
Engineered nanoparticles for systemic siRNA delivery to malignant brain tumours†
 *bc
and
Jordan J.
Green
*abcfgh
*bc
and
Jordan J.
Green
*abcfgh
Abstract
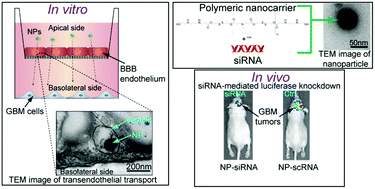
Improved delivery materials are needed to enable siRNA transport across biological barriers, including the blood–brain barrier (BBB), to treat diseases like brain cancer. We engineered bioreducible nanoparticles for systemic siRNA delivery to patient-derived glioblastoma cells in an orthotopic mouse tumor model. We first utilized a newly developed biomimetic in vitro model to evaluate and optimize the performance of the engineered bioreducible nanoparticles at crossing the brain microvascular endothelium. We performed transmission electron microscopy imaging which indicated that the engineered nanoparticles are able to cross the BBB endothelium via a vesicular mechanism. The nanoparticle formulation engineered to best cross the BBB model in vitro led to safe delivery across the BBB to the brain in vivo. The nanoparticles were internalized by human brain cancer cells, released siRNA to the cytosol via environmentally-triggered degradation, and gene silencing was obtained both in vitro and in vivo. This study opens new frontiers for the in vitro evaluation and engineering of nanomedicines for delivery to the brain, and reports a systemically administered biodegradable nanocarrier for oligonucleotide delivery to treat glioma.
- This article is part of the themed collection: Editor’s Choice: Recent breakthroughs in nanobiotechnology
 Please wait while we load your content...
Please wait while we load your content...

