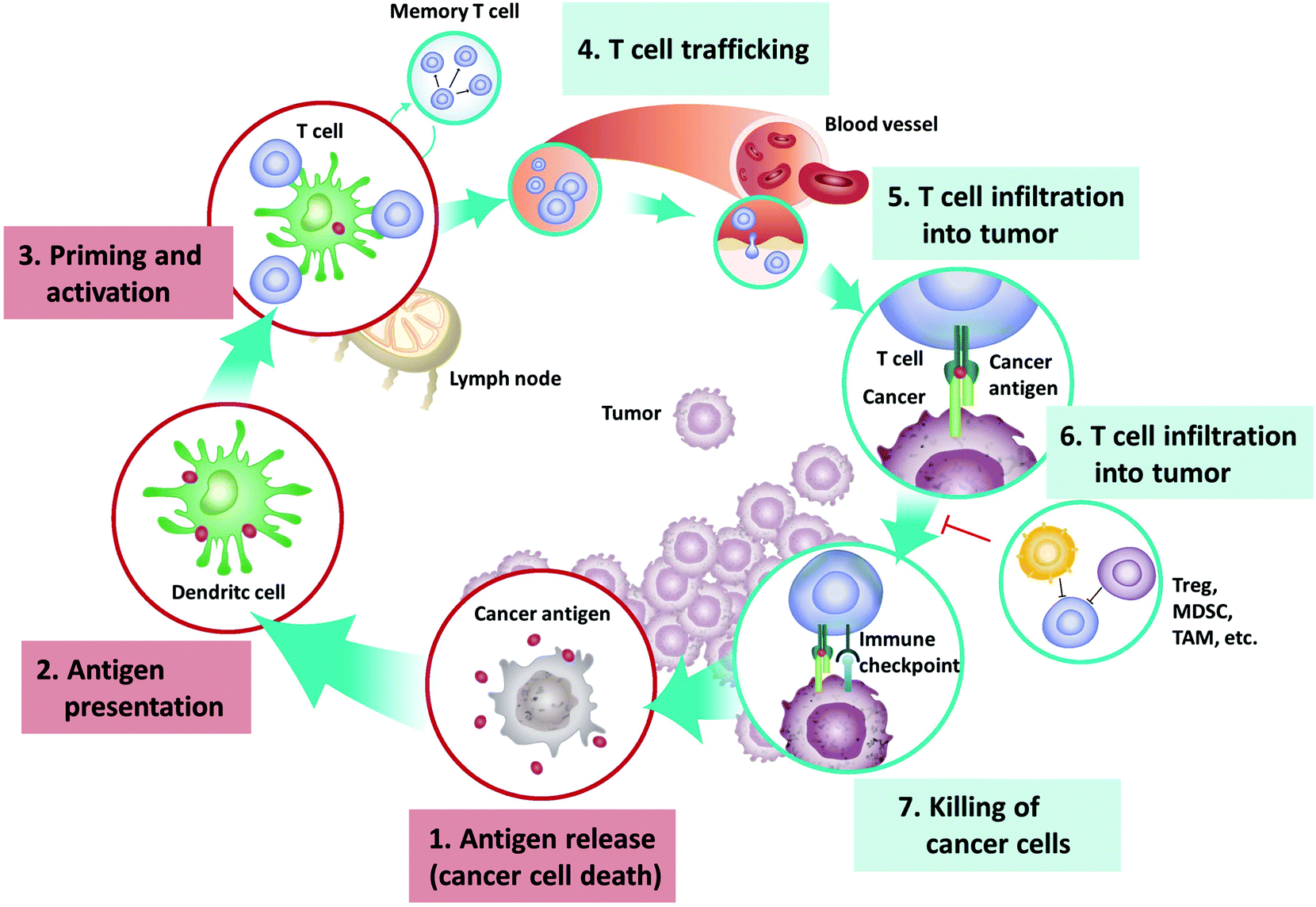 | ||
| Fig. 1 The cancer-immunity cycle that involves exposure or release of tumor antigens, tumor antigen processing and presentation by APCs, priming and activation of effective immune cells and formation of memory cells, trafficking and infiltration of T cells to tumor tissues, and the recognition and killing of tumor cells. Reprinted from ref. 10, Copyright (2019), with permission from Elsevier. | ||
Immune system activation towards cancer cells leads to a competition wherein immune cells inhibit fast cellular proliferation and mutation fosters tumor growth, but the formation of a detectable tumor is often the result of the failure of body immunity to tumor cells. If the immunity could not eradicate all the tumor cells at the beginning, it usually forms a selective force to tumor cell clones that can alter the composition of tumors, and gradually leave and promote the growth of tumor cell clones with the least immunogenicity. Ultimately, the immunoediting effect often gives rise to the most invasive and uncontrolled tumors formed by the most insensitive cells.13 Tumor cells have several mechanisms that assist in evading from being detected and eliminated by the immune system:
(1) Downregulation of MHC-I on tumor cells as a consequence of mutations of MHC genes or molecules involved in TAA antigen processing and presentation can reduce immunogenicity of tumor cells and evade cytotoxic T-lymphocyte mediated lysis.14
(2) Tumor cells could suppress immunity by expressing programmed death-ligand 1 (PD-L1) that binds to the negative co-stimulator programmed death (PD)-1 on lymphocyte's membrane, or by secreting immunosuppression cytokines that suppress the activation and differentiation of APCs and lymphocytes, such as transforming growth factor (TGF)-β.15,16
(3) Tumor cells could resist cytotoxic T-lymphocyte mediated killing effects by expressing granzyme serine proteases to inhibit the perforin/granzyme pathway that mediates tumor cell lysis, and decoy receptors for cell death including soluble Fas, osteoprotegerin and decoy receptor-3, 4. In addition, increased expression of oncogenic and antiapoptotic molecules such as the signal transducer and activator of transcription 3 and B-cell lymphoma 2 (Bcl2) also resists T-lymphocyte mediated lysis.17
(4) Immunoregulatory cells with immunosuppressive effects, such as myeloid-derived suppressor cells and M2 type tumor-associated macrophages, could be recruited by tumor cells in the tumor microenvironment, which could produce immunosuppressive cytokines, and also secrete the vascular endothelial growth factor and matrix metalloproteinases that favor tumor angiogenesis for tumor growth and invasion.18,19
(5) A physical barrier around the tumor could be generated by tumor cells through secreting a series of molecules such as collagen, thus forming an immune privilege area and preventing the lymphocytes and APCs from entering the tumor area.20
1.2. Introduction of nanotechnology in cancer immunotherapy
On the basis of these mechanisms, numerous novel molecular drugs aiming to enhance immunity against tumor cells have been rising in the past decade, and exhibited considerable progress in both lab experiments and clinical treatments.21–23 However, only a limited portion of the immunotherapeutic molecules could encounter and interact with the targets, as the majority will be eliminated by renal clearance, impeded by metabolism during the blood circulation or cannot penetrate biological barriers and successfully reach the target site.24,25 Under the circumstance, nanoparticles are utilized as suitable vehicles for immunotherapeutic molecules to counteract the pharmacokinetic shortages. Due to their inherent properties, nanoparticles could passively enrich within tumor tissue because of the immature tumor vasculature and damaged lymphatic drainage, which is also known as the enhanced permeation and retention (EPR) effect.26 The first generation nanoparticles principally depended on the EPR effect to enrich in tumor tissues, but the treatment effects varied among different types of cancer mainly because of variances in the tumor vascular structure and permeability.27 Similarly, effects of the passive enriched nanoparticles on metastatic tumor lesions were often insufficient also due to the different vascular beds of metastatic lesions.28 To overcome this limitation, positive target ability is equipped through modification of the nanoparticles with tumor-specific antibodies, and cellular uptake is also intensified, thus enhancing treatment efficacy and lowering off-target side-effects.29Appropriate selection and construction of the nanoparticle structure in the aspects of size, shape, coated ligands, loading method, zeta potential, hydrophilicity, elasticity and biocompatibility lead to suitable nano-vehicles for immunotherapeutic molecules, which generally ought to have a prolonged biological half-life period, protect payloads during circulation, penetrate through barriers or have target effects, controllably release payloads under specific microenvironments, and have low biotoxicity. Nanoparticles less than 5 nm in diameter tend to be excreted through renal filtration, and nanoparticles with diameter larger than 200 nm could be rapidly eliminated in spleen due to the size of inter-endothelial slits;30 thus the diameter in the range from 5 to 200 nm is the optimal size for nanoparticles. The shape of nanoparticles should also be elaborated as different shapes process different characteristics. For instance, worm-like nanoparticles exhibit preferable fluid dynamics compared with rod-, fingerprint-shaped or spherical nanoparticles;31 spherical nanoparticles are less likely to be accumulated in spleen, but flat cylindrical nanoparticles had the most retention in the liver, lung and spleen organs.32 Nanoparticles with slight negative charge had significantly lower retention in organs and had a prolonged circulation half-lifetime, but positive surface charge could assist the adherence to the anionic cellular membrane and is more advantageous in inducing cellular internalization.33,34 Structural stability in blood circulation is another critical aspect, which is needed to exercise caution especially for nanoparticles like polymeric micelles, as they are often not able to maintain the structural integrity well in the rapid bloodstream and will cause declines in delivery efficiency.35 Special designs including intensified structure stability or the environment-triggered releasing strategy are needed for these nanoparticles to achieve preferable transportation ability. Collectively, appropriate design of nanoparticles in accordance with the experimental purposes facilitates in optimizing the delivery efficiency and therapeutic effects of payloads.
Nanoparticles have been widely researched to be incorporated into tumor immunotherapy in recent years based on their inherent biophysical properties and multitudinous modifying potential, with tremendous progress made in preclinical and clinical trials. However, there are few reviews summarizing the advances in the fast-updating research area. In this review, we summarized and discussed the current status of nanoparticle-enhanced cancer immunotherapy strategies in the aspects of intensified delivery of tumor vaccines and immune adjuvants, ICI vehicles, targeting capacity to tumor-draining lymph nodes and immune cells, triggered releasing and regulating specific tumor microenvironments, and adoptive cell therapy enhancement effects.
2. Delivery of tumor vaccines by nanoparticles for tumor immunotherapy
Immune escape is often caused by the loss of valid TAA processing and presentation by immune cells; hence the additional provision of specific tumor antigens to immune cells could stimulate and enhance the anti-tumor immune process.36,37 Traditionally, most of the antigens are purified cytomembrane proteins, peptides, polysaccharides, or the DNA or RNA encoding these tumor antigens, which have medium reactogenicity but generally weak immunogenicity due to insufficient exogenous immune-provoking components. The emergence of whole-cell antitumor vaccines not only imported multiple tumor-specific antigens but also enhanced the antitumor immunoreaction. Accompanied by advanced delivery methods of nanoparticles that could precisely load the antitumor vaccines to targeted lymph nodes or immunocytes, the effects have been amplified in recent years.Table 1 summarizes the basic characteristics of recent studies focusing on the antitumor vaccine delivery by nanoparticles, and the therapeutic effects are listed in Table 2.38–51 By the form of payloads, it could be classified into three categories: tumor specific antigen delivery, immune bio-adjuvant delivery and co-delivery of antigens and adjuvants. In order to generate antitumor immunity in experiments, ovalbumin (OVA) was mostly selected as the model tumor antigen due to its exogeneity, easy feasibility and suitable immunogenicity. OVA was conjugated as a payload to various types of nanoparticles, including silica nanoparticles,38 liposomes,42,47,49 gold nanoparticles,41 alginate nanoparticles43 and polymeric nanoparticles,46 and injected into mice bearing OVA-expressing melanoma, lymphoma or thymoma-bearing animal models. Compared with single delivery of antigens, conjugated delivery of antigen–nanoparticle compounds induced significantly stronger antigen-specific antibody responses and CD8+ cytotoxic T lymphocyte responses accompanied by increased levels of antigen-specific IgG, interferon-γ and specific types of interleukins, inhibiting tumor growth and prolonging survival of mice (Table 2). On the other hand, delivery of multiple antigens instead of only single OVA may further enhance the anti-tumor immunoreaction. Zhang et al. conjugated two tumor specific peptides and one immunoadjuvant with layered double hydroxide nanoparticles and found a significantly stronger inhibition effect to melanoma compared with single antigen-delivering nanoparticles.40
| Studies | Nanoparticles | Size | Shape | Balanced surface charge | Payload | Experimental subject | Tumor model |
|---|---|---|---|---|---|---|---|
| Abbreviations: CpG, cytosine phosphorothioste-guanine; DC, dendritic cell; LCP, lipid–calcium–phosphate; MPLA, monophosphoryl lipid A; ODN, oligodeoxynucleotide; PLGA, poly(lactic-co-glycolic acid); PLP, proteolipid peptides; PTX, paclitaxel; TMC, trimethylchitosan; TLR, toll-like receptor; TRX, 3,5-didodecyloxybenzamidine. | |||||||
| Lu et al.38 | Organosilica NP | 200 nm | Sphere | Not given | Encapsulated: OVA and TLR agonist (CpG) | C57BL/6 mice | Murine B16-OVA melanoma |
| Yan et al.39 | Layered double hydroxide NP and nanosheet | 109.2 nm (NP), 176.8 nm (nanosheet) | Flat cylindrical and sheet | Not given | Coated: OVA and CpG | C57BL/6 mice | Murine E.G7-OVA T-lymphoma |
| Zhang et al.40 | Layered double hydroxide NP | 108.4 nm | Flat cylindrical | +39.0 mV | Coated: neo-epitopes and CpG | C57BL/6 mice | Murine B16F10 melanoma |
| Yata et al.41 | Gold NP | 50 nm | Not given | Not given | Coated: CpG DNA | C57BL/6 mice | Murine E.G7-OVA T-lymphoma |
| Thomas et al.42 | Liposome | 81–103/121–160 nm | Sphere | −6 to −8/+4–+6 mV | Coated: OVA | BALB/c, C57BL/6 mice | N/A |
| Zhang et al. (2)43 | Alginate NP | 310 nm | Sphere | −45.6 mV | Coated: OVA | BALB/c, C57BL/6 mice | Murine E.G7-OVA T-lymphoma |
| Sokolova et al.44 | LCP | 275 nm | Sphere | +20 mV | Coated: TRL ligand | BALB/c, C57BL/6 mice | N/A |
| Liu et al.45 | LCP | 58 nm | Sphere | +38 mV | Encapsulated: anti-CTLA-4 antibody and mRNA encoding tumor antigen MUC | BALB/c mice | Murine TNBC 4 T1 mammary carcinoma |
| Pearson et al.46 | PLGA | 72.2–656 nm | Sphere | −56.0 to −31.5 mV | Coated/encapsulated: OVA and PLP | SJL/J and C57BL/6 mice | N/A |
| Verbeke et al.47 | Cationic liposome | 160 nm | Sphere | +50 mV | Encapsulated: MPLA and mRNA encoding OVA | C57BL/6 mice | N/A |
| Takahashi et al.48 | Carbonate apatite NP | 30–40 nm | Sphere | Not given | Encapsulated: CpG and ODN | C57BL/6 mice | N/A |
| Yoshizaki et al.49 | Cationic liposome | 88–110 nm | Sphere | −65 to −60/−11 to −19 mV | Coated: TRX, CpG DNA; Encapsulated: OVA, CpG DNA | C57BL/6 mice | Murine E.G7-OVA T-lymphoma |
| Liang et al.50 | Liposome | 80–100 nm | Sphere | Not given | Encapsulated: mRNA encoding hemagglutinin | Not given | N/A |
| Xu et al.51 | TMC complex | 150 nm | Sphere | +28 mV | Encapsulated: PTX and DNA encoding (GM-CSF)-Fc | BALB/c and C57BL/6 mice | HeLa and B16 melanoma cells |
| Studies | Therapeutic effects |
|---|---|
| Abbreviations: APC, antigen presenting cell; CTL, cytotoxic T lymphocytes; DC, dendritic cell; LDH, layered double hydroxide; NPs, nanoparticles; OVA, ovalbumin; Tregs, regulatory T cells. | |
| Lu et al.38 | • APC's cellular uptake of both OVA and CpG was significantly enhanced in organosilica NP-encapsulated form than the free form |
| • OVA- and CpG-organosilica NP induced cellular endosome escape for antigen cross-presentation | |
| • OVA- and CpG-organosilica NP decreased the GSH level and increased the ROS level in vitro and in vivo, facilitating CTL proliferation, reducing tumor growth and prolonged survival in the melanoma mice model | |
| Yan et al.39 | • Nanomaterials induced much stronger humoral and cell-medicated immune responses, evidenced by higher levels of IgG1, IgG2a and interferon-γ |
| • LDH nanosheets showed higher activity to promote specific antibody response than LDH NPs but with a similar cell mediated immune response | |
| • Nanomaterials inhibited tumor growth and prolonged survival in vivo | |
| Zhang et al.40 | • LDH-NP induced significantly higher CTL activity and inhibited melanoma growth in vivo |
| Yata et al.41 | • Gold NPs increased tumor-associated antigen-specific IgG and interferon-γ expression in vivo |
| • Gold NPs significantly delayed tumor growth and extended the survival of mice | |
| Thomas et al.42 | • Mice immunized with OVA delivered by NPs produced much higher antibody titers than those without NPs |
| • 80 nm anionic nanoliposome is the most efficient type in eliciting a higher humoral and cellular immune response | |
| Zhang et al. (2)43 | • Alginate NPs facilitated OVA uptake and its cytosolic release in DC, and also enhanced the expression of surface co-stimulatory molecules of the DC |
| • Alginate NPs enhanced in vivo trafficking of OVA to draining lymph nodes and strengthened cross presentation of OVA to T cell hybridoma | |
| • Alginate NPs induced a major CTL response and the inhibition of tumor growth in vivo | |
| Sokolova et al.44 | • NP with Anti-146 could target the liver by intravenous injection |
| • The expression of IFN-α/β, TNF-α, IL-6 and IP-10 in hepatocytes, NPCs and LSECs was significantly increased by NP | |
| Liu et al.45 | • Nanoliposome targeted to mannose receptors on DCs, led to tumor antigen expression and induced a strong, antigen-specific CTL response against tumor cells |
| • Co-encapsulated anti-CTLA-4 antibody significantly enhanced the anti-tumor immune response compared to the mRNA alone-NPs | |
| Pearson et al.46 | • Immune cells’ cellular interaction induced by the 80 nm PLGA NP was significantly lower than the 400 nm PLGA NP |
| • CD25 expression on T cells was particle size independent | |
| • Induction of Tregs was particle size and concentration dependent | |
| Verbeke et al.47 | • Liposome induced high antigen expression and effective antigen-specific T cell immunity in vivo without provoking a type I IFN response |
| Takahashi et al.48 | • CA nanoparticles containing CpG ODNs induced significant IFN-α production by mouse dendritic cells and human peripheral blood mononuclear cells in vitro |
| • NP enhanced the production of interleukin-12 and IFN-γ | |
| • Treatment with NP resulted in higher cytokine production in draining lymph nodes, and induced higher antigen-specific antibody responses and CD8+ cytotoxic T lymphocyte responses in vivo in an interleukin-12- and type-I IFN-dependent manner | |
| Yoshizaki et al.49 | • Liposome promoted cytokine production from DCs and expression of co-stimulatory molecules in vitro |
| • Liposome induced antigen-specific immune responses, inhibited tumor growth and prolonged survival in vivo | |
| Liang et al.50 | • Nanoliposome induced rapid and local infiltration of neutrophils, monocytes and DCs to the site of administration and draining lymph nodes |
| • APCs efficiently internalized nanoliposomes, translated the encapsulated mRNA and upregulated key co-stimulatory receptors including CD80 and CD86 | |
| • Nanoliposome resulted in priming of H10-specific CD4+ T cells exclusively in draining lymph nodes, and induced type-1 IFN-polarized innate immune response | |
| Xu et al.51 | • Nano-complex enhanced intracellular uptake of DCs, upregulated the costimulatory molecules of CD80 and CD86, promoted the proliferation of CD4+ and CD8+ T cells, and reduced the generation of immunosuppressive FoxP3+ Treg cells |
| • Nano-complex has a synergistic effect on the maturation and function of DCs, increased immune stimulation and reduced immune escape, inhibited tumor growth and prolonged survival in vivo | |
Genetic antigens could also be the payload of nanoparticles to stimulate immunity against cancer. After delivered intracellularly, tumor specific antigens or immune adjuvants encoded by the genetic vaccine could be formed through transcription and translation, and continuously stimulate the immune system to generate long-term immunity49 (Fig. 2). They also have the advantages of easy manufacture, preserving, strong cellular immunity stimulation, being cross-immunity inducing and promising immunogenicity. Recent studies showed efficiency of the anti-tumor genetic vaccines in inducing both the antigen-specific cytotoxic T-lymphocyte response, and humoral immune response and tumor suppression effects.41,45,47,49,50 However, their safety concerns should still be noted for the possibility of genomic instability caused by the integration into the genome of the host cell, and the immunologic tolerance of the immune system after long-term overexpression of the encoded antigens.
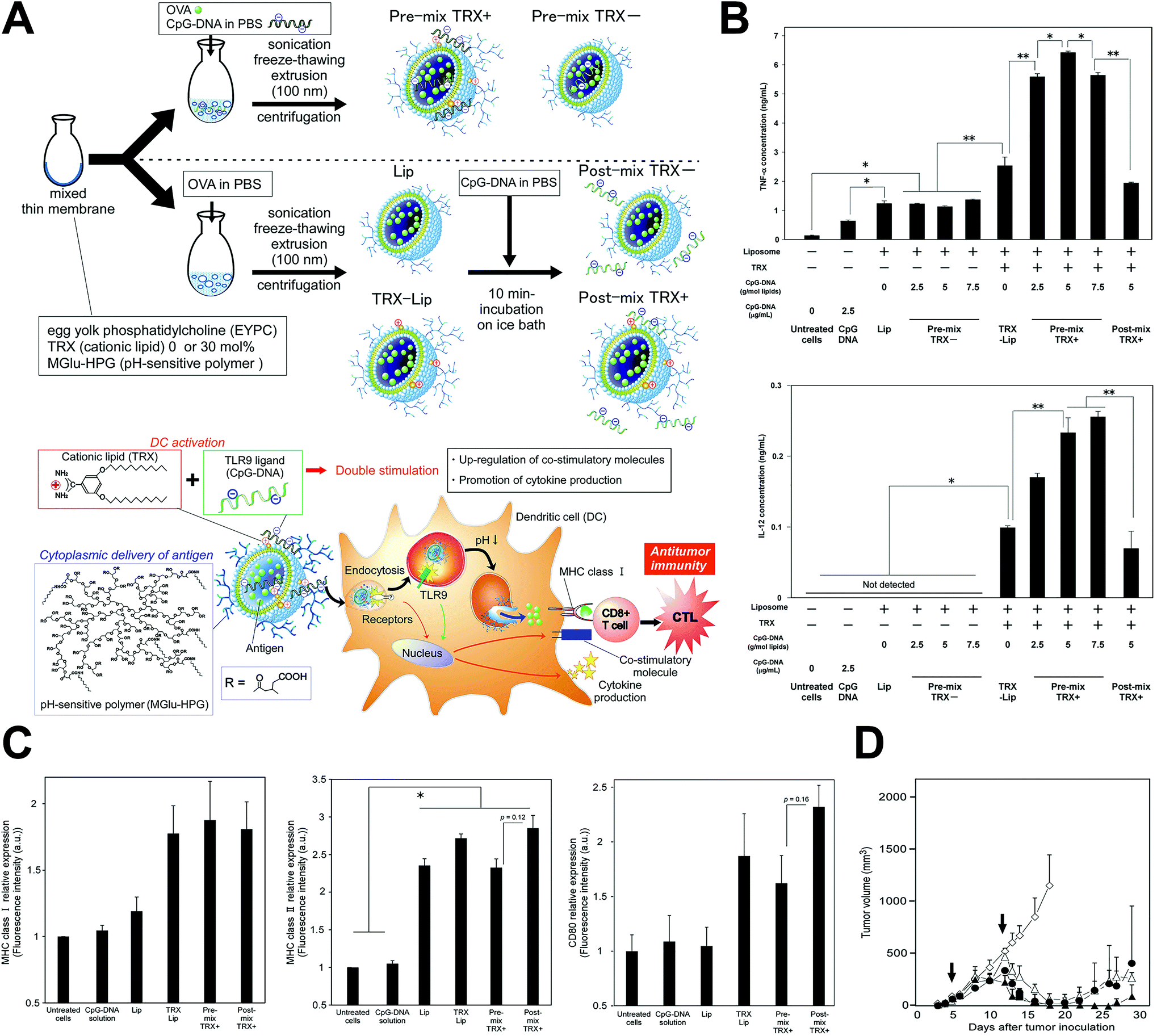 | ||
| Fig. 2 Genetic tumor nanovaccine enhanced immunotherapy. (A) Synthesis process and working mechanism of the nanoliposome. (B) Enhanced secretion of TNF-α and IL-12 after treatment. (C) Increased MHC-I, MHC-II and CD80 expression after treatment. (D) In vivo anti-tumor effects of the genetic liposome nanovaccine. Reprinted from ref. 49, Copyright (2017), with permission from Elsevier. | ||
Bio-adjuvants of cancer vaccine are usually ligands to pattern recognition receptors, including the toll-like receptor (TLR), C-type lectin receptor, NOD-like receptor and RIG-like receptor, which could assist the innate immune response, facilitate polarization of naive T lymphocytes to T helper cells, and accordingly induce the adaptive immunity.52–54 TCR agonists are the most studied immune adjuvant integrated into nanoparticles, and they showed significantly higher immune activation effects than the free soluble form. The co-delivery of adjuvants with antigens within the same nano-system achieved even better results, mainly through mechanisms of enhancing the immunogenicity of weak antigens, reducing the required amount of antigen for effective immune stimulation, and preserving payloads from biolysis. Among the studies, several types of TLR agonists (for example, CpG (TLR9 agonist) and MPLA (TLR4 agonist)) were usually simultaneously conjugated to nanoparticles accompanied by another TAA to realize stronger effects, as multiple TLRs were generally required to be stimulated by pathogens to further increase the levels of inflammatory chemokines and cytokines.55 In addition, the co-delivery of immune bio-adjuvants with immunosuppressive pathway inhibitors could also significantly suppress the proliferation of immunosuppressive cells and synergistically enhance the tumor-specific cellular immune response56 (Fig. 3).
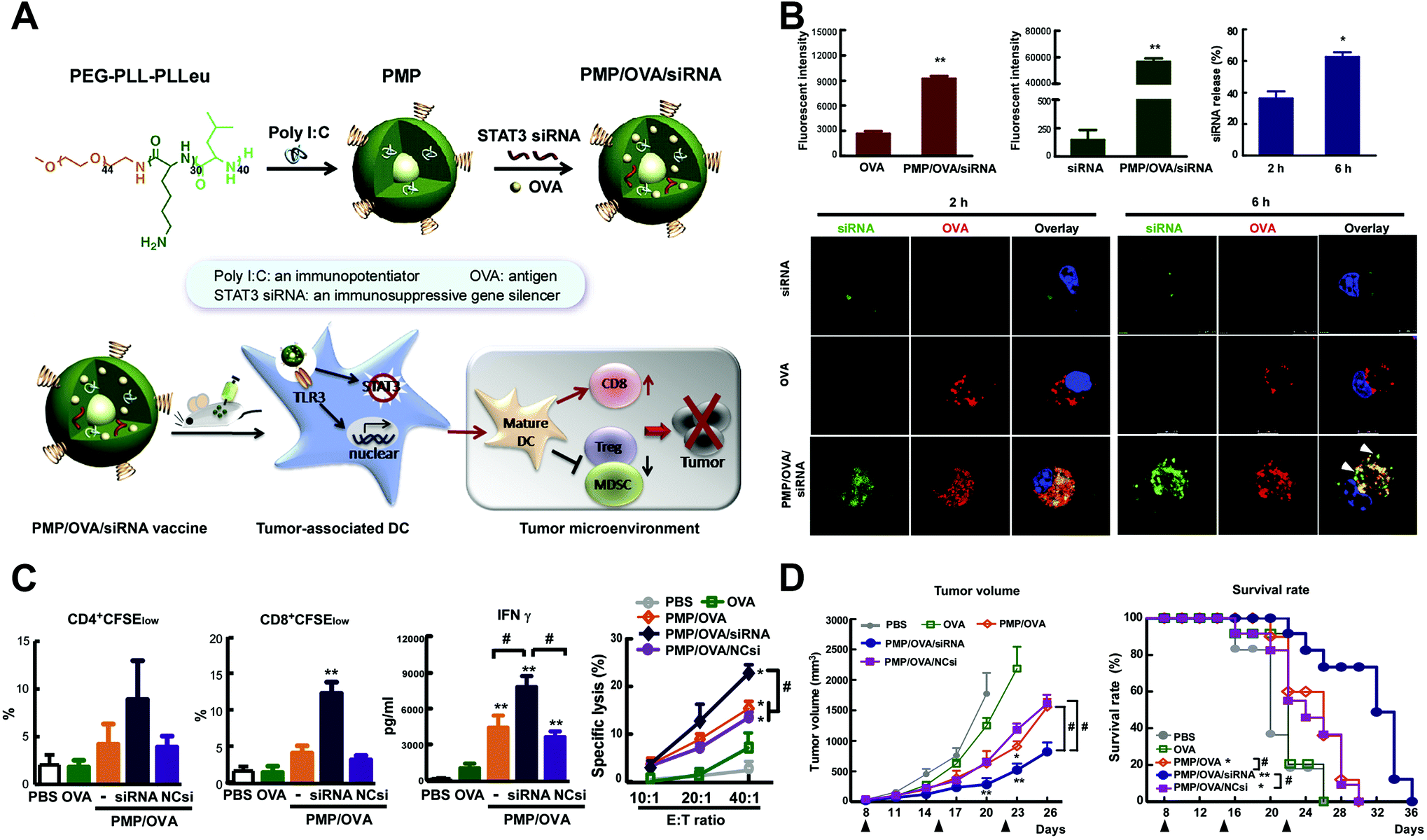 | ||
| Fig. 3 Tumor-specific cellular immune response induced by tumor nanovaccine co-delivering the bio-adjuvant and immunosuppressive pathway inhibitor. (A) Synthesis and working mechanism of the co-delivery nanovaccine. (B) Enhanced cellular uptake and intracellular localization of siRNA and tumor antigen (OVA) in mouse bone marrow dendritic cells. (C) Increased tumor antigen-specific CD4+ and CD8+ T-cell proliferation, IFN-γ production and CTL response by the nanovaccine. (D) The anti-tumor effects of the co-delivery nanovaccine and survival curves of tumor-bearing mice. Reprinted from ref. 56, Copyright (2015), with permission from Elsevier. | ||
The whole cell antitumor vaccine is mainly generated from two sources, whole tumor cell membrane or tumor cell lysates. Different from individual tumor specific antigens, the whole cell antitumor vaccine contains a full range of epitopes and could induce a multivalent immune response.57,58 Several groups fabricated core–shell PLGA nanostructures coated with cancer cell membrane antigens, and formed a robust platform towards multi-antigenic immune responses. For instance, Yang et al. loaded PLGA nanoparticles with a TLR7 agonist and an immunomodulator imiquimod, and then coated the compound with a cancer cell membrane to form an immune-adjuvant whole cell antitumor nanovaccine. In combination with checkpoint blockade therapy, the nanovaccine significantly activated anti-tumor immune cells and exhibited outstanding therapeutic efficacy against established tumor lesions.59 Lysates of resected tumor tissues were also conjugated to nanoparticles with immune adjuvants in other studies to direct immune responses against cancer.60,61 In addition, clay nanoparticles with antigens loaded were found to be able to form subcutaneous nodules with a loose structure at the site of injection, which acted as a depot for sustained antigen release and immune cell recruit, and induced continuous immune stimulation and memory T-cell proliferation for up to 35 days.62 Another strategy to develop a whole cell antitumor vaccine by nanoparticles is to generate the TAAs in situ, which means the generation of tumor cell lysates from damaged or dying cancer cells. It has been proved that the TAAs could be generated in situ after chemotherapy63 or radiotherapy,64 and more importantly, combined with photothermal therapy (PTT) or photodynamic therapy (PDT), specific nanoparticle platforms developed by Chen65 and Xu et al.66 were shown to have the ability to significantly further enhance the therapeutic efficacy of PTT and PDT, and reinforce tumor cell destruction and the subsequent TAA generation in situ, thus inducing strong antitumor immunity to inhibit the development of original and metastatic tumor lesions.
3. Nanoparticles delivering immune checkpoint inhibitors
Immune checkpoint inhibitors have emerged as the hot-spot in tumor immunotherapy recently. PD-1 is one of the most important immune checkpoint molecules and is overexpressed on activated T lymphocytes and has a tight correlation with tumor-associated immune suppression. In a normal situation, combination of PD-1 and PD-L1 can transmit inhibitory signals and reduce the proliferation of CD8+ T cells in lymph nodes, and PD-1 can also control the accumulation of antigen-specific T cells in lymph nodes by regulating the bcl-2 gene, thus protecting normal tissue from attacking under autoimmunity.67–69 However, tumor cells could also escape from immune attack by overexpressing the PD-L1. Similarly, CTLA-4 could induce T-lymphocytes to be non-reactive and negatively regulate the immune response after its incorporation with B7 expressed on activated APCs.70,71 The immune checkpoint inhibitors including anti-CTLA4 and anti-PD-1/PD-L1 antibodies have entered into clinical treatments and showed impressive curative effects; however, the efficacy is still limited by the relatively low rate of the treatment response and high incidence of side-effects. Therefore, nanoparticles are considered for the delivery of these drugs in a targeted and sustainable-releasing pattern, thus reducing the treatment dose and decreasing side-effects.Among the studies developing nanoparticles for the delivery of immune checkpoint inhibitors, although a variance in the nanoparticle types and sizes existed, the conjugation could enhance the immune response against tumors and decrease side effects of immune checkpoint inhibitors,72–78 which is mainly due to the accumulation of nanoparticles and the sustained release of inhibitors in the tumor tissue, as proven by Meir et al. that the amount of inhibitor accumulation within tumor tissues was linearly correlated with the intensity of the immune response against cancer.79 Furthermore, as the anti-PD-L1 and anti-CTLA4 treatments have been proven to have a synergistic treatment effect due to the different mechanisms, Chae et al. constructed a nano-system delivering both the inhibitors, and found a significantly slower tumor growth rate and longer survival of mice in the combined treatment group than either of the individual drug groups.80
In addition, there are studies combining the nanoparticle conjugated-immune checkpoint inhibitors with other treatment methods to further intensify therapeutic effects. Kuai et al. integrated lipid-doxorubicin (DOX) and anti-PD-1 antibody with synthetic high-density lipoprotein (sHDL) to form pH-responsive nanoparticles.81 The prolonged circulation enables adequate intratumoral delivery, followed by internalization by tumor cells and pH-related release of payloads in the endosomes and lysosomes. Released DOX killed tumor cells, and induced immunogenic cell death and release of danger signals, recruiting APCs to phagocytose the immunogenically dying tumor cells. The immune response would be further enhanced accompanied by the anti-PD-1 antibodies, leading to elimination of tumors and prevention of tumor relapse. As a result, the sHDL-DOX + anti-PD-1 nanoparticles significantly induced an antigen-specific cytotoxic T-cell response, suppressed tumor growth and prolonged survival than the free DOX + anti-PD-1 treatment or the single anti-PD-1 treatment (Fig. 4). Similar results were also found by Wang et al. when PD-L1 siRNA coated nanoparticles were integrated with a photosensitizer for photodynamic therapy that the combined nanosystem efficiently stimulated the immune response and silenced immune resistance, inhibiting tumor growth and preventing recurrence.82
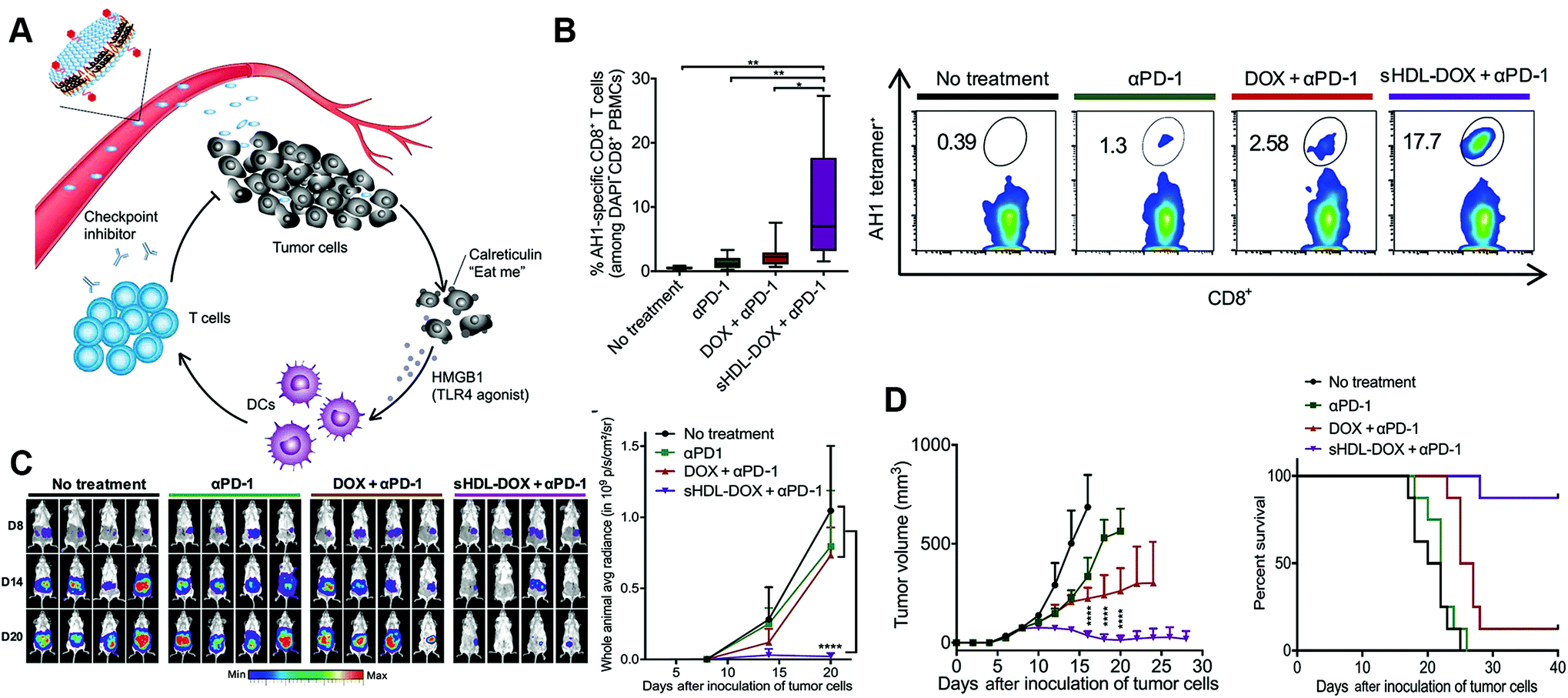 | ||
| Fig. 4 Elimination of tumors by chemoimmunotherapy with immune checkpoint inhibitor-conjugated nanoparticles. (A) Schematic of immune checkpoint inhibitor-combined nanoparticles for chemo-immunotherapy. (B) Percentage of tumor antigen-specific CD8+ T cells after treatment and the corresponding scatterplots. (C) Whole-animal in vivo imaging of tumors after treatment and quantification of the bioluminescence signal. (D) Tumor growth and survival curves after treatment. Reprinted from ref. 81, © The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. Distributed under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC), http://creativecommons.org/licenses/by-nc/4.0/. | ||
Another established negative feedback protein that suppresses the immune response and assists tumor cell proliferation is indoleamine 2,3-dioxygenase (IDO), which mediates the degradation of tryptophan to kynurenine and other metabolites, thus leading to intracellular accumulation, inducing cell cycle arrest and death of effector T lymphocytes, and promoting regulatory T cell proliferation with the immunosuppressive effects.83,84 Sun et al. coated the IDO inhibitor NLG919 and DOX with a redox-responsive immunostimulatory polymeric prodrug carrier. The nanosystem generated greater accumulation of NLG and DOX in tumor tissue than other organs, promoted apoptosis of breast and prostate cancer cells, and formed more significant immunoactivation effects compared with free DOX, liposomal DOX or free NLG groups.85 Other similar studies are also consistent with the reported results.86,87
4. Targeted delivery of nanoparticles to lymph nodes and immune cells
Tumor-draining lymph nodes (TDLNs), which majorly constitute tumor specific T-lymphocytes, B-lymphocytes and APCs, are located along the tumor-draining lymphatic channels.88,89 TDLNs play a crucial role in cellular and humoral immunity against cancer as they are antigen-encountered through drainage of TAAs, but they are often immune-suppressed.90–92 As nanoparticles have the potential for efficient draining and retention in TDLNs because of their size and particular structure which resembles that of pathogens, their potential effects to intensify activation and proliferation of APCs in TDLNs as the immuno-stimulatory agents have been studied by recent experiments. Jeanbart et al. coated both the adjuvant and TAA to form a nanosystem, and delivered it to the TDLN and non-TDLN of tumor-bearing mice.93 They found that the nanosystem delivered to the TDLN induced a substantially stronger cytotoxic CD8+ T-lymphocyte response both locally and systemically than the non-TDLN group, leading to tumor regression and a longer survival period. Nanoparticle coupling of both the adjuvant and antigen is necessary for the effective TDLN stimulation, and this delivering method could dramatically enhance the antigen presentation process of TDLN APCs, thus increasing the therapeutic efficacy. Similar results were also found by Kang et al. that nanoparticles co-delivering tumor membrane antigens, DAMP signal-augmenting element α-helix HSP70 functional peptide (αHSP70p) and CpG exhibited efficient TDLN trafficking, inducing not only the tumor-specific T-lymphocyte response, but also natural killer cells’ immune stimulation and effector memory T cells’ proliferation (Fig. 5).94 The distinction between the metastatic TDLN, the TDLN that has been colonized by metastatic tumor cells, and non-metastatic TDLN is very important for surgical resection and prognosis evaluation, and Cao et al. found that folate receptor-targeted trimodal polymer nanoparticles were efficient for the rapid and precise diagnosis of lymph node metastasis in vivo.95 The folate-functionalized nanoparticles were coated with PFBT, NIR775 and DTPA-BSA (Gd) for the near-infrared, photoacoustic and magnetic resonance imaging. The results showed that the nanoparticles rapidly accumulated in metastatic lymph nodes, which could effectively detect and distinguish metastatic lymph nodes at 1 h post-injection, and had excellent potential in real-time image-guided surgery.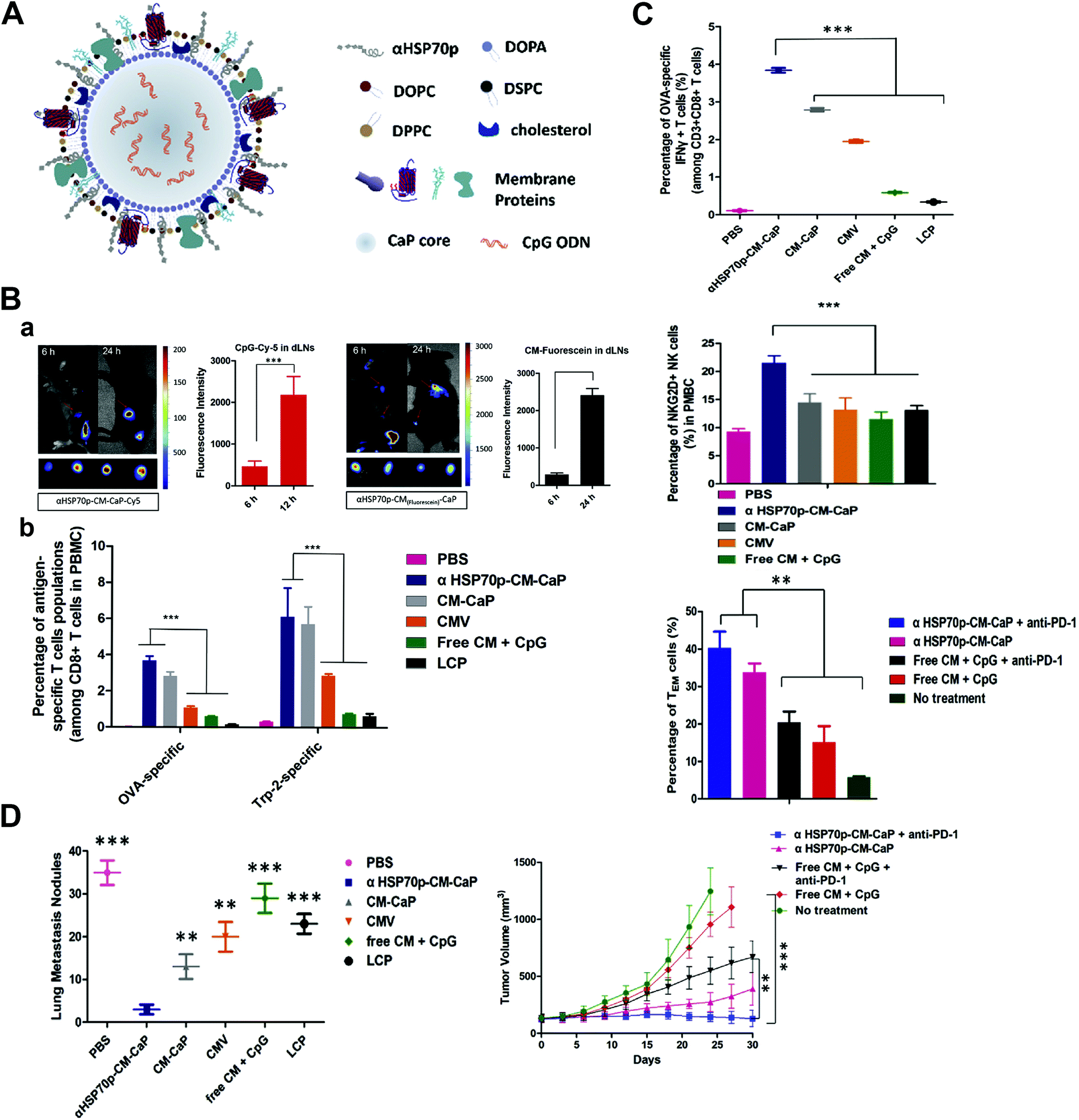 | ||
| Fig. 5 Tumor draining lymph node (TDLN) targeted nanoparticles boost the tumor-specific T-lymphocyte response, natural killer cells’ immune stimulation and effector memory T cells’ proliferation. (A) Structure of the TDLN targeted nanoparticle that enables co-delivery of three elements including tumor cell membrane proteins, adjuvant CpG and an a-helix peptide modified HSP70p. (B) Prolonged antigen delivery in TDLNs and multi-epitope T cells priming in vivo: a. fluorescence showing nanoparticle accumulation in the TDLNs and the corresponding fluorescence intensity; b. quantitative analysis of the frequency of the nanoparticle positive CD8α+ T cells in the TDLNs after treatment. (C) The nanoparticle activated antigen-specific CD8+ T cells, NKG2D+ NK cells, and induced effector memory T cell proliferation in vivo. (D) Elimination of primary and secondary tumors by the nanoparticle. Reprinted from ref. 94, Copyright (2018), with permission from Elsevier. | ||
Besides the TDLN, nanoparticles are also designed to target different types of immune cells, among which the DC is the most targeted cell because of its prominent role in antigen processing and presentation. The DC target is implemented by integrating ligands that combine with the membrane receptors on DCs, including the mannose, CD40, CD11c, DEC205 and DC-SIGN receptors. Studies found that the DC-targeted nanosystem could reinforce the endocytosis of DCs compared with the free form without nanoparticles.96–100 Wang et al. conjugated mannose on the lipid–calcium–phosphate nanoparticles and found improved DC engulfment in the targeting-delivered group.101 As a result, the co-encapsulated PD-L1 siRNA resulted in down-regulation of PD-L1 in DCs that presented tumor antigens, further significantly prompted T cell activation and proliferation, and exhibited profound inhibitory effects on tumor growth and metastasis. Similar results were reported in another study by Liu et al.102 In addition, targeting different molecules on the DC seemed to have different DC stimulation efficacy. Cruz et al. observed a small but significantly enhanced internalization of CD40-targeted nanoparticles in comparison with CD11c or DEC-205 targeted nanoparticles; however, all three targeted methods showed similar capacity to prime cytotoxic T-lymphocytes and subsequently induced tumor cell lysis.103 Different subsets of DCs had diverse specific functions and antigen presentation abilities based on types of immune responses and pathogens; however, no studies have been carried out concerning the immunity stimulation effects achieved by nanoparticles targeting the different subsets of DCs.
T-lymphocytes could also be targeted by the nanoparticles. Schmid et al. targeted CD8+ T-lymphocytes using PD-1 antibody after removing the Fc fragments and combining with PLGA nanoparticles.104 Meanwhile, the inhibitor of TGF-β which assists in immunosuppressive tumor microenvironment maintenance and immune adjuvants TLR7/8 agonists were also coated by the nanoparticle. The PD-1 positive T-lymphocytes were successfully targeted in blood and tumor tissue, and the TGF-β inhibitors and TLR7/8 agonists enhanced the immune responses, sensitized tumors to subsequent anti-PD-1 treatment and raised the proportion of tumor-infiltrating CD8+ T-lymphocytes, converting a “cold” tumor into a “hot” tumor, and extended the survival of mice in comparison with non-targeting compounds. In addition, they also found that by loading the drugs into targeted particles, the dose required decreased to less than one-tenth of the standard dose, thus mitigating potential toxicity.
He et al. developed a macrophage dual-targeted CpG-loading nanoparticle realized by mannosylated carboxymethyl chitosan (MCMC) and hyaluronan (HA), which resulted in a considerable shift of macrophages to activated M1 types and a significantly enhanced secretion of proinflammotory cytokines.105 Besides, the nanosystem also activated NF-κB and phosphoinositide 3-kinase/Akt signal pathways and induced Fas/FasL-mediated apoptosis in breast cancer models (Fig. 6). Cross-linking of B-lymphocyte receptors is the initiation of B-cell proliferation and differentiation to plasma cells secreting specific antibodies. In another study, biodegradable calcium phosphate nanoparticles decorated with model antigen hen egg lysozyme were preferentially bound and internalized by the tumor antigen specific B-lymphocytes, subsequently enhanced the surface expression of B-cell activation markers, and induced high humoral immunity, which was found to be approximately 100-fold more efficient in the activation of B-cells than the soluble form of tumor antigens.106
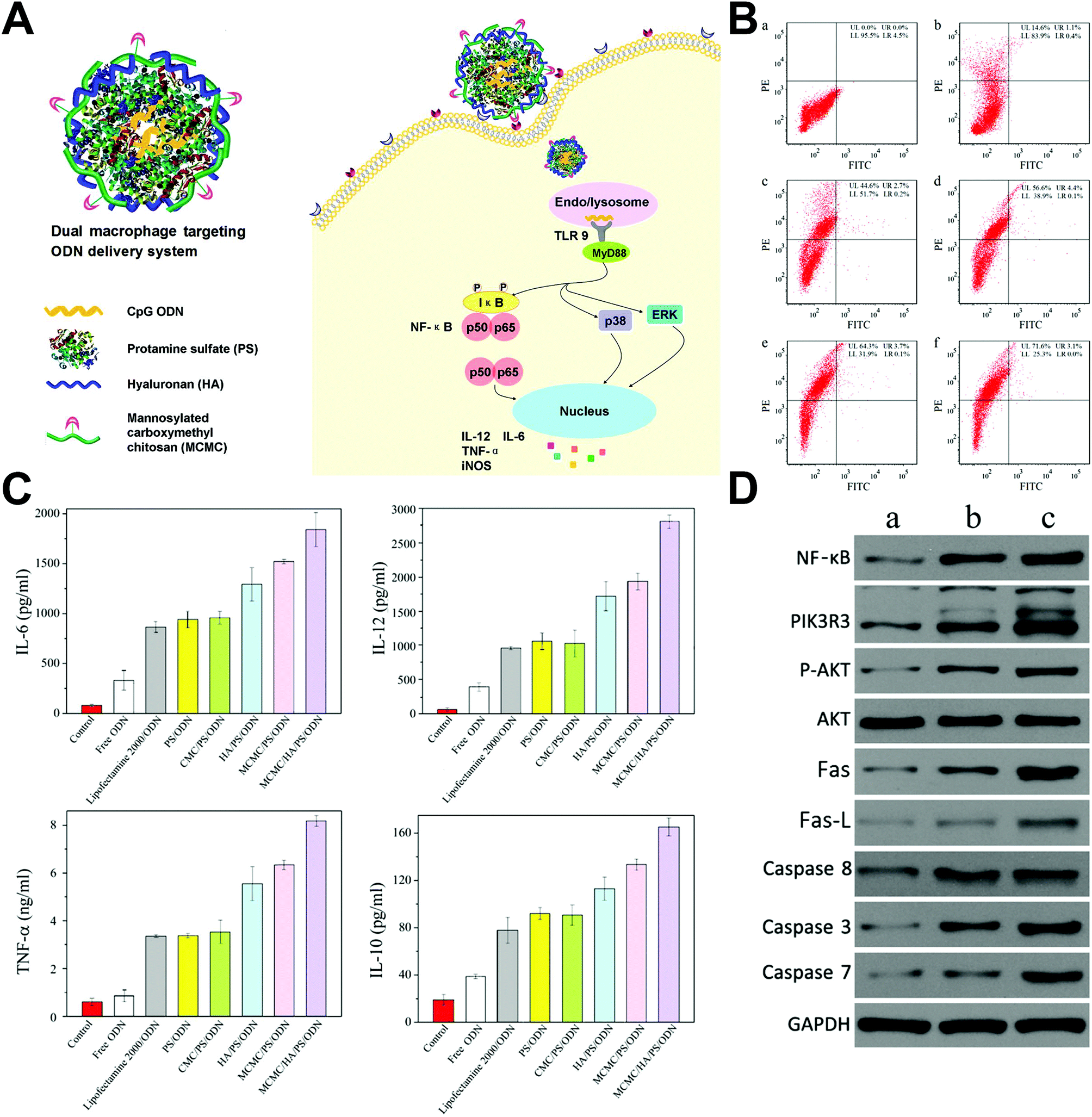 | ||
| Fig. 6 Macrophage dual-targeted nanoparticle for overcoming cancer-associated immunosuppression. (A) Structure of the dual-targeting delivery system inducing immunological stimulation in macrophages. (B) Flow cytometry showing the shift of the macrophage to activated M1 type that marked by CD80 (a. without treatment, and treated by b. naked nanoparticle, c. nontargeting system, d. mono-targeting system with HA, e. mono-targeting system with MCMC, and f. dual-targeting system with HA and MCMC). (C) Enhanced cytokine secretion of the macrophage after being treated. (D) Western blot analysis showing activated NF-κB, PI3 K/Akt and Fas/FasL signaling pathways after treatment (a. without treatment, b. treated by naked nanoparticles, and c. treated by the dual-targeting system). Reprinted with permission from ref. 105. Copyright (2017) American Chemical Society. | ||
5. Nanoparticles influencing the tumor microenvironment for immunotherapy enhancement
The tumor microenvironment refers to the surrounding complex scaffold around tumor cells, which is composed of the extracellular matrix, various signaling molecules, blood vessels, immune cells, fibroblasts and bone marrow-derived inflammatory cells.107 Tumor cells were closely related to the surrounding microenvironment, by releasing cellular signaling molecules and interact with immune and other related cells; tumors can influence the microenvironment to further promote the angiogenesis and induce immune tolerance.108,109 It has been reported that the extracellular matrix within the tumor microenvironment could play inhibitory roles to the adaptive anti-tumor immune response by inhibiting tumor specific T-lymphocyte proliferation through type I collagen ligation of LAIR receptors, and T-lymphocyte activation from its naive state.110 In addition, the abnormal tumor microenvironment could also provide critical biochemical and biomechanical cues to direct tumor cell growth, survival, proliferation and migration, and suppress the anti-tumor immunity function through suppressive cytokine release, cellular interaction, hypoxia condition, and hemodynamic obstruction of immunocytes.111,112 Thus, the regulation of the abnormal tumor microenvironment could largely relieve the inhibitory effects on the anti-tumor immunity and further increase therapeutic effects of immunotherapy. On the other hand, the tumor-specific stimuli in the abnormal tumor microenvironment including hypoxia, specific protease or proteins, and abnormal extracellular pH conditions could also be utilized to give nanoparticles the special conditions to accumulate and for orientating-release of payloads, thus achieving the regulation of the tumor microenvironment and enhancement of immunotherapy effects.Hypoxia was a commonly utilized tumor-specific condition for nanoparticle design with hypoxia-responsive chemical constructs coated, as the hypoxia is tightly associated with the immune suppressive tumor microenvironment and it is also a rare condition in normal tissues.113–115 The hypoxia environment could induce the polarization of macrophages from the immunosupportive M1 type to immunosuppressive M2 type, and was also responsible for recruitment of abundant regulatory T cells and release of immunosuppressive cytokines.116,117 Im et al. developed a photodynamic agent Chlorin e6 (Ce6)-loaded mesoporous silica nanoparticle with azobenzene used as the hypoxia-sensitive feasible cross-linker, which is also coated with the CpG/GC complex as the immune adjuvants.118 When the nanoparticles reached a hypoxic region within tumor tissue, the azobenzene cross-linkers were cleaved, and the silica core and the CpG/GC complex were released, leading to delivery of CpG for DC engulfment and activation. The authors also generate photodynamic therapy to induce both tumor cell death and recruit DCs to facilitate the release of tumor associated antigens and the uptake of DCs (Fig. 7). The nanoparticles combined with photodynamic therapy significantly enhanced the anti-tumor immune response and inhibited tumor growth in vivo. Yang et al. produced hollow manganese dioxide nanoparticles coated with PEG, Ce6 and DOX, which could be dissociated under the reduced pH microenvironment, hence releasing therapeutic drugs and meanwhile causing degradation of endogenous hydrogen peroxide to relieve intratumoral hypoxia.119 A remarkable synergistic effect of the nanoparticle with photodynamic therapy was achieved through intensifying the anti-tumor immune responses. Moreover, the additional combination of the therapy with immune checkpoint inhibitors led to the ablation of distant tumor metastasis.
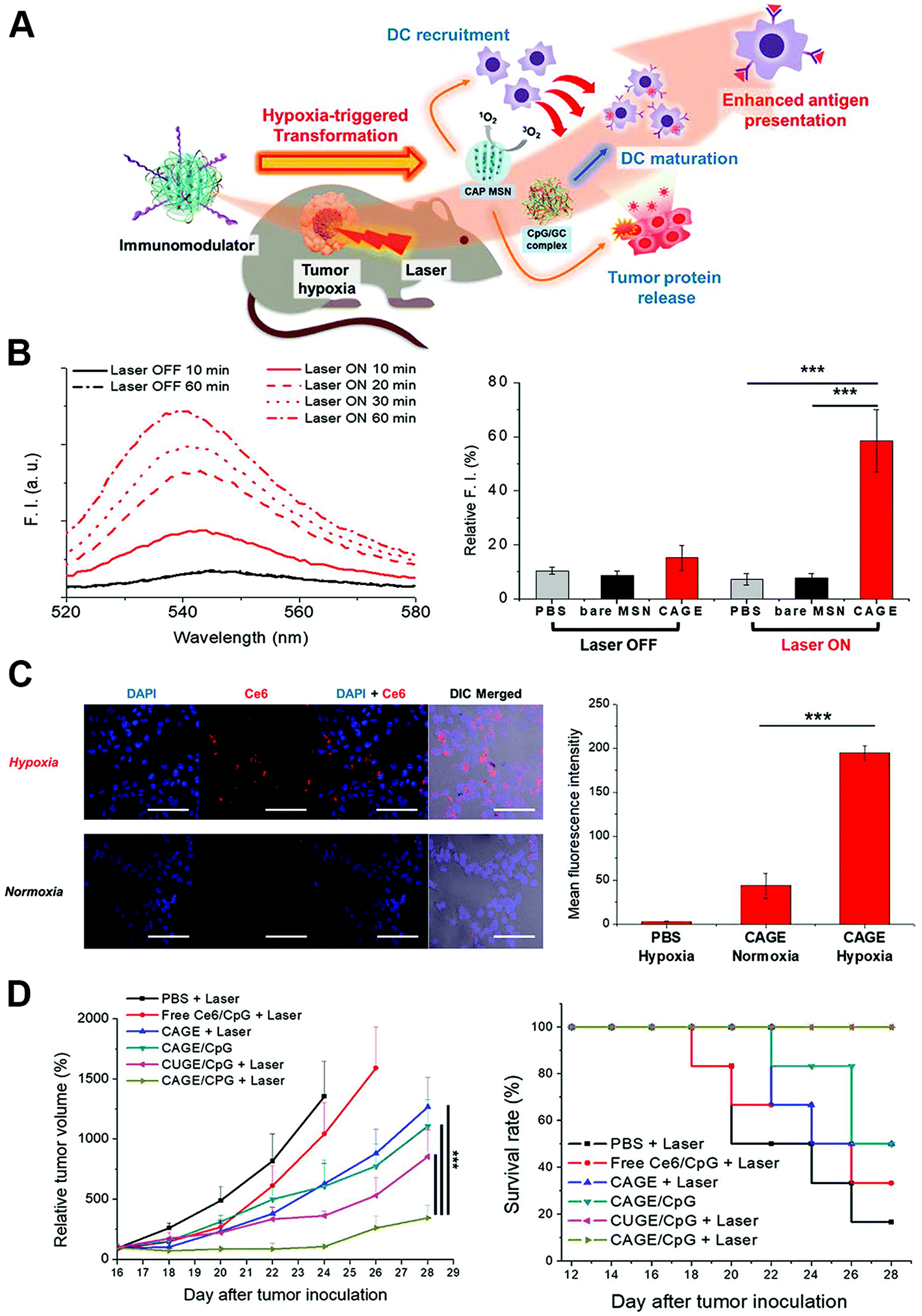 | ||
| Fig. 7 Hypoxia microenvironment-triggered transforming nanoparticles for cancer immunotherapy via photodynamically enhanced antigen presentation. (A) Schematic illustration of the working mechanism of the hypoxia-triggered transforming nanoparticle combined with photodynamic therapy. (B) Photoresponsive generation of singlet oxygen and the release profile of tumor proteins. (C) Hypoxia-responsive internalization of the nanoparticles into cells under hypoxic or normoxic conditions and the corresponding fluorescence intensity. (D) In vivo inhibition of tumor growth by the nanosystem and the survival curves. Reprinted with permission from ref. 118. Copyright (2019) American Chemical Society. | ||
The abnormal tumor vasculature featuring a heterogeneous vessel diameter, tortuosity and distribution, and insufficient blood and oxygen supply is closely correlated with hypoxia, reduced pH and immune suppressive features within the tumor microenvironment.120 As a result, normalization of tumor vasculature could be a way to enhance anti-tumor immunity.121 Accompanied by oral treatment of Erlotinib, an inhibitor of epidermal growth factor receptor (EGFR), Chen et al. found that the tumor uptake of immunostimulatory drug-loaded nanoparticles was significantly increased, and the combined therapy dramatically relieved the hypoxia status and altered the immunosuppressive tumor microenvironment into immunosupportive in three types of tumor models.122 The combined treatment also increased the tumor retention of anti-PD-L1 antibody, thus further inhibiting tumor growth and enhancing the treatment efficacy of anti-PD-L1 immunotherapy. On the other hand, the inhibition of copper trafficking could also have antiangiogenesis effects, as copper was required for the secretion of several angiogenic factors and stimulation of endothelial cell proliferation.123,124 Zhou et al. used a copper chelating coil-comb block copolymer, which has strong copper-chelating ability, to compose nanoparticles and conjugated it with TLR7/8 agonists and an immunomodulator Resiquimod.125 The nanoparticle exhibited accelerated releases in a reduced pH tumor microenvironment, displayed targeting ability and dramatically suppressed tumor growth in primary breast tumor and lung metastasis lesions, demonstrating considerable anti-tumor immune enhancement effects.
The relevant microenvironmental characteristics to differentiate malignancy from normal tissues also included specific enzymes such as matrix metalloproteinase (MMP), which functioned in degrading the extracellular matrix and assisted in carcinogenesis, tumor progression and metastasis, and its level is also found to be proportional to the malignancy of cancer.126–128 As a result, MMPs were harnessed as the key to trigger fragmentation of nanostructures and the release of payloads. Shin et al. used a MMP9-cleabable linker to trigger the detachment of the PEG corona in the tumor tissue, and this method significantly enhanced CD44 receptor-mediated endocytosis of nanoparticles, facilitated antigen presentation and inhibited tumor growth129 (Fig. 8). In another study, CpG DNA and anti-PD1 antibody were controllably released from nanoparticles in tumor tissue by the MMP9-dissociation method, and subsequently considerable immune responses were induced, and the risk of tumor relapse and metastasis after primary tumor resection was significantly reduced.130
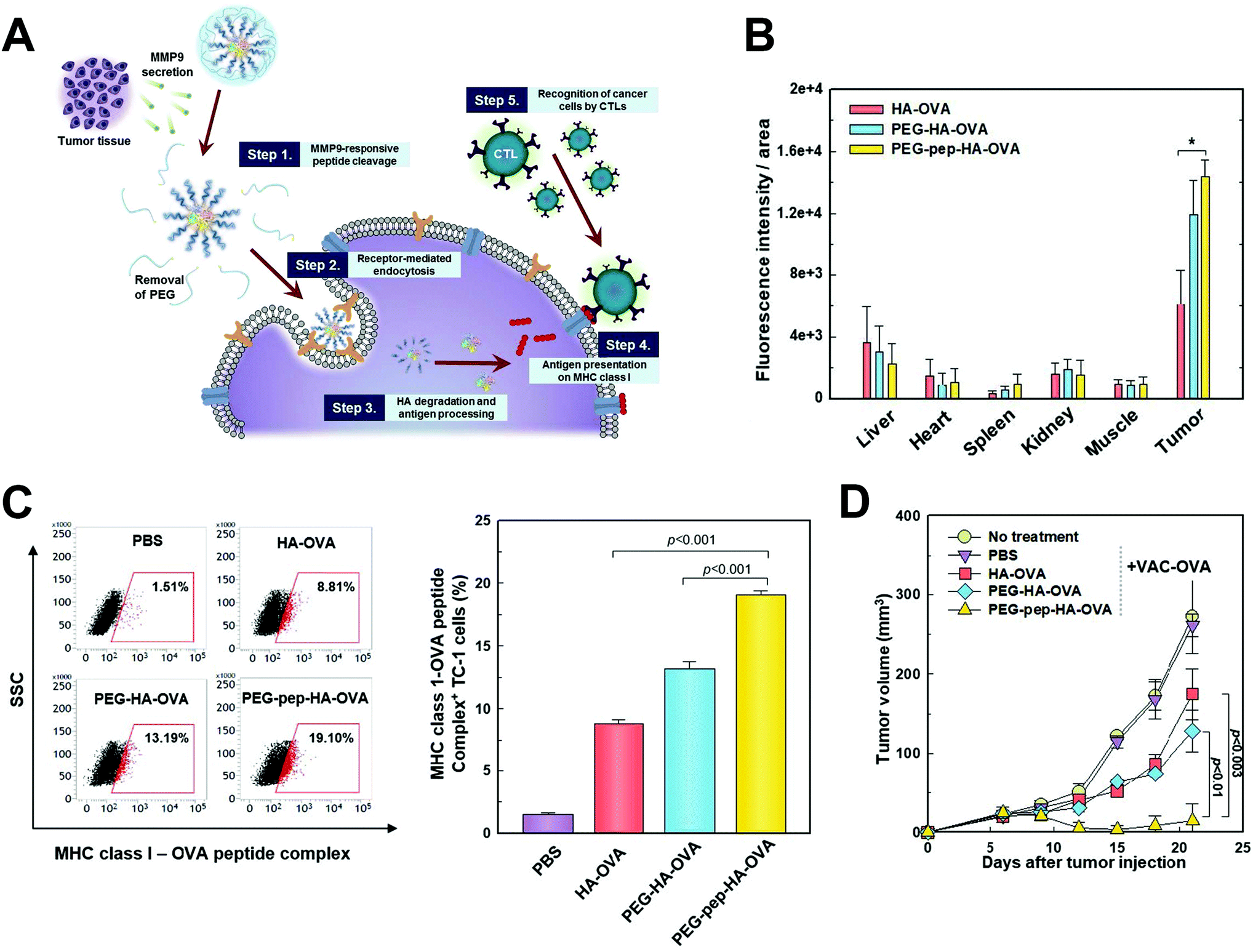 | ||
| Fig. 8 MMP9 responsive nanoparticle in enhancing cancer immunotherapy. (A) Schematic illustration depicting the working mechanism of the MMP9 responsive polymeric nanoparticle. (B) MMP9 responsive polymeric nanoparticle effectively accumulated in tumor tissue after systemic administration. (C) In vivo OVA antigen presentation in tumor cells after nanoparticle treatment. (D) In vivo therapeutic efficacy of the nanoparticle in tumor bearing mice. Reprinted from ref. 129, Copyright (2017), with permission from Elsevier. | ||
In addition, the redox-sensitive-based delivery strategy was often utilized as an effective way for controlled release of drugs into the tumor microenvironment. Generally, the nanoparticles contained redox-responsive disulfide bonds that could be degraded by intracellular glutathione. Along with reduction of the disulfide bonds, antigens could be released from the nanoparticle carrier and be cross-presented to induce cellular immunity.131,132 Xu et al. developed a redox-responsive nanoparticle platform and found that it had satisfactory features of long blood circulation, high tumor accumulation, fast payload release, and effective therapeutic effects.133 Wang et al. combined cell-penetrating peptides and redox-responsive disulfide bond cross-linking to design peptide carriers to form antigen delivery nanoparticles.134 The results showed that the redox-responsive nanoparticles could significantly induce the antigen-specific immune response by significantly increasing IgG titer and levels of cytokines including INF-γ, IL-12, IL-4, and IL-10, stimulating splenocyte proliferation and APC maturation, and could also enhance the immune memory function.
6. Nanoparticles in enhancing adoptive cell therapy
Adoptive cell therapy (ACT) is a crucial part of cancer immunotherapy, in which tumor-specific lymphocytes are isolated either from peripheral blood or tumor biopsies in patients, and then selected to recognize specific tumor antigens, or alternatively, polyclonal peripheral T-cells are artificially gene-modified to process desired tumor-targeting ligands. Then the cells are stimulated and expanded ex vivo, and finally infused back into patients.135–137 However, the therapeutic effects of ACT are impaired by insufficient ex vivo proliferation, inefficient trafficking of infused lymphocytes, and inadequate T-cell activity in the immunosuppressive tumor microenvironment due to immunosuppressive chemokines and abnormal tumor vasculature.138,139 Accompanied by specific designed nanoparticles, these limitations of ACT could be significantly altered. Targeting the lymphocytes with nanoliposomes conjugated with the inhibitor of TGF-β, an important immunosuppressive chemokine, prior to adoptive therapy could dramatically activate T-cell proliferation ex vivo and induce tumor regression after refusion.140 Stephan et al. developed poly-alginate microporous scaffolds which were loaded with proliferated T cells and VEGF antibody. After placing the scaffolds in tumor resection sites or close to inoperable tumors, the polymer could act as a reservoir for T-cell propagation and release, and the VEGF antibody could normalize tumor vasculature, assisting in T-cell infiltration and altering the immunosuppressive microenvironment. As a result, the polymer successively supported tumor-targeting T-lymphocytes throughout tumor resection beds and the draining lymph nodes, and inhibited tumor relapse, whereas the conventional delivery modalities or injection of tumor-targeting lymphocytes have limited therapeutic effects141 (Fig. 9).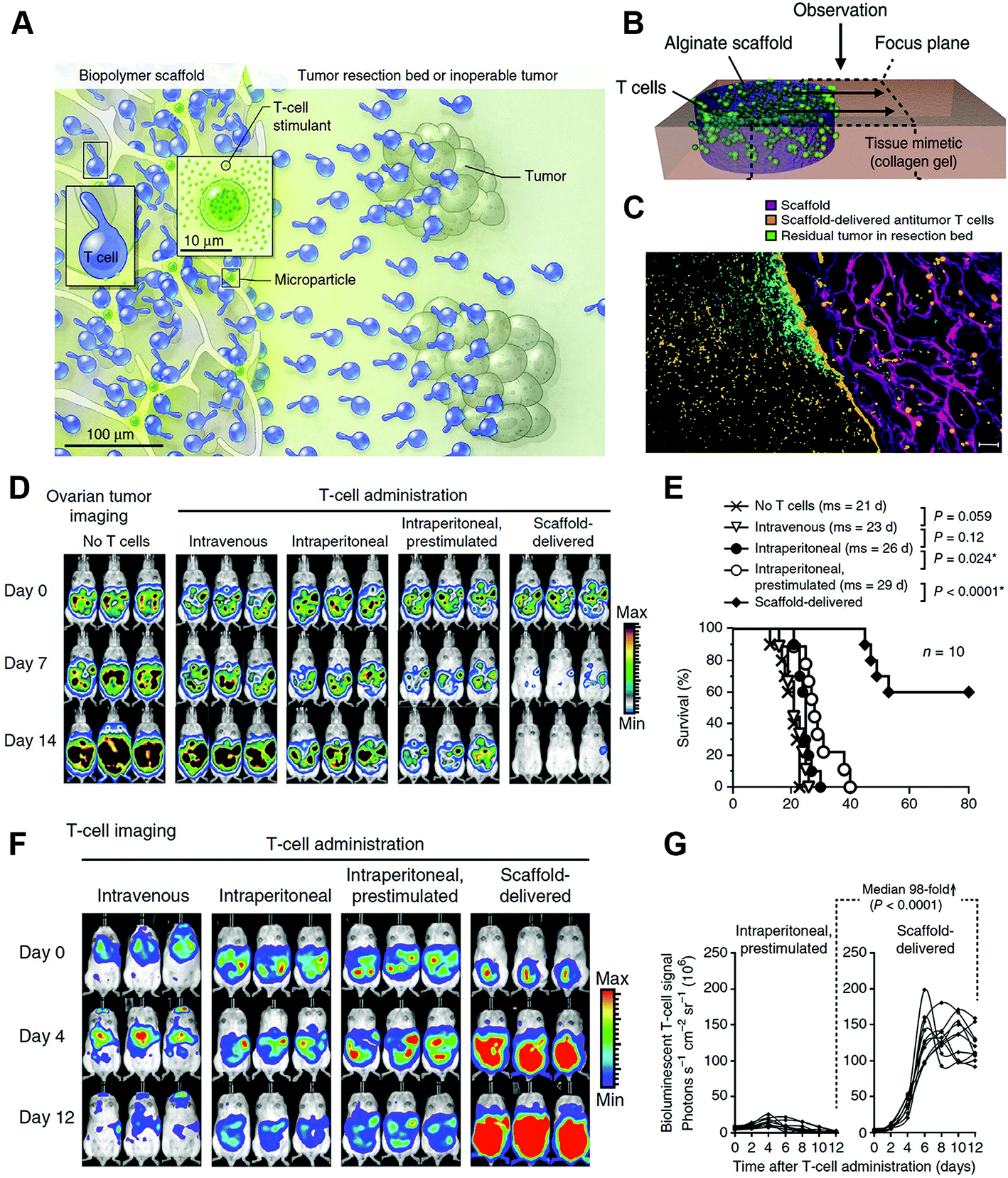 | ||
| Fig. 9 Nanostructured polyporous scaffold implants enhanced the efficacy of adoptive T-cell therapy. (A and B) Schematic diagram of the T cell-loaded scaffold surgically situated at a tumor site. Stimulatory microspheres incorporated into the device triggered cell expansion and promoted their egress into surrounding tissue. (C) Histological analysis of the scaffold and surrounding tissue 3 days after tumor-specific T cells (orange) were embedded in the scaffold (purple) and implanted into the tumor resection cavity. (D) Serial in vivo bioluminescence imaging of tumors. (E) Survival curves of tumor bearing mice following T-cell adoptive therapy. (F) In vivo bioluminescent imaging of T cells expressing luciferase. (G) Luciferase signal intensities after T-cell transfer, every line represented one animal and each dot reflected the whole animal photon count. Reprinted with permission from Springer Nature: Nature Biotechnology, ref. 141, Copyright (2014). | ||
Nanoparticles could also act on the ex vivo T-cell proliferation to the required quality, and reduce the time interval between cell acquirement and infusion. Integrin of T-lymphocytes played a crucial role in sustaining the immunological synapse with APCs through its binding to intercellular adhesion molecule-1, and the formation of immunological synapse led to reorganization of the cytoskeleton, which facilitated clustering and prevented migration of T-cells.142–144 Guasch et al. created a nanoparticle that cross-linked with peptides derived from fibronectin, an integrin signaling activator, and found that it was significantly more efficient in stimulating T-cell proliferation and increasing the expansion rate compared with nonfunctionalized nanoparticles.145 Perica et al. integrated paramagnetic iron-dextran nanoparticles with the MHC-Ig dimer as signal 1 and CD28 antibody as signal 2 to form an artificial APC.146 The nano-artificial APCs could selectively bind to antigen-specific T-lymphocyte rare naive precursors, and be retained in a magnetic column, thus achieving cell enrichment, followed by activation of the selected T-cells leading to rapid proliferation and expansion. The enrichment and expansion processes resulted in greater than 1000-fold proliferation of mouse and human antigen-specific T-lymphocytes in one week. The nano-artificial APCs could not only enhance T-cell proliferation in vivo culture but also after adoptive transfer, leading to a robust T-cell response (Fig. 10).
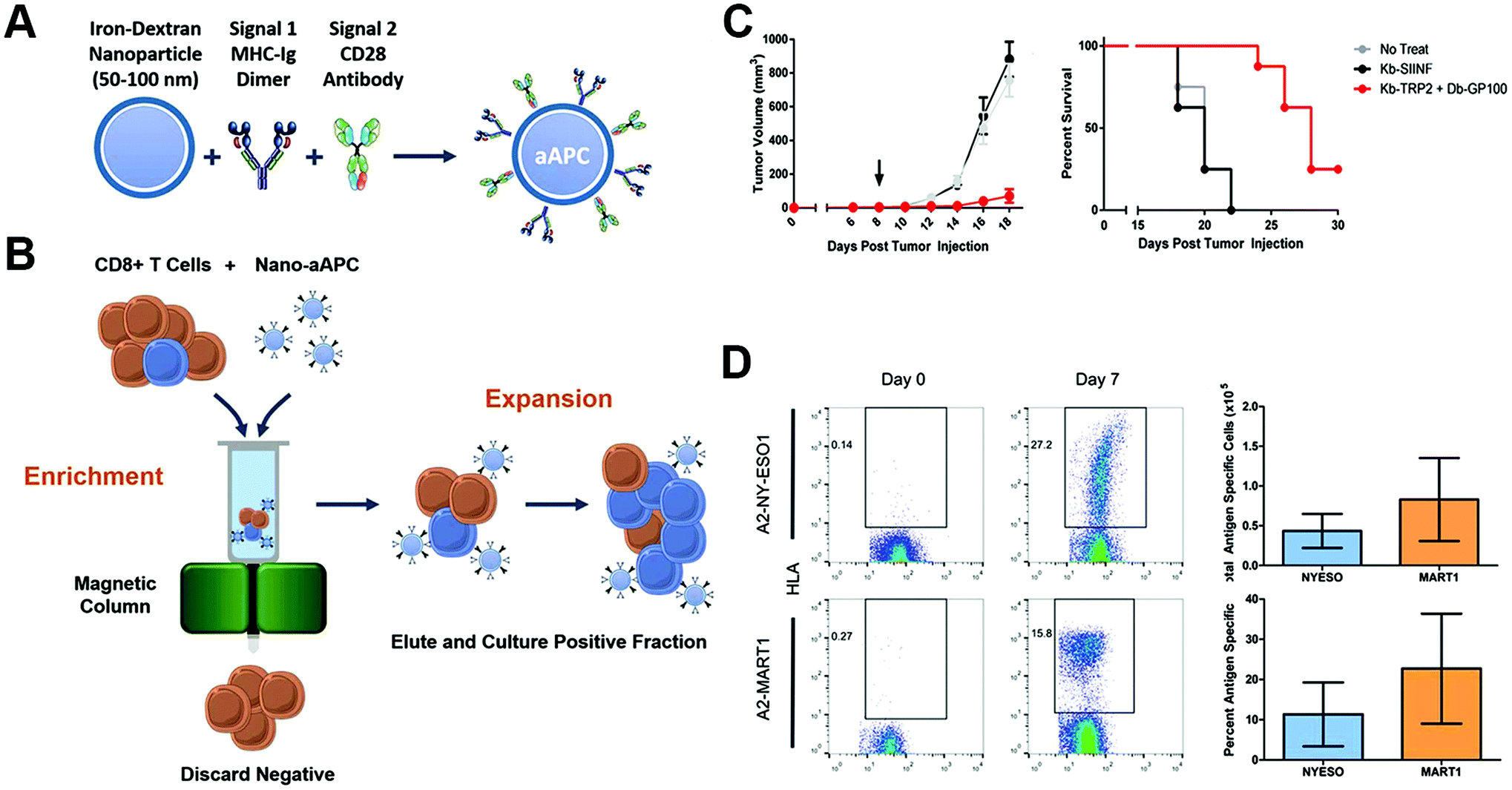 | ||
| Fig. 10 Enrichment and expansion with nano-artificial APCs for adoptive cell immunotherapy. (A) Nano-artificial APCs are synthesized by coupling the MHC-Ig dimer (signal 1) and anti-CD28 antibody (signal 2) to a 50–100 nm iron-dextran nanoparticle. (B) Schematic of magnetic enrichment. Antigen-specific CD8+ T cells (blue) bound to the nano-artificial APC are retained in a magnetic column in the enrichment step, while non-cognate (orange) cells are less likely to bind. Enriched T cells are then activated by the nano-artificial APC and proliferate in the expansion step. (C) Nano-artificial APC enriched and expanded lymphocytes inhibited melanoma growth after adoptive transfer. (D) Enrichment and expansion with the nano-artificial APC functionalized with human HLA-A2 and induced robust expansion against the tumor antigens. Reprinted with permission from ref. 146. Copyright (2015) American Chemical Society. | ||
7. Conclusion, challenges and perspective
Nanoparticle combined immunotherapy is at the initial stage of exploitation, but tremendous potential of the approach could be clearly witnessed. Compound nanoparticles have been developed in research studies as vehicles for concerted delivery of tumor antigens as vaccination and/or immune stimulatory adjuvants to DCs or other professional APCs. Nanoparticles carrying whole-cell tumor vaccines have multiple antigen immunity and could potentially stimulate cellular and humoral immune responses. Generally, in comparison with the traditional, non-nanoparticle combined tumor antigen vaccines, the nanoparticle loaded approaches could induce significantly stronger antigen-specific antibody responses and CD8+ cytotoxic T lymphocyte responses against cancer cells and efficaciously inhibited tumor growth. Based on the inherent physical properties of nanoparticles or the conjugation with targeted ligands, targeting transportation of tumor antigens or adjuvants to TDLNs or specific immune cells such as DCs, T-lymphocytes and macrophages could be realized, thus further enhancing the immune stimulatory effects. In addition, combination of the compound nanoparticles with molecules of immune checkpoint inhibitors or photodynamic therapy could complement each other and more efficiently suppress tumor proliferation at the primary and metastatic sites. Nanoparticles with specific features could also control the release of payloads in response to typical conditions in the tumor microenvironment including hypoxia, specific protease and decreased pH condition, and further alter the immunosuppressive microenvironment into immunosupportive features. Adoptive cell therapy could also be supported by nanoparticle approaches in the aspects of enhancing ex vivo T-cell proliferation, recovering the abnormal microenvironment, and intensifying the trafficking and activity of infused lymphocytes.Despite the impressive benefits of nanoparticles in cancer immunotherapy, there are still challenges that need to be treated with caution and investigated in future studies. First, the delivery ability of nanoparticles to the intended tissues or organs still needs to be strengthened. Currently, nanoparticles can only deliver a small portion of the administered payloads to the intended locations even with targeted ligands coated, which leads to the loss of delivered drugs and systemic cytotoxicity due to normal cell uptake of the nanoparticles. According to the study of Harrington et al. which included 17 patients with different types of locally advanced cancers, only less than 4% of the total administered dose could be accumulated in tumor tissues using pegylated liposome as the delivery method.28 In addition, considerable heterogeneity of uptake both between different tumor types and between different patients with the same tumor type was also observed, mainly caused by the various tumor vascular conditions.28 Similarly, because of the poor vascularization, nanoparticle accumulation is often insufficient in small metastatic lesions and the core of large tumors where usually exists risky dormant cancer cells. Other nanoparticles taken up by normal cells can cause toxicity including oxidative stress, pulmonary inflammation, and dysfunction of cells and organs through mechanisms of redox cycling, free radical formation and hydrophobic interaction.147,148
For the nanoparticles delivering payloads to the immune organs and cells, as they need to be transported in blood circulation and lymph vessels before reaching the targeted sites, the structural and colloidal stability is crucial to guarantee their final drainage into the immune organs and present payloads to immune cells.40 Unstable nanoparticles may form micrometer-sized aggregates that could plug the capillary bed of distal organs and cause serious complications to patients. Another challenge is how to efficiently regulate components within the whole immune system by the nanoparticles to optimize the enhancement effects on anti-tumor immunity. The innate and adaptive immune system works in a network, but currently, little is known about how the other immune components in the immune network are affected to influence the whole anti-tumor immunity when the intended component is regulated by the administered nanoparticles. Future efforts are still needed to investigate the inter-regulation mechanisms of immune components to maximize treatment effects of the nanoparticles.
Potential immunogenicity of nanoparticles is another crucial concern towards progress in clinical translations. In fact, the utilization of nanoparticles in cancer immunotherapy is based on the immunogenicity or the immune adjuvant effects of nanoparticles or the attached peptides, proteins or antibody fragments, but it could be detrimental if the immune response against un-intended components is activated. Nanoparticles can be antigenic themselves, and if the nanoparticles are recognized as foreign substances and opsonized by plasma protein, the complement pathway will be activated, resulting in rapid phagocytosis and clearance in liver and spleen. Complete activation of the immune response against nanoparticles may lead to serious complications including allergic reactions, hemolysis, thrombogenesis, and even disseminated intravascular coagulation (DIC).149,150 More importantly, when nanoparticles are applied for enhancing anti-tumor immunity, immune checkpoint inhibitors are often conjugated to the nanoparticles or systematically administered to form syncretistic treatment effects. However, one major risk of the immune checkpoint inhibitors is that the drugs can induce serious autoimmune diseases. As a consequence, the simultaneously administered nanoparticles for future clinical translations should be developed in weak antigenicity with the use of a strictly controlled dose, and the patients should be observed with caution as they are theoretically at higher risk of complications.
Collectively, clinical application of the nanoparticles combined with immunotherapy needs the optimization of nanostructures to overcome the several existing disadvantages including the insufficient targeted delivery capacity, systemic cytotoxicity due to normal cell's engulfment and the potential immunogenicity, and it will be crucial to ascertain the optimal physical and chemical properties of the nanoparticles in future investigations. It has been shown that the diameter of nanoparticles less than 100 nm displayed the most suitable surface chemistries, which could enable long-distance transportation within immune organs, strengthening internalization by immune cells, but other characteristics including the surface charge, shape, elasticity, hydrophilicity, ligands, and loading methods of encapsulating or conjugating payloads still need to be researched and determined to further improve the pharmacokinetics, toxicity, biocompatibility and biodistribution of the nanoparticles.
Only a small proportion of patients could achieve satisfactory survival after receiving one modality of immunotherapy, and there is a growing common view that combination of tumor vaccines with immune checkpoint inhibitors, adoptive T-cell therapy or tumor microenvironment regulatory treatments could form synergistic effects on inhibiting primary and abscopal tumor lesions. As a consequence, developing pleuripotent nanoparticles that integrate the multiple immunotherapy ingredients with targeted delivering and triggered releasing ability may could potentially attain comprehensive anti-tumor immunity enhancement and improved treatment effects. With further advances of nanotechnology and deeper understanding of interactions between the nanomaterials and immune system, the nanomedicine combined immunotherapy could be further consummated to achieve more effective therapeutic benefits.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors gratefully acknowledge the staff in the Krishnan Lab at the University of Texas MD Anderson Cancer Center for the valuable suggestions on the manuscript. The author C. A.-O. was supported by the National Institutes of Health (NIH) Translational Cancer Nanotechnology Postdoctoral Fellowship grant T32CA196561.References
- J. Mora, C. Mertens, J. K. Meier, D. C. Fuhrmann, B. Brune and M. Jung, Cells, 2019, 8 DOI:10.3390/cells8050445.
- M. D. Vesely, M. H. Kershaw, R. D. Schreiber and M. J. Smyth, Annu. Rev. Immunol., 2011, 29, 235–271 CrossRef CAS PubMed.
- H. J. Smith, T. R. McCaw, A. I. Londono, A. A. Katre, S. Meza-Perez, E. S. Yang, A. Forero, D. J. Buchsbaum, T. D. Randall, J. M. Straughn Jr., L. A. Norian and R. C. Arend, Cancer, 2018, 124, 4657–4666 CrossRef CAS PubMed.
- E. S. Nakasone, S. A. Hurvitz and K. E. McCann, Drugs Context, 2018, 7, 212520 Search PubMed.
- M. Dougan and G. Dranoff, Annu. Rev. Immunol., 2009, 27, 83–117 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- I. Marquez-Rodas, M. A. Aznar, A. Calles and I. Melero, Clin. Cancer Res., 2019, 25, 1127–1129 CrossRef PubMed.
- Y. Wang, C. Yao, L. Ding, C. Li, J. Wang, M. Wu and Y. Lei, J. Biomed. Nanotechnol., 2017, 13, 367–380 CrossRef CAS PubMed.
- T. Kieber-Emmons, B. Monzavi-Karbassi, L. F. Hutchins, A. Pennisi and I. Makhoul, Hum. Vaccines Immunother., 2017, 13, 323–331 CrossRef PubMed.
- Y. Yang, G. H. Nam, G. B. Kim, Y. K. Kim and I. S. Kim, Adv. Drug Delivery Rev., 2019 DOI:10.1016/j.addr.2019.05.007.
- R. Lotfi, J. Eisenbacher, G. Solgi, K. Fuchs, T. Yildiz, C. Nienhaus, M. T. Rojewski and H. Schrezenmeier, Eur. J. Immunol., 2011, 41, 2021–2028 CrossRef CAS.
- D. S. Chen and I. Mellman, Immunity, 2013, 39, 1–10 CrossRef CAS PubMed.
- L. A. Tashireva, V. M. Perelmuter, V. N. Manskikh, E. V. Denisov, O. E. Savelieva, E. V. Kaygorodova and M. V. Zavyalova, Biochemistry, 2017, 82, 542–555 CAS.
- A. Tarafdar, L. E. Hopcroft, P. Gallipoli, F. Pellicano, J. Cassels, A. Hair, K. Korfi, H. G. Jorgensen, D. Vetrie, T. L. Holyoake and A. M. Michie, Blood, 2017, 129, 199–208 CrossRef CAS PubMed.
- H. M. Guirgis, J. Immunother. Cancer, 2018, 6, 15 CrossRef PubMed.
- J. F. Jacobs, A. J. Idema, K. F. Bol, S. Nierkens, O. M. Grauer, P. Wesseling, J. A. Grotenhuis, P. M. Hoogerbrugge, I. J. de Vries and G. J. Adema, Neuro Oncol., 2009, 11, 394–402 CrossRef CAS PubMed.
- P. Sinha, V. K. Clements, S. Miller and S. Ostrand-Rosenberg, Cancer Immunol. Immunother., 2005, 54, 1137–1142 CrossRef CAS.
- D. I. Gabrilovich and S. Nagaraj, Nat. Rev. Immunol., 2009, 9, 162–174 CrossRef CAS PubMed.
- K. K. Goswami, T. Ghosh, S. Ghosh, M. Sarkar, A. Bose and R. Baral, Cell. Immunol., 2017, 316, 1–10 CrossRef CAS PubMed.
- M. W. Pickup, J. K. Mouw and V. M. Weaver, EMBO Rep., 2014, 15, 1243–1253 CrossRef CAS PubMed.
- P. A. Ott, F. S. Hodi and C. Robert, Clin. Cancer Res., 2013, 19, 5300–5309 CrossRef CAS PubMed.
- S. Baxi, A. Yang, R. L. Gennarelli, N. Khan, Z. Wang, L. Boyce and D. Korenstein, Br. Med. J., 2018, 360, k793 CrossRef PubMed.
- R. Wu, M. A. Forget, J. Chacon, C. Bernatchez, C. Haymaker, J. Q. Chen, P. Hwu and L. G. Radvanyi, Cancer J., 2012, 18, 160–175 CrossRef CAS PubMed.
- N. Mitsuiki, C. Schwab and B. Grimbacher, Immunol. Rev., 2019, 287, 33–49 CrossRef CAS PubMed.
- A. Ribas and J. D. Wolchok, Science, 2018, 359, 1350–1355 CrossRef CAS PubMed.
- J. M. Cha, D. G. You, E. J. Choi, S. J. Park, W. Um, J. Jeon, K. Kim, I. C. Kwon, J. C. Park, H. R. Kim and J. H. Park, J. Biomed. Nanotechnol., 2016, 12, 1724–1733 CrossRef CAS PubMed.
- H. Hashizume, P. Baluk, S. Morikawa, J. W. McLean, G. Thurston, S. Roberge, R. K. Jain and D. M. McDonald, Am. J. Pathol., 2000, 156, 1363–1380 CrossRef CAS PubMed.
- K. J. Harrington, S. Mohammadtaghi, P. S. Uster, D. Glass, A. M. Peters, R. G. Vile and J. S. Stewart, Clin. Cancer Res., 2001, 7, 243–254 CAS.
- J. K. Vasir and V. Labhasetwar, Technol. Cancer Res. Treat., 2005, 4, 363–374 CrossRef CAS PubMed.
- R. A. Petros and J. M. DeSimone, Nat. Rev. Drug Discovery, 2010, 9, 615–627 CrossRef CAS PubMed.
- Y. Wang, D. Wang, Q. Fu, D. Liu, Y. Ma, K. Racette, Z. He and F. Liu, Mol. Pharm., 2014, 11, 3766–3771 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- R. V. Benjaminsen, M. A. Mattebjerg, J. R. Henriksen, S. M. Moghimi and T. L. Andresen, Mol. Ther., 2013, 21, 149–157 CrossRef CAS PubMed.
- Y. Y. Yuan, C. Q. Mao, X. J. Du, J. Z. Du, F. Wang and J. Wang, Adv. Mater., 2012, 24, 5476–5480 CrossRef CAS PubMed.
- J. Lu, S. C. Owen and M. S. Shoichet, Macromolecules, 2011, 44, 6002–6008 CrossRef CAS PubMed.
- S. Terry, P. Savagner, S. Ortiz-Cuaran, L. Mahjoubi, P. Saintigny, J. Thiery and S. Chouaib, Mol. Oncol., 2017, 11, 824–846 CrossRef PubMed.
- G. Beatty and W. Gladney, Clin. Cancer Res., 2015, 21, 687–692 CrossRef CAS PubMed.
- Y. Lu, Y. Yang, Z. Gu, J. Zhang, H. Song, G. Xiang and C. Yu, Biomaterials, 2018, 175, 82–92 CrossRef CAS PubMed.
- S. Yan, W. Gu, B. Zhang, B. E. Rolfe and Z. P. Xu, Dalton Trans., 2018, 47, 2956–2964 RSC.
- L. X. Zhang, X. X. Xie, D. Q. Liu, Z. P. Xu and R. T. Liu, Biomaterials, 2018, 174, 54–66 CrossRef CAS.
- T. Yata, Y. Takahashi, M. Tan, H. Nakatsuji, S. Ohtsuki, T. Murakami, H. Imahori, Y. Umeki, T. Shiomi, Y. Takakura and M. Nishikawa, Biomaterials, 2017, 146, 136–145 CrossRef CAS PubMed.
- T. Courant, E. Bayon, H. L. Reynaud-Dougier, C. Villiers, M. Menneteau, P. N. Marche and F. P. Navarro, Biomaterials, 2017, 136, 29–42 CrossRef CAS PubMed.
- C. Zhang, G. Shi, J. Zhang, H. Song, J. Niu, S. Shi, P. Huang, Y. Wang, W. Wang, C. Li and D. Kong, J. Controlled Release, 2017, 256, 170–181 CrossRef CAS PubMed.
- V. Sokolova, Z. Shi, S. Huang, Y. Du, M. Kopp, A. Frede, T. Knuschke, J. Buer, D. Yang, J. Wu, A. M. Westendorf and M. Epple, Acta Biomater., 2017, 64, 401–410 CrossRef CAS PubMed.
- L. Liu, Y. Wang, L. Miao, Q. Liu, S. Musetti, J. Li and L. Huang, Mol. Ther., 2018, 26, 45–55 CrossRef CAS PubMed.
- R. M. Pearson, L. M. Casey, K. R. Hughes, L. Z. Wang, M. G. North, D. R. Getts, S. D. Miller and L. D. Shea, Mol. Ther., 2017, 25, 1655–1664 CrossRef CAS PubMed.
- R. Verbeke, I. Lentacker, L. Wayteck, K. Breckpot, M. Van Bockstal, B. Descamps, C. Vanhove, S. C. De Smedt and H. Dewitte, J. Controlled Release, 2017, 266, 287–300 CrossRef CAS PubMed.
- H. Takahashi, K. Misato, T. Aoshi, Y. Yamamoto, Y. Kubota, X. Wu, E. Kuroda, K. J. Ishii, H. Yamamoto and Y. Yoshioka, Front. Immunol., 2018, 9, 783 CrossRef PubMed.
- Y. Yoshizaki, E. Yuba, N. Sakaguchi, K. Koiwai, A. Harada and K. Kono, Biomaterials, 2017, 141, 272–283 CrossRef CAS PubMed.
- X. Liang, J. Duan, X. Li, X. Zhu, Y. Chen, X. Wang, H. Sun, D. Kong, C. Li and J. Yang, Nanoscale, 2018, 10, 9489–9503 RSC.
- M. Xu, Y. Chen, P. Banerjee, L. Zong and L. Jiang, AAPS PharmSciTech, 2017, 18, 2618–2625 CrossRef CAS PubMed.
- W. Long, J. Wang, J. Yang, H. Wu, J. Wang, X. Mu, H. He, Q. Liu, Y. M. Sun, H. Wang and X. D. Zhang, J. Biomed. Nanotechnol., 2019, 15, 62–76 CrossRef CAS PubMed.
- G. Zom, M. Willems, S. Khan, T. van der Sluis, J. Kleinovink, M. Camps, G. van der Marel, D. Filippov, C. Melief and F. Ossendorp, J. Immunother. Cancer, 2018, 6, 146 CrossRef PubMed.
- B. Seliger, C. Massa, B. Rini, J. Ko and J. Finke, Trends Mol. Med., 2010, 16, 184–192 CrossRef CAS PubMed.
- M. Ebrahimian, M. Hashemi, M. Maleki, G. Hashemitabar, K. Abnous, M. Ramezani and A. Haghparast, Front. Immunol., 2017, 8, 1077 CrossRef PubMed.
- Z. Luo, C. Wang, H. Yi, P. Li, H. Pan, L. Liu, L. Cai and Y. Ma, Biomaterials, 2015, 38, 50–60 CrossRef CAS PubMed.
- A. N. Alexander, M. K. Huelsmeyer, A. Mitzey, R. R. Dubielzig, I. D. Kurzman, E. G. Macewen and D. M. Vail, Cancer Immunol. Immunother., 2006, 55, 433–442 CrossRef CAS PubMed.
- M. Chen, R. Xiang, Y. Wen, G. Xu, C. Wang, S. Luo, T. Yin, X. Wei, B. Shao, N. Liu, F. Guo, M. Li, S. Zhang, M. Li, K. Ren, Y. Wang and Y. Wei, Sci. Rep., 2015, 5, 14421 CrossRef CAS PubMed.
- R. Yang, J. Xu, L. Xu, X. Sun, Q. Chen, Y. Zhao, R. Peng and Z. Liu, ACS Nano, 2018, 12, 5121–5129 CrossRef CAS PubMed.
- R. H. Fang, C. M. Hu, B. T. Luk, W. Gao, J. A. Copp, Y. Tai, D. E. O'Connor and L. Zhang, Nano Lett., 2014, 14, 2181–2188 CrossRef CAS PubMed.
- A. V. Kroll, R. H. Fang, Y. Jiang, J. Zhou, X. Wei, C. L. Yu, J. Gao, B. T. Luk, D. Dehaini, W. Gao and L. Zhang, Adv. Mater., 2017, 29 DOI:10.1002/adma.201703969.
- W. Chen, H. Zuo, B. Li, C. Duan, B. Rolfe, B. Zhang, T. J. Mahony and Z. P. Xu, Small, 2018, 14, e1704465 CrossRef PubMed.
- J. W. Hodge, C. T. Garnett, B. Farsaci, C. Palena, K. Y. Tsang, S. Ferrone and S. R. Gameiro, Int. J. Cancer, 2013, 133, 624–636 CrossRef CAS PubMed.
- S. C. Formenti and S. Demaria, Int. J. Radiat. Oncol., Biol., Phys., 2012, 84, 879–880 CrossRef PubMed.
- Q. Chen, L. Xu, C. Liang, C. Wang, R. Peng and Z. Liu, Nat. Commun., 2016, 7, 13193 CrossRef CAS PubMed.
- J. Xu, L. Xu, C. Wang, R. Yang, Q. Zhuang, X. Han, Z. Dong, W. Zhu, R. Peng and Z. Liu, ACS Nano, 2017, 11, 4463–4474 CrossRef CAS PubMed.
- V. A. Boussiotis, N. Engl. J. Med., 2016, 375, 1767–1778 CrossRef CAS PubMed.
- J. M. Kim and D. S. Chen, Ann. Oncol., 2016, 27, 1492–1504 CrossRef CAS PubMed.
- F. C. Santini and M. D. Hellmann, Cancer J., 2018, 24, 15–19 CrossRef CAS PubMed.
- Y. Liu and P. Zheng, Trends Immunol., 2018, 39, 953–956 CrossRef CAS PubMed.
- M. K. Callahan and J. D. Wolchok, Semin. Oncol., 2015, 42, 573–586 CrossRef CAS PubMed.
- N. Cheng, R. Watkins-Schulz, R. D. Junkins, C. N. David, B. M. Johnson, S. A. Montgomery, K. J. Peine, D. B. Darr, H. Yuan, K. P. McKinnon, Q. Liu, L. Miao, L. Huang, E. M. Bachelder, K. M. Ainslie and J. P. Ting, JCI Insight, 2018, 3 DOI:10.1172/jci.insight.120638.
- W. Yu, Y. Wang, J. Zhu, L. Jin, B. Liu, K. Xia, J. Wang, J. Gao, C. Liang and H. Tao, Biomaterials, 2019, 192, 128–139 CrossRef CAS PubMed.
- Y. Wu, W. Gu, L. Li, C. Chen and Z. P. Xu, Nanomaterials, 2019, 9 DOI:10.3390/nano9020159.
- Y. Wu, W. Gu, J. Li, C. Chen and Z. P. Xu, Nanomedicine, 2019, 14, 955–967 CrossRef CAS.
- C. Wang, Y. Ye, G. M. Hochu, H. Sadeghifar and Z. Gu, Nano Lett., 2016, 16, 2334–2340 CrossRef CAS.
- Y. Li, M. Fang, J. Zhang, J. Wang, Y. Song, J. Shi, W. Li, G. Wu, J. Ren, Z. Wang, W. Zou and L. Wang, OncoImmunology, 2016, 5, e1074374 CrossRef PubMed.
- P. Zhao, D. Atanackovic, S. Dong, H. Yagita, X. He and M. Chen, Mol. Pharm., 2017, 14, 1494–1500 CrossRef CAS PubMed.
- R. Meir, K. Shamalov, T. Sadan, M. Motiei, G. Yaari, C. J. Cohen and R. Popovtzer, ACS Nano, 2017, 11, 11127–11134 CrossRef CAS PubMed.
- Y. K. Chae, A. Arya, W. Iams, M. R. Cruz, S. Chandra, J. Choi and F. Giles, J. Immunother. Cancer, 2018, 6, 39 CrossRef PubMed.
- R. Kuai, W. M. Yuan, S. Son, J. Nam, Y. Xu, Y. C. Fan, A. Schwendeman and J. J. Moon, Sci. Adv., 2018, 4, eaao1736 CrossRef PubMed.
- D. Wang, T. Wang, J. Liu, H. Yu, S. Jiao, B. Feng, F. Zhou, Y. Fu, Q. Yin, P. Zhang, Z. Zhang, Z. Zhou and Y. Li, Nano Lett., 2016, 16, 5503–5513 CrossRef CAS PubMed.
- S. Lob, A. Konigsrainer, H. G. Rammensee, G. Opelz and P. Terness, Nat. Rev. Cancer, 2009, 9, 445–452 CrossRef PubMed.
- C. Sheridan, Nat. Biotechnol., 2015, 33, 321–322 CrossRef CAS PubMed.
- J. J. Sun, Y. C. Chen, Y. X. Huang, W. C. Zhao, Y. H. Liu, R. Venkataramanan, B. F. Lu and S. Li, Acta Pharmacol. Sin., 2017, 38, 823–834 CrossRef CAS PubMed.
- N. Wang, Z. Wang, Z. Xu, X. Chen and G. Zhu, Angew. Chem., Int. Ed., 2018, 57, 3426–3430 CrossRef CAS PubMed.
- Y. Chen, R. Xia, Y. Huang, W. Zhao, J. Li, X. Zhang, P. Wang, R. Venkataramanan, J. Fan, W. Xie, X. Ma, B. Lu and S. Li, Nat. Commun., 2016, 7, 13443 CrossRef CAS PubMed.
- A. Takahashi, K. Kono, F. Ichihara, H. Sugai, H. Amemiya, H. Iizuka, H. Fujii and Y. Matsumoto, Int. J. Cancer, 2003, 104, 393–399 CrossRef CAS PubMed.
- V. Carriere, R. Colisson, C. Jiguet-Jiglaire, E. Bellard, G. Bouche, T. Al Saati, F. Amalric, J. P. Girard and C. M'Rini, Cancer Res., 2005, 65, 11639–11648 CrossRef CAS PubMed.
- D. H. Munn and A. L. Mellor, Immunol. Rev., 2006, 213, 146–158 CrossRef PubMed.
- K. Imai, Y. Minamiya, S. Koyota, M. Ito, H. Saito, Y. Sato, S. Motoyama, T. Sugiyama and J. Ogawa, J. Exp. Clin. Cancer Res., 2012, 31, 3 CrossRef CAS PubMed.
- L. M. Habenicht, T. C. Albershardt, B. M. Iritani and A. Ruddell, OncoImmunology, 2016, 5, e1204505 CrossRef PubMed.
- L. Jeanbart, M. Ballester, A. de Titta, P. Corthesy, P. Romero, J. A. Hubbell and M. A. Swartz, Cancer Immunol. Res., 2014, 2, 436–447 CrossRef CAS PubMed.
- T. Kang, Y. Huang, Q. Zhu, H. Cheng, Y. Pei, J. Feng, M. Xu, G. Jiang, Q. Song, T. Jiang, H. Chen, X. Gao and J. Chen, Biomaterials, 2018, 164, 80–97 CrossRef CAS PubMed.
- F. W. Cao, Y. X. Guo, Y. Li, S. Y. Tang, Y. D. Yang, H. Yang and L. Q. Xiong, Adv. Funct. Mater., 2018, 28 DOI:10.1002/adfm.201707174.
- N. H. Cho, T. C. Cheong, J. H. Min, J. H. Wu, S. J. Lee, D. Kim, J. S. Yang, S. Kim, Y. K. Kim and S. Y. Seong, Nat. Nanotechnol., 2011, 6, 675–682 CrossRef CAS PubMed.
- K. Sehgal, R. Ragheb, T. M. Fahmy, M. V. Dhodapkar and K. M. Dhodapkar, J. Immunol., 2014, 193, 2297–2305 CrossRef CAS PubMed.
- T. Z. Chang, S. S. Stadmiller, E. Staskevicius and J. A. Champion, Biomater. Sci., 2017, 5, 223–233 RSC.
- R. Liang, J. Xie, J. Li, K. Wang, L. Liu, Y. Gao, M. Hussain, G. Shen, J. Zhu and J. Tao, Biomaterials, 2017, 149, 41–50 CrossRef CAS.
- C. Zhang, J. Zhang, G. Shi, H. Song, S. Shi, X. Zhang, P. Huang, Z. Wang, W. Wang, C. Wang, D. Kong and C. Li, Mol. Pharm., 2017, 14, 1760–1770 CrossRef CAS PubMed.
- Y. Wang, L. Zhang, Z. Xu, L. Miao and L. Huang, Mol. Ther., 2018, 26, 420–434 CrossRef CAS.
- Q. Liu, H. Zhu, Y. Liu, S. Musetti and L. Huang, Cancer Immunol. Immunother., 2018, 67, 299–310 CrossRef CAS PubMed.
- L. J. Cruz, R. A. Rosalia, J. W. Kleinovink, F. Rueda, C. W. Lowik and F. Ossendorp, J. Controlled Release, 2014, 192, 209–218 CrossRef CAS.
- D. Schmid, C. G. Park, C. A. Hartl, N. Subedi, A. N. Cartwright, R. B. Puerto, Y. Zheng, J. Maiarana, G. J. Freeman, K. W. Wucherpfennig, D. J. Irvine and M. S. Goldberg, Nat. Commun., 2017, 8, 1747 CrossRef.
- X. Y. He, B. Y. Liu, J. L. Wu, S. L. Ai, R. X. Zhuo and S. X. Cheng, ACS Appl. Mater. Interfaces, 2017, 9, 42566–42576 CrossRef CAS.
- V. V. Temchura, D. Kozlova, V. Sokolova, K. Uberla and M. Epple, Biomaterials, 2014, 35, 6098–6105 CrossRef CAS.
- O. Meurette and P. Mehlen, Cancer Cell, 2018, 34, 536–548 CrossRef CAS.
- L. M. Zahn, Science, 2017, 355, 1386–1388 Search PubMed.
- W. Tomaszewski, L. Sanchez-Perez, T. F. Gajewski and J. H. Sampson, Clin. Cancer Res., 2019, 25, 4202–4210 CrossRef.
- L. Meyaard, J. Leukocyte Biol., 2008, 83, 799–803 CrossRef CAS PubMed.
- H. C. Hope and R. J. Salmond, Eur. J. Immunol., 2019, 49, 1147–1152 CAS.
- G. M. Rodriguez, K. J. C. Galpin, C. W. McCloskey and B. C. Vanderhyden, Cancer, 2018, 10 DOI:10.3390/cancers10080242.
- J. S. Fang, R. D. Gillies and R. A. Gatenby, Semin. Cancer Biol., 2008, 18, 330–337 CrossRef CAS PubMed.
- B. Keith and M. C. Simon, Cell, 2007, 129, 465–472 CrossRef CAS PubMed.
- G. Lin, S. Chen and P. Mi, J. Biomed. Nanotechnol., 2018, 14, 1189–1207 CrossRef CAS PubMed.
- J. E. Park, B. Dutta, S. W. Tse, N. Gupta, C. F. Tan, J. K. Low, K. W. Yeoh, O. L. Kon, J. P. Tam and S. K. Sze, Oncogene, 2019, 38, 5158–5173 CrossRef CAS PubMed.
- Y. Li, S. P. Patel, J. Roszik and Y. Qin, Front. Immunol., 2018, 9, 1591 CrossRef PubMed.
- S. Im, J. Lee, D. Park, A. Park, Y. M. Kim and W. J. Kim, ACS Nano, 2019, 13, 476–488 CrossRef CAS PubMed.
- G. B. Yang, L. G. Xu, Y. Chao, J. Xu, X. Q. Sun, Y. F. Wu, R. Peng and Z. Liu, Nat. Commun., 2017, 8, 902 CrossRef PubMed.
- G. Molema, B. Kroesen, W. Helfrich, D. Meijer and L. de Leij, J. Controlled Release, 2000, 64, 229–239 CrossRef CAS.
- E. Lanitis, M. Irving and G. Coukos, Curr. Opin. Immunol., 2015, 33, 55–63 CrossRef CAS PubMed.
- Q. Chen, L. Xu, J. Chen, Z. Yang, C. Liang, Y. Yang and Z. Liu, Biomaterials, 2017, 148, 69–80 CrossRef CAS PubMed.
- A. Gupte and R. J. Mumper, Cancer Treat. Rev., 2009, 35, 32–46 CrossRef CAS PubMed.
- H. Xie and Y. J. Kang, Curr. Med. Chem., 2009, 16, 1304–1314 CrossRef CAS PubMed.
- P. Zhou, J. Qin, C. Zhou, G. Wan, Y. Liu, M. Zhang, X. Yang, N. Zhang and Y. Wang, Biomaterials, 2019, 195, 86–99 CrossRef CAS PubMed.
- J. E. Rundhaug, J. Cell. Mol. Med., 2005, 9, 267–285 CrossRef CAS PubMed.
- K. Kessenbrock, V. Plaks and Z. Werb, Cell, 2010, 141, 52–67 CrossRef CAS.
- C. M. Overall and C. Lopez-Otin, Nat. Rev. Cancer, 2002, 2, 657–672 CrossRef CAS PubMed.
- J. M. Shin, S. J. Oh, S. Kwon, V. G. Deepagan, M. Lee, S. H. Song, H. J. Lee, S. Kim, K. H. Song, T. W. Kim and J. H. Park, J. Controlled Release, 2017, 267, 181–190 CrossRef CAS PubMed.
- C. Wang, W. Sun, G. Wright, A. Z. Wang and Z. Gu, Adv. Mater., 2016, 28, 8912–8920 CrossRef CAS PubMed.
- M. J. Heffernan and N. Murthy, Ann. Biomed. Eng., 2009, 37, 1993–2002 CrossRef PubMed.
- D. Xiao, J. J. Hu, J. Y. Zhu, S. B. Wang, R. X. Zhuo and X. Z. Zhang, Nanoscale, 2016, 8, 16702–16709 RSC.
- X. Xu, J. Wu, S. Liu, P. E. Saw, W. Tao, Y. Li, L. Krygsman, S. Yegnasubramanian, A. M. De Marzo, J. Shi, C. J. Bieberich and O. C. Farokhzad, Small, 2018, 14, e1802565 CrossRef PubMed.
- K. Wang, Y. Yang, W. Xue and Z. Liu, Mol. Pharm., 2018, 15, 975–984 CrossRef CAS PubMed.
- E. Verdegaal, Curr. Opin. Immunol., 2016, 39, 90–95 CrossRef CAS PubMed.
- S. Rosenberg and M. Dudley, Curr. Opin. Immunol., 2009, 21, 233–240 CrossRef CAS PubMed.
- J. Cohen, S. Merims, S. Frank, R. Engelstein, T. Peretz and M. Lotem, Immunotherapy, 2017, 9, 183–196 CrossRef CAS PubMed.
- S. Mardiana, B. Solomon, P. Darcy and P. Beavis, Sci. Transl. Med., 2019, 11 DOI:10.1126/scitranslmed.aaw2293.
- A. Wallace, V. Kapoor, J. Sun, P. Mrass, W. Weninger, D. Heitjan, C. June, L. Kaiser, L. Ling and S. Albelda, Clin. Cancer Res., 2008, 14, 3966–3974 CrossRef CAS PubMed.
- Y. Zheng, L. Tang, L. Mabardi, S. Kumari and D. J. Irvine, ACS Nano, 2017, 11, 3089–3100 CrossRef CAS PubMed.
- S. B. Stephan, A. M. Taber, I. Jileaeva, E. P. Pegues, C. L. Sentman and M. T. Stephan, Nat. Biotechnol., 2015, 33, 97–101 CrossRef CAS PubMed.
- A. Grakoui, S. K. Bromley, C. Sumen, M. M. Davis, A. S. Shaw, P. M. Allen and M. L. Dustin, Science, 1999, 285, 221–227 CrossRef CAS PubMed.
- F. D. Brown and S. J. Turley, J. Immunol., 2015, 194, 1389–1394 CrossRef CAS PubMed.
- E. H. Neto, A. L. Coelho, A. L. Sampaio, M. Henriques, C. Marcinkiewicz, M. S. De Freitas and C. Barja-Fidalgo, Biochim. Biophys. Acta, 2007, 1773, 176–184 CrossRef PubMed.
- J. Guasch, C. A. Muth, J. Diemer, H. Riahinezhad and J. P. Spatz, Nano Lett., 2017, 17, 6110–6116 CrossRef CAS PubMed.
- K. Perica, J. G. Bieler, C. Schutz, J. C. Varela, J. Douglass, A. Skora, Y. L. Chiu, M. Oelke, K. Kinzler, S. Zhou, B. Vogelstein and J. P. Schneck, ACS Nano, 2015, 9, 6861–6871 CrossRef CAS PubMed.
- A. Nel, T. Xia, L. Madler and N. Li, Science, 2006, 311, 622–627 CrossRef CAS PubMed.
- C. Medina, M. J. Santos-Martinez, A. Radomski, O. I. Corrigan and M. W. Radomski, Br. J. Pharmacol., 2007, 150, 552–558 CrossRef CAS PubMed.
- M. A. Dobrovolskaia, P. Aggarwal, J. B. Hall and S. E. McNeil, Mol. Pharm., 2008, 5, 487–495 CrossRef CAS PubMed.
- K. Greish, G. Thiagarajan, H. Herd, R. Price, H. Bauer, D. Hubbard, A. Burckle, S. Sadekar, T. Yu, A. Anwar, A. Ray and H. Ghandehari, Nanotoxicology, 2012, 6, 713–723 CrossRef CAS PubMed.
Footnote |
| † The authors contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2019 |